
Role of Sulphur and Heavier Chalcogens on the Antioxidant Power and Bioactivity of Natural Phenolic Compounds

 ,
,  ,
,  ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Natural Phenolic Compounds as a Valuable Source of Molecular Scaffolds Endowed with Diverse Biological Activities
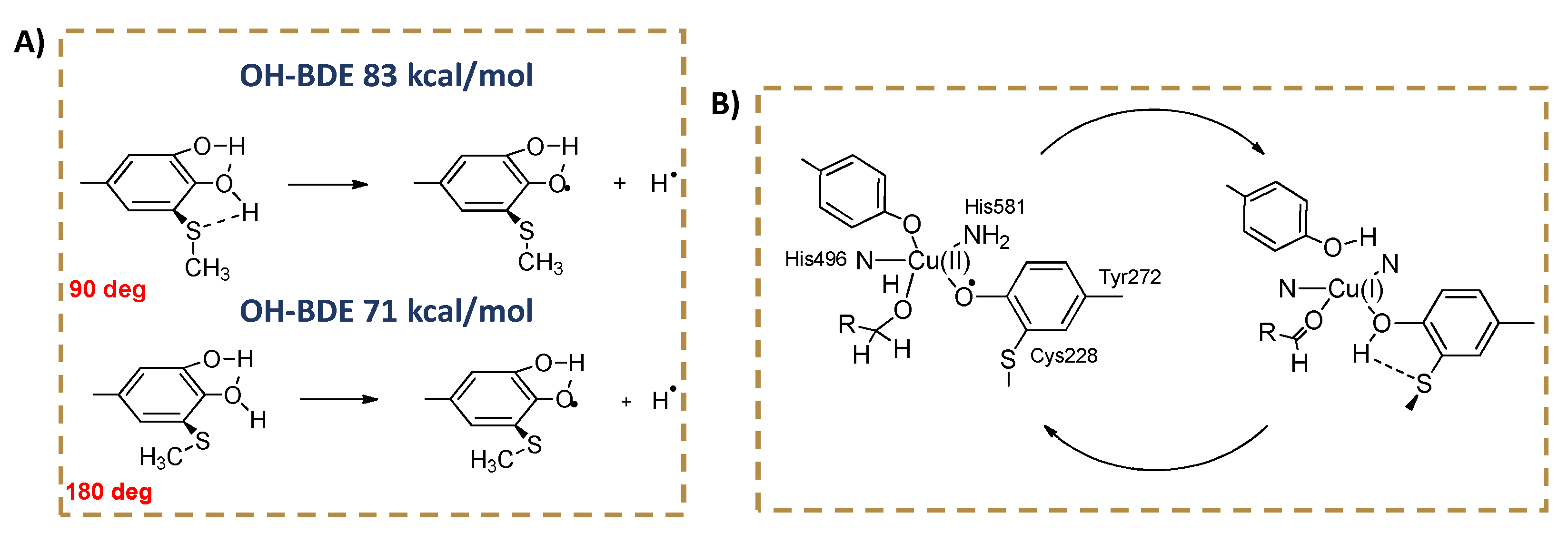
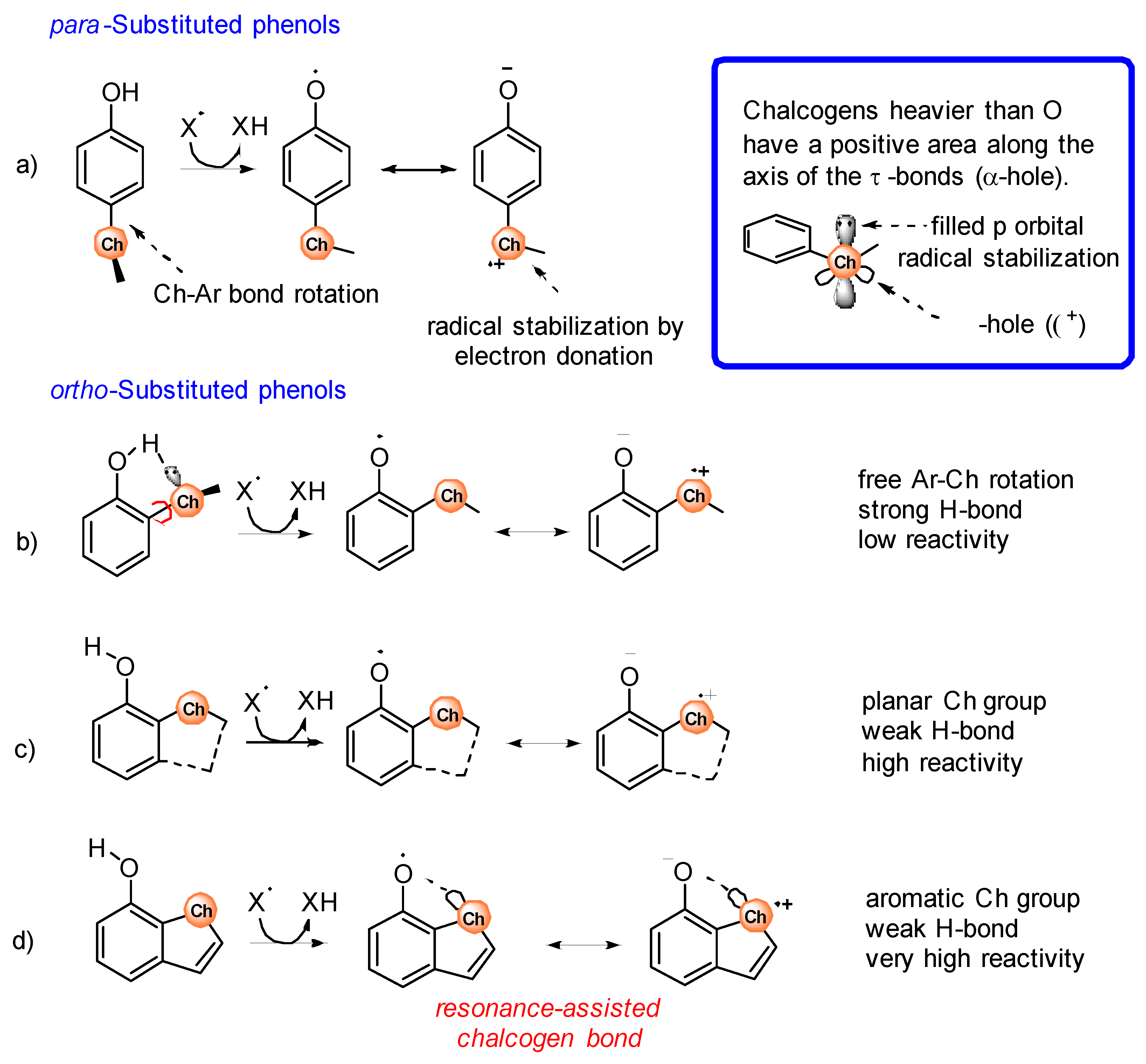
2. Electronic and Stereo-Electronic Effect and H-Bonding of Chalcogens as Substituents in the Phenolic Ring of Simple Molecules
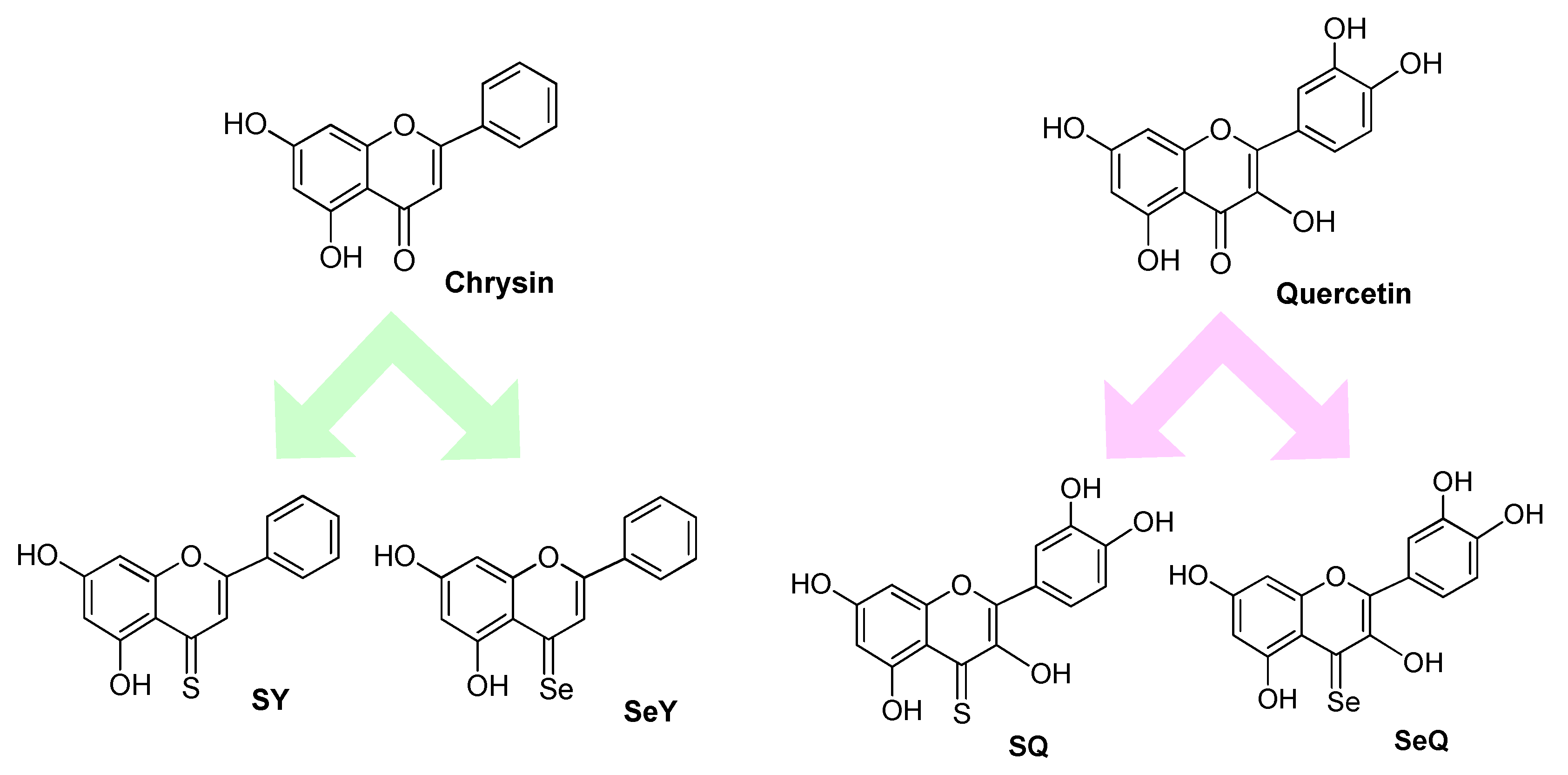
3. Isosteric O to S or Se Replacement
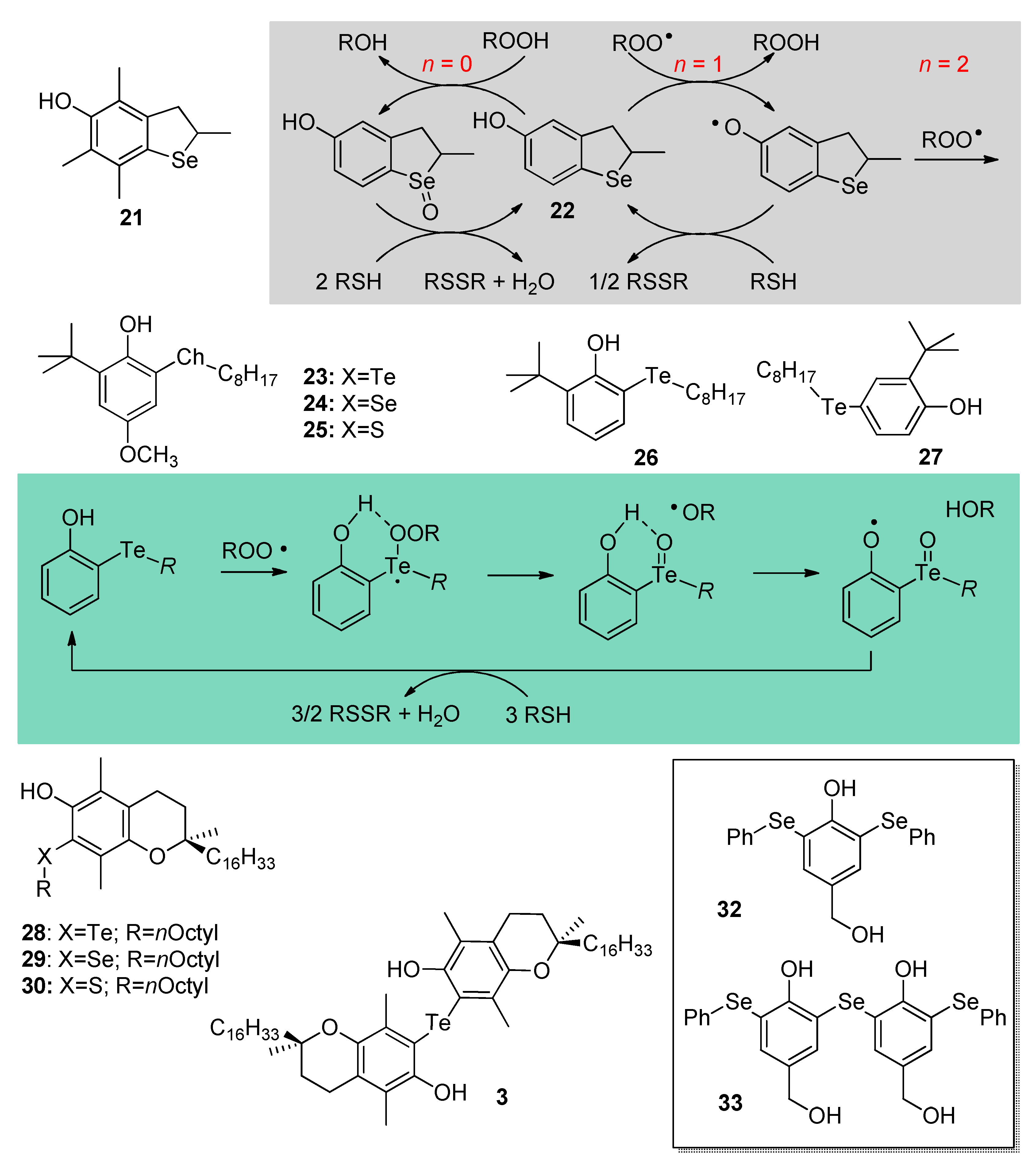
4. Chalcogen-Substituted Phenolic Antioxidants Inspired by Vitamin E and Flavonoids
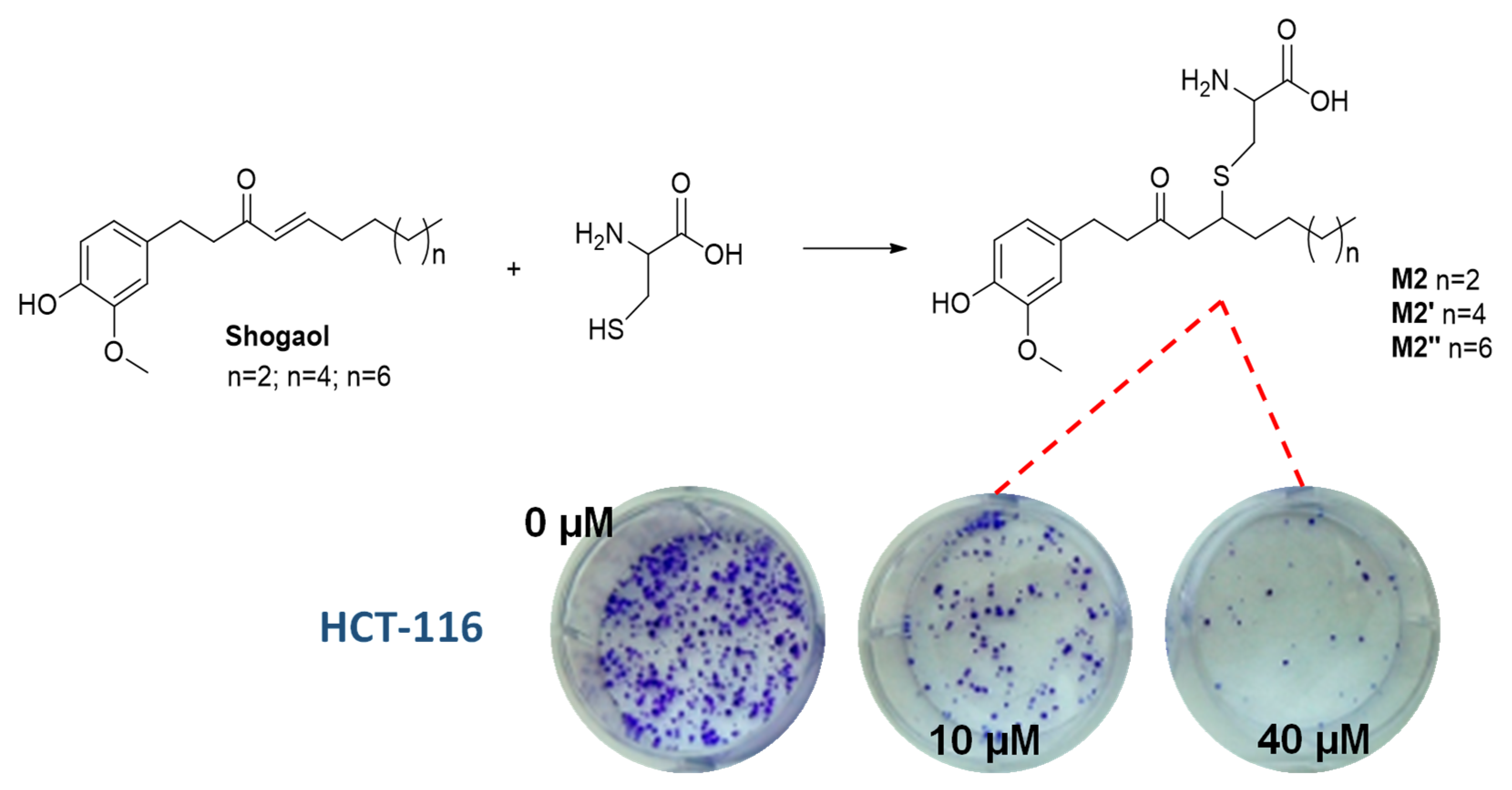
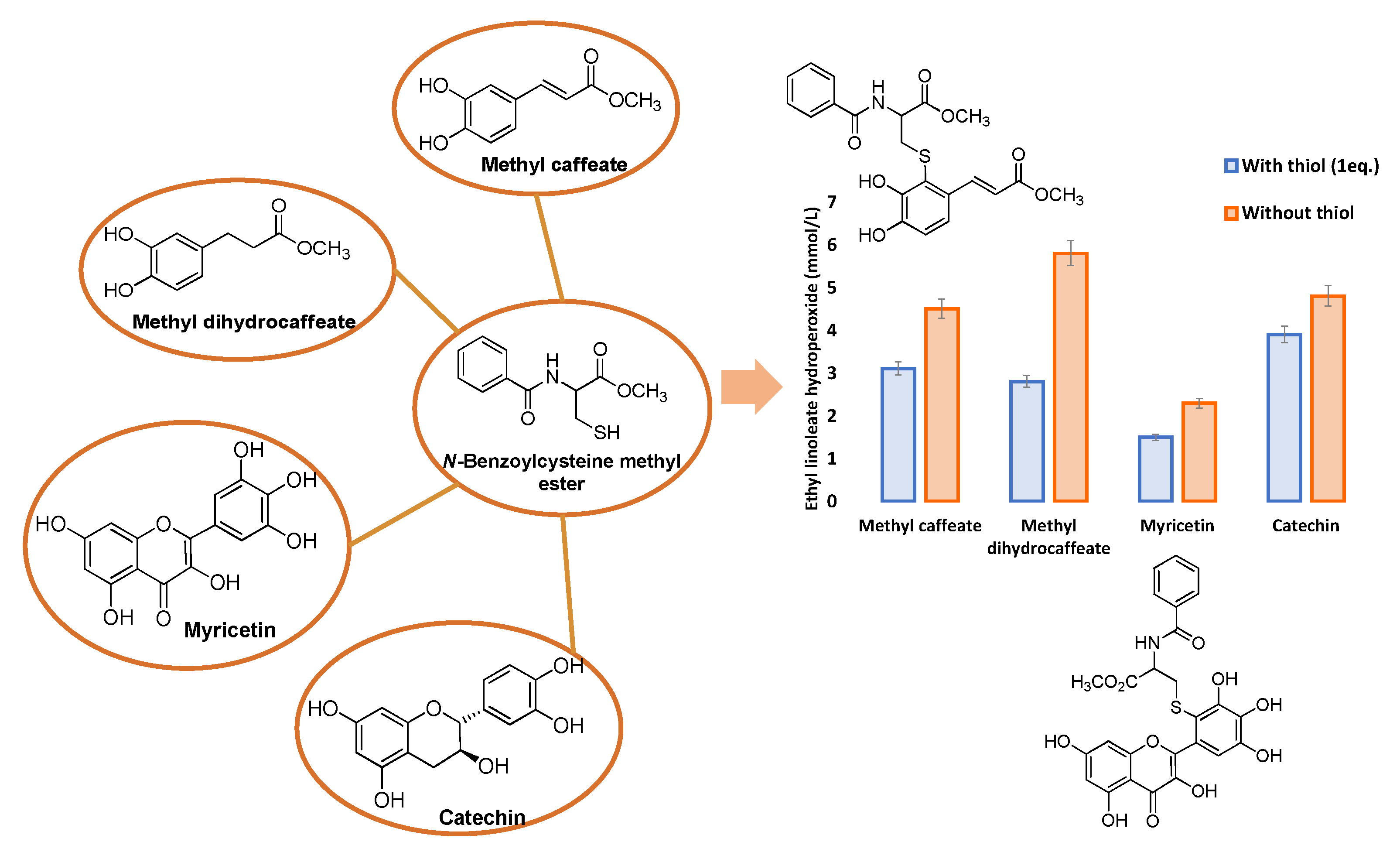
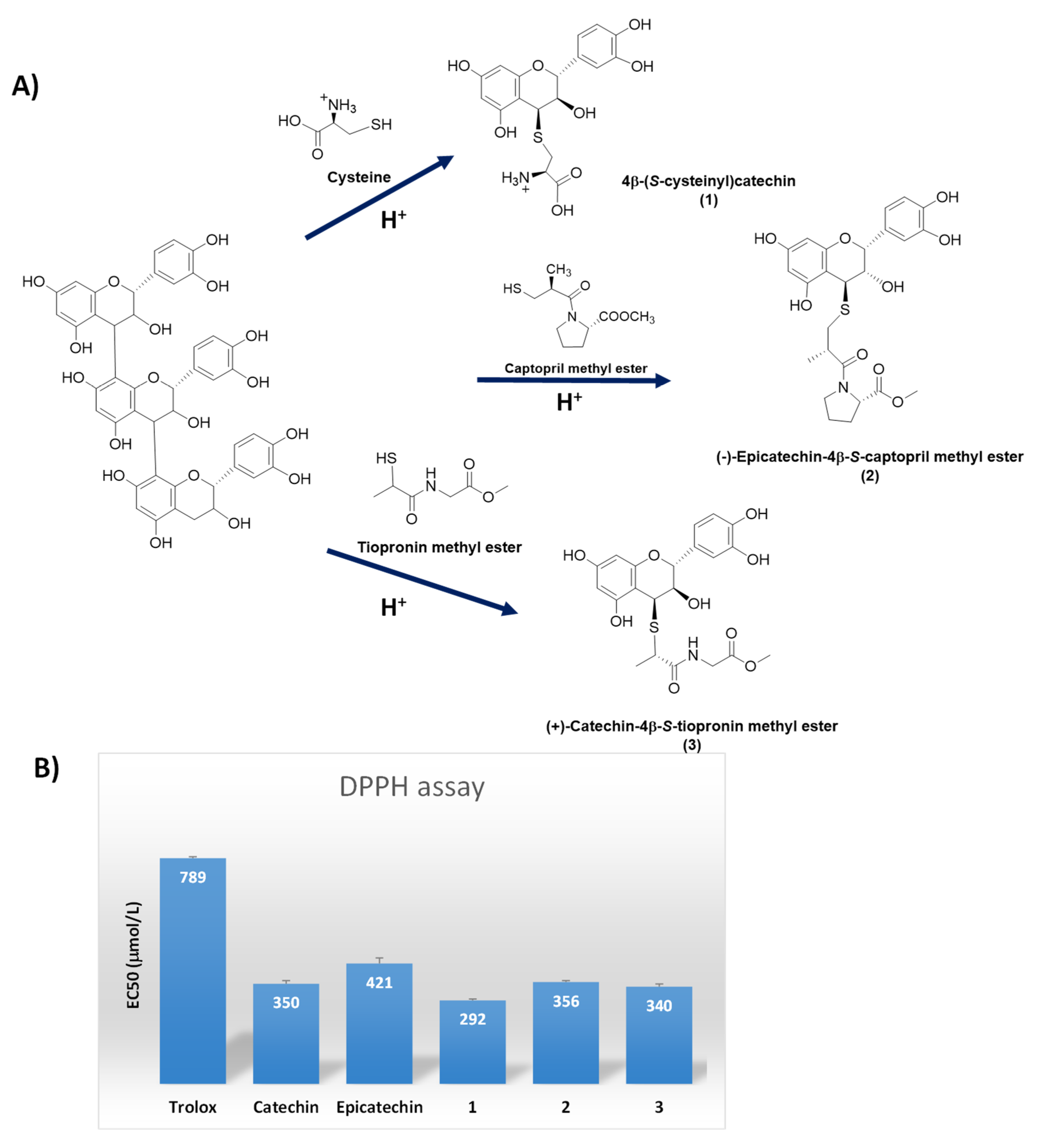
5. Conjugation with Biological Thiols
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Russo, G.L.; Tedesco, I.; Spagnuolo, C.; Russo, M. Antioxidant Polyphenols in Cancer Treatment: Friend, Foe or Foil? Semin. Cancer Biol. 2017, 46, 1–13. [Google Scholar] [CrossRef]
- Oliviero, F.; Scanu, A.; Zamudio-Cuevas, Y.; Punzi, L.; Spinella, P. Anti-Inflammatory Effects of Polyphenols in Arthritis. J. Sci. Food Agric. 2018, 98, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Vicente, M.; Barrajon-Catalan, E.; Herranz-Lopez, M.; Segura-Carretero, A.; Joven, J.; Encinar, J.A.; Micol, V. Plant-Derived Polyphenols in Human Health: Biological Activity, Metabolites and Putative Molecular Targets. Curr. Drug Metabol. 2018, 19, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Gao, Y.; Tu, Y. Mechanisms of Body Weight Reduction by Black Tea Polyphenols. Molecules 2016, 21, 1659. [Google Scholar] [CrossRef]
- de Lima Cherubim, D.J.; Buzanello Martins, C.V.; Oliveira Fariña, L.; da Silva de Lucca, R.A. Polyphenols as Natural Antioxidants in Cosmetics Applications. J. Cosmet. Dermatol. 2020, 19, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Heilmann, J.; König, B. Natural Phenolic Metabolites with Anti-Angiogenic Properties—A Review from the Chemical Point of View. Beilstein J. Org. Chem. 2015, 11, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yong, H.; Yao, X.; Hu, H.; Yun, D.; Xiao, L. Recent Advances in Phenolic–Protein Conjugates: Synthesis, Characterization, Biological Activities and Potential Applications. RSC Adv. 2019, 9, 35825–35840. [Google Scholar] [CrossRef]
- Groussin, A.L.; Antoniotti, S. Valuable Chemicals by the Enzymatic Modification of Molecules of Natural Origin: Terpenoids, Steroids, Phenolics and Related Compounds. Bioresour. Technol. 2012, 115, 237–243. [Google Scholar] [CrossRef]
- Pinheiro, P.F.; Menini, L.A.P.; Bernardes, P.C.; Saraiva, S.H.; Carneiro, J.W.M.; Costa, A.V.; Arruda, T.R.; Lage, M.R.; Gonçalves, P.M.; Bernardes, C.D.O.; et al. Semisynthetic Phenol Derivatives Obtained from Natural Phenols: Antimicrobial Activity and Molecular Properties. J. Agric. Food Chem. 2018, 66, 323–330. [Google Scholar] [CrossRef]
- Floris, B.; Galloni, P.; Conte, V.; Sabuzi, F. Tailored Functionalization of Natural Phenols to Improve Biological Activity. Biomolecules 2021, 11, 1325. [Google Scholar] [CrossRef]
