
Dual Role of ACBD6 in the Acylation Remodeling of Lipids and Proteins
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
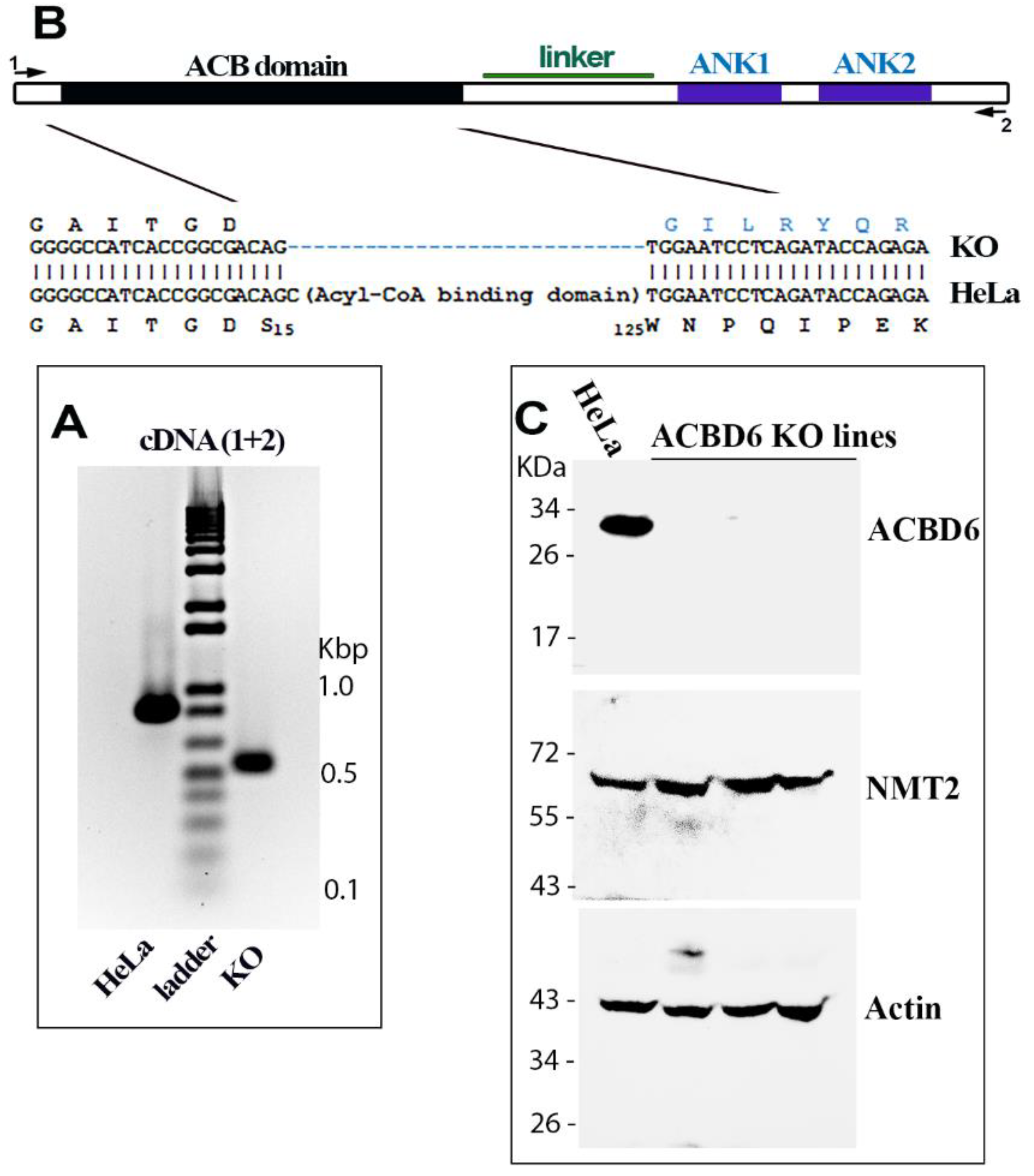
2.1. Construction and Characterization of the ACBD6.KO Cells
2.2. Cell Culture and Growth Experiments
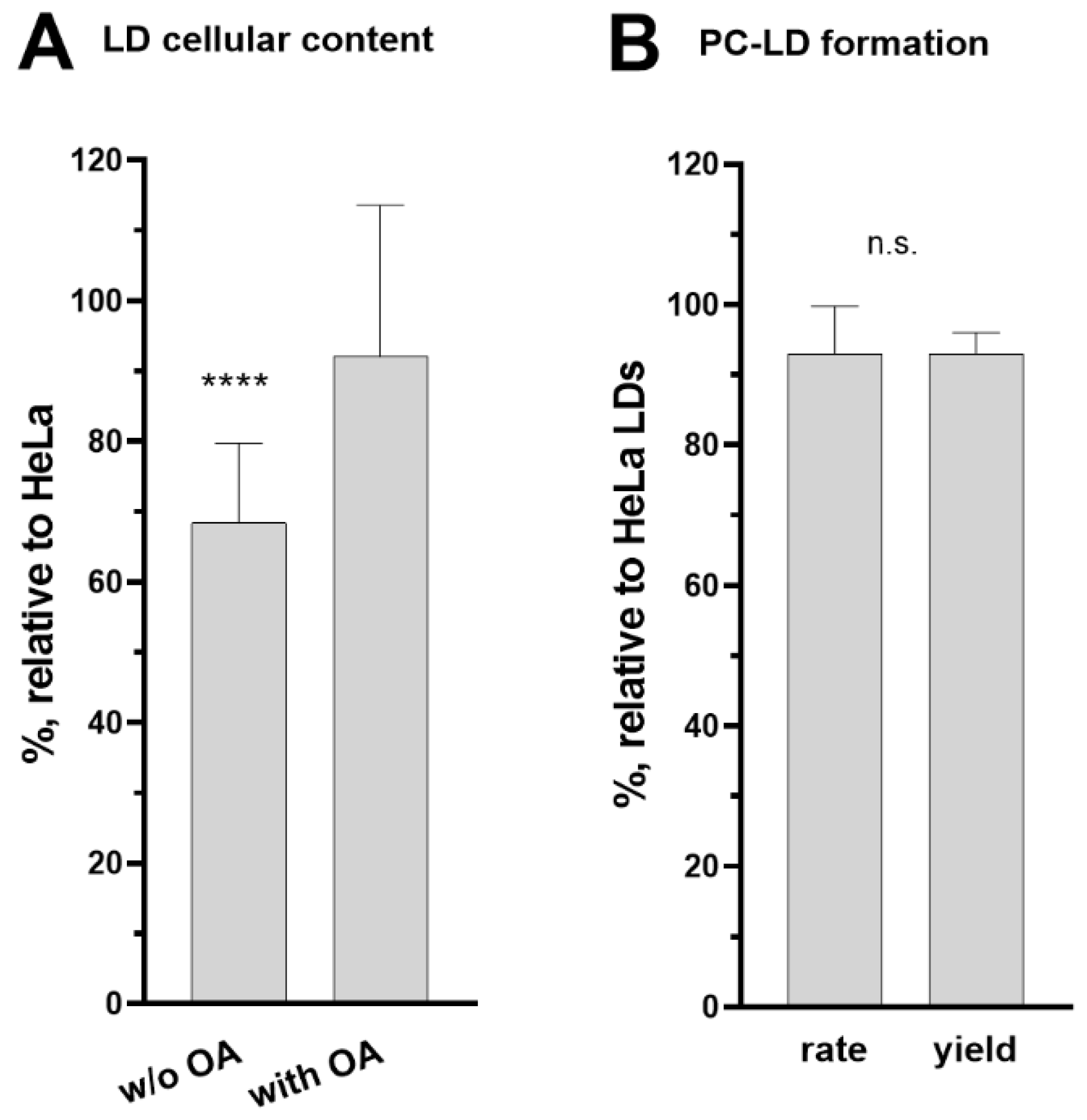
2.3. Lipid Droplet Quantification and Isolation
2.4. N-myristoyltransferase Activity Measurements
2.5. Protein Palmitoylation Quantification
2.6. Fatty Acid Incorporation
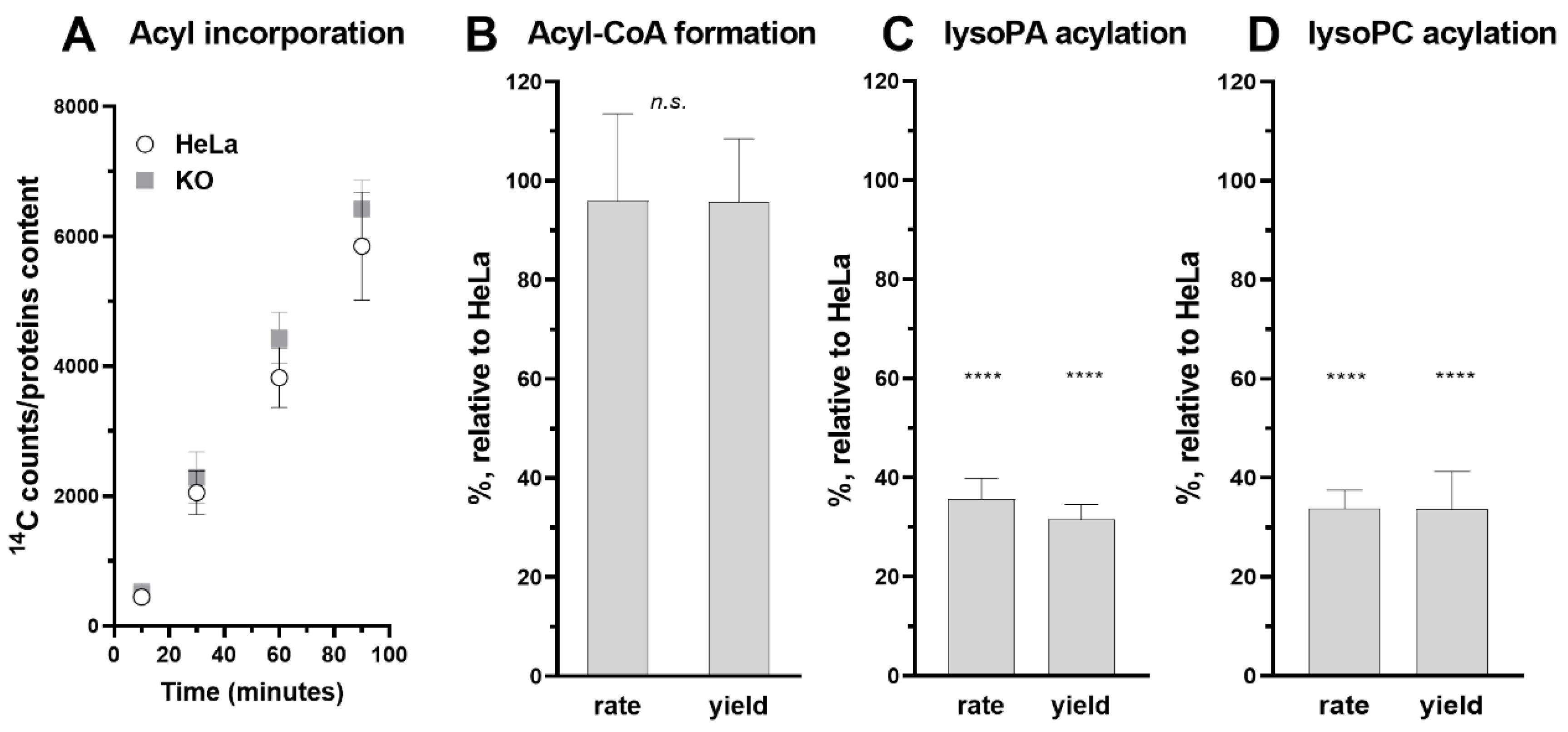
2.7. Acyl-CoA Synthetase and lysoPL Acyltransferase Assays
3. Results
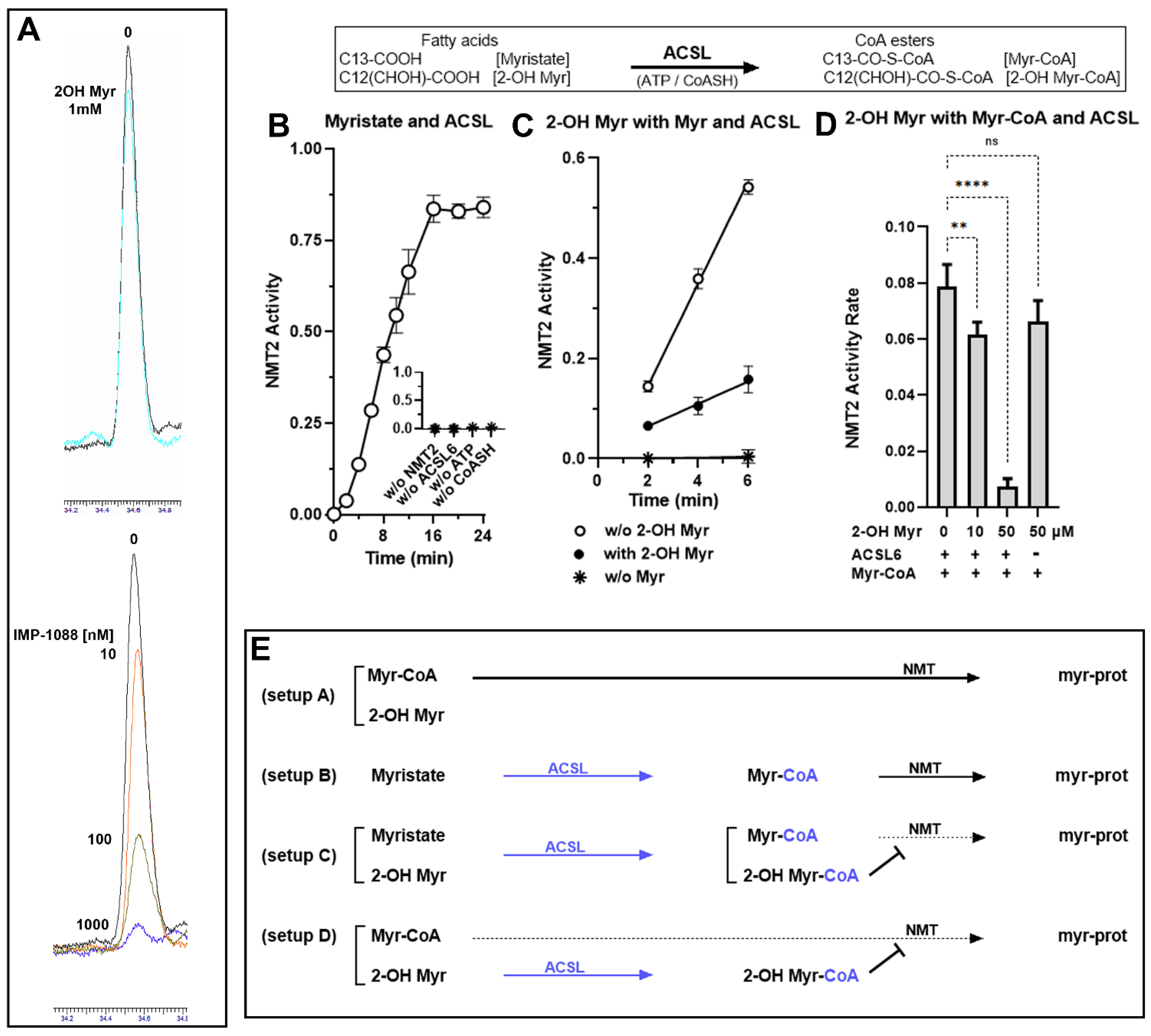
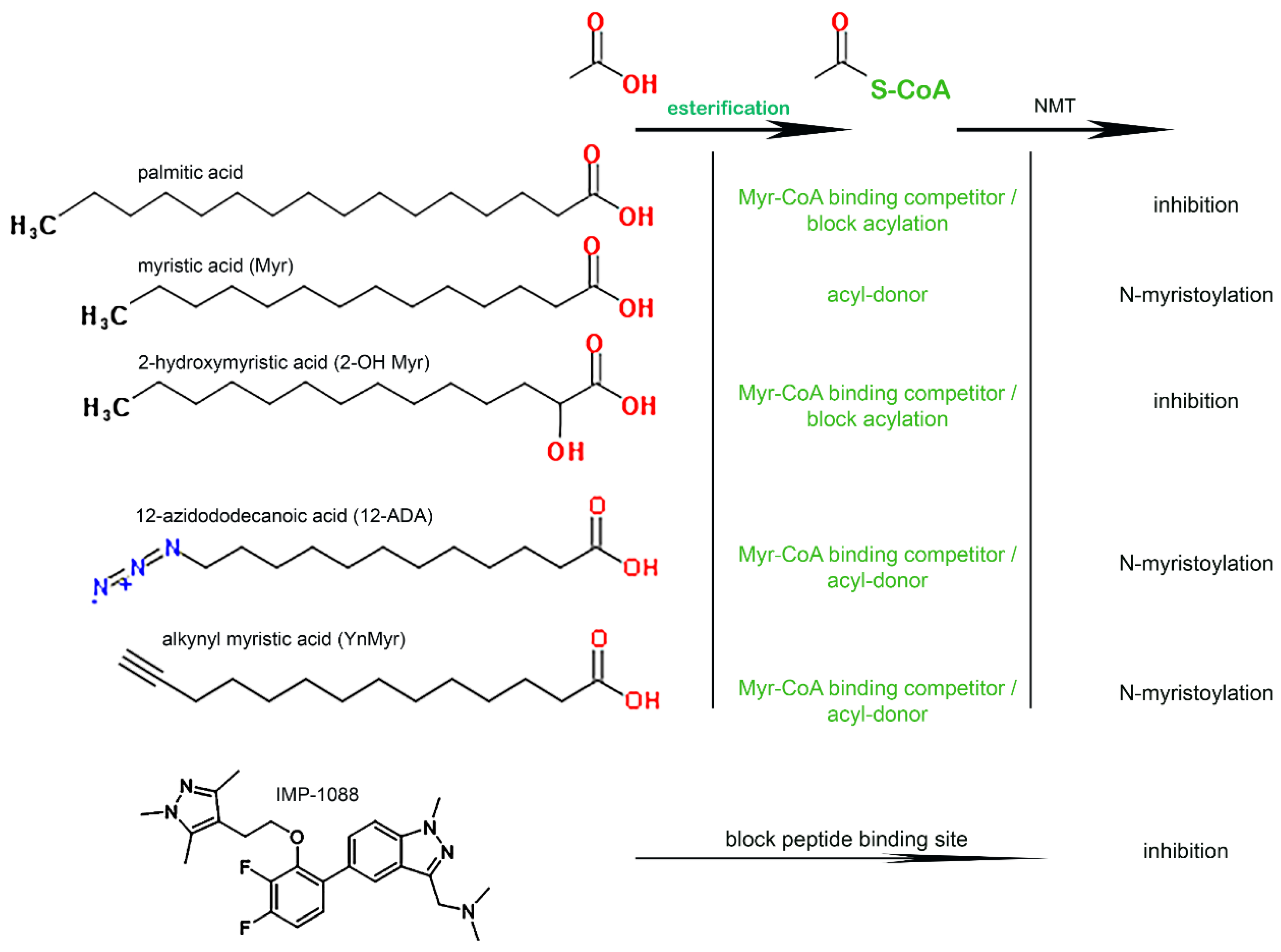
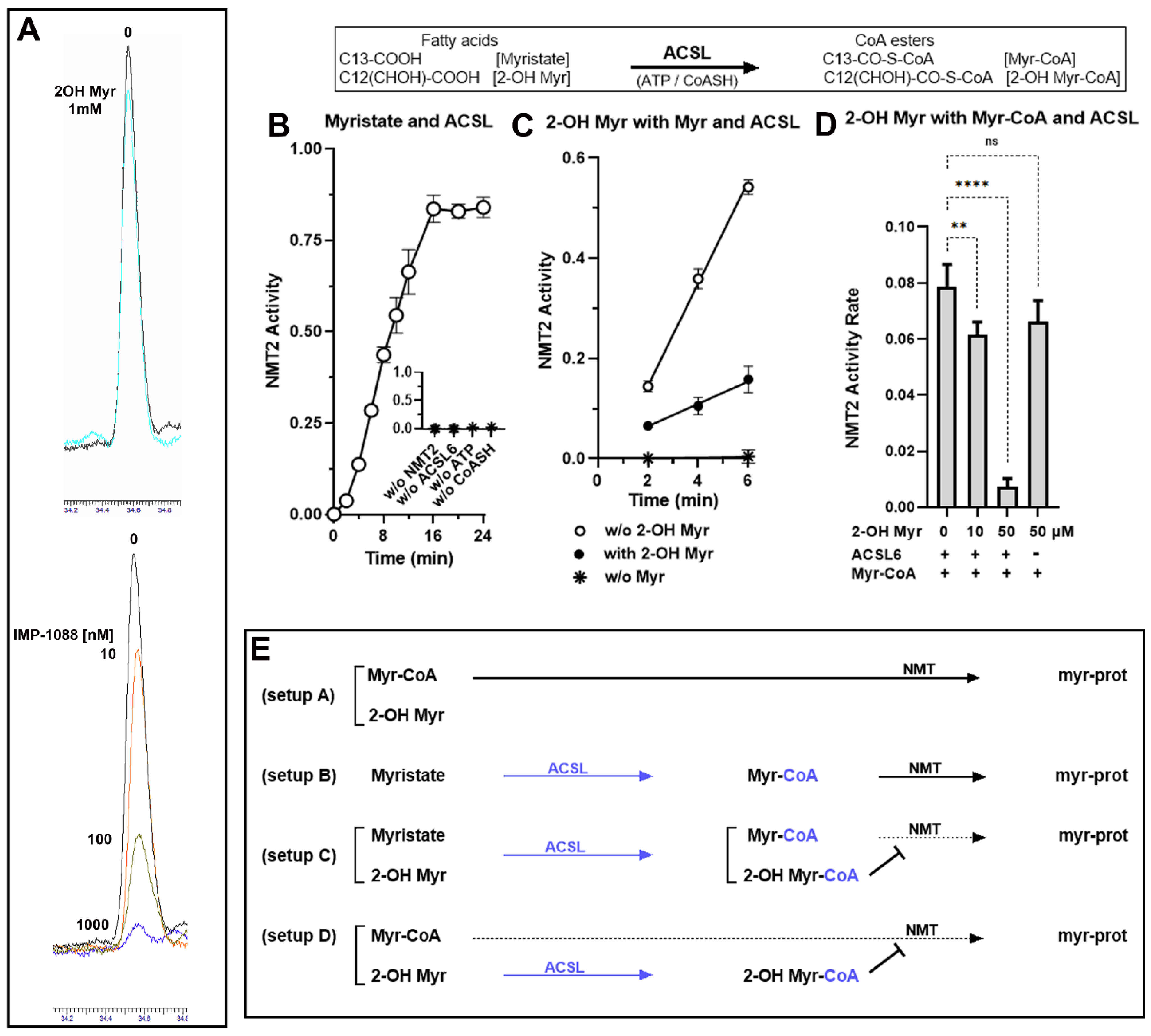
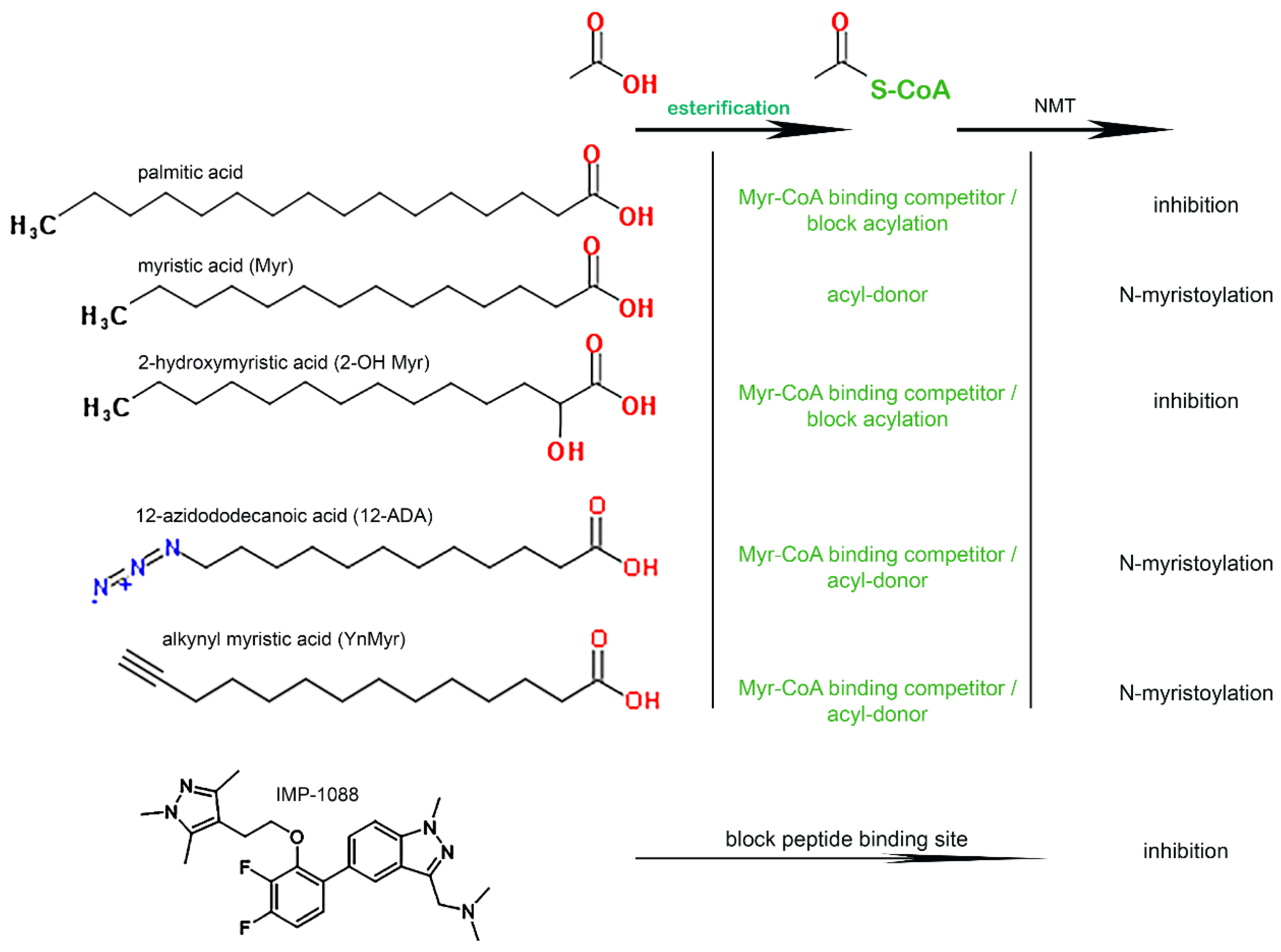
3.1. Role of the Acyl-CoA Synthetase Enzymes in the Myristoylation Reaction
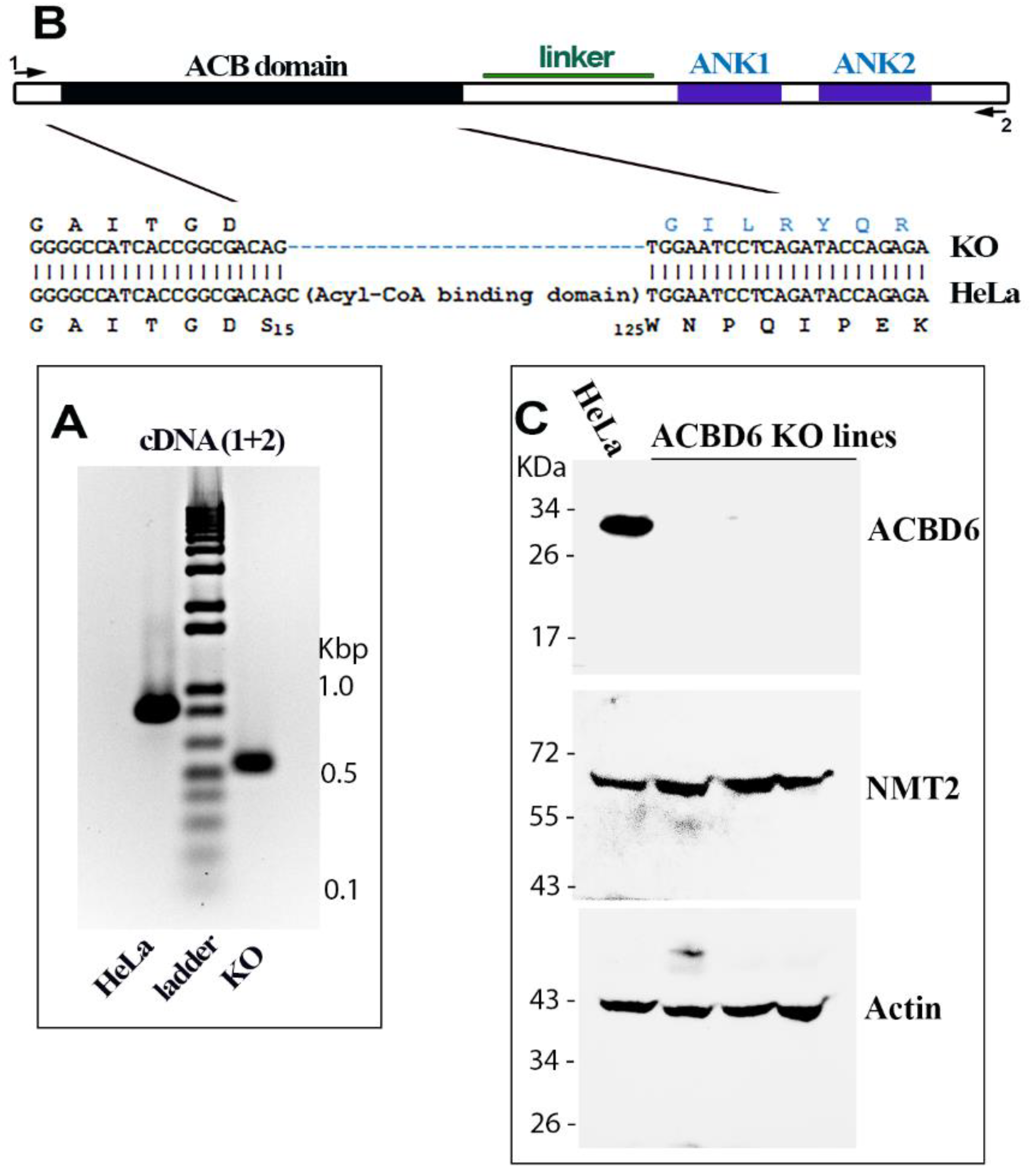
3.2. Disruption of ACBD6 in Human Cells
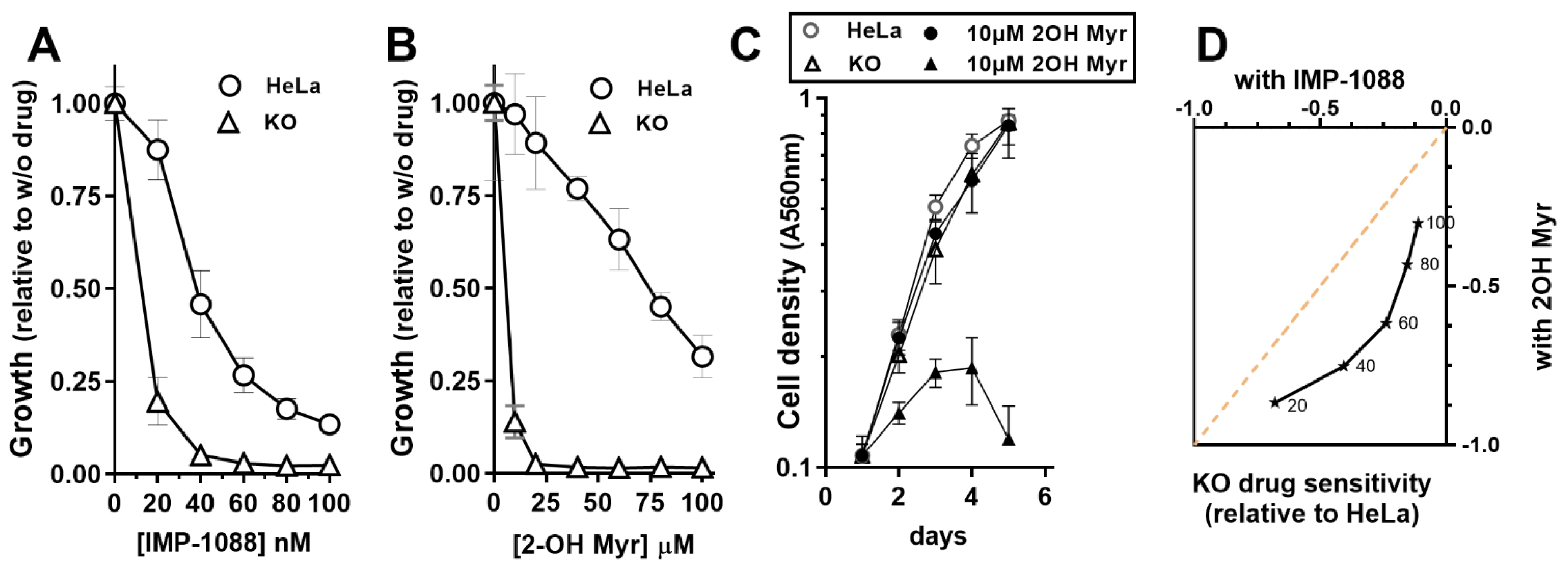
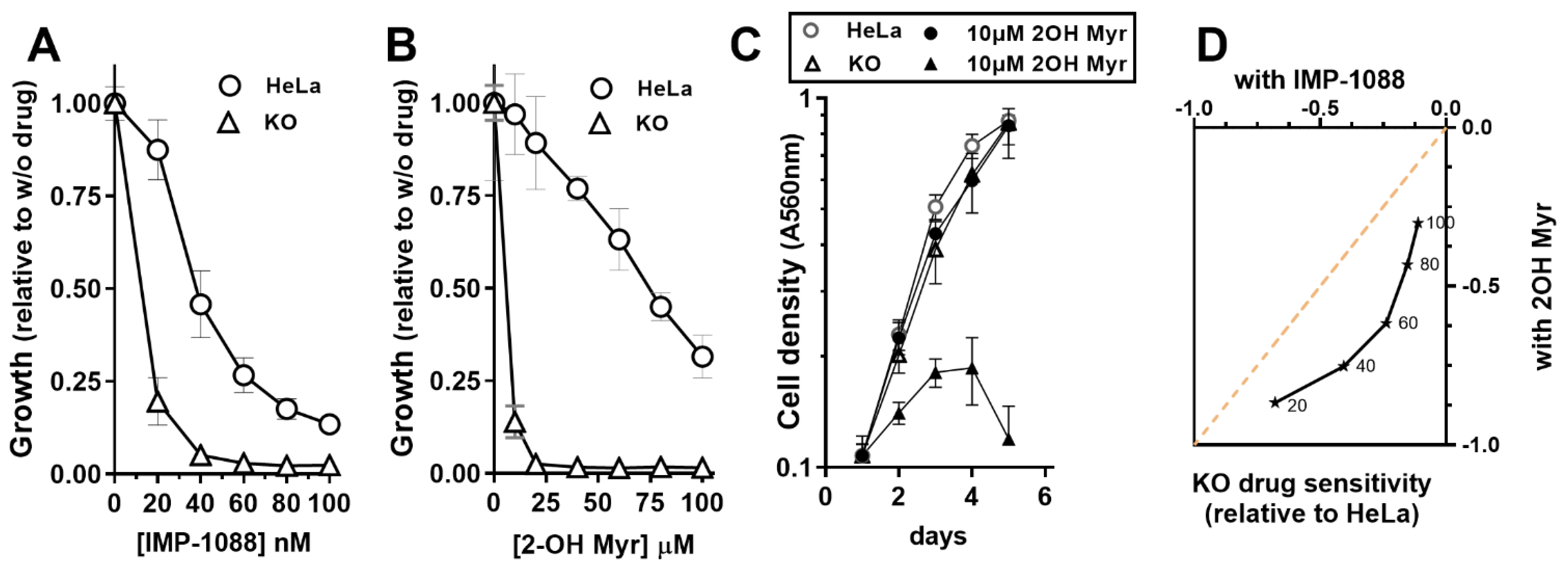
3.3. NMT Activity Deficiency in the Absence of ACBD6
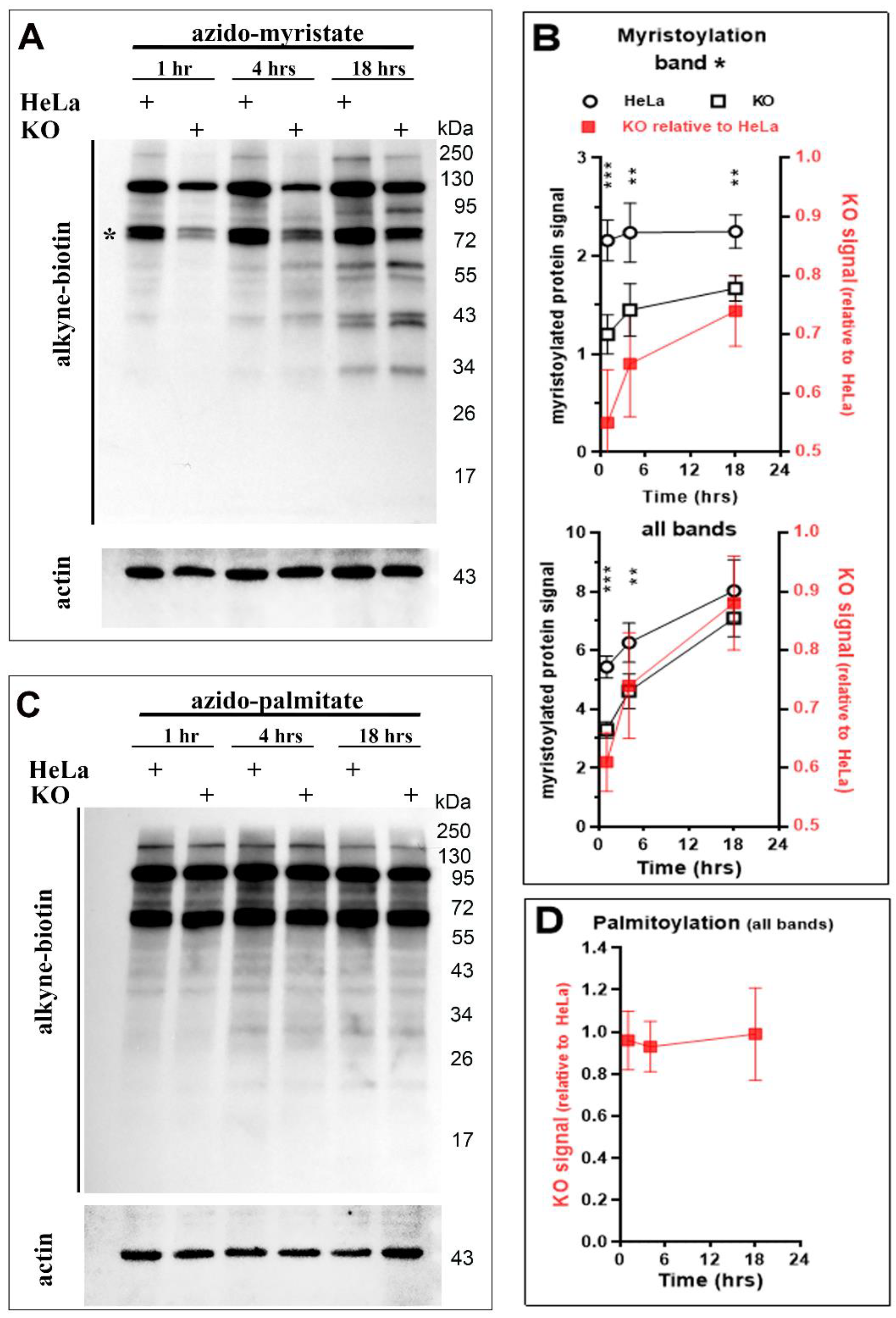
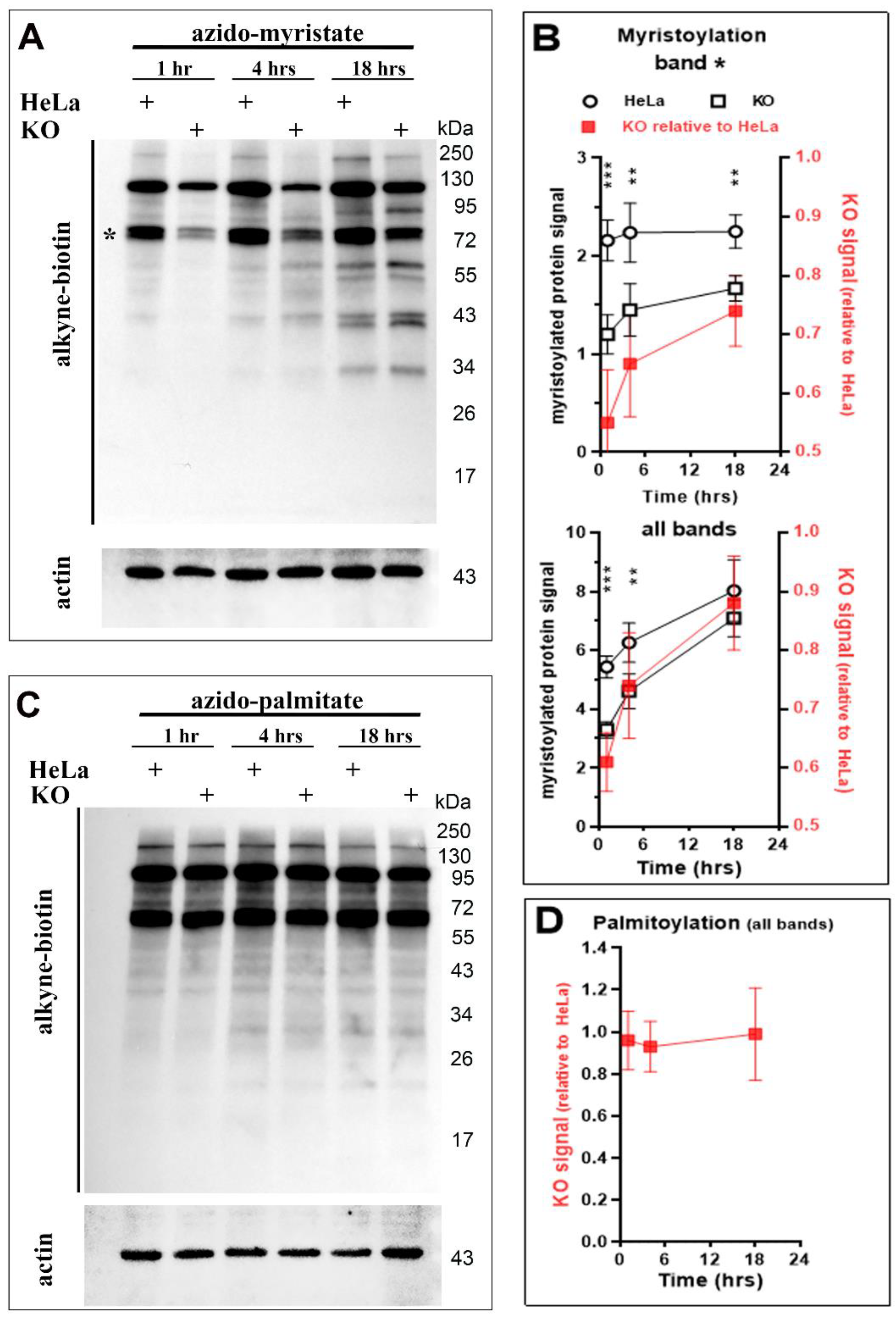
3.4. Protein N-Myristoylation Deficiency in the Absence of ACBD6
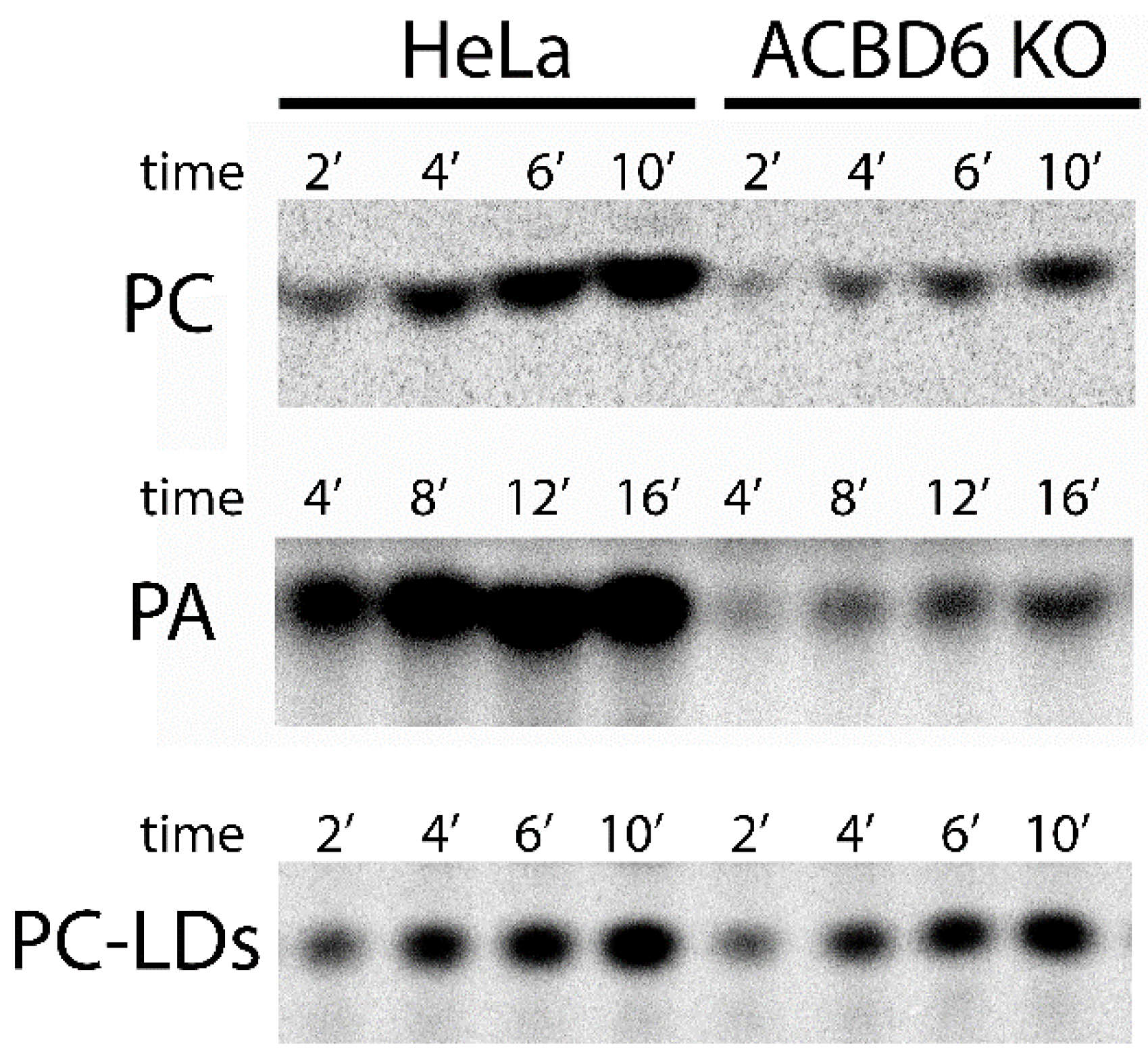
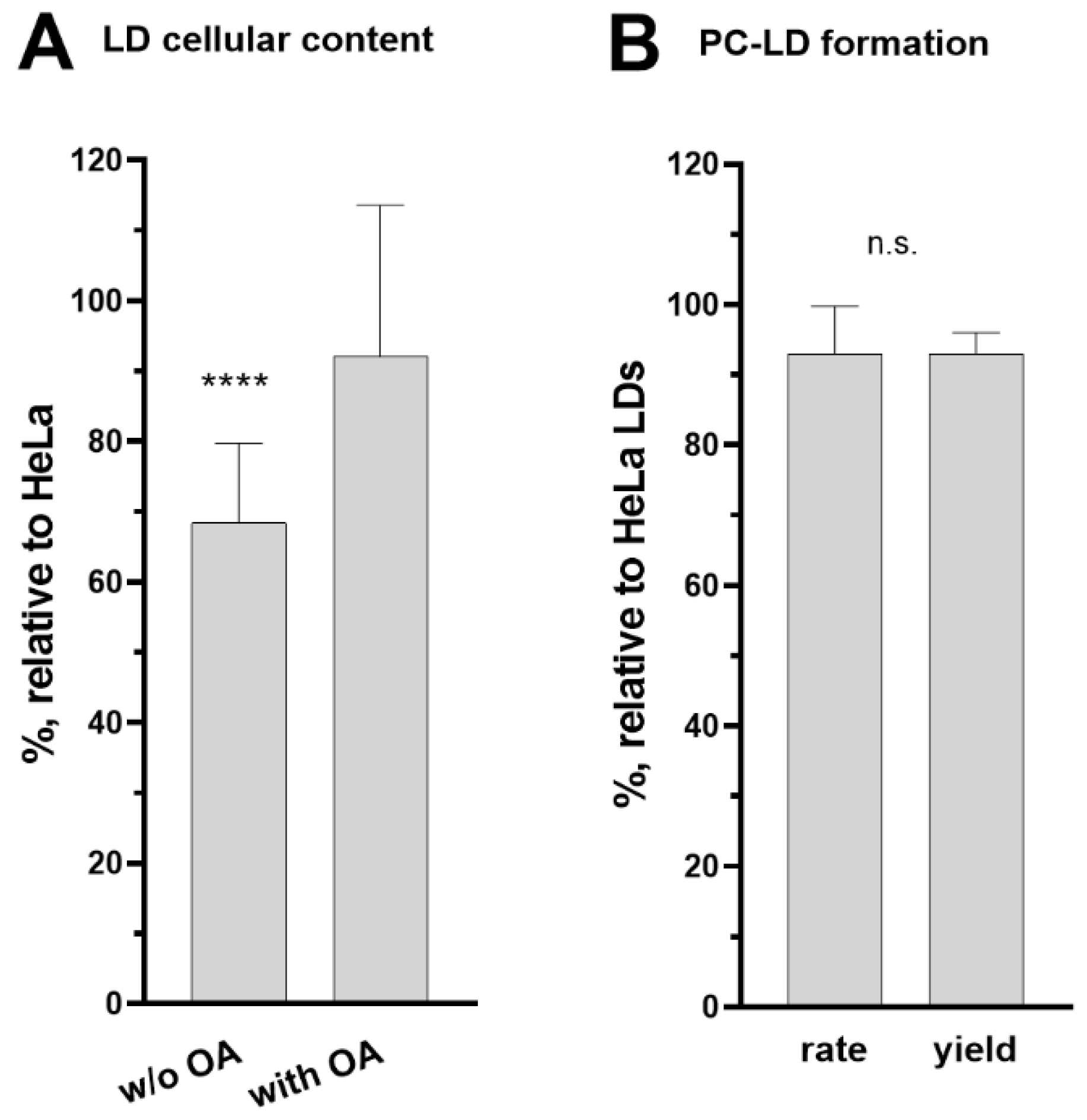
3.5. Role of ACBD6 in the Acyl-CoA Dependent Acylation of Lipids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Soupene, E.; Kao, J.; Cheng, D.H.; Wang, D.; Greninger, A.L.; Knudsen, G.M.; DeRisi, J.L.; Kuypers, F.A. Association of NMT2 with the acyl-CoA carrier ACBD6 protects the N-myristoyltransferase reaction from palmitoyl-CoA. J. Lipid Res. 2016, 57, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soupene, E.; Kuypers, F.A. ACBD6 protein controls acyl chain availability and specificity of the N-myristoylation modification of proteins. J. Lipid Res. 2019, 60, 624–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soupene, E.; Schatz, U.A.; Rudnik-Schoneborn, S.; Kuypers, F.A. Requirement of the acyl-CoA carrier ACBD6 in myristoylation of proteins: Activation by ligand binding and protein interaction. PLoS ONE 2020, 15, e0229718. [Google Scholar] [CrossRef] [PubMed]
