
DNA End Joining: G0-ing to the Core


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
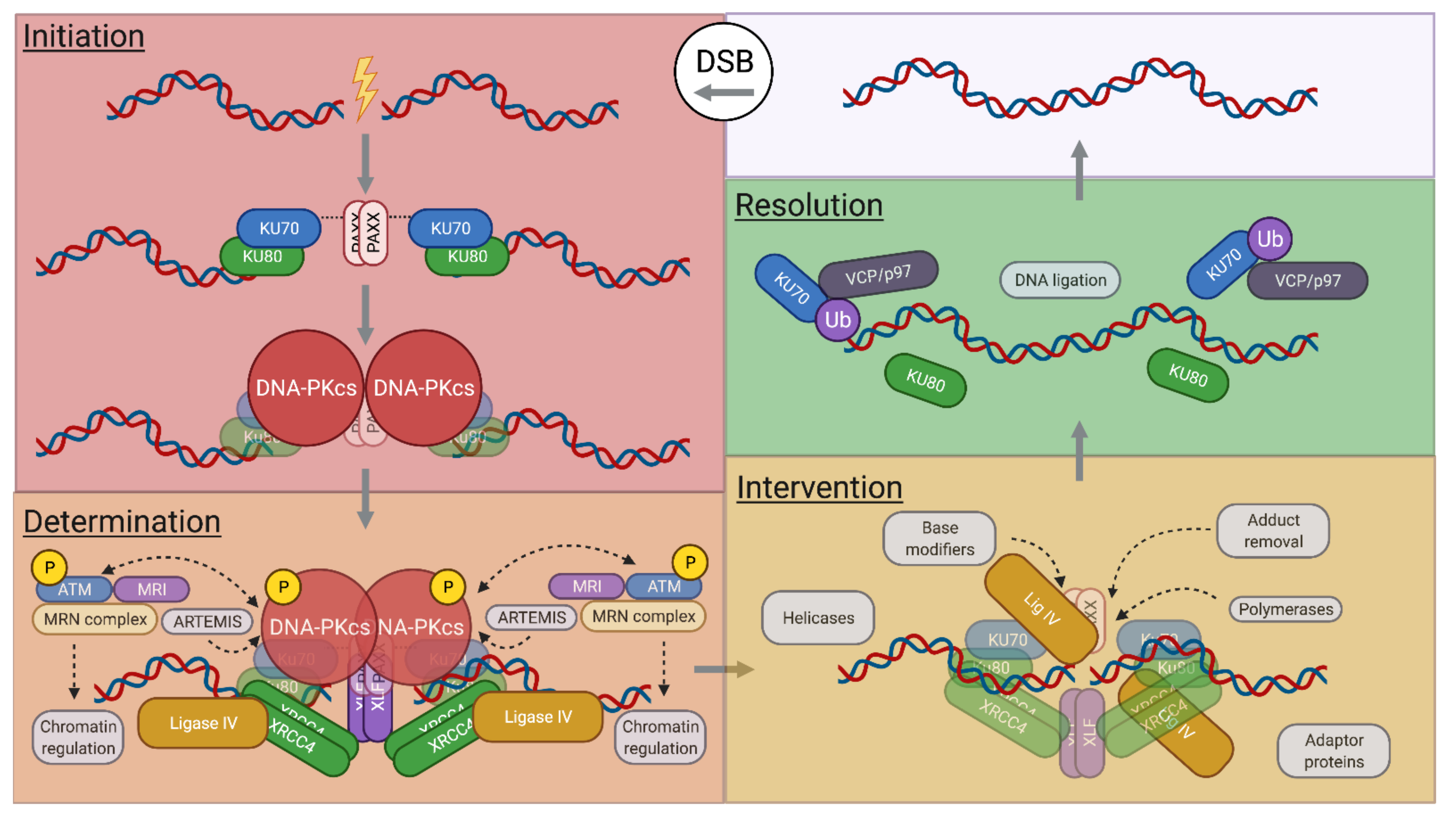
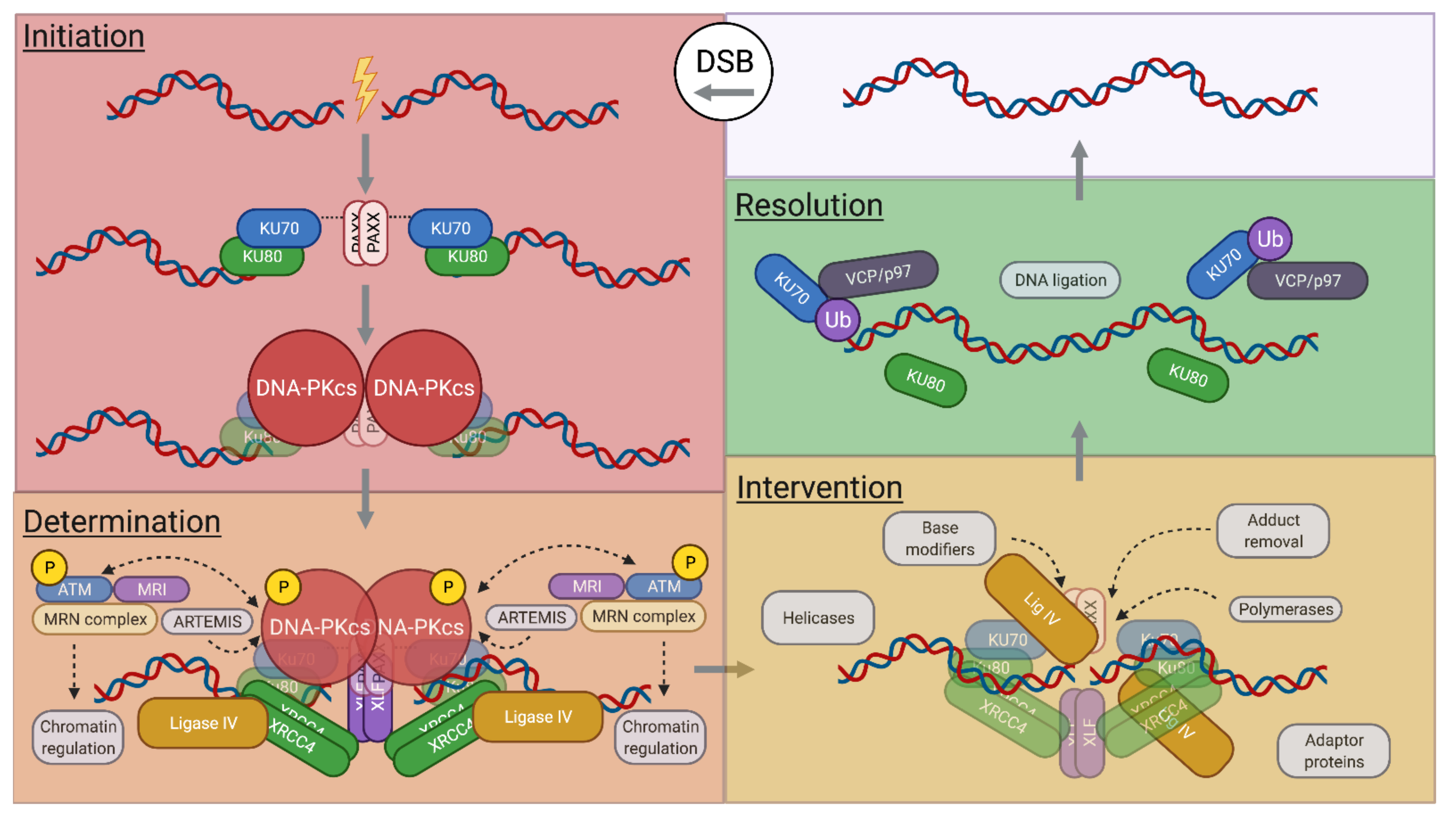
2. NHEJ Overview
2.1. NHEJ Initiation
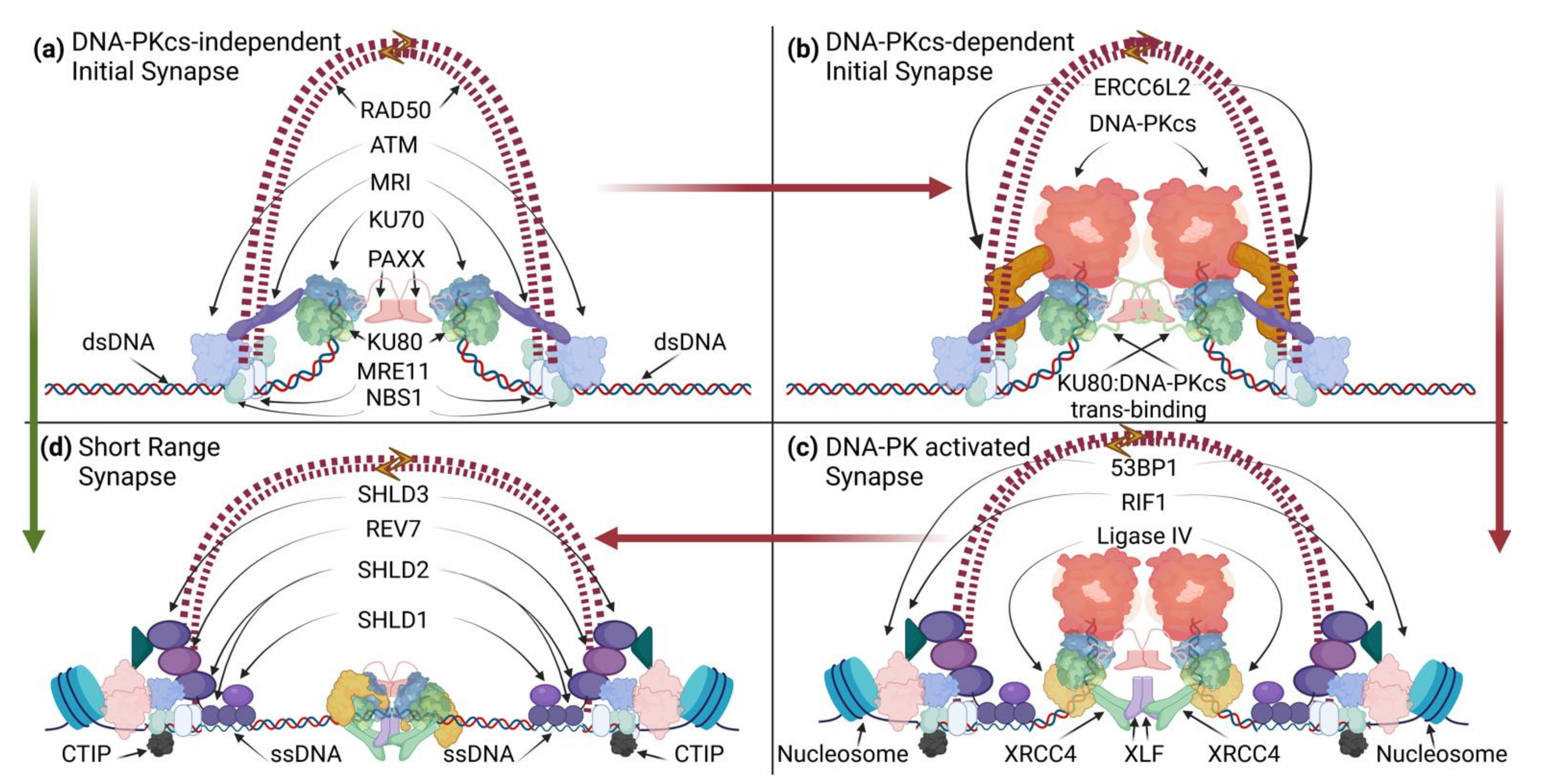
2.1.1. NHEJ “Long-Range” Synapsis
2.1.2. NHEJ Filaments
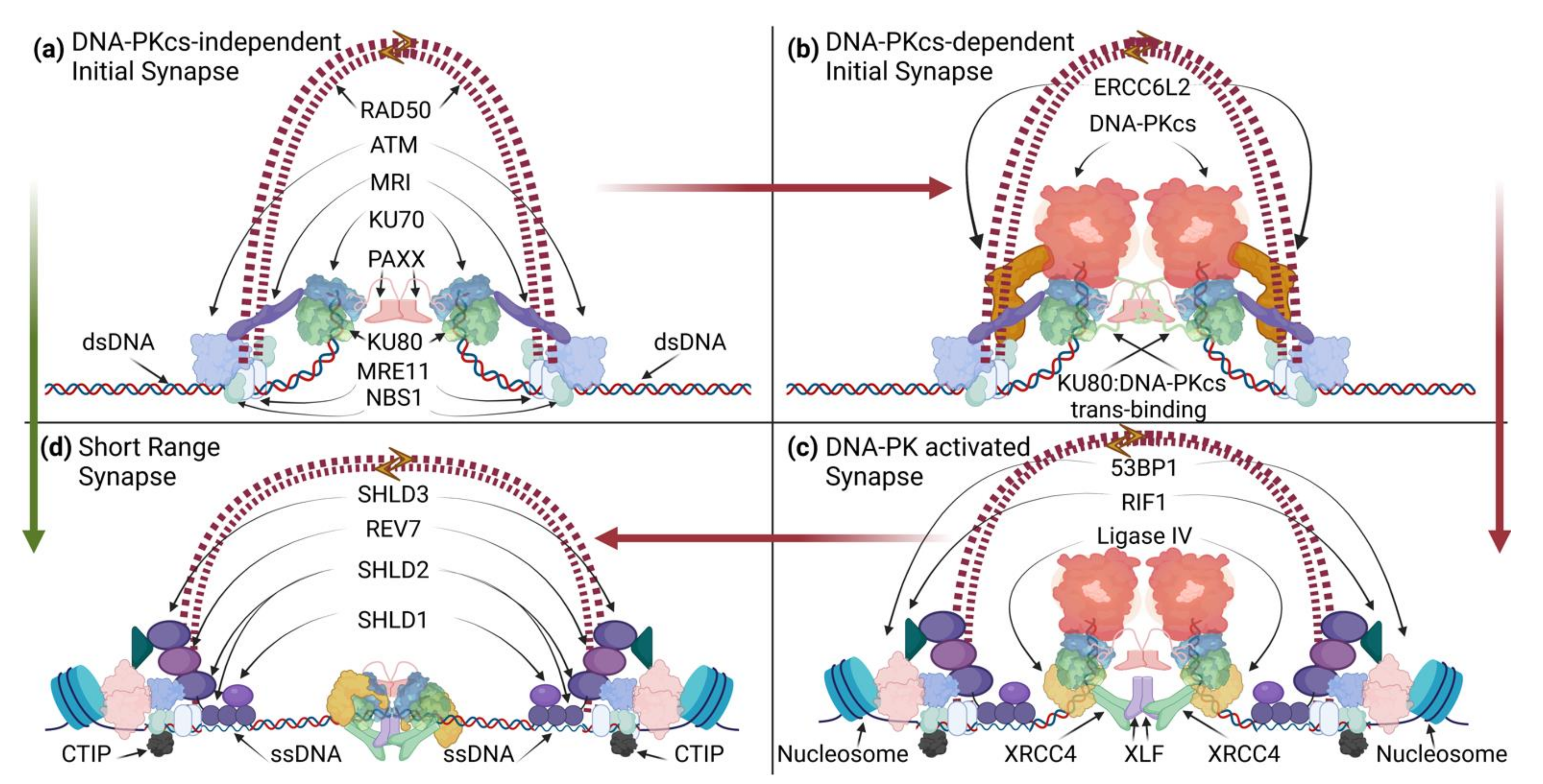
2.1.3. NHEJ/DDR-Mediated Tethering
2.2. NHEJ Determination
2.3. NHEJ Intervention
2.4. NHEJ Resolution
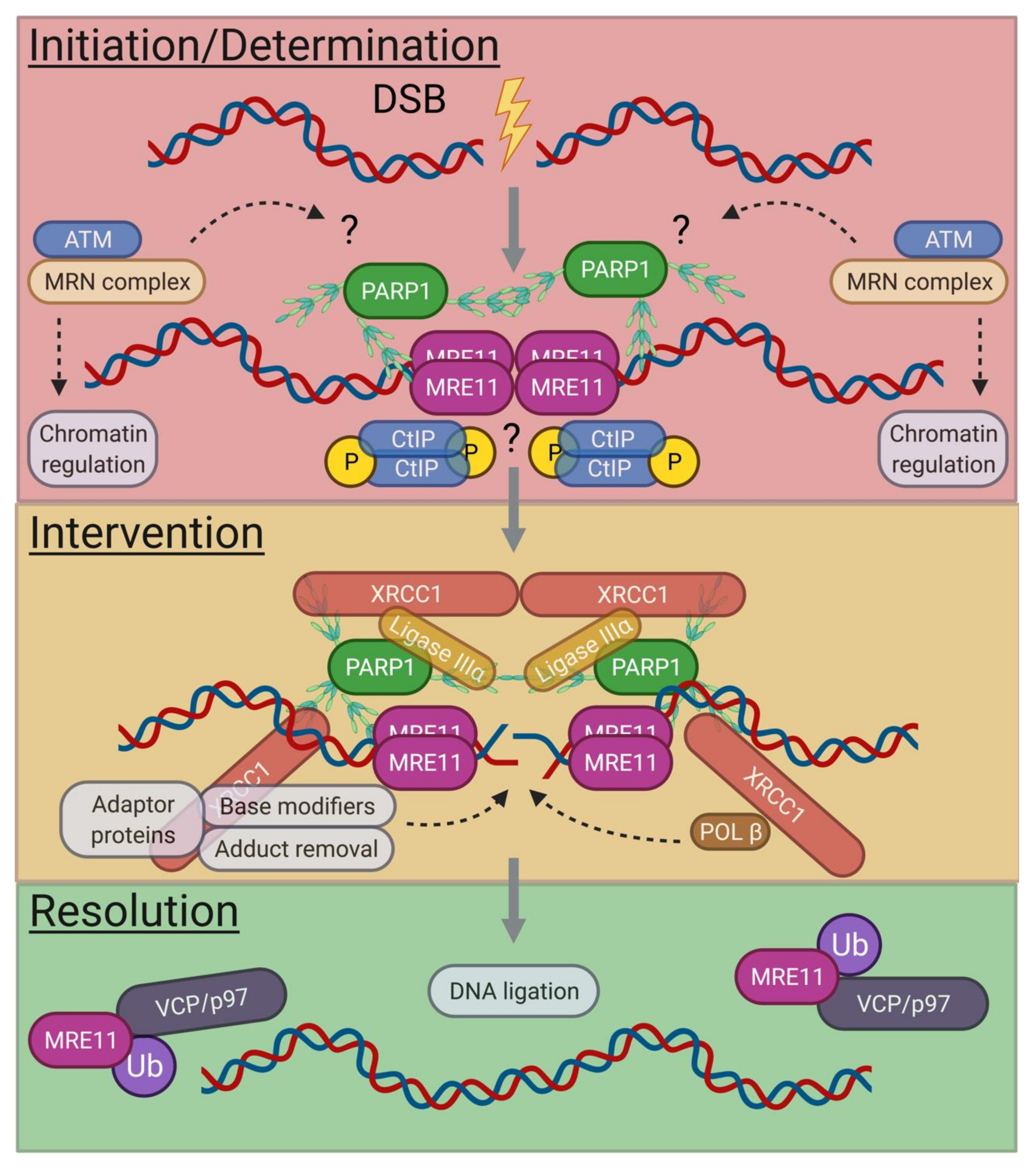
3. A-EJ Overview
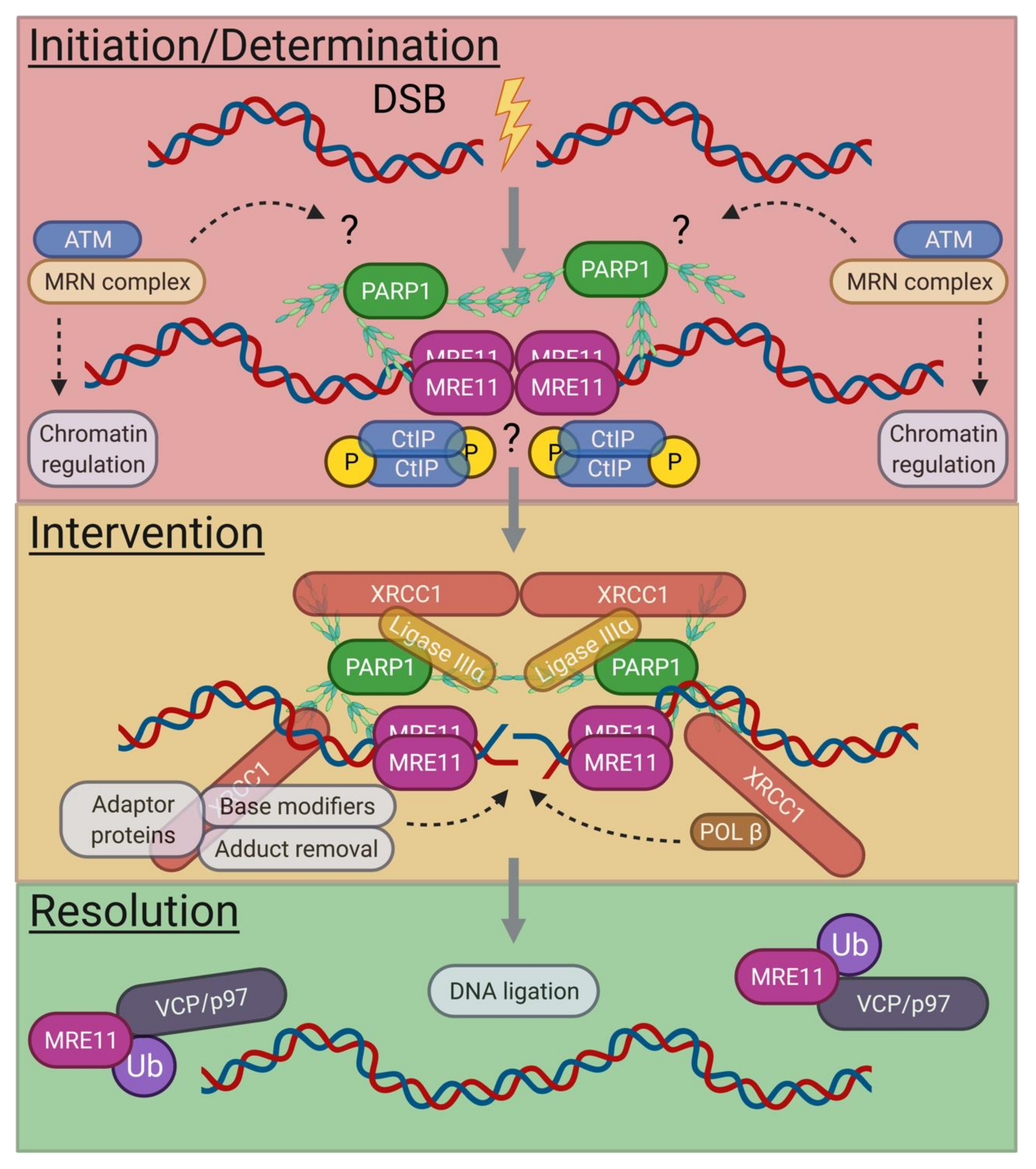
3.1. A-EJ Pathways and Cell Cycle Dependence
3.2. G0-Phase A-EJ Initiation and Determination
3.3. G0-Phase A-EJ Intervention and Resolution
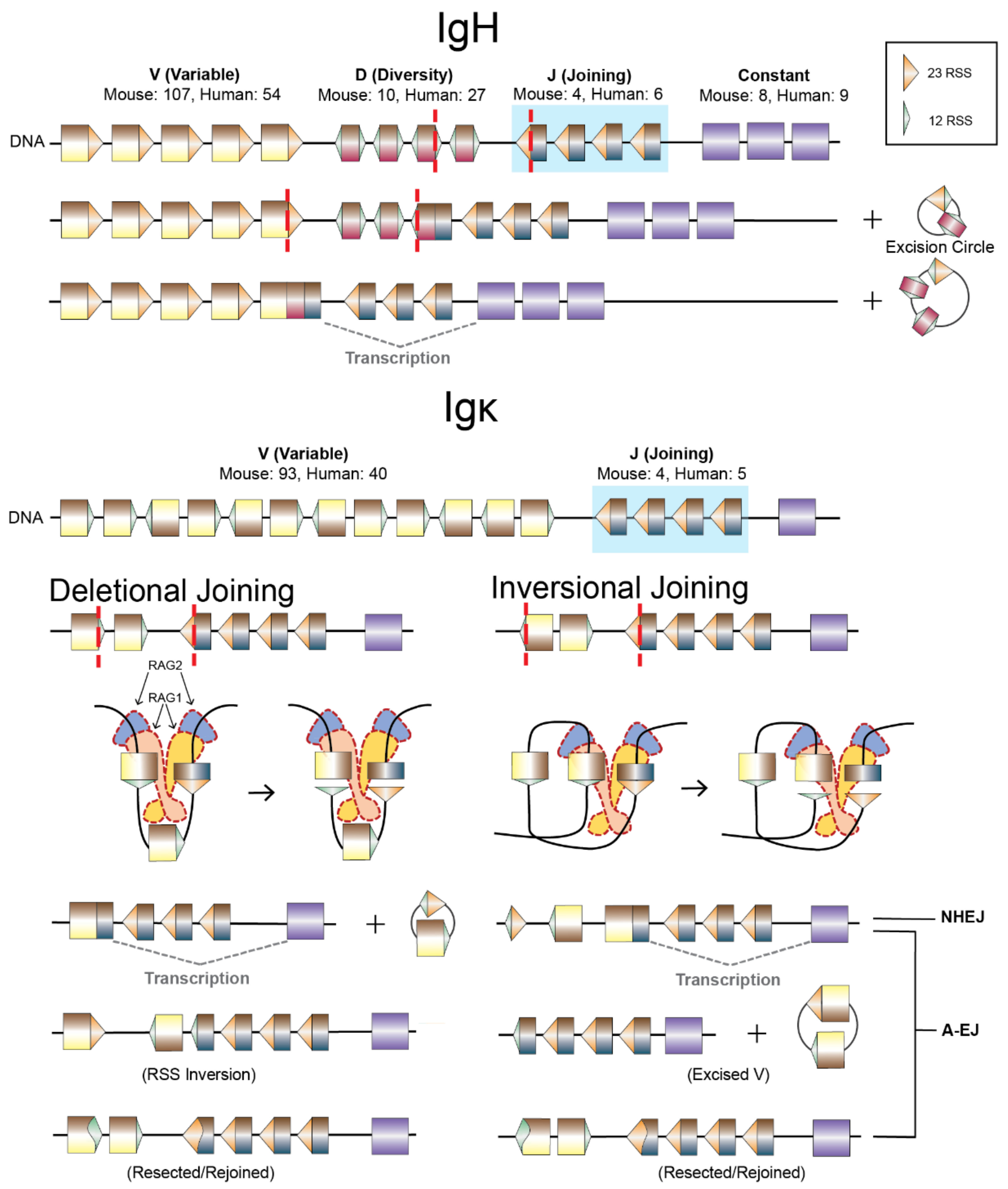
4. End-Joining Pathway Utilization during V(D)J Recombination
4.1. V(D)J Recombination
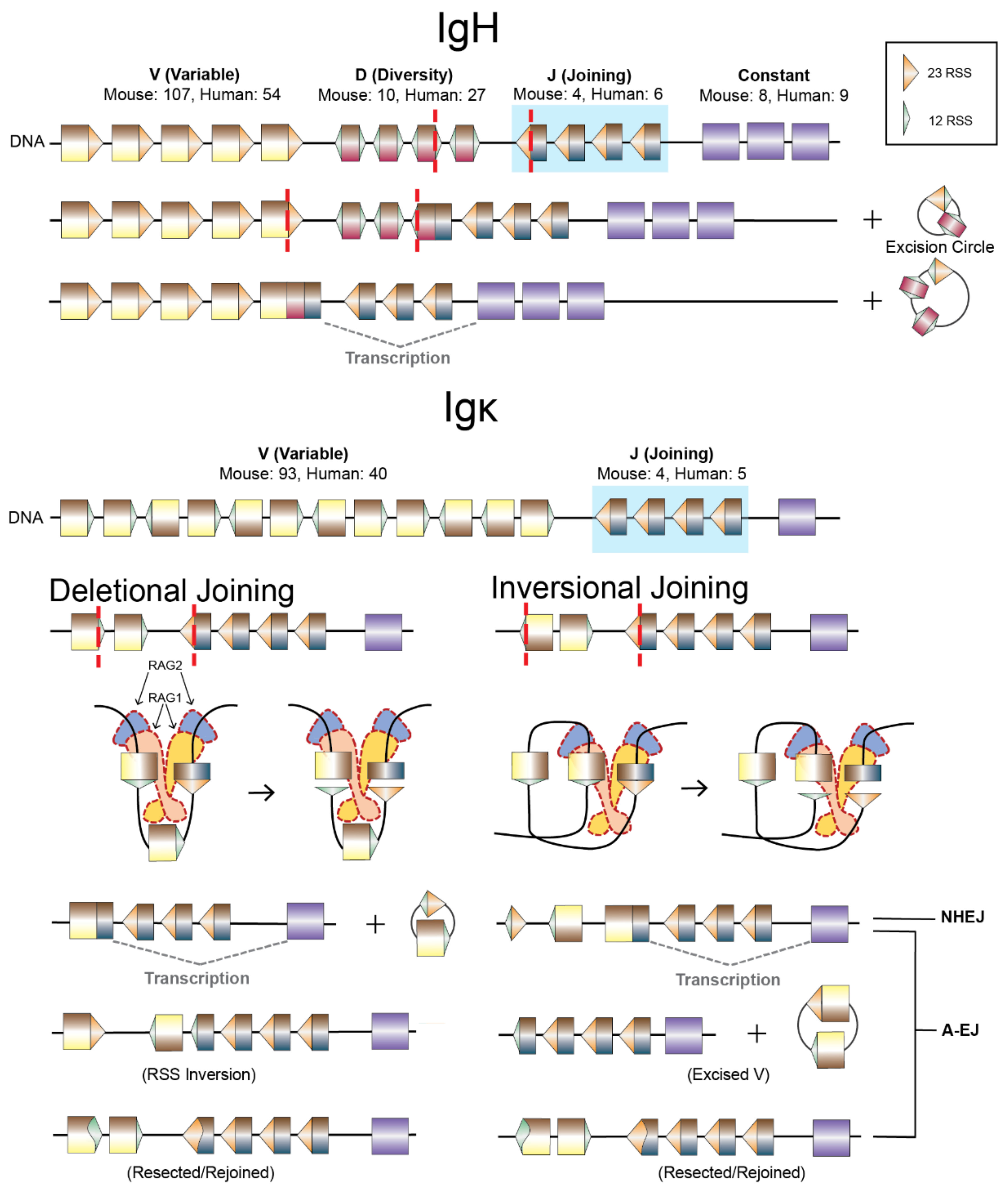
4.2. A-EJ of RAG DSEs, RAG2 Functions, and Repair Pathway Choice
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. Mechanisms of human lymphoid chromosomal translocations. Nat. Rev. Cancer 2016, 16, 387–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, P.; Blasco, M.A. Telomere-driven diseases and telomere-targeting therapies. J. Cell Biol. 2017, 216, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Menon, V.; Povirk, L.F. End-processing nucleases and phosphodiesterases: An elite supporting cast for the non-homologous end joining pathway of DNA double-strand break repair. DNA Repair 2016, 43, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Chanut, P.; Britton, S.; Coates, J.; Jackson, S.P.; Calsou, P. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 2016, 7, 12889. [Google Scholar] [CrossRef] [Green Version]
- Mirman, Z.; de Lange, T. 53BP1: A DSB escort. Genes Dev. 2020, 34, 7–23. [Google Scholar] [CrossRef] [Green Version]
- Willis, N.A.; Frock, R.L.; Menghi, F.; Duffey, E.E.; Panday, A.; Camacho, V.; Hasty, E.P.; Liu, E.T.; Alt, F.W.; Scully, R. Mechanism of tandem duplication formation in BRCA1-mutant cells. Nature 2017, 551, 590–595. [Google Scholar] [CrossRef]
- Marshall, C.J.; Santangelo, T.J. Archaeal DNA Repair Mechanisms. Biomolecules 2020, 10, 1472. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [Green Version]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 2012, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Sharda, M.; Badrinarayanan, A.; Seshasayee, A.S.N. Evolutionary and Comparative Analysis of Bacterial Nonhomologous End Joining Repair. Genome Biol. Evol. 2020, 12, 2450–2466. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; de Lange, T. Removal of shelterin reveals the telomere end-protection problem. Science 2012, 336, 593–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.Y.; Watanabe, G.; Gerodimos, C.A.; Ochi, T.; Blundell, T.L.; Jackson, S.P.; Lieber, M.R. Different DNA End Configurations Dictate Which NHEJ Components Are Most Important for Joining Efficiency. J. Biol. Chem. 2016, 291, 24377–24389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemos, B.R.; Kaplan, A.C.; Bae, J.E.; Ferrazzoli, A.E.; Kuo, J.; Anand, R.P.; Waterman, D.P.; Haber, J.E. CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles. Proc. Natl. Acad. Sci. USA 2018, 115, E2040–E2047. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Kumar, V.; Le Bouteiller, M.; Zurita, J.; Kenrick, J.; Lin, S.G.; Lou, J.; Hu, J.; Ye, A.Y.; Boboila, C.; et al. Ku70 suppresses alternative end joining in G1-arrested progenitor B cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2103630118. [Google Scholar] [CrossRef]
- Pryor, J.M.; Waters, C.A.; Aza, A.; Asagoshi, K.; Strom, C.; Mieczkowski, P.A.; Blanco, L.; Ramsden, D.A. Essential role for polymerase specialization in cellular nonhomologous end joining. Proc. Natl. Acad. Sci. USA 2015, 112, E4537–E4545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, C.A.; Strande, N.T.; Pryor, J.M.; Strom, C.N.; Mieczkowski, P.; Burkhalter, M.D.; Oh, S.; Qaqish, B.F.; Moore, D.T.; Hendrickson, E.A.; et al. The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining. Nat. Commun. 2014, 5, 4286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Palomero, L.; Moore, J.; Guix, I.; Espin, R.; Aytes, A.; Mao, J.H.; Paulovich, A.G.; Whiteaker, J.R.; Ivey, R.G.; et al. Loss of TGFbeta signaling increases alternative end-joining DNA repair that sensitizes to genotoxic therapies across cancer types. Sci. Transl. Med. 2021, 13, eabc4465. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.E.; White, T.B.; Streva, V.A.; DeFreece, C.B.; Hedges, D.J.; Deininger, P.L. The contribution of alu elements to mutagenic DNA double-strand break repair. PLoS Genet. 2015, 11, e1005016. [Google Scholar] [CrossRef]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Re, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Chaplin, A.K.; Hardwick, S.W.; Liang, S.; Kefala Stavridi, A.; Hnizda, A.; Cooper, L.R.; De Oliveira, T.M.; Chirgadze, D.Y.; Blundell, T.L. Dimers of DNA-PK create a stage for DNA double-strand break repair. Nat. Struct. Mol. Biol. 2021, 28, 13–19. [Google Scholar] [CrossRef]
- Chaplin, A.K.; Hardwick, S.W.; Stavridi, A.K.; Buehl, C.J.; Goff, N.J.; Ropars, V.; Liang, S.; De Oliveira, T.M.; Chirgadze, D.Y.; Meek, K.; et al. Cryo-EM of NHEJ supercomplexes provides insights into DNA repair. Mol. Cell 2021, 81, 3400–3409.e3. [Google Scholar] [CrossRef]
- Chen, B.R.; Wang, Y.; Tubbs, A.; Zong, D.; Fowler, F.C.; Zolnerowich, N.; Wu, W.; Bennett, A.; Chen, C.C.; Feng, W.; et al. LIN37-DREAM prevents DNA end resection and homologous recombination at DNA double-strand breaks in quiescent cells. eLife 2021, 10, e68466. [Google Scholar] [CrossRef]
- Chen, S.; Lee, L.; Naila, T.; Fishbain, S.; Wang, A.; Tomkinson, A.E.; Lees-Miller, S.P.; He, Y. Structural basis of long-range to short-range synaptic transition in NHEJ. Nature 2021, 593, 294–298. [Google Scholar] [CrossRef]
- Chen, X.; Xu, X.; Chen, Y.; Cheung, J.C.; Wang, H.; Jiang, J.; de Val, N.; Fox, T.; Gellert, M.; Yang, W. Structure of an activated DNA-PK and its implications for NHEJ. Mol. Cell 2021, 81, 801–810.e3. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinson, B.M.; Moreno, A.T.; Walter, J.C.; Loparo, J.J. A Mechanism to Minimize Errors during Non-homologous End Joining. Mol. Cell 2020, 77, 1080–1091.e8. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Lescale, C.; Babin, L.; Bedora-Faure, M.; Lenden-Hasse, H.; Baron, L.; Demangel, C.; Yelamos, J.; Brunet, E.; Deriano, L. Repair of G1 induced DNA double-strand breaks in S-G2/M by alternative NHEJ. Nat. Commun. 2020, 11, 5239. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, T.C.; Huang, G.; Lu, Q.; Surleac, M.D.; Mandell, J.D.; Pontarotti, P.; Petrescu, A.J.; Xu, A.; Xiong, Y.; et al. Transposon molecular domestication and the evolution of the RAG recombinase. Nature 2019, 569, 79–84. [Google Scholar] [CrossRef]
- Zhao, B.; Watanabe, G.; Morten, M.J.; Reid, D.A.; Rothenberg, E.; Lieber, M.R. The essential elements for the noncovalent association of two DNA ends during NHEJ synapsis. Nat. Commun. 2019, 10, 3588. [Google Scholar] [CrossRef] [Green Version]
