Abstract
B lymphocyte development has two DNA recombination processes: V(D)J recombination of the immunoglobulin (Igh) gene variable region, and class switching of the Igh constant regions from IgM to IgG, IgA, or IgE. V(D)J recombination is required for the successful maturation of B cells from pro-B to pre-B to immature-B and then to mature B cells in the bone marrow. CSR occurs outside of the bone marrow when mature B cells migrate to peripheral lymphoid organs, such as spleen and lymph nodes. Both V(D)J recombination and CSR depend on an open chromatin state that makes DNA accessible to specific enzymes, recombination activating gene (RAG), and activation-induced cytidine deaminase (AID). Acetyltransferases GCN5 and PCAF possess redundant functions acetylating histone H3 lysine 9 (H3K9). Here, we generated a mouse model that lacked both GCN5 and PCAF in B cells. Double-deficient mice possessed low levels of mature B cells in the bone marrow and peripheral organs, an accumulation of pro-B cells in bone marrow, and reduced CSR levels. We concluded that both GCN5 and PCAF are required for B-cell development in vivo.
1. Introduction
The development of B lymphocytes starts in the bone marrow where progenitor (pro)-B cells using recombination-activating genes (RAG) generate DNA double-strand breaks (DSBs) and initiate V(D)J recombination [1]. In maturating B cells, the V(D)J recombination process is genetic recombination of variable (V), diversity (D), and joining (J) gene segments arranging into a newly formed VDJ part of immunoglobulin gene (Ig) [2,3,4,5,6]. Following V(D)J recombination, B cells develop from pro-B cells expressing specific markers cluster of differentiation 19 (CD19), B220/CD45, and CD43 (CD19+B220+CD43+) to pre-B cells (CD19+B220+CD43−), immature B (CD19+B220+IgM+, low immunoglobulin M, IgM) and mature B (CD19+B220+IgM+, high IgM) cells in bone marrow [2]. Mature B lymphocytes leave the bone marrow and migrate to periphery-populating spleen and lymph nodes through the blood.
Then, mature B cells initiate another DNA recombination process to change the constant regions of immunoglobulin genes, referred to as class switch recombination (CSR). During the CSR in mice, IgM is replaced by IgG3, IgG1, IgG2a, IgG2b, IgE, or IgA [2]. The CSR is initiated by nonproductive transcription known as a germline transcription (GLT), which is needed to separate two DNA strands. Single-stranded DNA is then targeted by activation-induced cytidine deaminase (AID), a B lymphocyte-specific enzyme deaminating cytosine to uracil (C to U). Then, uracil DNA N-glycosylase (UNG) removes uracil from DNA, leading to single-strand break formation (SSB) [7]. Two SSBs facing each other then form a DSB and allow for recombination [2,3,4,5,8]. Both V(D)J and CSR can be regarded as processes following fundamentally similar strategies of genomic recombination [9,10].
DSBs formed during the V(D)J recombination and CSR are recognized, processed, and repaired by the nonhomologous end-joining pathway (NHEJ), and initiate a more complex signaling and chromatin modification pathway known as DNA damage response (DDR) [2,3,4,5]. The NHEJ is initiated when the Ku70 and Ku80 heterodimer (Ku) recognizes and binds the DSBs. Ku serves as a platform for downstream factors including DNA-dependent protein kinase catalytic subunit (DNA-PKcs), X-ray repair cross-complementing protein 4 (XRCC4), XRCC4-like factor (XLF), the paralogue of XRCC4 and XLF (PAXX), a modulator of retroviral infection (MRI), and DNA ligase 4 (LIG4) [3,4,5,11]. There are additional factors that are sometimes optional for NHEJ, including Artemis with nuclease activity required for processing hairpin-sealed DNA ends and overhangs [2,5,12].
One type of DDR pathway acts downstream of the ataxia telangiectasia mutated (ATM) protein kinase, which is activated by DSBs and then phosphorylates multiple substrates, including NHEJ and DDR factors. ATM phosphorylates histone H2AX, which in turn recruits the mediator of DNA damage checkpoint 1 (MDC1) and facilitates the accumulation of really interesting new gene (RING) finger motif (RNF) 8 and RNF168 ubiquitin ligases, and then the p53-binding protein (53BP1). The phosphorylation of H2AX is related to the acetylation of histones, including histone H3K9. In particular, histone acetylation relies on ATM-dependent H2AX phosphorylation and SWI/SNF chromatin remodeling factors [13]. The acetylation of histone H3K9 is mediated by GCN5 and PCAF [13,14].
There is a complex genetic interaction associated with the functional redundancy between NHEJ factors [3,4], including the following pairs: DNA-PKcs/XLF [5,15,16,17], PAXX/XLF [18,19,20,21,22,23,24], and MRI/XLF [25,26]. Moreover, there is a genetic interaction between NHEJ and DDR pathway factors ATM/XLF and H2AX/XLF [27], MDC1/XLF [28], RNF8/XLF and RNF168/XLF [29], 53BP1/XLF [30,31], and others [3,4]. Moreover, acetyltransferases GCN5 and PCAF are redundant in promoting histone H3 lysine K9 acetylation [32].