- Castrec, B.; Dian, C.; Ciccone, S.; Ebert, C.L.; Bienvenut, W.V.; Le Caer, J.P.; Steyaert, J.M.; Giglione, C.; Meinnel, T. Structural and genomic decoding of human and plant myristoylomes reveals a definitive recognition pattern. Nat. Chem. Biol. 2018, 14, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Giglione, C.; Meinnel, T. Mapping the myristoylome through a complete understanding of protein myristoylation biochemistry. Prog. Lipid Res. 2022, 85, 101139. [Google Scholar] [CrossRef]
- Jiang, H.; Zhang, X.; Chen, X.; Aramsangtienchai, P.; Tong, Z.; Lin, H. Protein Lipidation: Occurrence, Mechanisms, Biological Functions, and Enabling Technologies. Chem. Rev. 2018, 118, 919–988. [Google Scholar] [CrossRef]
- Kallemeijn, W.W.; Lanyon-Hogg, T.; Panyain, N.; Goya Grocin, A.; Ciepla, P.; Morales-Sanfrutos, J.; Tate, E.W. Proteome-wide analysis of protein lipidation using chemical probes: In-gel fluorescence visualization, identification and quantification of N-myristoylation, N- and S-acylation, O-cholesterylation, S-farnesylation and S-geranylgeranylation. Nat. Protoc. 2021, 16, 5083–5122. [Google Scholar] [CrossRef]
- Resh, M.D. Fatty acylation of proteins: The long and the short of it. Prog. Lipid Res. 2016, 63, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Udenwobele, D.I.; Su, R.C.; Good, S.V.; Ball, T.B.; Varma Shrivastav, S.; Shrivastav, A. Myristoylation: An Important Protein Modification in the Immune Response. Front. Immunol. 2017, 8, 751. [Google Scholar] [CrossRef] [Green Version]
- Varland, S.; Osberg, C.; Arnesen, T. N-terminal modifications of cellular proteins: The enzymes involved, their substrate specificities and biological effects. Proteomics 2015, 15, 2385–2401. [Google Scholar] [CrossRef]
- Meinnel, T. Comment on “Binding Affinity Determines Substrate Specificity and Enables Discovery of Substrates for N-Myristoyltransferases”. ACS Catal. 2022, 12, 8195–8201. [Google Scholar] [CrossRef]
- Meinnel, T.; Dian, C.; Giglione, C. Myristoylation, an Ancient Protein Modification Mirroring Eukaryogenesis and Evolution. Trends Biochem. Sci. 2020, 45, 619–632. [Google Scholar] [CrossRef]
- Bhatnagar, R.S.; Futterer, K.; Waksman, G.; Gordon, J.I. The structure of myristoyl-CoA:protein N-myristoyltransferase. Biochim. Biophys. Acta 1999, 1441, 162–172. [Google Scholar] [CrossRef]
