4.2. Synthesis
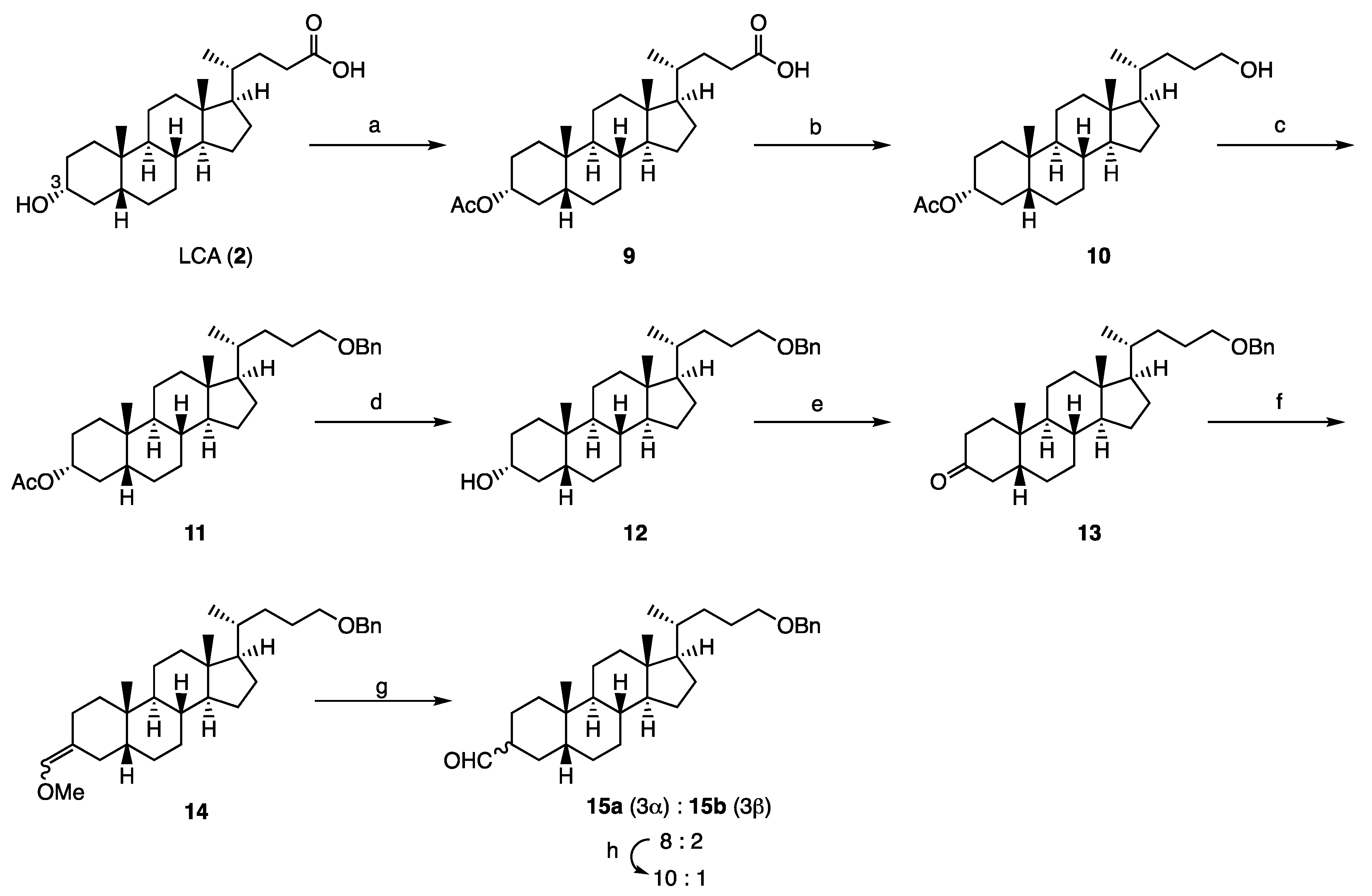
Synthesis of compound 9: Acetic anhydride (6.174 g, 60.48 mmol) and 4-dimethylaminopyridine (97 mg, 0.80 mmol) were added to a solution of lithocholic acid (1.522 g, 4.04 mmol) in dry pyridine (40 mL). The mixture was stirred for 20 h at room temperature, then quenched with water and extracted with a mixture of ethyl acetate and n-hexane (1:1). The organic layer was washed with 2 M hydrochloric acid and brine, dried over sodium sulfate, and filtered. The filtrate was concentrated to give 9 (1.757 g, quant.) as a yellow solid. 1H NMR (600 MHz, CDCl3) δ 4.74-4.70 (m, 1 H), 2.40 (ddd, J = 15.6, 10.2, 5.4 Hz, 1 H), 2.26 (ddd, J = 15.6, 9.6, 6.6 Hz, 1 H), 2.03 (s, 3 H), 1.98-1.95 (m, 1 H), 1.85-1.80 (m, 5 H), 1.68-1.00 (m, 20 H), 0.92 (d, J = 6.6 Hz, 3 H), 0.92 (s, 3 H), 0.64 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 178.33, 170.73, 74.39, 56.43, 55.90, 42.70, 41.82, 40.33, 40.08, 35.72, 35.28, 34.98, 34.54, 32.18, 30.71, 30.60, 28.16, 26.96, 26.57, 26.27, 24.14, 23.30, 21.50, 20.78, 18.20, 12.01; HRMS calcd for C26H42NaO4 (M + Na)+ 441.2975, found 441,2970.