General control nondepressible 5 (GCN5) acetyltransferase is also known as lysine acetyltransferase (KAT) 2A. The germline inactivation of GCN5 in mice resulted in early embryonic lethality due to the role of the protein in neurogenesis [32]. GCN5 is functionally redundant with another acetyltransferase, KAT2B, also referred to as p300/CBP-associated factor (PCAF). While the inactivation of the Pcaf gene in mice has no detectable phenotype, the double knockout of Gcn5/Pcaf genes resulted in even earlier embryonic lethality than that in Gcn5−/− mice [32]. Because histone H3K9 acetylation works downstream of ATM and H2AX in DDR [13], one could propose that GCN5, PCAF, or both enzymes are required for lymphocyte development in vivo. However, the embryonic lethality of Gcn5−/− and Gcn5−/−Pcaf−/− mice [32] challenged studies. To overcome the obstacle, we developed a complex mouse model when Pcaf gene was germline-inactivated [32] while floxed Gcn5 gene [33] was conditionally inactivated in B-cell lineages by CRE recombinase expressed under Cd19 promoter [34]. To sort out CRE-positive and -negative cells, we used Rosa26-stop-YFP knockin, which only expressed YFP following CRE activation [35].
Here, we found that GCN5 and PCAF acetyltransferases are functionally redundant during early B-cell maturation, while GCN5 is required for robust CSR.
2. Materials and Methods
2.1. Mice
Gcn5f/f [33], Pcaf+/− [32], Cd19Cre+ [34] (no. 006785; The Jackson Laboratory, Bar Harbor, ME, USA), Rosa26-stop-YFP+ [35] (no. 006148, The Jackson Laboratory, Bar Harbor, ME, USA), and Aid−/− [36] mice were previously described. Mice used for experiments were between 8 and 12 weeks of age. All experiments were performed in compliance with the Danish Working Environment Authority, the Danish Animal Experiment Inspectorate, the Department of Experimental Medicine (University of Copenhagen, Copenhagen, Denmark), and the Animal Resources Care Facility of Norwegian University of Science and Technology (NTNU, Trondheim, Norway).
2.2. Flow Cytometry
Flow cytometry experiments were performed as we described earlier [25,28,37,38,39]. In particular, we used fluorescent antibodies recognizing the proteins described below. B220 (PE-CF594, FITC, Alexa Fluor 700; all clone RA3-6B2, BD Bioscience, Franklin Lakes, NJ, USA). IgM (PerCP-eFluor 710, APC-eFluor 780, APC, FITC; all clone II/41, eBioscience, Santa Clara, CA, USA). IgG1 (PE, MOPC-21, Biolegend, SanDiego, CA, USA). IgG3 (PE, MG3-35, Biolegend, San Diego, CA, USA). CD3-APC (Biolegends, USA, #100312). CD19 (Alexa Fluor 700, APC eFluor 780, both clone 1D3, eBioscience, Santa Clara, CA, USA). CD43 (APC and PE-Cy7, both clones S7, BD Bioscience, Franklin Lakes, NJ, USA).
2.3. Class Switch Recombination
The CSR was performed as we described earlier [15,27,38,39,40,41,42].
2.4. Western Blot
The Western blot procedures were performed as we described earlier [28,37,38,43]. Briefly, cells were lysed for 30 min on ice in a radioimmunoprecipitation assay (RIPA) buffer (Sigma Aldrich, St. Louis, MO, USA, #R0278) supplemented with cOmplete™ EDTA-free Protease Inhibitor Cocktail (Sigma Aldrich, #11873580001). Proteins were analyzed by 4%–12% Bis-Tris NuPAGE gels (Invitrogen, Carlsbad, CA, USA, #NP0322), transferred to PDVF membranes (GE Healthcare, Boston, MA, USA, #GE10600023), and probed with indicated antibodies. Rat anti-AID (1:500, Active Motif, Carlsbad, CA, USA, #39886); mouse anti-GCN5, clone A-11 (1:500, Santa Cruz Biotechnology, Dallas, TX, USA, #sc-365321); rabbit anti-PCAF, clone C14G9 (1:1000, Cell Signaling Technology, Leiden, The Netherlands, #3378); rabbit antihistone H3 (1:1000, Abcam, Cambridge, UK, #ab1791); rabbit antihistone H3 acetyl K9 (1:500, Abcam, Cambridge, UK, #ab32129).
2.5. Statistics
We performed statistical analyses with one-way ANOVA using GraphPad Prism 8.0.1.244 (San Diego, CA, USA). In the tests, p values less than 0.05 were defined as significant, i.e., * p < 0.05; ** p < 0.01; *** p < 0.001; and **** p < 0.0001.
3. Results
3.1. Generation of Mice Lacking GCN5 and PCAF in B Cells
The combined inactivation of Gcn5 and Pcaf genes in mice results in embryonic lethality [32]. To overcome this challenge, we designed a complex genetic model when the floxed Gcn5 gene is conditionally inactivated in B-cell lineages by the CRE enzyme under the Cd19 promoter (Cd19Cre+) [34]. To sort out cells with activated CRE, we used a model with knocked-in yellow fluorescent protein gene (YFP) into the ROSA-26 locus. YFP is inactive until CRE removes the “STOP” signal (Rosa-26-YFP+) [35]. Thus, we obtained Gcn5f/fPcaf−/−Cd19+/CreYFP+ mice and simpler controls. Further in the text, we skip Cd19+/Cre and YFP+ for simplicity in most cases, and refer to mice on the basis of the status of Gcn5 and Pcaf genes, i.e., as Gcn5f/fPcaf−/−, Gcn5f/f, Pcaf−/−, and WT. When the CRE is active and describing sorted B cells, we indicate Gcn5−/−, a knockout status of the gene. The lack of GCN5 and PCAF, and H3K9 acetylation, were validated using Western blot (Figure S1).