- Bhatnagar, R.S.; Schall, O.F.; Jackson-Machelski, E.; Sikorski, J.A.; Devadas, B.; Gokel, G.W.; Gordon, J.I. Titration calorimetric analysis of AcylCoA recognition by myristoylCoA:protein N-myristoyltransferase. Biochemistry 1997, 36, 6700–6708. [Google Scholar] [CrossRef]
- DeMar, J.C., Jr.; Anderson, R.E. Identification and quantitation of the fatty acids composing the CoA ester pool of bovine retina, heart, and liver. J. Biol. Chem. 1997, 272, 31362–31368. [Google Scholar] [CrossRef] [Green Version]
- Heuckeroth, R.O.; Glaser, L.; Gordon, J.I. Heteroatom-substituted fatty acid analogs as substrates for N-myristoyltransferase: An approach for studying both the enzymology and function of protein acylation. Proc. Natl. Acad. Sci. USA 1988, 85, 8795–8799. [Google Scholar] [CrossRef] [Green Version]
- Kishore, N.S.; Wood, D.C.; Mehta, P.P.; Wade, A.C.; Lu, T.; Gokel, G.W.; Gordon, J.I. Comparison of the acyl chain specificities of human myristoyl-CoA synthetase and human myristoyl-CoA:protein N-myristoyltransferase. J. Biol. Chem. 1993, 268, 4889–4902. [Google Scholar] [CrossRef]
- Rudnick, D.A.; Lu, T.; Jackson-Machelski, E.; Hernandez, J.C.; Li, Q.; Gokel, G.W.; Gordon, J.I. Analogs of palmitoyl-CoA that are substrates for myristoyl-CoA:protein N-myristoyltransferase. Proc. Natl. Acad. Sci. USA 1992, 89, 10507–10511. [Google Scholar] [CrossRef] [Green Version]
- Bhatnagar, R.S.; Jackson-Machelski, E.; McWherter, C.A.; Gordon, J.I. Isothermal titration calorimetric studies of Saccharomyces cerevisiae myristoyl-CoA:protein N-myristoyltransferase. Determinants of binding energy and catalytic discrimination among acyl-CoA and peptide ligands. J. Biol. Chem. 1994, 269, 11045–11053. [Google Scholar] [CrossRef]
- Kishore, N.S.; Lu, T.B.; Knoll, L.J.; Katoh, A.; Rudnick, D.A.; Mehta, P.P.; Devadas, B.; Huhn, M.; Atwood, J.L.; Adams, S.P.; et al. The substrate specificity of Saccharomyces cerevisiae myristoyl-CoA:protein N-myristoyltransferase. Analysis of myristic acid analogs containing oxygen, sulfur, double bonds, triple bonds, and/or an aromatic residue. J. Biol. Chem. 1991, 266, 8835–8855. [Google Scholar] [CrossRef]