- Hartlerode, A.J.; Morgan, M.J.; Wu, Y.; Buis, J.; Ferguson, D.O. Recruitment and activation of the ATM kinase in the absence of DNA-damage sensors. Nat. Struct. Mol. Biol. 2015, 22, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Barton, O.; Naumann, S.C.; Diemer-Biehs, R.; Kunzel, J.; Steinlage, M.; Conrad, S.; Makharashvili, N.; Wang, J.; Feng, L.; Lopez, B.S.; et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J. Cell Biol. 2014, 206, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhasz, S.; Kunzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Lobrich, M. DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell 2017, 65, 671–684.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyama, T.; Wilson 3rd, D.M. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryor, J.M.; Conlin, M.P.; Carvajal-Garcia, J.; Luedeman, M.E.; Luthman, A.J.; Small, G.W.; Ramsden, D.A. Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining. Science 2018, 361, 1126–1129. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Strande, N.; Burkhalter, M.D.; Strom, C.; Havener, J.M.; Hasty, P.; Ramsden, D.A. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature 2010, 464, 1214–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Wilson, T.E. Rejoining of DNA double-strand breaks as a function of overhang length. Mol. Cell Biol. 2005, 25, 896–906. [Google Scholar] [CrossRef] [Green Version]
- Frank, K.M.; Sekiguchi, J.M.; Seidl, K.J.; Swat, W.; Rathbun, G.A.; Cheng, H.L.; Davidson, L.; Kangaloo, L.; Alt, F.W. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase, I.V. Nature 1998, 396, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Sun, Y.; Frank, K.M.; Dikkes, P.; Fujiwara, Y.; Seidl, K.J.; Sekiguchi, J.M.; Rathbun, G.A.; Swat, W.; Wang, J.; et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell 1998, 95, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Seidl, K.J.; Rathbun, G.A.; Zhu, C.; Manis, J.P.; van der Stoep, N.; Davidson, L.; Cheng, H.L.; Sekiguchi, J.M.; Frank, K.; et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity 1997, 7, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Nussenzweig, A.; Chen, C.; da Costa Soares, V.; Sanchez, M.; Sokol, K.; Nussenzweig, M.C.; Li, G.C. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 1996, 382, 551–555. [Google Scholar] [CrossRef]
- Rooney, S.; Sekiguchi, J.; Zhu, C.; Cheng, H.L.; Manis, J.; Whitlow, S.; DeVido, J.; Foy, D.; Chaudhuri, J.; Lombard, D.; et al. Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol. Cell 2002, 10, 1379–1390. [Google Scholar] [CrossRef]
- Taccioli, G.E.; Rathbun, G.; Oltz, E.; Stamato, T.; Jeggo, P.A.; Alt, F.W. Impairment of V(D)J recombination in double-strand break repair mutants. Science 1993, 260, 207–210. [Google Scholar] [CrossRef]
- Zhu, C.; Bogue, M.A.; Lim, D.S.; Hasty, P.; Roth, D.B. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell 1996, 86, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.S.; Lee, B.J.; Zha, S. The recent advances in non-homologous end-joining through the lens of lymphocyte development. DNA Repair 2020, 94, 102874. [Google Scholar] [CrossRef]
- Frit, P.; Ropars, V.; Modesti, M.; Charbonnier, J.B.; Calsou, P. Plugged into the Ku-DNA hub: The NHEJ network. Prog. Biophys. Mol. Biol. 2019, 147, 62–76. [Google Scholar] [CrossRef]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Yoo, S.; Dynan, W.S. Geometry of a complex formed by double strand break repair proteins at a single DNA end: Recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Res. 1999, 27, 4679–4686. [Google Scholar] [CrossRef]
- Krasner, D.S.; Daley, J.M.; Sung, P.; Niu, H. Interplay between Ku and Replication Protein A in the Restriction of Exo1-mediated DNA Break End Resection. J. Biol. Chem. 2015, 290, 18806–18816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oz, R.; Wang, J.L.; Guerois, R.; Goyal, G.; Kk, S.; Ropars, V.; Sharma, R.; Koca, F.; Charbonnier, J.B.; Modesti, M.; et al. Dynamics of Ku and bacterial non-homologous end-joining characterized using single DNA molecule analysis. Nucleic Acids Res. 2021, 49, 2629–2641. [Google Scholar] [CrossRef] [PubMed]
- Gell, D.; Jackson, S.P. Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res. 1999, 27, 3494–3502. [Google Scholar] [CrossRef] [PubMed]
- Singleton, B.K.; Torres-Arzayus, M.I.; Rottinghaus, S.T.; Taccioli, G.E.; Jeggo, P.A. The C terminus of Ku80 activates the DNA-dependent protein kinase catalytic subunit. Mol. Cell Biol. 1999, 19, 3267–3277. [Google Scholar] [CrossRef] [Green Version]
- Graham, T.G.; Walter, J.C.; Loparo, J.J. Two-Stage Synapsis of DNA Ends during Non-homologous End Joining. Mol. Cell 2016, 61, 850–858. [Google Scholar] [CrossRef] [Green Version]
- Graham, T.G.W.; Carney, S.M.; Walter, J.C.; Loparo, J.J. A single XLF dimer bridges DNA ends during nonhomologous end joining. Nat. Struct. Mol. Biol. 2018, 25, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Duboc, C.; Wu, Q.; Ochi, T.; Liang, S.; Tsutakawa, S.E.; Lees-Miller, S.P.; Nadal, M.; Tainer, J.A.; Blundell, T.L.; et al. Dissection of DNA double-strand-break repair using novel single-molecule forceps. Nat. Struct. Mol. Biol. 2018, 25, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadi, S.K.; Tellier-Lebegue, C.; Nemoz, C.; Drevet, P.; Audebert, S.; Roy, S.; Meek, K.; Charbonnier, J.B.; Modesti, M. PAXX Is an Accessory c-NHEJ Factor that Associates with Ku70 and Has Overlapping Functions with XLF. Cell Rep. 2016, 17, 541–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, M.; Yang, M.; Huo, W.; Feng, F.; Wei, L.; Jiang, W.; Ning, S.; Yan, Z.; Li, W.; Wang, Q.; et al. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat. Commun. 2015, 6, 6233. [Google Scholar] [CrossRef] [Green Version]
- Carney, S.M.; Moreno, A.T.; Piatt, S.C.; Cisneros-Aguirre, M.; Lopezcolorado, F.W.; Stark, J.M.; Loparo, J.J. XLF acts as a flexible connector during non-homologous end joining. eLife 2020, 9, e61920. [Google Scholar] [CrossRef]
- Grundy, G.J.; Rulten, S.L.; Arribas-Bosacoma, R.; Davidson, K.; Kozik, Z.; Oliver, A.W.; Pearl, L.H.; Caldecott, K.W. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat. Commun. 2016, 7, 11242. [Google Scholar] [CrossRef]
- Hammel, M.; Yu, Y.; Radhakrishnan, S.K.; Chokshi, C.; Tsai, M.S.; Matsumoto, Y.; Kuzdovich, M.; Remesh, S.G.; Fang, S.; Tomkinson, A.E.; et al. An Intrinsically Disordered APLF Links Ku, DNA-PKcs, and XRCC4-DNA Ligase IV in an Extended Flexible Non-homologous End Joining Complex. J. Biol. Chem. 2016, 291, 26987–27006. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Mahaney, B.L.; Yano, K.; Ye, R.; Fang, S.; Douglas, P.; Chen, D.J.; Lees-Miller, S.P. DNA-PK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA Repair 2008, 7, 1680–1692. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Wang, W.; Ding, Q.; Ye, R.; Chen, D.; Merkle, D.; Schriemer, D.; Meek, K.; Lees-Miller, S.P. DNA-PK phosphorylation sites in XRCC4 are not required for survival after radiation or for V(D)J recombination. DNA Repair 2003, 2, 1239–1252. [Google Scholar] [CrossRef]