3.2. Mice Lacking GCN5 and PCAF in B Cells Possess Small Spleens
We obtained mice with the germline inactivation of Pcaf gene and conditional inactivation of Gcn5 in B cells under the Cd19 promoter (Figure 1). The germline inactivation of Pcaf gene alone had no detectable effect on mouse development, in line with a previous observation [32]. The conditional inactivation of the Gcn5 gene in B cells had no visible effect on sizes of WT and Pcaf-deficient mice, which were 15 to 19 g on average (p > 0.1433) (Figure 1A). However, the inactivation of Gcn5 resulted in smaller spleens in mice (Gcn5−/−, 54 mg) when compared to WT (69 mg) and Pcaf−/− (72 mg) mice. The combined inactivation of Pcaf and Gcn5 in B cells resulted in even smaller spleens (Gcn5−/−Pcaf−/−, 29 mg, p < 0.0001). The spleens of mice without CRE activity with the Gcn5 gene being floxed and functional (Gcn5f/fPcaf−/−, 67 mg) were comparable in size to the ones of WT mice (Figure 1B,C).
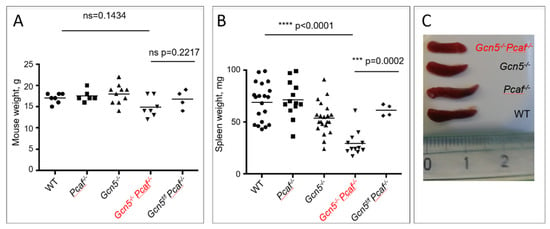
Figure 1.
Generation of mice with germline inactivation of Pcaf and conditional inactivation of Gcn5 in B-cell lineages. (A) Sizes of 8-week-old mice of indicated genotype were similar (p > 0.1433). (B) Size of spleens from mice of indicated genotypes. WT vs. Pcaf−/−, n.s. p = 0.9559; WT vs. Gcn5−/−, * p = 0.0192; WT vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Pcaf−/− vs. Gcn5−/−, * p = 0.0119; Pcaf−/− vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Gcn5−/− vs. Gcn5−/−Pcaf−/−, *** p = 0.0008. (C) Example of spleens of indicated genotypes. Gcn5−/− indicates Cd19Cre-dependent inactivation of Gcn5 in B-cell lineages.
3.3. Mice Lacking GCN5 and PCAF in B Cells Possess Delayed B Lymphocyte Development
To detect mature Gcn5−/−Pcaf−/− B cells, we identified B220+IgM+ cells in the spleen using flow cytometry (Figure 2A,B). The inactivation of the Pcaf gene alone did not affect B-cell proportions in the spleen (58%) when compared to WT mice (52%, p = 0.4777) (Figure 2A). The inactivation of Gcn5 alone resulted in an insignificant reduction in mature splenocytes when compared to WT mice (Gcn5−/−, 46%, p < 0.2532), although Gcn5−/−Pcaf−/− mice had significantly less B-cell frequency in the spleen (21%, p < 0.0001). Similarly, the number of Gcn5−/−Pcaf−/− B splenocytes was the lowest (3, 4 million), while the number of Gcn5−/− B cells (27 million) was also reduced when compared to Pcaf−/− (54 million, ** p = 0.0065) and WT (44 million, * p = 0.0214) controls (Figure 2B).
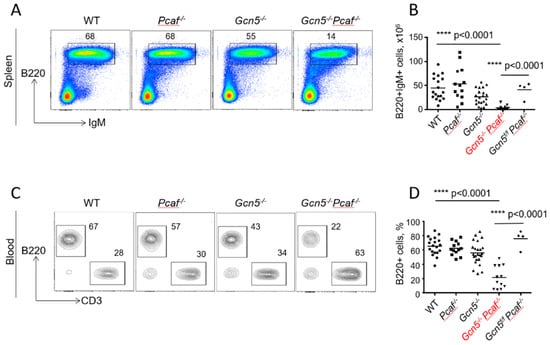
Figure 2.
Reduced levels of mature B lymphocytes in spleens and blood of 8–12-week-old mice of indicated genotypes. (A) Proportions of B220+IgM+ mature B cells in spleen. WT vs. Pcaf−/−, n.s. p = 0.4777; WT vs. Gcn5−/−, ns, p = 0.2532; WT vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Pcaf−/− vs. Gcn5−/−, * p = 0.0119; Pcaf−/− vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Gcn5−/− vs. Gcn5−/−Pcaf−/−, *** p < 0.0001. (B) Summary of experiments as shown in (A), number of cells. WT vs. Pcaf−/−, n.s., p = 0.9212; WT vs. Gcn5−/−, * p = 0.0214; WT vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Pcaf−/− vs. Gcn5−/−, ** p = 0.0053; Pcaf−/− vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Gcn5−/− vs. Gcn5−/−Pcaf−/−, * p < 0.0269. (C) Proportions of B220+ B cells and CD3+ T cells in blood. (D) Summary of several experiments from (C) reflecting proportions of B cells in blood. WT vs. Pcaf−/−, n.s. p = 0.9196; WT vs. Gcn5−/−, n.s., p = 0.1204; WT vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Pcaf−/− vs. Gcn5−/−, n.s., p = 0.5398; Pcaf−/− vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Gcn5−/− vs. Gcn5−/−Pcaf−/−, *** p < 0.0001. Gcn5−/− and Gcn5−/−Pcaf−/− indicate Cd19Cre-dependent inactivation of Gcn5 in B-cell lineages.