- Zheng, G.Q.; Hu, X.; Cassady, J.M.; Paige, L.A.; Geahlen, R.L. Synthesis of myristoyl CoA analogues and myristoyl peptides as inhibitors of myristoyl CoA:protein N-myristoyltransferase. J. Pharm. Sci. 1994, 83, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Paige, L.A.; Zheng, G.Q.; DeFrees, S.A.; Cassady, J.M.; Geahlen, R.L. Metabolic activation of 2-substituted derivatives of myristic acid to form potent inhibitors of myristoyl CoA:protein N-myristoyltransferase. Biochemistry 1990, 29, 10566–10573. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Eppinger, E.; Schuster, I.G.; Weigand, L.U.; Liang, X.; Kremmer, E.; Peschel, C.; Krackhardt, A.M. Formin-like 1 (FMNL1) is regulated by N-terminal myristoylation and induces polarized membrane blebbing. J. Biol. Chem. 2009, 284, 33409–33417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, D.R.; Gilbert, R.L.; Blunt, C.; McIlhinney, R.A. Inhibition of varicella-zoster virus replication by an inhibitor of protein myristoylation. J. Gen. Virol. 1993, 74 Pt 6, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Nadler, M.J.; Harrison, M.L.; Ashendel, C.L.; Cassady, J.M.; Geahlen, R.L. Treatment of T cells with 2-hydroxymyristic acid inhibits the myristoylation and alters the stability of p56lck. Biochemistry 1993, 32, 9250–9255. [Google Scholar] [CrossRef]
- Perez, M.; Greenwald, D.L.; de la Torre, J.C. Myristoylation of the RING finger Z protein is essential for arenavirus budding. J. Virol. 2004, 78, 11443–11448. [Google Scholar] [CrossRef] [Green Version]
- Renna, L.; Stefano, G.; Majeran, W.; Micalella, C.; Meinnel, T.; Giglione, C.; Brandizzi, F. Golgi traffic and integrity depend on N-myristoyl transferase-1 in Arabidopsis. Plant Cell 2013, 25, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Urata, S.; Yasuda, J.; de la Torre, J.C. The z protein of the new world arenavirus tacaribe virus has bona fide budding activity that does not depend on known late domain motifs. J. Virol. 2009, 83, 12651–12655. [Google Scholar] [CrossRef] [Green Version]
- Vaandrager, A.B.; Ehlert, E.M.; Jarchau, T.; Lohmann, S.M.; de Jonge, H.R. N-terminal myristoylation is required for membrane localization of cGMP-dependent protein kinase type II. J. Biol. Chem. 1996, 271, 7025–7029. [Google Scholar] [CrossRef] [Green Version]