- Normanno, D.; Negrel, A.; de Melo, A.J.; Betzi, S.; Meek, K.; Modesti, M. Mutational phospho-mimicry reveals a regulatory role for the XRCC4 and XLF C-terminal tails in modulating DNA bridging during classical non-homologous end joining. eLife 2017, 6, e22900. [Google Scholar] [CrossRef] [PubMed]
- Andres, S.N.; Vergnes, A.; Ristic, D.; Wyman, C.; Modesti, M.; Junop, M. A human XRCC4-XLF complex bridges DNA. Nucleic Acids Res. 2012, 40, 1868–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammel, M.; Rey, M.; Yu, Y.; Mani, R.S.; Classen, S.; Liu, M.; Pique, M.E.; Fang, S.; Mahaney, B.L.; Weinfeld, M.; et al. XRCC4 protein interactions with XRCC4-like factor (XLF) create an extended grooved scaffold for DNA ligation and double strand break repair. J. Biol. Chem. 2011, 286, 32638–32650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammel, M.; Yu, Y.; Fang, S.; Lees-Miller, S.P.; Tainer, J.A. XLF regulates filament architecture of the XRCC4.ligase IV complex. Structure 2010, 18, 1431–1442. [Google Scholar] [CrossRef] [Green Version]
- Mahaney, B.L.; Hammel, M.; Meek, K.; Tainer, J.A.; Lees-Miller, S.P. XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. BioChem. Cell Biol. 2013, 91, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Ropars, V.; Drevet, P.; Legrand, P.; Baconnais, S.; Amram, J.; Faure, G.; Marquez, J.A.; Pietrement, O.; Guerois, R.; Callebaut, I.; et al. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proc. Natl. Acad. Sci. USA 2011, 108, 12663–12668. [Google Scholar] [CrossRef] [Green Version]
- Reid, D.A.; Keegan, S.; Leo-Macias, A.; Watanabe, G.; Strande, N.T.; Chang, H.H.; Oksuz, B.A.; Fenyo, D.; Lieber, M.R.; Ramsden, D.A.; et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2015, 112, E2575–E2584. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, I.; Sitters, G.; Candelli, A.; Heerema, S.J.; Heller, I.; de Melo, A.J.; Zhang, H.; Normanno, D.; Modesti, M.; Peterman, E.J.; et al. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature 2016, 535, 566–569. [Google Scholar] [CrossRef]
- Li, W.; Bai, X.; Li, J.; Zhao, Y.; Liu, J.; Zhao, H.; Liu, L.; Ding, M.; Wang, Q.; Shi, F.Y.; et al. The nucleoskeleton protein IFFO1 immobilizes broken DNA and suppresses chromosome translocation during tumorigenesis. Nat. Cell Biol. 2019, 21, 1273–1285. [Google Scholar] [CrossRef]
- Rober, R.A.; Weber, K.; Osborn, M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: A developmental study. Development 1989, 105, 365–378. [Google Scholar] [CrossRef]
- Bell, E.S.; Lammerding, J. Causes and consequences of nuclear envelope alterations in tumour progression. Eur. J. Cell Biol. 2016, 95, 449–464. [Google Scholar] [CrossRef] [Green Version]
- Lucas, J.S.; Zhang, Y.; Dudko, O.K.; Murre, C. 3D trajectories adopted by coding and regulatory DNA elements: First-passage times for genomic interactions. Cell 2014, 158, 339–352. [Google Scholar] [CrossRef] [Green Version]
- Setiaputra, D.; Durocher, D. Shieldin—The protector of DNA ends. EMBO Rep. 2019, 20, e47560. [Google Scholar] [CrossRef]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef]
- Bredemeyer, A.L.; Sharma, G.G.; Huang, C.Y.; Helmink, B.A.; Walker, L.M.; Khor, K.C.; Nuskey, B.; Sullivan, K.E.; Pandita, T.K.; Bassing, C.H.; et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 2006, 442, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Callen, E.; Jankovic, M.; Difilippantonio, S.; Daniel, J.A.; Chen, H.T.; Celeste, A.; Pellegrini, M.; McBride, K.; Wangsa, D.; Bredemeyer, A.L.; et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell 2007, 130, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, N.; Bai, P.; Buchek, G.; Korza, G.; Weller, S.K. Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J. Virol. 2010, 84, 12504–12514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Goldstein, M.; Alexander, P.; Wakeman, T.P.; Sun, T.; Feng, J.; Lou, Z.; Kastan, M.B.; Wang, X.F. Rad17 recruits the MRE11-RAD50-NBS1 complex to regulate the cellular response to DNA double-strand breaks. EMBO J. 2014, 33, 862–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, P.J.; Johnson, B.; Chen, B.R.; Byrum, A.K.; Bredemeyer, A.L.; Yewdell, W.T.; Johnson, T.E.; Lee, B.J.; Deivasigamani, S.; Hindi, I.; et al. MRI Is a DNA Damage Response Adaptor during Classical Non-homologous End Joining. Mol. Cell 2018, 71, 332–342.e8. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, N.; Correia, A.; Ma, J.; Merlo, A.; Garcia-Gomez, S.; Maric, M.; Tognetti, M.; Benner, C.W.; Boulton, S.J.; Saghatelian, A.; et al. Regulation of DNA repair pathway choice in S and G2 phases by the NHEJ inhibitor CYREN. Nature 2017, 549, 548–552. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Alt, F.W.; Frock, R.L. PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proc. Natl. Acad. Sci. USA 2016, 113, 10619–10624. [Google Scholar] [CrossRef] [Green Version]
- Musilli, S.; Abramowski, V.; Roch, B.; de Villartay, J.P. An in vivo study of the impact of deficiency in the DNA repair proteins PAXX and XLF on development and maturation of the hemolymphoid system. J. Biol. Chem. 2020, 295, 2398–2406. [Google Scholar] [CrossRef]
- Francica, P.; Mutlu, M.; Blomen, V.A.; Oliveira, C.; Nowicka, Z.; Trenner, A.; Gerhards, N.M.; Bouwman, P.; Stickel, E.; Hekkelman, M.L.; et al. Functional Radiogenetic Profiling Implicates ERCC6L2 in Non-homologous End Joining. Cell Rep. 2020, 32, 108068. [Google Scholar] [CrossRef]
- Liu, X.; Liu, T.; Shang, Y.; Dai, P.; Zhang, W.; Lee, B.J.; Huang, M.; Yang, D.; Wu, Q.; Liu, L.D.; et al. ERCC6L2 promotes DNA orientation-specific recombination in mammalian cells. Cell Res. 2020, 30, 732–744. [Google Scholar] [CrossRef]
- Olivieri, M.; Cho, T.; Alvarez-Quilon, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef]
- Zhang, S.; Pondarre, C.; Pennarun, G.; Labussiere-Wallet, H.; Vera, G.; France, B.; Chansel, M.; Rouvet, I.; Revy, P.; Lopez, B.; et al. A nonsense mutation in the DNA repair factor Hebo causes mild bone marrow failure and microcephaly. J. Exp. Med. 2016, 213, 1011–1028. [Google Scholar] [CrossRef] [PubMed]
- Tummala, H.; Dokal, A.D.; Walne, A.; Ellison, A.; Cardoso, S.; Amirthasigamanipillai, S.; Kirwan, M.; Browne, I.; Sidhu, J.K.; Rajeeve, V.; et al. Genome instability is a consequence of transcription deficiency in patients with bone marrow failure harboring biallelic ERCC6L2 variants. Proc. Natl. Acad. Sci. USA 2018, 115, 7777–7782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravind, L.; Walker, D.R.; Koonin, E.V. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res. 1999, 27, 1223–1242. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tomkinson, A.E. Yeast Nej1 is a key participant in the initial end binding and final ligation steps of nonhomologous end joining. J. Biol. Chem. 2011, 286, 4931–4940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, T.T. 20 Years of Mre11 Biology: No End in Sight. Mol. Cell 2018, 71, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Myler, L.R.; Gallardo, I.F.; Soniat, M.M.; Deshpande, R.A.; Gonzalez, X.B.; Kim, Y.; Paull, T.T.; Finkelstein, I.J. Single-Molecule Imaging Reveals How Mre11-Rad50-Nbs1 Initiates DNA Break Repair. Mol. Cell 2017, 67, 891–898.e4. [Google Scholar] [CrossRef] [Green Version]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [Green Version]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Ann. Rev. BioChem. 2018, 87, 263–294. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, H.; Lim, C.T.; Konno, H.; Shiozaki, K.; Shinohara, A.; Uchihashi, T.; Furukohri, A. Rad50 zinc hook functions as a constitutive dimerization module interchangeable with SMC hinge. Nat. Commun. 2020, 11, 370. [Google Scholar] [CrossRef]