- Karas, D.; Ulrichová, J.; Valentová, K. Galloylation of Polyphenols Alters Their Biological Activity. Food Chem. Toxicol. 2017, 105, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Correia-da-Silva, M.; Sousa, E.; Pinto, M.M.M. Emerging Sulfated Flavonoids and Other Polyphenols as Drugs: Nature as an Inspiration. Med. Res. Rev. 2014, 34, 223–279. [Google Scholar] [CrossRef] [PubMed]
- van der Horst, M.A.; Hartog, A.F.; el Morabet, R.; Marais, A.; Kircz, M.; Wever, R. Enzymatic Sulfation of Phenolic Hydroxy Groups of Various Plant Metabolites by an Arylsulfotransferase. Eur. J. Org. Chem. 2015, 2015, 534–541. [Google Scholar] [CrossRef]
- Chen, X.; Gu, H.; Lyu, Z.; Liu, X.; Wang, L.; Chen, H.; Brash, J.L. Sulfonate Groups and Saccharides as Essential Structural Elements in Heparin-Mimicking Polymers Used as Surface Modifiers: Optimization of Relative Contents for Antithrombogenic Properties. ACS Appl. Mater. Interfaces 2018, 10, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, M.L.; Panzella, L.; Duarte, B.; Goncalves-Monteiro, S.; Marques, F.; Morato, M.; Correia-da-Silva, M.; Verotta, L.; Napolitano, A. Sulfated Oligomers of Tyrosol: Toward a New Class of Bioinspired Nonsaccharidic Anticoagulants. Biomacromolecules 2021, 22, 399–409. [Google Scholar] [CrossRef]
- Bastos, P.; Gomes, T.; Ribeiro, L. Catechol-O-Methyltransferase (COMT): An Update on Its Role in Cancer, Neurological and Cardiovascular Diseases. In Reviews of Physiology, Biochemistry and Pharmacology; Nilius, B., de Tombe, P., Gudermann, T., Jahn, R., Lill, R., Petersen, O., Eds.; Springer: Cham, Switzerland, 2017; Volume 173, pp. 1–39. [Google Scholar]
- Murota, K.; Nakamura, Y.; Uehara, M. Flavonoid Metabolism: The Interaction of Metabolites and Gut Microbiota. Biosci. Biotechnol. Biochem. 2018, 82, 600–610. [Google Scholar] [CrossRef]
- Amorati, R.; Valgimigli, L. Methods to Measure the Antioxidant Activity of Phytochemicals and Plant Extracts. J. Agric. Food Chem. 2018, 66, 3324–3329. [Google Scholar] [CrossRef]
- Lucarini, M.; Pedulli, G.F.; Valgimigli, L.; Amorati, R.; Minisci, F. Thermochemical and Kinetic Studies of a Bisphenol Antioxidant. J. Org. Chem. 2001, 66, 5456–5462. [Google Scholar] [CrossRef]
- Mulder, P.; Korth, H.G.; Pratt, D.A.; DiLabio, G.A.; Valgimigli, L.; Pedulli, G.F.; Ingold, K.U. Critical Re-Evaluation of the O-H Bond Dissociation Enthalpy in Phenol. J. Phys. Chem. A 2005, 109, 2647–2655. [Google Scholar] [CrossRef]
- Amorati, R.; Valgimigli, L. Modulation of the Antioxidant Activity of Phenols by Non-Covalent Interactions. Org. Biomol. Chem. 2012, 10, 4147–4158. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, L.; Pratt, D.A. Maximizing the Reactivity of Phenolic and Aminic Radical-Trapping Antioxidants: Just Add Nitrogen! Acc. Chem. Res. 2015, 48, 966–975. [Google Scholar] [CrossRef]
- Shanks, D.; Amorati, R.; Fumo, M.G.; Pedulli, G.F.; Valgimigli, L.; Engman, L. Synthesis and Antioxidant Profile of All-Rac-α-Selenotocopherol. J. Org. Chem. 2006, 71, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen Bonding: An Overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Carugo, O.; Resnati, G.; Metrangolo, P. Chalcogen Bonds Involving Selenium in Protein Structures. ACS Chem. Biol. 2021, 16, 1622–1627. [Google Scholar] [CrossRef]