- Van’t Hof, W.; Resh, M.D. Dual fatty acylation of p59(Fyn) is required for association with the T cell receptor zeta chain through phosphotyrosine-Src homology domain-2 interactions. J. Cell Biol. 1999, 145, 377–389. [Google Scholar] [CrossRef]
- Vilas, G.L.; Corvi, M.M.; Plummer, G.J.; Seime, A.M.; Lambkin, G.R.; Berthiaume, L.G. Posttranslational myristoylation of caspase-activated p21-activated protein kinase 2 (PAK2) potentiates late apoptotic events. Proc. Natl. Acad. Sci. USA 2006, 103, 6542–6547. [Google Scholar] [CrossRef] [Green Version]
- Galbiati, F.; Guzzi, F.; Magee, A.I.; Milligan, G.; Parenti, M. Chemical inhibition of myristoylation of the G-protein Gi1 alpha by 2-hydroxymyristate does not interfere with its palmitoylation or membrane association. Evidence that palmitoylation, but not myristoylation, regulates membrane attachment. Biochem. J. 1996, 313 Pt 3, 717–720. [Google Scholar] [CrossRef]
- Kallemeijn, W.W.; Lueg, G.A.; Faronato, M.; Hadavizadeh, K.; Goya Grocin, A.; Song, O.R.; Howell, M.; Calado, D.P.; Tate, E.W. Validation and Invalidation of Chemical Probes for the Human N-myristoyltransferases. Cell Chem. Biol. 2019, 26, 892–900.e894. [Google Scholar] [CrossRef] [Green Version]
- Charron, G.; Zhang, M.M.; Yount, J.S.; Wilson, J.; Raghavan, A.S.; Shamir, E.; Hang, H.C. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J. Am. Chem. Soc. 2009, 131, 4967–4975. [Google Scholar] [CrossRef]
- Ritzefeld, M.; Wright, M.H.; Tate, E.W. New developments in probing and targeting protein acylation in malaria, leishmaniasis and African sleeping sickness. Parasitology 2018, 145, 157–174. [Google Scholar] [CrossRef]
- Wright, M.H.; Clough, B.; Rackham, M.D.; Rangachari, K.; Brannigan, J.A.; Grainger, M.; Moss, D.K.; Bottrill, A.R.; Heal, W.P.; Broncel, M.; et al. Validation of N-myristoyltransferase as an antimalarial drug target using an integrated chemical biology approach. Nat. Chem. 2014, 6, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Broncel, M.; Dominicus, C.; Vigetti, L.; Nofal, S.D.; Bartlett, E.J.; Touquet, B.; Hunt, A.; Wallbank, B.A.; Federico, S.; Matthews, S.; et al. Profiling of myristoylation in Toxoplasma gondii reveals an N-myristoylated protein important for host cell penetration. Elife 2020, 9. [Google Scholar] [CrossRef]