- De Jager, M.; van Noort, J.; van Gent, D.C.; Dekker, C.; Kanaar, R.; Wyman, C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol. Cell 2001, 8, 1129–1135. [Google Scholar] [CrossRef]
- Hohl, M.; Kwon, Y.; Galvan, S.M.; Xue, X.; Tous, C.; Aguilera, A.; Sung, P.; Petrini, J.H. The Rad50 coiled-coil domain is indispensable for Mre11 complex functions. Nat. Struct. Mol. Biol. 2011, 18, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Herrero, F.; de Jager, M.; Dekker, N.H.; Kanaar, R.; Wyman, C.; Dekker, C. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature 2005, 437, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Castaneda-Zegarra, S.; Fernandez-Berrocal, M.; Tkachov, M.; Yao, R.; Upfold, N.L.E.; Oksenych, V. Genetic interaction between the non-homologous end-joining factors during B and T lymphocyte development: In vivo mouse models. Scand. J. Immunol. 2020, 92, e12936. [Google Scholar] [CrossRef]
- Lescale, C.; Lenden Hasse, H.; Blackford, A.N.; Balmus, G.; Bianchi, J.J.; Yu, W.; Bacoccina, L.; Jarade, A.; Clouin, C.; Sivapalan, R.; et al. Specific Roles of XRCC4 Paralogs PAXX and XLF during V(D)J Recombination. Cell Rep. 2016, 16, 2967–2979. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shao, Z.; Jiang, W.; Lee, B.J.; Zha, S. PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat. Commun. 2017, 8, 13816. [Google Scholar] [CrossRef]
- Oksenych, V.; Kumar, V.; Liu, X.; Guo, C.; Schwer, B.; Zha, S.; Alt, F.W. Functional redundancy between the XLF and DNA-PKcs DNA repair factors in V(D)J recombination and nonhomologous DNA end joining. Proc. Natl. Acad. Sci. USA 2013, 110, 2234–2239. [Google Scholar] [CrossRef] [Green Version]
- Zha, S.; Guo, C.; Boboila, C.; Oksenych, V.; Cheng, H.L.; Zhang, Y.; Wesemann, D.R.; Yuen, G.; Patel, H.; Goff, P.H.; et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2011, 469, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Arnould, C.; Rocher, V.; Finoux, A.L.; Clouaire, T.; Li, K.; Zhou, F.; Caron, P.; Mangeot, P.E.; Ricci, E.P.; Mourad, R.; et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 2021, 590, 660–665. [Google Scholar] [CrossRef]
- Li, K.; Bronk, G.; Kondev, J.; Haber, J.E. Yeast ATM and ATR kinases use different mechanisms to spread histone H2A phosphorylation around a DNA double-strand break. Proc. Natl. Acad. Sci. USA 2020, 117, 21354–21363. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Clouaire, T.; Legube, G. A Snapshot on the Cis Chromatin Response to DNA Double-Strand Breaks. Trends Genet. 2019, 35, 330–345. [Google Scholar] [CrossRef] [Green Version]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Williams, G.J.; Limbo, O.; Williams, R.S.; Kuhnlein, J.; Lee, J.H.; Classen, S.; Guenther, G.; Russell, P.; Tainer, J.A.; et al. ATP-driven Rad50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 2014, 33, 482–500. [Google Scholar] [CrossRef] [PubMed]
- Lammens, K.; Bemeleit, D.J.; Mockel, C.; Clausing, E.; Schele, A.; Hartung, S.; Schiller, C.B.; Lucas, M.; Angermuller, C.; Soding, J.; et al. The Mre11:Rad50 structure shows an ATP-dependent molecular clamp in DNA double-strand break repair. Cell 2011, 145, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.S.; Kim, J.S.; Park, Y.B.; Gwon, G.H.; Cho, Y. Crystal structure of the Mre11-Rad50-ATPgammaS complex: Understanding the interplay between Mre11 and Rad50. Genes Dev. 2011, 25, 1091–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, G.J.; Williams, R.S.; Williams, J.S.; Moncalian, G.; Arvai, A.S.; Limbo, O.; Guenther, G.; SilDas, S.; Hammel, M.; Russell, P.; et al. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat. Struct. Mol. Biol. 2011, 18, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, T.T.; Gellert, M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell 1998, 1, 969–979. [Google Scholar] [CrossRef]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, J.; Chapman, J.R.; Clapperton, J.A.; Haire, L.F.; Hartsuiker, E.; Li, J.; Carr, A.M.; Jackson, S.P.; Smerdon, S.J. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell 2009, 139, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.N.; Iwasaki, H.; Russell, P.; et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.A.; Lee, J.H.; Arora, S.; Paull, T.T. Nbs1 Converts the Human Mre11/Rad50 Nuclease Complex into an Endo/Exonuclease Machine Specific for Protein-DNA Adducts. Mol. Cell 2016, 64, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, T.; Baer, R.; Gottesman, M.; Gautier, J. MRN, CtIP, and BRCA1 mediate repair of topoisomerase II-DNA adducts. J. Cell Biol. 2016, 212, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, S.E.; Li, Y.; Wu-Baer, F.; Chait, B.T.; Baer, R.; Yan, H.; Gottesman, M.E.; Gautier, J. Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol. Cell 2013, 49, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmink, B.A.; Bredemeyer, A.L.; Lee, B.S.; Huang, C.Y.; Sharma, G.G.; Walker, L.M.; Bednarski, J.J.; Lee, W.L.; Pandita, T.K.; Bassing, C.H.; et al. MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. J. Exp. Med. 2009, 206, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.S.; Neiditch, M.B.; Salus, S.S.; Roth, D.B. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell 2004, 117, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Mages, C.F.; Wintsche, A.; Bernhart, S.H.; Muller, G.A. The DREAM complex through its subunit Lin37 cooperates with Rb to initiate quiescence. eLife 2019, 6, e26876. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Alimirah, F.; Desprez, P.Y.; Campisi, J.; Wiley, C. Context-dependent effects of cellular senescence in cancer development. Br. J. Cancer 2016, 114, 1180–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Zglinicki, T.; Wan, T.; Miwa, S. Senescence in Post-Mitotic Cells: A Driver of Aging? Antioxid. Redox Signal. 2021, 34, 308–323. [Google Scholar] [CrossRef]
- Zhou, Y.; Lee, J.H.; Jiang, W.; Crowe, J.L.; Zha, S.; Paull, T.T. Regulation of the DNA Damage Response by DNA-PKcs Inhibitory Phosphorylation of ATM. Mol. Cell 2017, 65, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [Green Version]
- Weterings, E.; Verkaik, N.S.; Keijzers, G.; Florea, B.I.; Wang, S.Y.; Ortega, L.G.; Uematsu, N.; Chen, D.J.; van Gent, D.C. The Ku80 carboxy terminus stimulates joining and artemis-mediated processing of DNA ends. Mol. Cell Biol. 2009, 29, 1134–1142. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.; Watanabe, G.; Lieber, M.R. Unifying the DNA end-processing roles of the artemis nuclease: Ku-dependent artemis resection at blunt DNA ends. J. Biol. Chem. 2015, 290, 24036–24050. [Google Scholar] [CrossRef] [Green Version]