3.4. Inactivation of Gcn5 and Pcaf Results in a Reduced Proportion of B Cells in the Blood
To detect mature B cells in the blood, we used B220 markers (Figure 2C,D). The inactivation of Pcaf alone resulted in 62% of B cells after red blood cells had been lysed, which was comparable to WT mice with 65% of B cells in the blood (p = 0.92). The inactivation of the Gcn5 gene alone resulted in a modest reduction of B-cell proportion to 56% (p = 0.12), while the combined inactivation of Gcn5 and Pcaf led to even lower B-cell levels in the blood (22%, p < 0.0001). The levels of B cells in the blood of control mice without CRE recombinase expression when the Gcn5 gene was functional (Gcn5f/fPcaf−/−) were comparable to those of WT mice (78%). GCN5 and PCAF are thus both required and functionally redundant for B-cell development in mice.
One reason for the low Gcn5−/−Pcaf−/− B-cell count in spleen and blood in mice could be cell death following the normal development of B cells in bone marrow and migration to the periphery. Another option could be the blocked or delayed maturation of B cells in bone marrow during the earlier developmental stages. To test the latter possibility, we analyzed B cells in the bone marrow of the mice (Figure 3).
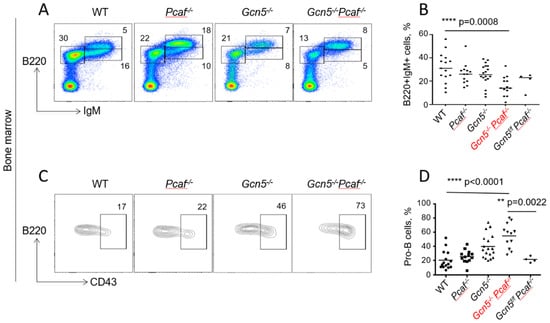
Figure 3.
Delayed development of GCN5/PCAF-deficient B cells in bone marrow. Flow cytometry analyses of developing B lymphocytes in bone marrow of 8–12-week-old mice indicated genotypes. (A) Examples of B220+IgM− (pro-B and pre-B), B220+IgM+low (immature B), and B220+IgM+high (mature B) cell populations. (B) Summary of several experiments from (A) indicating B220+IgM+ cells. WT vs. Pcaf−/−, n.s. p = 0.6445; WT vs. Gcn5−/−, n.s., p = 0.4432; WT vs. Gcn5−/−Pcaf−/−, *** p = 0.0008; Pcaf−/− vs. Gcn5−/−, n.s., p = 0.9997; Pcaf−/− vs. Gcn5−/−Pcaf−/−, * p < 0.0138; Gcn5−/− vs. Gcn5−/−Pcaf−/−, * p < 0.0211 (C) CD43+ (pro-B cells) and CD43- (pre-B cells) gated from B220+IgM− populations in (A). (D) Summary of several experiments detecting B220+IgM−CD43+ (pro-B) cells. WT vs. Pcaf−/−, n.s. p = 0.7825; WT vs. Gcn5−/−, ** p = 0.0022; WT vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Pcaf−/− vs. Gcn5−/−, n.s., p = 0.0505; Pcaf−/− vs. Gcn5−/−Pcaf−/−, **** p < 0.0001; Gcn5−/− vs. Gcn5−/−Pcaf−/−, * p < 0.0322 Gcn5−/− and Gcn5−/−Pcaf−/− indicate Cd19Cre-dependent inactivation of Gcn5 in B-cell lineages.
3.5. Inactivation of Gcn5 and Pcaf Results in Accumulation of Pro-B Cells in Bone Marrow
To characterize B-cell maturation in bone marrow, we followed the expression of B220 (B220+IgM−) and IgM (B220+IgM+) on the lymphocyte surface. The inactivation of Gcn5 or Pcaf resulted in an insignificant decline in the B220+IgM+ population (26%, 5–6 million) when compared to WT (32%, 8 million, p = 0.44) (Figure 3 A,B). The combined inactivation of Gcn5 and Pcaf resulted in an additional reduction in mature B cells in bone marrow (3 million, 16%) (Figure 3A,B).
We further focused on B220+IgM− populations by determining the CD43+ (pro-B cells) and CD43- (pre-B cells). The proportion of early-stage pro-B cells increased from WT mice (20% on average) and Pcaf−/− mice (26%, p = 0.78) to Gcn5−/− (39%, ** p = 0.0022) to Gcn5−/−Pcaf−/− (55%, p < 0.0001) (Figure 3C,D). However, the number of pro-B cells in bone marrow, estimated using our method of cell extraction, was rather stable, with 0.8 million for WT (n = 13) and about 1.3 million for Gcn5−/− (n = 13), Pcaf−/− (n = 13), and Gcn5−/−Pcaf−/− (n = 5) cells (n.s., p > 0.3830). This suggested that the proportion of Gcn5−/−Pcaf−/− pro-B cells was increased because the total number of mature B cells was reduced (Supplemental Figure S3 and 3B).