- Thinon, E.; Serwa, R.A.; Broncel, M.; Brannigan, J.A.; Brassat, U.; Wright, M.H.; Heal, W.P.; Wilkinson, A.J.; Mann, D.J.; Tate, E.W. Global profiling of co- and post-translationally N-myristoylated proteomes in human cells. Nat. Commun. 2014, 5, 4919. [Google Scholar] [CrossRef] [Green Version]
- Witten, A.J.; Ejendal, K.F.K.; Gengelbach, L.M.; Traore, M.A.; Wang, X.; Umulis, D.M.; Calve, S.; Kinzer-Ursem, T.L. Fluorescent imaging of protein myristoylation during cellular differentiation and development. J. Lipid Res. 2017, 58, 2061–2070. [Google Scholar] [CrossRef] [Green Version]
- Su, D.; Kosciuk, T.; Lin, H. Reply to Comment on “Binding Affinity Determines Substrate Specificity and Enables Discovery of substrates for N-Myristoyltransferases”. ACS Catal. 2022, 12, 8829–8832. [Google Scholar] [CrossRef]
- Islinger, M.; Costello, J.L.; Kors, S.; Soupene, E.; Levine, T.P.; Kuypers, F.A.; Schrader, M. The diversity of ACBD proteins—From lipid binding to protein modulators and organelle tethers. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118675. [Google Scholar] [CrossRef] [PubMed]
- Soupene, E.; Kuypers, F.A. Ligand binding to the ACBD6 protein regulates the acyl-CoA transferase reactions in membranes. J. Lipid Res. 2015, 56, 1961–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soupene, E.; Wang, D.; Kuypers, F.A. Remodeling of host phosphatidylcholine by Chlamydia acyltransferase is regulated by acyl-CoA binding protein ACBD6 associated with lipid droplets. Microbiologyopen 2015, 4, 235–251. [Google Scholar] [CrossRef] [PubMed]
- Soupene, E.; Kuypers, F.A. Phosphatidylserine decarboxylase CT699, lysophospholipid acyltransferase CT775, and acyl-ACP synthase CT776 provide membrane lipid diversity to Chlamydia trachomatis. Sci. Rep. 2017, 7, 15767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brannigan, J.A.; Smith, B.A.; Yu, Z.; Brzozowski, A.M.; Hodgkinson, M.R.; Maroof, A.; Price, H.P.; Meier, F.; Leatherbarrow, R.J.; Tate, E.W.; et al. N-myristoyltransferase from Leishmania donovani: Structural and functional characterisation of a potential drug target for visceral leishmaniasis. J. Mol. Biol. 2010, 396, 985–999. [Google Scholar] [CrossRef] [Green Version]