- Meek, K. Activation of DNA-PK by hairpinned DNA ends reveals a stepwise mechanism of kinase activation. Nucleic Acids Res. 2020, 48, 9098–9108. [Google Scholar] [CrossRef] [PubMed]
- Nemoz, C.; Ropars, V.; Frit, P.; Gontier, A.; Drevet, P.; Yu, J.; Guerois, R.; Pitois, A.; Comte, A.; Delteil, C.; et al. XLF and APLF bind Ku80 at two remote sites to ensure DNA repair by non-homologous end joining. Nat. Struct. Mol. Biol. 2018, 25, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs kinases-the lessons from the mouse models: Inhibition not equal deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Saha, J.; Sun, J.; Fattah, K.R.; Wang, S.C.; Jakob, B.; Chi, L.; Wang, S.Y.; Taucher-Scholz, G.; Davis, A.J.; et al. Phosphorylation of Ku dictates DNA double-strand break (DSB) repair pathway choice in S phase. Nucleic Acids Res. 2016, 44, 1732–1745. [Google Scholar] [CrossRef] [Green Version]
- Ishida, N.; Nakagawa, T.; Iemura, S.I.; Yasui, A.; Shima, H.; Katoh, Y.; Nagasawa, Y.; Natsume, T.; Igarashi, K.; Nakayama, K. Ubiquitylation of Ku80 by RNF126 Promotes Completion of Nonhomologous End Joining-Mediated DNA Repair. Mol. Cell Biol. 2017, 37, e00347-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, L.; Chen, J. The E3 ligase RNF8 regulates KU80 removal and NHEJ repair. Nat. Struct. Mol. Biol. 2012, 19, 201–206. [Google Scholar] [CrossRef]
- Postow, L.; Ghenoiu, C.; Woo, E.M.; Krutchinsky, A.N.; Chait, B.T.; Funabiki, H. Ku80 removal from DNA through double strand break-induced ubiquitylation. J. Cell Biol. 2008, 182, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Tsai, L.J.; Lopezcolorado, F.W.; Bhargava, R.; Mendez-Dorantes, C.; Jahanshir, E.; Stark, J.M. RNF8 has both KU-dependent and independent roles in chromosomal break repair. Nucleic Acids Res. 2020, 48, 6032–6052. [Google Scholar] [CrossRef]
- Brown, J.S.; Lukashchuk, N.; Sczaniecka-Clift, M.; Britton, S.; le Sage, C.; Calsou, P.; Beli, P.; Galanty, Y.; Jackson, S.P. Neddylation promotes ubiquitylation and release of Ku from DNA-damage sites. Cell Rep. 2015, 11, 704–714. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; Gagne, J.P.; Genois, M.M.; Strickfaden, H.; McDonald, D.; Xu, Z.; Poirier, G.G.; Masson, J.Y.; Hendzel, M.J. The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol. 2015, 17, 1446–1457. [Google Scholar] [CrossRef]
- Schmidt, C.K.; Galanty, Y.; Sczaniecka-Clift, M.; Coates, J.; Jhujh, S.; Demir, M.; Cornwell, M.; Beli, P.; Jackson, S.P. Systematic E2 screening reveals a UBE2D-RNF138-CtIP axis promoting DNA repair. Nat. Cell Biol. 2015, 17, 1458–1470. [Google Scholar] [CrossRef]
- Van den Boom, J.; Wolf, M.; Weimann, L.; Schulze, N.; Li, F.; Kaschani, F.; Riemer, A.; Zierhut, C.; Kaiser, M.; Iliakis, G.; et al. VCP/p97 Extracts Sterically Trapped Ku70/80 Rings from DNA in Double-Strand Break Repair. Mol. Cell 2016, 64, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhang, H.; Jiang, Y.N.; Wang, Z.Q.; Sun, L.; Zhou, Z.W. Post-Translational Modification of MRE11: Its Implication in DDR and Diseases. Genes 2021, 12, 1158. [Google Scholar] [CrossRef] [PubMed]
- Kilgas, S.; Singh, A.N.; Paillas, S.; Then, C.K.; Torrecilla, I.; Nicholson, J.; Browning, L.; Vendrell, I.; Konietzny, R.; Kessler, B.M.; et al. p97/VCP inhibition causes excessive MRE11-dependent DNA end resection promoting cell killing after ionizing radiation. Cell Rep. 2021, 35, 109153. [Google Scholar] [CrossRef]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boboila, C.; Jankovic, M.; Yan, C.T.; Wang, J.H.; Wesemann, D.R.; Zhang, T.; Fazeli, A.; Feldman, L.; Nussenzweig, A.; Nussenzweig, M.; et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc. Natl. Acad. Sci. USA 2010, 107, 3034–3039. [Google Scholar] [CrossRef] [Green Version]
- Boboila, C.; Oksenych, V.; Gostissa, M.; Wang, J.H.; Zha, S.; Zhang, Y.; Chai, H.; Lee, C.S.; Jankovic, M.; Saez, L.M.; et al. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1). Proc. Natl. Acad. Sci. USA 2012, 109, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Boboila, C.; Yan, C.; Wesemann, D.R.; Jankovic, M.; Wang, J.H.; Manis, J.; Nussenzweig, A.; Nussenzweig, M.; Alt, F.W. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J. Exp. Med. 2010, 207, 417–427. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Haber, J.E. Alternative endings. Proc. Natl. Acad. Sci. USA 2008, 105, 405–406. [Google Scholar] [CrossRef] [Green Version]
- Howard, S.M.; Yanez, D.A.; Stark, J.M. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015, 11, e1004943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.J.; Yan, C.T. Regulation of DNA repair in the absence of classical non-homologous end joining. DNA Repair 2018, 68, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Ma, L.; Jones, T.; Palomero, L.; Pujana, M.A.; Martinez-Ruiz, H.; Ha, P.K.; Murnane, J.; Cuartas, I.; Seoane, J.; et al. Subjugation of TGFbeta Signaling by Human Papilloma Virus in Head and Neck Squamous Cell Carcinoma Shifts DNA Repair from Homologous Recombination to Alternative End Joining. Clin. Cancer Res. 2018, 24, 6001–6014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, W.Y.; Borgmann, K.; Petersen, C.; Dikomey, E.; Dahm-Daphi, J. The absence of Ku but not defects in classical non-homologous end-joining is required to trigger PARP1-dependent end-joining. DNA Repair 2013, 12, 1134–1142. [Google Scholar] [CrossRef] [PubMed]
- Masani, S.; Han, L.; Meek, K.; Yu, K. Redundant function of DNA ligase 1 and 3 in alternative end-joining during immunoglobulin class switch recombination. Proc. Natl. Acad. Sci. USA 2016, 113, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, V.; Halabelian, L.; Balagere, S.; Samaniego-Castruita, D.; Feldman, D.E.; Arrowsmith, C.H.; Rao, A.; Aravind, L. HMCES Functions in the Alternative End-Joining Pathway of the DNA DSB Repair during Class Switch Recombination in B Cells. Mol. Cell 2020, 77, 1154. [Google Scholar] [CrossRef]