We concluded that GCN5 and PCAF are required for the maturation of B cells from the pro-B to the pre-B cell stage, and later to mature B cells.
3.6. GCN5 Is Required for Robust Class Switch Recombination
The CSR relies on the ATM-dependent DDR [2,4,5,27]. Because H3K9 acetylation works downstream of H2AX phosphorylation, and GCN5/PCAF might work downstream of ATM/ATR/DNA-PKcs, we tested if the CSR depends on GCN5 and PCAF (Figure 4). We purified B splenocytes from 8- to 12-week-old mice and stimulated the CSR from IgM to IgG3 using established protocols [40,42]. We focused on matched pairs of Gcn5f/f (the functional equivalent of WT cells) and Gcn5−/−, as well as Gcn5f/fPcaf−/− (the functional equivalent of Pcaf−/−) and Gcn5−/−Pcaf−/− cells. We used Aid−/− cells as a CSR-deficient control to detect an experimental background (Figure 4). The inactivation of Pcaf alone had no effect on CSR levels (WT vs. Gcn5f/fPcaf−/−, p > 0.96). On the other hand, the inactivation of Gcn5 resulted in a reduction in CSR from about 14% in WT and Gcn5f/f cells to 6% in Gcn5−/− cells, * p < 0.0008 (Figure 4). The combined deletion of Pcaf and Gcn5 resulted in a similar reduction from 12% in Gcn5f/fPcaf−/− cells to 6% in Gcn5−/−Pcaf−/− cells, ** p = 0.0080. We concluded that GCN5 is required for robust CSR to IgG3 because the additional inactivation of Pcaf did not affect CSR levels when compared to Gcn5−/− and Gcn5−/−Pcaf−/− B cells, p > 0.9999 (Figure 4).
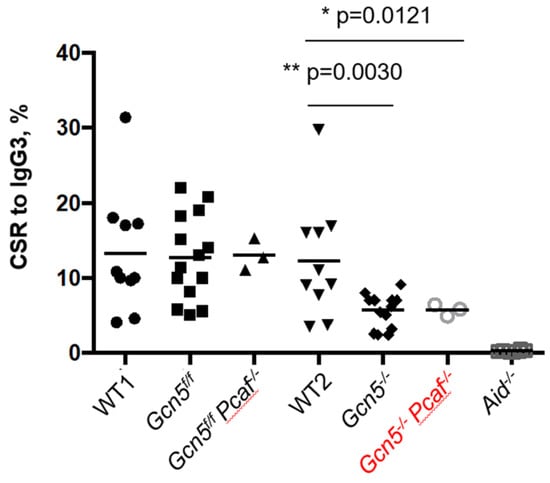
Figure 4.
Class switch recombination of stimulated mature B cells from IgM to IgG3. Gcn5−/− and Gcn5−/−Pcaf−/− indicate Cd19Cre-dependent inactivation of Gcn5 in B-cell lineages. Levels of CSR for WT, Gcn5f/f, Gcn5f/fPcaf−/− were not significantly different (n.s.); levels for Gcn5−/− and Gcn5−/−Pcaf−/− were lower than those of the three former groups (** p = 0.0030 and * p = 0.0121, correspondently, when compared to WT2); Gcn5−/− vs. Gcn5−/−Pcaf−/− levels were similar (n.s., p = 0.9997), and Aid−/− only had background levels, WT2 vs. Aid−/− is (**** p > 0.0001).
4. Discussion
Both GCN5 and PCAF are involved in chromatin modification and DDR response, which made them relevant candidates to facilitate lymphocyte development [13,14]. One challenge was the lack of a relevant in vivo model because GCN5 and PCAF have certain redundant functions in acetylating H3K9, and because the germline inactivation of Gcn5 results in early embryonic lethality in mice [32]. Here, we generated and analyzed a complex mouse model that allowed for studying of double-deficient Gcn5−/−Pcaf−/− B cells development in vivo and ex vivo. We used a germline knockout of Pcaf [32], a conditional knockout of Gcn5f/f, a knockin of CRE recombinase expressed under the B-cell-specific Cd19 promoter [34], and a knockin of YFP to track the activity of CRE recombinase [35].
For such a complex mouse model (Gcn5f/fPcaf−/−Cd19+/creRosa-26-YFP+), multiple controls were used. In one line of the controls, mice lacking PCAF and having a floxed Gcn5 gene but expressing no CRE recombinase were considered (Gcn5f/fPcaf−/−Rosa-26-YFP+, Figure S2). The GCN5-deficient and GCN5/PCAF double-deficient B cells possessed developmental delay, with lower levels of mature B cells in thee spleen and blood, and the accumulation of progenitor B cells in bone marrow (Figure 1, Figure 2, Figure 3 and Figure 4). Control mice without CRE expression, on the other hand, demonstrated WT levels of B-cell development in all groups, i.e., WT levels of B220+IgM+ mature B cells in the spleen (Figure S2A,B) and blood (Figure S2C,D). In addition, these mice possessed high levels of B220+IgM+ cells (Figure S2E,F), and stable and low levels of pro-B cells (B220+IgM−CD43+) in bone marrow (Figure S2G,H).