- Dian, C.; Perez-Dorado, I.; Riviere, F.; Asensio, T.; Legrand, P.; Ritzefeld, M.; Shen, M.; Cota, E.; Meinnel, T.; Tate, E.W.; et al. High-resolution snapshots of human N-myristoyltransferase in action illuminate a mechanism promoting N-terminal Lys and Gly myristoylation. Nat. Commun. 2020, 11, 1132. [Google Scholar] [CrossRef] [Green Version]
- Mousnier, A.; Bell, A.S.; Swieboda, D.P.; Morales-Sanfrutos, J.; Perez-Dorado, I.; Brannigan, J.A.; Newman, J.; Ritzefeld, M.; Hutton, J.A.; Guedan, A.; et al. Fragment-derived inhibitors of human N-myristoyltransferase block capsid assembly and replication of the common cold virus. Nat. Chem. 2018, 10, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.J.; Ludwig, E.H. Analysis of fatty acids of continuously cultured mammalian cells by gas-liquid chromatography. Nature 1962, 196, 893–894. [Google Scholar] [CrossRef]
- Shindou, H.; Hishikawa, D.; Harayama, T.; Eto, M.; Shimizu, T. Generation of membrane diversity by lysophospholipid acyltransferases. J. Biochem. 2013, 154, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Valentine, W.J.; Yanagida, K.; Kawana, H.; Kono, N.; Noda, N.N.; Aoki, J.; Shindou, H. Update and nomenclature proposal for mammalian lysophospholipid acyltransferases, which create membrane phospholipid diversity. J. Biol. Chem. 2022, 298, 101470. [Google Scholar] [CrossRef]
- Soupene, E.; Kuypers, F.A. Phosphatidylcholine formation by LPCAT1 is regulated by Ca(2+) and the redox status of the cell. BMC Biochem. 2012, 13, 8. [Google Scholar] [CrossRef] [Green Version]
- Lands, W.E. Metabolism of glycerolipids. 2. The enzymatic acylation of lysolecithin. J. Biol. Chem. 1960, 235, 2233–2237. [Google Scholar] [CrossRef]
- Moessinger, C.; Kuerschner, L.; Spandl, J.; Shevchenko, A.; Thiele, C. Human lysophosphatidylcholine acyltransferases 1 and 2 are located in lipid droplets where they catalyze the formation of phosphatidylcholine. J. Biol. Chem. 2011, 286, 21330–21339. [Google Scholar] [CrossRef] [Green Version]