- Simsek, D.; Brunet, E.; Wong, S.Y.; Katyal, S.; Gao, Y.; McKinnon, P.J.; Lou, J.; Zhang, L.; Li, J.; Rebar, E.J.; et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS Genet. 2011, 7, e1002080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Perrault, A.R.; Takeda, Y.; Qin, W.; Wang, H.; Iliakis, G. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res. 2003, 31, 5377–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.S.; Zhao, J.; Wu-Baer, F.; Shao, Z.; Lee, B.J.; Cupo, O.M.; Rabadan, R.; Gautier, J.; Baer, R.; Zha, S. CtIP-mediated DNA resection is dispensable for IgH class switch recombination by alternative end-joining. Proc. Natl. Acad. Sci. USA 2020, 117, 25700–25711. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D. A Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Xie, A.; Kwok, A.; Scully, R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.; Bjoras, M.; Daniel, J.A.; Alt, F.W.; Oksenych, V. Synthetic lethality between murine DNA repair factors XLF and DNA-PKcs is rescued by inactivation of Ku70. DNA Repair 2017, 57, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Du, Z.; Wang, Y.; Feng, Z.; Fan, P.; Yan, C.; Willers, H.; Zhang, J. 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic Acids Res. 2015, 43, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi-Iwai, Y.; Sonoda, E.; Sasaki, M.S.; Morrison, C.; Haraguchi, T.; Hiraoka, Y.; Yamashita, Y.M.; Yagi, T.; Takata, M.; Price, C.; et al. Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 1999, 18, 6619–6629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.T.; Boboila, C.; Souza, E.K.; Franco, S.; Hickernell, T.R.; Murphy, M.; Gumaste, S.; Geyer, M.; Zarrin, A.A.; Manis, J.P.; et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature 2007, 449, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Wyatt, D.W.; Takata, K.; Mu, Y.X.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublie, S.; Johansson, E.; Ramsden, D.A.; et al. Mechanism of Suppression of Chromosomal Instability by DNA Polymerase POLQ. PLoS Genet. 2014, 10, e1004654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Britton, S.; Delteil, C.; Coates, J.; Jackson, S.P.; Barboule, N.; Frit, P.; Calsou, P. Single-stranded DNA oligomers stimulate error-prone alternative repair of DNA double-strand breaks through hijacking Ku protein. Nucleic Acids Res. 2015, 43, 10264–10276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zan, H.; Tat, C.; Qiu, Z.; Taylor, J.R.; Guerrero, J.A.; Shen, T.; Casali, P. Rad52 competes with Ku70/Ku86 for binding to S-region DSB ends to modulate antibody class-switch DNA recombination. Nat. Commun. 2017, 8, 14244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Jasin, M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol. 2011, 18, 80–84. [Google Scholar] [CrossRef]
- Zhu, C.; Mills, K.D.; Ferguson, D.O.; Lee, C.; Manis, J.; Fleming, J.; Gao, Y.; Morton, C.C.; Alt, F.W. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 2002, 109, 811–821. [Google Scholar] [CrossRef] [Green Version]
- Chandramouly, G.; Zhao, J.; McDevitt, S.; Rusanov, T.; Hoang, T.; Borisonnik, N.; Treddinick, T.; Lopezcolorado, F.W.; Kent, T.; Siddique, L.A.; et al. Poltheta reverse transcribes RNA and promotes RNA-templated DNA repair. Sci. Adv. 2021, 7, eabf1771. [Google Scholar] [CrossRef]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase theta in Cancer Therapy. Trends Cancer 2021, 7, 98–111. [Google Scholar] [CrossRef]
- Britton, S.; Chanut, P.; Delteil, C.; Barboule, N.; Frit, P.; Calsou, P. ATM antagonizes NHEJ proteins assembly and DNA-ends synapsis at single-ended DNA double strand breaks. Nucleic Acids Res. 2020, 48, 9710–9723. [Google Scholar] [CrossRef]
- Willis, N.A.; Panday, A.; Duffey, E.E.; Scully, R. Rad51 recruitment and exclusion of non-homologous end joining during homologous recombination at a Tus/Ter mammalian replication fork barrier. PLoS Genet. 2018, 14, e1007486. [Google Scholar] [CrossRef]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.; Fousteri, M.I. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, H.; Bednar, T.; Wang, M.; Paul, K.; Mladenov, E.; Bencsik-Theilen, A.A.; Iliakis, G. Functional redundancy between DNA ligases I and III in DNA replication in vertebrate cells. Nucleic Acids Res. 2012, 40, 2599–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, D.S.; McKenna, A.E.; Motycka, T.A.; Matsumoto, Y.; Tomkinson, A.E. Interaction between PCNA and DNA ligase I is critical for joining of Okazaki fragments and long-patch base-excision repair. Curr. Biol. 2000, 10, 919–922. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.; Pena-Diaz, J.; Andersen, S.; Liabakk, N.B.; Otterlei, M.; Krokan, H.E. Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair 2009, 8, 834–843. [Google Scholar] [CrossRef]
- Ferrari, G.; Rossi, R.; Arosio, D.; Vindigni, A.; Biamonti, G.; Montecucco, A. Cell cycle-dependent phosphorylation of human DNA ligase I at the cyclin-dependent kinase sites. J. Biol. Chem. 2003, 278, 37761–37767. [Google Scholar] [CrossRef] [Green Version]
- Montecucco, A.; Biamonti, G.; Savini, E.; Focher, F.; Spadari, S.; Ciarrocchi, G. DNA ligase I gene expression during differentiation and cell proliferation. Nucleic Acids Res. 1992, 20, 6209–6214. [Google Scholar] [CrossRef] [Green Version]
- Rossi, R.; Villa, A.; Negri, C.; Scovassi, I.; Ciarrocchi, G.; Biamonti, G.; Montecucco, A. The replication factory targeting sequence/PCNA-binding site is required in G(1) to control the phosphorylation status of DNA ligase, I. EMBO J. 1999, 18, 5745–5754. [Google Scholar] [CrossRef] [Green Version]
- Karanjawala, Z.E.; Adachi, N.; Irvine, R.A.; Oh, E.K.; Shibata, D.; Schwarz, K.; Hsieh, C.L.; Lieber, M.R. The embryonic lethality in DNA ligase IV-deficient mice is rescued by deletion of Ku: Implications for unifying the heterogeneous phenotypes of NHEJ mutants. DNA Repair 2002, 1, 1017–1026. [Google Scholar] [CrossRef]
- Gao, Y.; Ferguson, D.O.; Xie, W.; Manis, J.P.; Sekiguchi, J.; Frank, K.M.; Chaudhuri, J.; Horner, J.; DePinho, R.A.; Alt, F.W. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 2000, 404, 897–900. [Google Scholar] [CrossRef]
- Gu, Y.; Sekiguchi, J.; Gao, Y.; Dikkes, P.; Frank, K.; Ferguson, D.; Hasty, P.; Chun, J.; Alt, F.W. Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc. Natl. Acad. Sci. USA 2000, 97, 2668–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Liu, C.; Chen, S.H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Perina, D.; Mikoc, A.; Ahel, J.; Cetkovic, H.; Zaja, R.; Ahel, I. Distribution of protein poly(ADP-ribosyl)ation systems across all domains of life. DNA Repair 2014, 23, 4–16. [Google Scholar] [CrossRef] [Green Version]
- Henrie, M.S.; Kurimasa, A.; Burma, S.; Menissier-de Murcia, J.; de Murcia, G.; Li, G.C.; Chen, D.J. Lethality in PARP-1/Ku80 double mutant mice reveals physiological synergy during early embryogenesis. DNA Repair 2003, 2, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Menisser-de Murcia, J.; Mark, M.; Wendling, O.; Wynshaw-Boris, A.; de Murcia, G. Early embryonic lethality in PARP-1 Atm double-mutant mice suggests a functional synergy in cell proliferation during development. Mol. Cell Biol. 2001, 21, 1828–1832. [Google Scholar] [CrossRef] [Green Version]
- Menissier de Murcia, J.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Ame, J.C.; Dierich, A.; LeMeur, M.; et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef] [Green Version]
- Prasad, R.; Horton, J.K.; Wilson, S.H. Requirements for PARP-1 covalent crosslinking to DNA (PARP-1 DPC). DNA Repair 2020, 90, 102850. [Google Scholar] [CrossRef]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Andres, S.N.; Li, Z.M.; Erie, D.A.; Williams, R.S. Ctp1 protein-DNA filaments promote DNA bridging and DNA double-strand break repair. J. Biol. Chem. 2019, 294, 3312–3320. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, O.J.; Martin-Gonzalez, A.; Kang, H.; Northall, S.J.; Wigley, D.B.; Moreno-Herrero, F.; Dillingham, M.S. CtIP forms a tetrameric dumbbell-shaped particle which bridges complex DNA end structures for double-strand break repair. eLife 2019, 8, e42129. [Google Scholar] [CrossRef]