- Amorati, R.; Pedulli, G.F.; Valgimigli, L.; Johansson, H.; Engman, L. Organochalcogen Substituents in Phenolic Antioxidants. Org. Lett. 2010, 12, 2326–2329. [Google Scholar] [CrossRef]
- Viglianisi, C.; Amorati, R.; di Pietro, L.; Menichetti, S. A Straightforward Route to Potent Phenolic Chain-Breaking Antioxidants by Acid-Promoted Transposition of 1,4-Benzo[b]Oxathiines to Dihydrobenzo[b]Thiophenes. Chem. Eur. J. 2015, 21, 16639–16645. [Google Scholar] [CrossRef] [PubMed]
- Viglianisi, C.; Menichetti, S. Chain Breaking Antioxidant Activity of Heavy (S, Se, Te) Chalcogens Substituted Polyphenols. Antioxidants 2019, 8, 487. [Google Scholar] [CrossRef]
- Menichetti, S.; Amorati, R.; Meoni, V.; Tofani, L.; Caminati, G.; Viglianisi, C. Role of Noncovalent Sulfur·Oxygen Interactions in Phenoxyl Radical Stabilization: Synthesis of Super Tocopherol-like Antioxidants. Org. Lett. 2016, 18, 5464–5467. [Google Scholar] [CrossRef]
- Tanini, D.; Panzella, L.; Amorati, R.; Capperucci, A.; Pizzo, E.; Napolitano, A.; Menichetti, S.; d’Ischia, M. Resveratrol-Based Benzoselenophenes with an Enhanced Antioxidant and Chain Breaking Capacity. Org. Biomol. Chem. 2015, 13, 5757–5764. [Google Scholar] [CrossRef]
- Domazetovic, V.; Fontani, F.; Tanini, D.; D’Esopo, V.; Viglianisi, C.; Marcucci, G.; Panzella, L.; Napolitano, A.; Brandi, M.L.; Capperucci, A.; et al. Protective Role of Benzoselenophene Derivatives of Resveratrol on the Induced Oxidative Stress in Intestinal Myofibroblasts and Osteocytes. Chem.-Biol. Interact. 2017, 275, 13–21. [Google Scholar] [CrossRef]
- Angeli, A.; Tanini, D.; Viglianisi, C.; Panzella, L.; Capperucci, A.; Menichetti, S.; Supuran, C.T. Evaluation of Selenide, Diselenide and Selenoheterocycle Derivatives as Carbonic Anhydrase I, II, IV, VII and IX Inhibitors. Bioorg. Med. Chem. 2017, 25, 2518–2523. [Google Scholar] [CrossRef]
- Amorati, R.; Valgimigli, L.; Panzella, L.; Napolitano, A.; d’Ischia, M. 5-S-Lipoylhydroxytyrosol, a Multidefense Antioxidant Featuring a Solvent-Tunable Peroxyl Radical-Scavenging 3-Thio-1,2-Dihydroxybenzene Motif. J. Org. Chem. 2013, 78, 9857–9864. [Google Scholar] [CrossRef] [PubMed]
- Amorati, R.; Catarzi, F.; Menichetti, S.; Pedulli, G.F.; Viglianisi, C. Effect of Ortho-SR Groups on O-H Bond Strength and H-Atom Donating Ability of Phenols: A Possible Role for the Tyr-Cys Link in Galactose Oxidase Active Site? J. Am. Chem. Soc. 2008, 130, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, L.; Ingold, K.U.; Lusztyk, J. Solvent Effects on the Reactivity and Free Spin Distribution of 2,2-Diphenyl-1-Picrylhydrazyl Radicals1. J. Org. Chem. 1996, 61, 7947–7950. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.Y.; Yang, J.; Dai, F.; Ding, D.J.; Kang, Y.F.; Wang, F.; Li, X.Z.; Liu, G.Y.; Yu, S.S.; Jin, X.L.; et al. Extraordinary Radical Scavengers: 4-Mercaptostilbenes. Chem. Eur. J. 2012, 18, 5898–5905. [Google Scholar] [CrossRef]
- Chavarria, D.; Fernandes, C.; Silva, T.; Garrido, J.; Remião, F.; Oliveira, P.J.; Borges, F. Bioisosteric OH- to SH-Replacement Changes the Antioxidant Profile of Ferulic Acid. Org. Biomol. Chem. 2019, 17, 9646–9654. [Google Scholar] [CrossRef]