- Beilstein, F.; Lemasson, M.; Pène, V.; Rainteau, D.; Demignot, S.; Rosenberg, A.R. Lysophosphatidylcholine acyltransferase 1 is downregulated by hepatitis C virus: Impact on production of lipo-viro-particles. Gut 2017, 66, 2160–2169. [Google Scholar] [CrossRef] [Green Version]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangère, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; de Barros, J.P.; Hillon, P.; et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, J.; Mandrup, S.; Rasmussen, J.T.; Andreasen, P.H.; Poulsen, F.; Kristiansen, K. The function of acyl-CoA-binding protein (ACBP)/diazepam binding inhibitor (DBI). Mol. Cell. Biochem. 1993, 123, 129–138. [Google Scholar] [CrossRef]
- Rasmussen, J.T.; Rosendal, J.; Knudsen, J. Interaction of acyl-CoA binding protein (ACBP) on processes for which acyl-CoA is a substrate, product or inhibitor. Biochem. J. 1993, 292 Pt 3, 907–913. [Google Scholar] [CrossRef] [Green Version]
- Elle, I.C.; Simonsen, K.T.; Olsen, L.C.; Birck, P.K.; Ehmsen, S.; Tuck, S.; Le, T.T.; Faergeman, N.J. Tissue- and paralogue-specific functions of acyl-CoA-binding proteins in lipid metabolism in Caenorhabditis elegans. Biochem. J. 2011, 437, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Heal, W.P.; Wickramasinghe, S.R.; Bowyer, P.W.; Holder, A.A.; Smith, D.F.; Leatherbarrow, R.J.; Tate, E.W. Site-specific N-terminal labelling of proteins in vitro and in vivo using N-myristoyl transferase and bioorthogonal ligation chemistry. Chem. Commun. 2008, 4, 480–482. [Google Scholar] [CrossRef]
- Heal, W.P.; Wickramasinghe, S.R.; Leatherbarrow, R.J.; Tate, E.W. N-Myristoyl transferase-mediated protein labelling in vivo. Org. Biomol. Chem. 2008, 6, 2308–2315. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soupene, E.; Kuypers, F.A. Dual Role of ACBD6 in the Acylation Remodeling of Lipids and Proteins. Biomolecules 2022, 12, 1726. https://doi.org/10.3390/biom12121726
Soupene E, Kuypers FA. Dual Role of ACBD6 in the Acylation Remodeling of Lipids and Proteins. Biomolecules. 2022; 12(12):1726. https://doi.org/10.3390/biom12121726
Chicago/Turabian StyleSoupene, Eric, and Frans A. Kuypers. 2022. "Dual Role of ACBD6 in the Acylation Remodeling of Lipids and Proteins" Biomolecules 12, no. 12: 1726. https://doi.org/10.3390/biom12121726
APA StyleSoupene, E., & Kuypers, F. A. (2022). Dual Role of ACBD6 in the Acylation Remodeling of Lipids and Proteins. Biomolecules, 12(12), 1726. https://doi.org/10.3390/biom12121726