- Oz, R.; Howard, S.M.; Sharma, R.; Tornkvist, H.; Ceppi, I.; Kk, S.; Kristiansson, E.; Cejka, P.; Westerlund, F. Phosphorylated CtIP bridges DNA to promote annealing of broken ends. Proc. Natl. Acad. Sci. USA 2020, 117, 21403–21412. [Google Scholar] [CrossRef]
- Gamper, A.M.; Rofougaran, R.; Watkins, S.C.; Greenberger, J.S.; Beumer, J.H.; Bakkenist, C.J. ATR kinase activation in G1 phase facilitates the repair of ionizing radiation-induced DNA damage. Nucleic Acids Res. 2013, 41, 10334–10344. [Google Scholar] [CrossRef]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Wang, W.; Li, S.; Zhan, J.; Li, H.; Zhao, M.; Zhou, X.A.; Li, S.; Li, X.; Huo, Y.; et al. C1QBP Promotes Homologous Recombination by Stabilizing MRE11 and Controlling the Assembly and Activation of MRE11/RAD50/NBS1 Complex. Mol. Cell 2019, 75, 1299–1314.e6. [Google Scholar] [CrossRef]
- Broderick, R.; Nieminuszczy, J.; Baddock, H.T.; Deshpande, R.; Gileadi, O.; Paull, T.T.; McHugh, P.J.; Niedzwiedz, W. EXD2 promotes homologous recombination by facilitating DNA end resection. Nat. Cell Biol. 2016, 18, 271–280. [Google Scholar] [CrossRef] [Green Version]
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Tkac, J.; Xu, G.; Adhikary, H.; Young, J.T.F.; Gallo, D.; Escribano-Diaz, C.; Krietsch, J.; Orthwein, A.; Munro, M.; Sol, W.; et al. HELB Is a Feedback Inhibitor of DNA End Resection. Mol. Cell 2016, 61, 405–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, K.L.; Kelliher, J.L.; Xu, Z.; An, L.; Reed, M.R.; Eoff, R.L.; Wang, J.; Huen, M.S.Y.; Leung, J.W.C. LC8/DYNLL1 is a 53BP1 effector and regulates checkpoint activation. Nucleic Acids Res. 2019, 47, 6236–6249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Kim, W.; Gao, H.; Liu, C.; Zhang, Y.; Chen, Y.; Deng, M.; Zhou, Q.; Huang, J.; Hu, Q.; et al. ASTE1 promotes shieldin-complex-mediated DNA repair by attenuating end resection. Nat. Cell Biol. 2021, 23, 894–904. [Google Scholar] [CrossRef]
- Polo, L.M.; Xu, Y.; Hornyak, P.; Garces, F.; Zeng, Z.; Hailstone, R.; Matthews, S.J.; Caldecott, K.W.; Oliver, A.W.; Pearl, L.H. Efficient Single-Strand Break Repair Requires Binding to Both Poly(ADP-Ribose) and DNA by the Central BRCT Domain of XRCC1. Cell Rep. 2019, 26, 573–581.e5. [Google Scholar] [CrossRef] [Green Version]
- Demin, A.A.; Hirota, K.; Tsuda, M.; Adamowicz, M.; Hailstone, R.; Brazina, J.; Gittens, W.; Kalasova, I.; Shao, Z.; Zha, S.; et al. XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 2021, 81, 3018–3030.e5. [Google Scholar] [CrossRef]
- Bienstock, R.J.; Beard, W.A.; Wilson, S.H. Phylogenetic analysis and evolutionary origins of DNA polymerase X-family members. DNA Repair 2014, 22, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Pedersen, L.C.; Kirby, T.W.; DeRose, E.F.; London, R.E. Characterization of the APLF FHA-XRCC1 phosphopeptide interaction and its structural and functional implications. Nucleic Acids Res. 2017, 45, 12374–12387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aceytuno, R.D.; Piett, C.G.; Havali-Shahriari, Z.; Edwards, R.A.; Rey, M.; Ye, R.; Javed, F.; Fang, S.; Mani, R.; Weinfeld, M.; et al. Structural and functional characterization of the PNKP-XRCC4-LigIV DNA repair complex. Nucleic Acids Res. 2017, 45, 6238–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherry, A.L.; Nott, T.J.; Kelly, G.; Rulten, S.L.; Caldecott, K.W.; Smerdon, S.J. Versatility in phospho-dependent molecular recognition of the XRCC1 and XRCC4 DNA-damage scaffolds by aprataxin-family FHA domains. DNA Repair 2015, 35, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osheroff, W.P.; Jung, H.K.; Beard, W.A.; Wilson, S.H.; Kunkel, T.A. The fidelity of DNA polymerase beta during distributive and processive DNA synthesis. J. Biol. Chem. 1999, 274, 3642–3650. [Google Scholar] [CrossRef] [Green Version]
- Chagovetz, A.M.; Sweasy, J.B.; Preston, B.D. Increased activity and fidelity of DNA polymerase beta on single-nucleotide gapped DNA. J. Biol. Chem. 1997, 272, 27501–27504. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.W.; Liu, T.C.; Liang, R.Y.; Chu, L.Y.; Cheng, H.L.; Chu, J.W.; Hsiao, Y.Y. Structural basis for overhang excision and terminal unwinding of DNA duplexes by TREX1. PLoS Biol. 2018, 16, e2005653. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Kim, Y.S.; Yoon, J.; Wang, H.; Suzuki, T.; Morse 3rd, H.C. The 3′-5′ DNA exonuclease TREX1 directly interacts with poly(ADP-ribose) polymerase-1 (PARP1) during the DNA damage response. J. Biol. Chem. 2014, 289, 32548–32558. [Google Scholar] [CrossRef] [Green Version]
- Frank, K.M.; Sharpless, N.E.; Gao, Y.; Sekiguchi, J.M.; Ferguson, D.O.; Zhu, C.; Manis, J.P.; Horner, J.; DePinho, R.A.; Alt, F.W. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell 2000, 5, 993–1002. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 2005, 3, e181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, E.C.; Vicari, C.; Tsakou-Ngouafo, L.; Pontarotti, P.; Petrescu, A.J.; Schatz, D.G. Identification of RAG-like transposons in protostomes suggests their ancient bilaterian origin. Mob. DNA 2020, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Carmona, L.M.; Fugmann, S.D.; Schatz, D.G. Collaboration of RAG2 with RAG1-like proteins during the evolution of V(D)J recombination. Genes Dev. 2016, 30, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatz, D.G.; Swanson, P.C. V(D)J recombination: Mechanisms of initiation. Ann. Rev. Genet. 2011, 45, 167–202. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, M.P. Nomenclature of the human immunoglobulin kappa (IGK) genes. Exp. Clin. Immunogenet. 2001, 18, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, M.P. Nomenclature of the human immunoglobulin heavy (IGH) genes. Exp. Clin. Immunogenet. 2001, 18, 100–116. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.G.; Ba, Z.; Alt, F.W.; Zhang, Y. RAG Chromatin Scanning During V(D)J Recombination and Chromatin Loop Extrusion are Related Processes. Adv. Immunol. 2018, 139, 93–135. [Google Scholar] [PubMed]
- Zhang, Y.; Zhang, X.; Ba, Z.; Liang, Z.; Dring, E.W.; Hu, H.; Lou, J.; Kyritsis, N.; Zurita, J.; Shamim, M.S.; et al. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature 2019, 573, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Schatz, D.G.; Ji, Y. Recombination centres and the orchestration of V(D)J recombination. Nat. Rev. Immunol. 2011, 11, 251–263. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Y.; Zhao, L.; Frock, R.L.; Du, Z.; Meyers, R.M.; Meng, F.L.; Schatz, D.G.; Alt, F.W. Chromosomal Loop Domains Direct the Recombination of Antigen Receptor Genes. Cell 2015, 163, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Frock, R.L.; Du, Z.; Hu, J.; Chen, L.; Krangel, M.S.; Alt, F.W. Orientation-specific RAG activity in chromosomal loop domains contributes to Tcrd V(D)J recombination during T cell development. J. Exp. Med. 2016, 213, 1921–1936. [Google Scholar] [CrossRef]