- Chavarria, D.; Fernandes, C.; Aguiar, B.; Silva, T.; Garrido, J.; Remião, F.; Oliveira, P.J.; Uriarte, E.; Borges, F. Insights into the Discovery of Novel Neuroprotective Agents: A Comparative Study between Sulfanylcinnamic Acid Derivatives and Related Phenolic Analogues. Molecules 2019, 24, 4405. [Google Scholar] [CrossRef]
- Carreon-Gonzalez, M.; Vivier-Bunge, A.; Alvarez-Idaboy, J.R. Thiophenols, Promising Scavengers of Peroxyl Radicals: Mechanisms and Kinetics. J. Comput. Chem. 2019, 40, 2103–2110. [Google Scholar] [CrossRef]
- Castañeda-Arriaga, R.; Marino, T.; Russo, N.; Alvarez-Idaboy, J.R.; Galano, A. Chalcogen Effects on the Primary Antioxidant Activity of Chrysin and Quercetin. New J. Chem. 2020, 44, 9073–9082. [Google Scholar] [CrossRef]
- Valgimigli, L.; Amorati, R. CHAPTER 11: Vitamin E Inspired Synthetic Antioxidants. Food Chem. Funct. Anal. 2019, 2019, 151–164. [Google Scholar] [CrossRef]
- Amorati, R.; Baschieri, A.; Valgimigli, L. The Role of Sulfur and Heavier Chalcogens in the Chemistry of Antioxidants. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 638–642. [Google Scholar] [CrossRef]
- Zahalka, H.A.; Robillard, B.; Hughes, L.; Lusztyk, J.; Burton, G.W.; Ingold, K.U.; Janzen, E.G.; Kotake, Y. Antioxidant Activity of 1-Thio-Alpha-Tocopherol and Related Compounds. EPR, ENDOR, and UV-Visible Absorption Spectra of Some of the Derived Phenoxyl Radicals. J. Org. Chem. 1988, 53, 3739–3745. [Google Scholar] [CrossRef]
- Menichetti, S.; Amorati, R.; Bartolozzi, M.G.; Pedulli, G.F.; Salvini, A.; Viglianisi, C. A Straightforward Hetero-Diels–Alder Approach to (2-Ambo,4′R,8′R)-α/β/γ/δ-4-Thiatocopherol. Eur. J. Org. Chem. 2010, 2010, 2218–2225. [Google Scholar] [CrossRef]
- Amorati, R.; Fumo, M.G.; Pedulli, G.F.; Menichetti, S.; Pagliuca, C.; Viglianisi, C. Antioxidant and Antiradical Activity of Hydroxy-Substituted 4-Thiaflavanes. Helv. Chim. Acta 2006, 89, 2462–2472. [Google Scholar] [CrossRef]
- Amorati, R.; Cavalli, A.; Fumo, M.G.; Masetti, M.; Menichetti, S.; Pagliuca, C.; Pedulli, G.F.; Viglianisi, C. Kinetic and Thermochemical Study of the Antioxidant Activity of Sulfur-Containing Analogues of Vitamin E. Chem. Eur. J. 2007, 13, 8223–8230. [Google Scholar] [CrossRef] [PubMed]
- Viglianisi, C.; Bartolozzi, M.G.; Pedulli, G.F.; Amorati, R.; Menichetti, S. Optimization of the Antioxidant Activity of Hydroxy-Substituted 4-Thiaflavanes: A Proof-of-Concept Study. Chem. Eur. J. 2011, 17, 12396–12404. [Google Scholar] [CrossRef]
- Burton, G.W.; Doba, T.; Gabe, E.J.; Hughes, L.; Lee, F.L.; Prasad, L.; Ingoldo, K.U. Autoxidation of Biological Molecules. 4. Maximizing the Antioxidant Activity of Phenols. J. Am. Chem. Soc. 1985, 107, 7053–7065. [Google Scholar] [CrossRef]
- Viglianisi, C.; Vasa, K.; Tanini, D.; Capperucci, A.; Amorati, R.; Valgimigli, L.; Baschieri, A.; Menichetti, S. Ditocopheryl Sulfides and Disulfides: Synthesis and Antioxidant Profile. Chem. Eur. J. 2019, 25, 9108–9116. [Google Scholar] [CrossRef]
- Kumar, S.; Johansson, H.; Engman, L.; Valgimigli, L.; Amorati, R.; Fumo, M.G.; Pedulli, G.F. Regenerable Chain-Breaking 2,3-Dihydrobenzo[b]Selenophene-5-Ol Antioxidants. J. Org. Chem. 2007, 72, 2583–2595. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Shanks, D.; Engman, L.; Amorati, R.; Pedulli, G.F.; Valgimigli, L. Long-Lasting Antioxidant Protection: A Regenerable BHA Analogue. J. Org. Chem. 2010, 75, 7535–7541. [Google Scholar] [CrossRef]