Mice lacking PCAF and with conditional knockout of Gcn5 in B cells were alive and resembled WT littermates (Figure 1A). One clear feature that the Gcn5f/fPcaf−/−Cd19+/creRosa-26-YFP+ mice had was a small spleen (Figure 1B,C), which was also the case in mice lacking only Gcn5 in B cells. The small spleen could indicate a defect in B-cell development, and we indeed found low numbers of mature B cells in the spleen, blood, and bone marrow. One could propose that mature B cells lacking GCN5 or both GCN5 and PCAF possess low proliferation speed or tend to trigger apoptosis. Alternatively, GCN5 and PCAF might be required for the V(D)J recombination. This option could be tested by, for example, using vAbl pre-B cell lines, as we and others did before [4,15,18,23,27,28,29,30,31]. Another intriguing question is whether the physical presence or enzymatic activity of GCN5 and PCAF are required for the observed phenotypes, i.e., abrogated B-cell maturation and reduced levels of CSR. To investigate this question, one could use specific inhibitors of GCN5 and PCAF enzymes, or enzyme-dead mutations introduced to the Gcn5 and Pcaf genes.
The inactivation of Gcn5 in murine B cells also resulted in reduced lymphomagenesis in mice overexpressing MYC oncoprotein [44]. Our findings further highlight this observation, suggesting that GCN5 and potentially also PCAF enzymes are attractive targets for cancer therapy [44].
CSR levels were reduced in B cells lacking GCN5 (Figure 4). The challenge in this set of experiments was that mice of the Gcn5f/fPcaf−/−Cd19+/creRosa-26-YFP+ genotype were rather rare and possessed a very low number of suitable B splenocytes (Figure 1 and Figure 2). Although our data on IgG3 are sufficient, one could extend the study by generating knockout cell lines lacking GCN5 and PCAF, and suitable for CSR. One possible model system is CH12F3 cells capable of supporting CSR to IgA [45], which were used for this kind of experiment [24,37,42]. The CSR itself is a complex multistage process. Generating relevant cell lines also provides tools to determine specific stages of CSR affected in GCN5-deficient mice, i.e., germline transcription, AID recruitment, generation of DSBs, or DNA repair.
5. Conclusions
Acetyltransferases GCN5 and PCAF possess redundant functions in B-cell maturation. GCN5 is required for robust class switch recombination ex vivo.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biom12010061/s1, Figure S1: Detection of GCN5, PCAF and histones; Figure S2: Detection of developing B cells in mice of indicated genotypes using flow cytometry; Figure S3: Count of WT, Pcaf-deficient, Gcn5-deficient, and Gcn5/Pcaf double-deficient pro-B cells in bone marrow.
Author Contributions
Conceptualization, methodology, validation, formal analysis, investigation, resources, and data curation, J.A.D., D.S. and V.O.; writing—original draft preparation, V.O.; writing—review and editing, V.O. and J.A.D.; supervision, J.A.D.; project administration, J.A.D. and V.O.; funding acquisition, J.A.D. and V.O. All authors have read and agreed to the published version of the manuscript.
Funding
This research was mainly supported by the Novo Nordisk Foundation Grant NNF14CC0001 to J.A.D. In addition, V.O. was supported by The Research Council of Norway (nos. 249774, 270491, and 291217), the Norwegian Cancer Society (no. 182355), The Health Authority of Central Norway (nos. 13477 and 38811), and The Outstanding Academic Fellow Program at NTNU 2017–2021. The final part of the work was supported by the Stiftelsen Kristian Gerhard Jebsen (grant SKGJ-MED-019).