- Ba, Z.; Lou, J.; Ye, A.Y.; Dai, H.Q.; Dring, E.W.; Lin, S.G.; Jain, S.; Kyritsis, N.; Kieffer-Kwon, K.R.; Casellas, R.; et al. CTCF orchestrates long-range cohesin-driven V(D)J recombinational scanning. Nature 2020, 586, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.Q.; Hu, H.; Lou, J.; Ye, A.Y.; Ba, Z.; Zhang, X.; Zhang, Y.; Zhao, L.; Yoon, H.S.; Chapdelaine-Williams, A.M.; et al. Loop extrusion mediates physiological Igh locus contraction for RAG scanning. Nature 2021, 590, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Hill, L.; Ebert, A.; Jaritz, M.; Wutz, G.; Nagasaka, K.; Tagoh, H.; Kostanova-Poliakova, D.; Schindler, K.; Sun, Q.; Bonelt, P.; et al. Wapl repression by Pax5 promotes V gene recombination by Igh loop extrusion. Nature 2020, 584, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Ba, Z.; Zhang, Y.; Dai, H.Q.; Alt, F.W. CTCF-Binding Elements Mediate Accessibility of RAG Substrates During Chromatin Scanning. Cell 2018, 174, 102–116.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovely, G.A.; Braikia, F.-Z.; Singh, A.; Schatz, D.G.; Murre, C.; Liu, Z.; Sen, R. Direct observation of RAG recombinase recruitment to chromatin and the IgH locus in live pro-B cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Helmink, B.A.; Sleckman, B.P. The response to and repair of RAG-mediated DNA double-strand breaks. Ann. Rev. Immunol. 2012, 30, 175–202. [Google Scholar] [CrossRef] [Green Version]
- Deriano, L.; Stracker, T.H.; Baker, A.; Petrini, J.H.; Roth, D.B. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Mol. Cell 2009, 34, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Chaumeil, J.; Micsinai, M.; Ntziachristos, P.; Roth, D.B.; Aifantis, I.; Kluger, Y.; Deriano, L.; Skok, J.A. The RAG2 C-terminus and ATM protect genome integrity by controlling antigen receptor gene cleavage. Nat. Commun. 2013, 4, 2231. [Google Scholar] [CrossRef] [Green Version]
- Corneo, B.; Wendland, R.L.; Deriano, L.; Cui, X.; Klein, I.A.; Wong, S.Y.; Arnal, S.; Holub, A.J.; Weller, G.R.; Pancake, B.A.; et al. Rag mutations reveal robust alternative end joining. Nature 2007, 449, 483–486. [Google Scholar] [CrossRef] [PubMed]
- Deriano, L.; Chaumeil, J.; Coussens, M.; Multani, A.; Chou, Y.; Alekseyenko, A.V.; Chang, S.; Skok, J.A.; Roth, D.B. The RAG2 C terminus suppresses genomic instability and lymphomagenesis. Nature 2011, 471, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Gigi, V.; Lewis, S.; Shestova, O.; Mijuskovic, M.; Deriano, L.; Meng, W.; Luning Prak, E.T.; Roth, D.B. RAG2 mutants alter DSB repair pathway choice in vivo and illuminate the nature of ‘alternative NHEJ’. Nucleic Acids Res. 2014, 42, 6352–6364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Chang, F.C.; Ross, A.E.; Lee, J.; Nakayama, K.; Nakayama, K.; Desiderio, S. Ubiquitylation of RAG-2 by Skp2-SCF links destruction of the V(D)J recombinase to the cell cycle. Mol. Cell 2005, 18, 699–709. [Google Scholar] [CrossRef]
- Lee, J.; Desiderio, S. Cyclin A/CDK2 regulates V(D)J recombination by coordinating RAG-2 accumulation and DNA repair. Immunity 1999, 11, 771–781. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Dordai, D.I.; Lee, J.; Desiderio, S. A conserved degradation signal regulates RAG-2 accumulation during cell division and links V(D)J recombination to the cell cycle. Immunity 1996, 5, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Coussens, M.A.; Wendland, R.L.; Deriano, L.; Lindsay, C.R.; Arnal, S.M.; Roth, D.B. RAG2’s acidic hinge restricts repair-pathway choice and promotes genomic stability. Cell Rep. 2013, 4, 870–878. [Google Scholar] [CrossRef]
- Lu, C.; Ward, A.; Bettridge, J.; Liu, Y.; Desiderio, S. An autoregulatory mechanism imposes allosteric control on the V(D)J recombinase by histone H3 methylation. Cell Rep. 2015, 10, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.; Kumari, G.; Sen, R.; Desiderio, S. The RAG-2 Inhibitory Domain Gates Accessibility of the V(D)J Recombinase to Chromatin. Mol. Cell Biol. 2018, 38, e00159-18. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Resch, W.; Corbett, E.; Yamane, A.; Casellas, R.; Schatz, D.G. The in vivo pattern of binding of RAG1 and RAG2 to antigen receptor loci. Cell 2010, 141, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Teng, G.; Maman, Y.; Resch, W.; Kim, M.; Yamane, A.; Qian, J.; Kieffer-Kwon, K.R.; Mandal, M.; Ji, Y.; Meffre, E.; et al. RAG Represents a Widespread Threat to the Lymphocyte Genome. Cell 2015, 162, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Akamatsu, Y.; Monroe, R.; Dudley, D.D.; Elkin, S.K.; Gartner, F.; Talukder, S.R.; Takahama, Y.; Alt, F.W.; Bassing, C.H.; Oettinger, M.A. Deletion of the RAG2 C terminus leads to impaired lymphoid development in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1209–1214. [Google Scholar] [CrossRef] [Green Version]
- Fattah, F.; Lee, E.H.; Weisensel, N.; Wang, Y.; Lichter, N.; Hendrickson, E.A. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010, 6, e1000855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattah, F.J.; Kweon, J.; Wang, Y.; Lee, E.H.; Kan, Y.; Lichter, N.; Weisensel, N.; Hendrickson, E.A. A role for XLF in DNA repair and recombination in human somatic cells. DNA Repair 2014, 15, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnie, N.J.; Gottlieb, T.M.; Blunt, T.; Jeggo, P.A.; Jackson, S.P. DNA-dependent protein kinase activity is absent in xrs-6 cells: Implications for site-specific recombination and DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 1995, 92, 320–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghezraoui, H.; Piganeau, M.; Renouf, B.; Renaud, J.B.; Sallmyr, A.; Ruis, B.; Oh, S.; Tomkinson, A.E.; Hendrickson, E.A.; Giovannangeli, C.; et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 2014, 55, 829–842. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Nelsen, C.; Hendrickson, E.A. Ku86 is essential in human somatic cells. Proc. Natl. Acad. Sci. USA 2002, 99, 832–837. [Google Scholar] [CrossRef] [Green Version]
- Ruis, B.; Molan, A.; Takasugi, T.; Hendrickson, E.A. Absence of XRCC4 and its paralogs in human cells reveal differences in outcomes for DNA repair and V(D)J recombination. DNA Repair 2020, 85, 102738. [Google Scholar] [CrossRef]
- Wang, Y.; Ghosh, G.; Hendrickson, E.A. Ku86 represses lethal telomere deletion events in human somatic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12430–12435. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frock, R.L.; Sadeghi, C.; Meng, J.; Wang, J.L. DNA End Joining: G0-ing to the Core. Biomolecules 2021, 11, 1487. https://doi.org/10.3390/biom11101487
Frock RL, Sadeghi C, Meng J, Wang JL. DNA End Joining: G0-ing to the Core. Biomolecules. 2021; 11(10):1487. https://doi.org/10.3390/biom11101487
Chicago/Turabian StyleFrock, Richard L., Cheyenne Sadeghi, Jodie Meng, and Jing L. Wang. 2021. "DNA End Joining: G0-ing to the Core" Biomolecules 11, no. 10: 1487. https://doi.org/10.3390/biom11101487
APA StyleFrock, R. L., Sadeghi, C., Meng, J., & Wang, J. L. (2021). DNA End Joining: G0-ing to the Core. Biomolecules, 11(10), 1487. https://doi.org/10.3390/biom11101487