- Amorati, R.; Valgimigli, L.; Dinér, P.; Bakhtiari, K.; Saeedi, M.; Engman, L. Multi-Faceted Reactivity of Alkyltellurophenols towards Peroxyl Radicals: Catalytic Antioxidant versus Thiol-Depletion Effect. Chem. Eur. J. 2013, 19, 7510–7522. [Google Scholar] [CrossRef]
- Poon, J.F.; Singh, V.P.; Yan, J.; Engman, L. Regenerable Antioxidants—Introduction of Chalcogen Substituents into Tocopherols. Chem. Eur. J. 2015, 21, 2447–2457. [Google Scholar] [CrossRef] [PubMed]
- Poon, J.-f.; Yan, J.; Singh, V.P.; Gates, P.J.; Engman, L. Regenerable Radical-Trapping Tellurobistocopherol Antioxidants. J. Org. Chem. 2016, 81, 12540–12544. [Google Scholar] [CrossRef]
- Jamier, V.; Ba, L.A.; Jacob, C. Selenium- and Tellurium-Containing Multifunctional Redox Agents as Biochemical Redox Modulators with Selective Cytotoxicity. Chem. Eur. J. 2010, 16, 10920–10928. [Google Scholar] [CrossRef] [PubMed]
- Estevam, E.C.; Witek, K.; Faulstich, L.; Nasim, M.J.; Latacz, G.; Domínguez-Álvarez, E.; Kieć-Kononowicz, K.; Demasi, M.; Handzlik, J.; Jacob, C. Aspects of a Distinct Cytotoxicity of Selenium Salts and Organic Selenides in Living Cells with Possible Implications for Drug Design. Molecules 2015, 20, 13894–13912. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, C.W.; Barbosa, N.V.; Rocha, J.B.T. Toxicology and Pharmacology of Synthetic Organoselenium Compounds: An Update. Arch. Toxicol. 2021, 95, 1179–1226. [Google Scholar] [CrossRef]
- Singh, V.P.; Poon, J.-F.; Butcher, R.J.; Engman, L. Pyridoxine-Derived Organoselenium Compounds with Glutathione Peroxidase-like and Chain-Breaking Antioxidant Activity. Chem. Eur. J. 2014, 20, 12563–12571. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Johansson, H.; Kanda, T.; Engman, L.; Müller, T.; Jonsson, M.; Pedulli, G.F.; Petrucci, S.; Valgimigli, L. Catalytic Chain-Breaking Pyridinol Antioxidants. Org. Lett. 2008, 10, 4895–4898. [Google Scholar] [CrossRef]
- Fu, J.; Chen, H.; Soroka, D.N.; Warin, R.F.; Sang, S. Cysteine-Conjugated Metabolites of Ginger Components, Shogaols, Induce Apoptosis through Oxidative Stress-Mediated P53 Pathway in Human Colon Cancer Cells. J. Agric. Food Chem. 2014, 62, 4632–4642. [Google Scholar] [CrossRef]
- Miura, Y.; Inai, M.; Honda, S.; Masuda, A.; Masuda, T. Reducing Effects of Polyphenols on Metmyoglobin and the in Vitro Regeneration of Bright Meat Color by Polyphenols in the Presence of Cysteine. J. Agric. Food Chem. 2014, 62, 9472–9478. [Google Scholar] [CrossRef]
- Fujimoto, A.; Inai, M.; Masuda, T. Chemical Evidence for the Synergistic Effect of a Cysteinyl Thiol on the Antioxidant Activity of Caffeic and Dihydrocaffeic Esters. Food Chem. 2013, 138, 1483–1492. [Google Scholar] [CrossRef]
- Masuda, T.; Miura, Y.; Inai, M.; Masuda, A. Enhancing Effect of a Cysteinyl Thiol on the Antioxidant Activity of Flavonoids and Identification of the Antioxidative Thiol Adducts of Myricetin. Biosci. Biotechnol. Biochem. 2013, 77, 1753–1758. [Google Scholar] [CrossRef]
- Figueroa-Espinoza, M.C.; Rouau, X. Effect of Cysteinyl Caffeic Acid, Caffeic Acid, and l-Dopa on the Oxidative Cross-Linking of Feruloylated Arabinoxylans by a Fungal Laccase. J. Agric. Food Chem. 1999, 47, 497–503. [Google Scholar] [CrossRef]