Institutional Review Board Statement
All experiments were performed in compliance with the Danish Working Environment Authority, the Danish Animal Experiment Inspectorate, the Department of Experimental Medicine (University of Copenhagen), and the Animal Resources Care Facility of Norwegian University of Science and Technology (NTNU, Trondheim, Norway), FOTS #11931 (2017) and FOTS #13405 (2018).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the main text, figures, tables and Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Lescale, C.; Deriano, L. The RAG recombinase: Beyond breaking. Mech. Ageing Dev. 2017, 165, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Alt, F.W.; Zhang, Y.; Meng, F.L.; Guo, C.; Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013, 152, 417–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.S.; Lee, B.J.; Zha, S. The recent advances in non-homologous end-joining through the lens of lymphocyte development. DNA Repair (Amst.) 2020, 94, 102874. [Google Scholar] [CrossRef]
- Castaneda-Zegarra, S.; Fernandez-Berrocal, M.; Tkachov, M.; Yao, R.; Upfold, N.L.E.; Oksenych, V. Genetic interaction between the non-homologous end-joining factors during B and T lymphocyte development: In vivo mouse models. Scand. J. Immunol. 2020, 92, e12936. [Google Scholar] [CrossRef]
- Kumar, V.; Alt, F.W.; Oksenych, V. Functional overlaps between XLF and the ATM-dependent DNA double strand break response. DNA Repair (Amst.) 2014, 16, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Frock, R.L.; Sadeghi, C.; Meng, J.; Wang, J.L. DNA End Joining: G0-ing to the Core. Biomolecules 2021, 11, 1487. [Google Scholar] [CrossRef]
- Bordin, D.L.; Lirussi, L.; Nilsen, H. Cellular response to endogenous DNA damage: DNA base modifications in gene expression regulation. DNA Repair (Amst.) 2021, 99, 103051. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Sundaravinayagam, D.; Di Virgilio, M. Charting a DNA Repair Roadmap for Immunoglobulin Class Switch Recombination. Trends Biochem. Sci. 2021, 46, 184–199. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Ba, Z.; Kyritsis, N.; Casellas, R.; Alt, F.W. Fundamental roles of chromatin loop extrusion in antibody class switching. Nature 2019, 575, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Ba, Z.; Liang, Z.; Dring, E.W.; Hu, H.; Lou, J.; Kyritsis, N.; Zurita, J.; Shamim, M.S.; et al. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature 2019, 573, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Menolfi, D.; Zha, S. DNA-PKcs kinase activity orchestrates both end-processing and end-ligation. Trends Cell Biol. 2021, 12, 1–3. [Google Scholar] [CrossRef]
- Ragunathan, K.; Upfold, N.L.E.; Oksenych, V. Interaction between Fibroblasts and Immune Cells Following DNA Damage Induced by Ionizing Radiation. Int. J. Mol. Sci. 2020, 21, 8635. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, J.H.; Kim, S.J.; Kwon, S.J.; Kwon, J. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J. 2010, 29, 1434–1445. [Google Scholar] [CrossRef]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef]
- Oksenych, V.; Kumar, V.; Liu, X.; Guo, C.; Schwer, B.; Zha, S.; Alt, F.W. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proc. Natl. Acad. Sci. USA 2013, 110, 2234–2239. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.; Bjoras, M.; Daniel, J.A.; Alt, F.W.; Oksenych, V. Synthetic lethality between murine DNA repair factors XLF and DNA-PKcs is rescued by inactivation of Ku70. DNA Repair (Amst.) 2017, 57, 133–138. [Google Scholar] [CrossRef]
- Xing, M.; Oksenych, V. Genetic interaction between DNA repair factors PAXX, XLF, XRCC4 and DNA-PKcs in human cells. FEBS Open Bio 2019, 9, 1315–1326. [Google Scholar] [CrossRef] [Green Version]
- Abramowski, V.; Etienne, O.; Elsaid, R.; Yang, J.; Berland, A.; Kermasson, L.; Roch, B.; Musilli, S.; Moussu, J.P.; Lipson-Ruffert, K.; et al. PAXX and Xlf interplay revealed by impaired CNS development and immunodeficiency of double KO mice. Cell Death Differ. 2018, 25, 444–452. [Google Scholar] [CrossRef] [Green Version]
- Balmus, G.; Barros, A.C.; Wijnhoven, P.W.; Lescale, C.; Hasse, H.L.; Boroviak, K.; le Sage, C.; Doe, B.; Speak, A.O.; Galli, A.; et al. Synthetic lethality between PAXX and XLF in mammalian development. Genes Dev. 2016, 30, 2152–2157. [Google Scholar] [CrossRef] [Green Version]
- Castaneda-Zegarra, S.; Xing, M.; Gago-Fuentes, R.; Saeterstad, S.; Oksenych, V. Synthetic lethality between DNA repair factors Xlf and Paxx is rescued by inactivation of Trp53. DNA Repair (Amst.) 2019, 73, 164–169. [Google Scholar] [CrossRef]
- Lescale, C.; Lenden Hasse, H.; Blackford, A.N.; Balmus, G.; Bianchi, J.J.; Yu, W.; Bacoccina, L.; Jarade, A.; Clouin, C.; Sivapalan, R.; et al. Specific Roles of XRCC4 Paralogs PAXX and XLF during V(D)J Recombination. Cell Rep. 2016, 16, 2967–2979. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shao, Z.; Jiang, W.; Lee, B.J.; Zha, S. PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Hung, P.J.; Chen, B.R.; George, R.; Liberman, C.; Morales, A.J.; Colon-Ortiz, P.; Tyler, J.K.; Sleckman, B.P.; Bredemeyer, A.L. Deficiency of XLF and PAXX prevents DNA double-strand break repair by non-homologous end joining in lymphocytes. Cell Cycle 2017, 16, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Alt, F.W.; Frock, R.L. PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc. Natl. Acad. Sci. USA 2016, 113, 10619–10624. [Google Scholar] [CrossRef] [Green Version]