- Napolitano, A.; Panzella, L.; Leone, L.; d’Ischia, M. Red Hair Benzothiazines and Benzothiazoles: Mutation-Inspired Chemistry in the Quest for Functionality. Acc. Chem. Res. 2012, 46, 519–528. [Google Scholar] [CrossRef]
- Napolitano, A.; Memoli, S.; Nappi, A.J.; d’Ischia, M.; Prota, G. 5-S-Cysteinyldopa, a Diffusible Product of Melanocyte Activity, Is an Efficient Inhibitor of Hydroxylation/Oxidation Reactions Induced by the Fenton System. Biochim. Biophys. Acta Gen. Subj. 1996, 1291, 75–82. [Google Scholar] [CrossRef]
- Memoli, S.; Napolitano, A.; d’Ischia, M.; Misuraca, G.; Palumbo, A.; Prota, G. Diffusible Melanin-Related Metabolites Are Potent Inhibitors of Lipid Peroxidation. Biochim. Biophys. Acta Lipids Lipid Metabol. 1997, 1346, 61–68. [Google Scholar] [CrossRef]
- Nezirević Dernroth, D.; Årstrand, K.; Greco, G.; Panzella, L.; Napolitano, A.; Kågedal, B. Pheomelanin-Related Benzothiazole Isomers in the Urine of Patients with Diffuse Melanosis of Melanoma. Clin. Chim. Acta 2010, 411, 1195–1203. [Google Scholar] [CrossRef]
- Panzella, L.; Wakamatsu, K.; Monfrecola, G.; Ito, S.; Ayala, F.; Napolitano, A. Increased Cysteinyldopa Plasma Levels Hint to Melanocyte as Stress Sensor in Psoriasis. Exp. Dermatol. 2011, 20, 288–290. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ito, S.; Wakamatsu, K.; Zecca, L. Structure and Function of Neuromelanin. In Catecholamine Research. Advances in Behavioral Biology; Nagatsu, T., Nabeshima, T.M.R., Goldstein, D.S., Eds.; Springer: Boston, MA, USA, 2002; Volume 43, pp. 269–272. [Google Scholar]
- Vauzour, D.; Ravaioli, G.; Vafeiadou, K.; Rodriguez-Mateos, A.; Angeloni, C.; Spencer, J.P.E. Peroxynitrite Induced Formation of the Neurotoxins 5-S-Cysteinyl-Dopamine and DHBT-1: Implications for Parkinson’s Disease and Protection by Polyphenols. Arch. Biochem. Biophys. 2008, 476, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Greco, G.; Panzella, L.; Gentile, G.; Errico, M.E.; Carfagna, C.; Napolitano, A.; d’Ischia, M. A Melanin-Inspired pro-Oxidant System for Dopa(Mine) Polymerization: Mimicking the Natural Casing Process. Chem. Comm. 2011, 47, 10308–10310. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Ohtara, K.; Ito, S. Chemical Analysis of Late Stages of Pheomelanogenesis: Conversion of Dihydrobenzothiazine to a Benzothiazole Structure. Pigm. Cell Res. 2009, 22, 474–486. [Google Scholar] [CrossRef]
- Lee, H.A.; Ma, Y.; Zhou, F.; Hong, S.; Lee, H. Material-Independent Surface Chemistry beyond Polydopamine Coating. Acc. Chem. Res. 2019, 52, 704–713. [Google Scholar] [CrossRef]
- d’Ischia, M.; Napolitano, A.; Pezzella, A.; Meredith, P.; Buehler, M. Melanin Biopolymers: Tailoring Chemical Complexity for Materials Design. Angew. Chem. Int. Ed. 2020, 132, 11292–11301. [Google Scholar] [CrossRef]
- Ryu, J.H.; Messersmith, P.B.; Lee, H. Polydopamine Surface Chemistry: A Decade of Discovery. ACS Appl. Mater. Interfaces 2018, 10, 7523–7540. [Google Scholar] [CrossRef]
- Della Vecchia, N.F.; Marega, R.; Ambrico, M.; Iacomino, M.; Micillo, R.; Napolitano, A.; Bonifazi, D.; d’Ischia, M. Tailoring Melanins for Bioelectronics: Polycysteinyldopamine as an Ion Conducting Redox-Responsive Polydopamine Variant for pro-Oxidant Thin Films. J. Mater. Chem. C 2015, 3, 6525–6531. [Google Scholar] [CrossRef]
- Ambrico, M.; Della Vecchia, N.F.; della Ambrico, P.F.; Cardone, A.; Cicco, S.R.; Ligonzo, T.; Avolio, R.; Napolitano, A.; d’Ischia, M. A Photoresponsive Red-Hair-Inspired Polydopamine-Based Copolymer for Hybrid Photocapacitive Sensors. Adv. Funct. Mater. 2014, 24, 7161–7172. [Google Scholar] [CrossRef]
- Tian, R.; Suo, H.; Zhang, S.; Sun, B. Separation of a Family of Antioxidants Flavan-3-Ol Thio-Conjugates from Procyanidins by High-Speed Counter-Current Chromatography. Eur. Food Res. Technol. 2020, 246, 1017–1029. [Google Scholar] [CrossRef]
- Torres, J.L.; Lozano, C.; Julià, L.; Sánchez-Baeza, F.J.; Anglada, J.M.; Centelles, J.J.; Cascante, M. Cysteinyl-Flavan-3-Ol Conjugates from Grape Procyanidins. Antioxidant and Antiproliferative Properties. Bioorg. Med. Chem. 2002, 10, 2497–2509. [Google Scholar] [CrossRef]
- Torres, J.L.; Lozano, C.; Maher, P. Conjugation of Catechins with Cysteine Generates Antioxidant Compounds with Enhanced Neuroprotective Activity. Phytochemistry 2005, 66, 2032–2037. [Google Scholar] [CrossRef] [PubMed]
- Bernini, R.; Barontini, M.; Spatafora, C. New Lipophilic Piceatannol Derivatives Exhibiting Antioxidant Activity Prepared by Aromatic Hydroxylation with 2-Iodoxybenzoic Acid (IBX). Molecules 2009, 14, 4669–4681. [Google Scholar] [CrossRef]
- Panzella, L.; Napolitano, A.; d’Ischia, M. Oxidative Conjugation of Chlorogenic Acid with Glutathione: Structural Characterization of Addition Products and a New Nitrite-Promoted Pathway. Bioorg. Med. Chem. 2003, 11, 4797–4805. [Google Scholar] [CrossRef]
- Panzella, L.; De Lucia, M.; Napolitano, A.; d’Ischia, M. The First Expedient Entry to the Human Melanogen 2-S-Cysteinyldopa Exploiting the Anomalous Regioselectivity of 3,4-Dihydroxycinnamic Acid–Thiol Conjugation. Tetrahedron Lett. 2007, 48, 7650–7652. [Google Scholar] [CrossRef]
- Bassil, D.; Makris, D.P.; Kefalas, P. Oxidation of Caffeic Acid in the Presence of L-Cysteine: Isolation of 2-S-Cysteinylcaffeic Acid and Evaluation of Its Antioxidant Properties. Food Res. Int. 2005, 38, 395–402. [Google Scholar] [CrossRef]
- Ferreira-Lima, N.; Vallverdú-Queralt, A.; Meudec, E.; Mazauric, J.P.; Sommerer, N.; Bordignon-Luiz, M.T.; Cheynier, V.; le Guernevé, C. Synthesis, Identification, and Structure Elucidation of Adducts Formed by Reactions of Hydroxycinnamic Acids with Glutathione or Cysteinylglycine. J. Nat. Prod. 2016, 79, 2211–2222. [Google Scholar] [CrossRef] [PubMed]
- Cheynier, V.F.; Trousdale, E.K.; Singleton, V.L.; Salgues, M.J.; Wylde, R. Characterization of 2-S-Glutathionyl Caftaric Acid and Its Hydrolysis in Relation to Grape Wines. J. Agric. Food Chem. 1986, 34, 217–221. [Google Scholar] [CrossRef]
- d’Ischia, M.; Napolitano, A.; Manini, P.; Panzella, L. Secondary Targets of Nitrite-Derived Reactive Nitrogen Species: Nitrosation/Nitration Pathways, Antioxidant Defense Mechanisms and Toxicological Implications. Chem. Res. Toxicol. 2011, 24, 2071–2092. [Google Scholar] [CrossRef]
- d’Ischia, M.; Panzella, L.; Manini, P.; Napolitano, A. The Chemical Basis of the Antinitrosating Action of Polyphenolic Cancer Chemopreventive Agents. Curr. Med. Chem. 2006, 13, 3133–3144. [Google Scholar] [CrossRef]