- Castaneda-Zegarra, S.; Zhang, Q.; Alirezaylavasani, A.; Fernandez-Berrocal, M.; Yao, R.; Oksenych, V. Leaky severe combined immunodeficiency in mice lacking non-homologous end joining factors XLF and MRI. Aging (Albany NY) 2020, 12, 23578–23597. [Google Scholar] [CrossRef]
- Hung, P.J.; Johnson, B.; Chen, B.R.; Byrum, A.K.; Bredemeyer, A.L.; Yewdell, W.T.; Johnson, T.E.; Lee, B.J.; Deivasigamani, S.; Hindi, I.; et al. MRI Is a DNA Damage Response Adaptor during Classical Non-homologous End Joining. Mol. Cell 2018, 71, 332–342. [Google Scholar] [CrossRef]
- Zha, S.; Guo, C.; Boboila, C.; Oksenych, V.; Cheng, H.L.; Zhang, Y.; Wesemann, D.R.; Yuen, G.; Patel, H.; Goff, P.H.; et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2011, 469, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Beck, C.; Castaneda-Zegarra, S.; Huse, C.; Xing, M.; Oksenych, V. Mediator of DNA Damage Checkpoint Protein 1 Facilitates V(D)J Recombination in Cells Lacking DNA Repair Factor XLF. Biomolecules 2019, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.R.; Wang, Y.; Shen, Z.J.; Bennett, A.; Hindi, I.; Tyler, J.K.; Sleckman, B.P. The RNF8 and RNF168 Ubiquitin Ligases Regulate Pro- and Anti-Resection Activities at Broken DNA Ends During Non-Homologous End Joining. DNA Repair (Amst.) 2021, 108, 103217. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, W.; Dubois, R.L.; Yamamoto, K.; Wolner, Z.; Zha, S. Overlapping functions between XLF repair protein and 53BP1 DNA damage response factor in end joining and lymphocyte development. Proc. Natl. Acad. Sci. USA 2012, 109, 3903–3908. [Google Scholar] [CrossRef] [Green Version]
- Oksenych, V.; Alt, F.W.; Kumar, V.; Schwer, B.; Wesemann, D.R.; Hansen, E.; Patel, H.; Su, A.; Guo, C. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc. Natl. Acad. Sci. USA 2012, 109, 2455–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Edmondson, D.G.; Evrard, Y.A.; Wakamiya, M.; Behringer, R.R.; Roth, S.Y. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat. Genet. 2000, 26, 229–232. [Google Scholar] [CrossRef]
- Lin, W.; Srajer, G.; Evrard, Y.A.; Phan, H.M.; Furuta, Y.; Dent, S.Y. Developmental potential of Gcn5(-/-) embryonic stem cells in vivo and in vitro. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2007, 236, 1547–1557. [Google Scholar] [CrossRef]
- Rickert, R.C.; Roes, J.; Rajewsky, K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997, 25, 1317–1318. [Google Scholar] [CrossRef]
- Srinivas, S.; Watanabe, T.; Lin, C.S.; William, C.M.; Tanabe, Y.; Jessell, T.M.; Costantini, F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001, 1, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Dewan, A.; Xing, M.; Lundbaek, M.B.; Gago-Fuentes, R.; Beck, C.; Aas, P.A.; Liabakk, N.B.; Saeterstad, S.; Chau, K.T.P.; Kavli, B.M.; et al. Robust DNA repair in PAXX-deficient mammalian cells. FEBS Open Bio 2018, 8, 442–448. [Google Scholar] [CrossRef]
- Gago-Fuentes, R.; Xing, M.; Saeterstad, S.; Sarno, A.; Dewan, A.; Beck, C.; Bradamante, S.; Bjoras, M.; Oksenych, V. Normal development of mice lacking PAXX, the paralogue of XRCC4 and XLF. FEBS Open Bio 2018, 8, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Zegarra, S.; Huse, C.; Rosand, O.; Sarno, A.; Xing, M.; Gago-Fuentes, R.; Zhang, Q.; Alirezaylavasani, A.; Werner, J.; Ji, P.; et al. Generation of a Mouse Model Lacking the Non-Homologous End-Joining Factor Mri/Cyren. Biomolecules 2019, 9, 798. [Google Scholar] [CrossRef] [Green Version]
- Starnes, L.M.; Su, D.; Pikkupeura, L.M.; Weinert, B.T.; Santos, M.A.; Mund, A.; Soria, R.; Cho, Y.W.; Pozdnyakova, I.; Kubec Hojfeldt, M.; et al. A PTIP-PA1 subcomplex promotes transcription for IgH class switching independently from the associated MLL3/MLL4 methyltransferase complex. Genes Dev. 2016, 30, 149–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, D.; Vanhee, S.; Soria, R.; Gyllenback, E.J.; Starnes, L.M.; Hojfeldt, M.K.; Pedersen, G.K.; Yuan, J.; Daniel, J.A. PTIP chromatin regulator controls development and activation of B cell subsets to license humoral immunity in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E9328–E9337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boboila, C.; Oksenych, V.; Gostissa, M.; Wang, J.H.; Zha, S.; Zhang, Y.; Chai, H.; Lee, C.S.; Jankovic, M.; Saez, L.M.; et al. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1). Proc. Natl. Acad. Sci. USA 2012, 109, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Gago-Fuentes, R.; Oksenych, V. Non-Homologous End Joining Factors XLF, PAXX and DNA-PKcs Maintain the Neural Stem and Progenitor Cell Population. Biomolecules 2020, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Farria, A.T.; Plummer, J.B.; Salinger, A.P.; Shen, J.; Lin, K.; Lu, Y.; McBride, K.M.; Koutelou, E.; Dent, S.Y.R. Transcriptional Activation of MYC-Induced Genes by GCN5 Promotes B-cell Lymphomagenesis. Cancer Res. 2020, 80, 5543–5553. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Kondo, S.; Sugai, M.; Nazarea, M.; Imamura, S.; Honjo, T. High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int. Immunol. 1996, 8, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).