- De Lucia, M.; Panzella, L.; Pezzella, A.; Napolitano, A.; d’Ischia, M. Plant Catechols and Their S-Glutathionyl Conjugates as Antinitrosating Agents: Expedient Synthesis and Remarkable Potency of 5-S-Glutathionylpiceatannol. Chem. Res. Toxicol. 2008, 21, 2407–2413. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, M.; Panzella, L.; Crescenzi, O.; Napolitano, A.; Barone, V.; d’Ischia, M. The Catecholic Antioxidant Piceatannol Is an Effective Nitrosation Inhibitor via an Unusual Double Bond Nitration. Bioorg. Med. Chem. Lett. 2006, 16, 2238–2242. [Google Scholar] [CrossRef]
- Panzella, L.; Verotta, L.; Goya, L.; Ramos, S.; Martín, M.A.; Bravo, L.; Napolitano, A.; d’Ischia, M. Synthesis and Bioactivity Profile of 5-S-Lipoylhydroxytyrosol-Based Multidefense Antioxidants with a Sizeable (Poly)Sulfide Chain. J. Agric. Food Chem. 2013, 61, 1710–1717. [Google Scholar] [CrossRef]
- Anwar, A.; Gould, E.; Tinson, R.; Groom, M.; Hamilton, C.J. Think Yellow and Keep Green—Role of Sulfanes from Garlic in Agriculture. Antioxidants 2016, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Munday, R. Harmful and Beneficial Effects of Organic Monosulfides, Disulfides, and Polysulfides in Animals and Humans. Chem. Res. Toxicol. 2011, 25, 47–60. [Google Scholar] [CrossRef]
- Officioso, A.; Panzella, L.; Tortora, F.; Alfieri, M.L.; Napolitano, A.; Manna, C. Comparative Analysis of the Effects of Olive Oil Hydroxytyrosol and Its 5-S-Lipoyl Conjugate in Protecting Human Erythrocytes from Mercury Toxicity. Oxidative Med. Cell. Longev. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Micillo, R.; Sirés-Campos, J.; García-Borrón, J.C.; Panzella, L.; Napolitano, A.; Olivares, C. Conjugation with Dihydrolipoic Acid Imparts Caffeic Acid Ester Potent Inhibitory Effect on Dopa Oxidase Activity of Human Tyrosinase. Int. J. Mol. Sci. 2018, 19, 2156. [Google Scholar] [CrossRef] [PubMed]
- Micillo, R.; Pistorio, V.; Pizzo, E.; Panzella, L.; Napolitano, A.; d’Ischia, M. 2-S-Lipoylcaffeic Acid, a Natural Product-Based Entry to Tyrosinase Inhibition via Catechol Manipulation. Biomimetics 2017, 2, 15. [Google Scholar] [CrossRef] [PubMed]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfieri, M.L.; Panzella, L.; Amorati, R.; Cariola, A.; Valgimigli, L.; Napolitano, A. Role of Sulphur and Heavier Chalcogens on the Antioxidant Power and Bioactivity of Natural Phenolic Compounds. Biomolecules 2022, 12, 90. https://doi.org/10.3390/biom12010090
Alfieri ML, Panzella L, Amorati R, Cariola A, Valgimigli L, Napolitano A. Role of Sulphur and Heavier Chalcogens on the Antioxidant Power and Bioactivity of Natural Phenolic Compounds. Biomolecules. 2022; 12(1):90. https://doi.org/10.3390/biom12010090
Chicago/Turabian StyleAlfieri, Maria Laura, Lucia Panzella, Riccardo Amorati, Alice Cariola, Luca Valgimigli, and Alessandra Napolitano. 2022. "Role of Sulphur and Heavier Chalcogens on the Antioxidant Power and Bioactivity of Natural Phenolic Compounds" Biomolecules 12, no. 1: 90. https://doi.org/10.3390/biom12010090
APA StyleAlfieri, M. L., Panzella, L., Amorati, R., Cariola, A., Valgimigli, L., & Napolitano, A. (2022). Role of Sulphur and Heavier Chalcogens on the Antioxidant Power and Bioactivity of Natural Phenolic Compounds. Biomolecules, 12(1), 90. https://doi.org/10.3390/biom12010090