Synthesis of compound 10: Triethylamine (533 mg, 5.27 mmol) and ethyl chloroformate (616 mg, 5.680 mmol) were added to a solution of 9 (1.757 g, 4.04 mmol) in distilled THF (40 mL). The mixture was stirred for 2 h at room temperature, then cooled to 0 °C, and sodium borohydride (737 mg, 19.5 mmol) and dry methanol (20 mL) were added to it. The reaction mixture was stirred for 2 h 15 min at 0 °C and then quenched with water. After removal of the solvent in vacuo, the residue was extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1:3) to give 10 (1.672 g, quant.) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 4.74-4.69 (m, 1 H), 3.65-3.58 (m, 2 H), 2.03 (s, 3 H), 1.99-1.96 (m, 1 H), 1.97-1.80 (m, 4 H), 1.70-1.02 (m, 23H), 0.93 (s, 3 H), 0.92 (d, J = 6.6 Hz, 3 H), 0.65 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 170.68, 74.37, 63.58, 56.43, 56.10, 42.62, 41.81, 40.32, 40.08, 35.70, 35.51, 34.95, 34.51, 32.16, 31.74, 29.31, 28.25, 26.95, 26.55, 26.26, 24.13, 23.28, 21.47, 20.76, 18.57, 11.98; HRMS calcd for C26H44NaO3 (M + Na)+ 427.3183, found 427.3174.
Synthesis of compound 11: Trifluoromethanesulfonic acid (0.25 mL, 2.85 mmol) was added to a solution of 10 (1.309 g, 3.24 mmol) in dry 1,4-dioxane (40.0 ml) and benzyl 2,2,2-trichloroacetimidate (2.206 g, 8.74 mmol) at 0 °C under an argon atmosphere. The reaction mixture was stirred for 3 h at room temperature, then cooled to 0 °C, quenched with saturated sodium hydrogen carbonate, and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1:14) to give compound 11 (1.297 g, 81%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.35-7.32 (m, 4 H), 7.30-7.27 (m, 1 H), 4.74-4.70 (m, 1 H), 4.51 (d, J = 12.0 Hz, 1 H), 4.49 (d, J = 12.0, 1 H), 3.46-3.40 (m, 2 H), 2.03 (s, 3 H), 1.98-1.96 (m, 1 H), 1.84-1.79 (m, 4 H), 1.68-1.66 (m, 2 H), 1.56-1.02 (m, 21 H), 0.92 (s, 3 H), 0.91 (d, J = 6.6 Hz, 3 H), 0.63 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 170.71, 138.61, 128.30, 127.60, 127.43, 74.41, 72.29, 71.01, 56.44, 56.14, 42.63, 41.83, 40.33, 40.10, 35.73, 35.53, 34.97, 34.53, 32.18, 32.12, 28.24, 26.98, 26.56, 26.28, 26.25, 24.16, 23.30, 21.48, 20.77, 18.55, 11.99; HRMS calcd for C33H50NaO3 (M + Na)+ 517.3652, found 517.3638.
Synthesis of compound 12: Potassium carbonate (1.464 g, 10.6 mmol) was added to a solution of 11 (1.344 g, 2.72 mmol) in dry methanol (30 mL) and distilled THF (5 mL). The mixture was stirred for 6 h 20 min at room temperature under an argon atmosphere and then quenched with acetic acid. After removal of the solvent in vacuo, the residue was extracted with ethyl acetate. The organic layer was washed with water and brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1:3) to give 12 (1.190 g, 97%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.36-7.33 (m, 4 H), 7.30-7.27 (m, 1 H), 4.51 (d, J = 13.2 Hz, 1 H), 4.49 (d, J = 12.0 Hz, 1 H), 3.64-3.60 (m, 1 H), 3.46-3.40 (m, 2 H), 1.96 (dt, J = 12.0, 3.0 Hz, 1 H), 1.86-1.66 (m, 7 H), 1.59-0.94 (m, 20 H), 0.91 (s, 3 H), 0.91 (d, J = 6.6 Hz, 3 H), 0.63 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 138.65, 128.32, 127.61, 172.43, 72.80, 71.88, 71.02, 56.46, 56.13, 42.64, 42.04, 40.37, 40.13, 36.39, 35.79, 35.57, 35.29, 34.53, 32.13, 30.50, 28.27, 27.16, 26.39, 26.28, 24.20, 23.35, 20.78, 18.56, 12.00; HRMS calcd for C31H48NaO2 (M + Na)+ 475.3547, found 475.3541.
Synthesis of compound 13: Sulfuric acid (0.23 mL) was added to a cooled solution of chromium (VI) oxide (267 mg) in water (0.77 mL) just prior to use. An aliquot (0.7 mL) of this Jones reagent was added to a solution of 12 (1.173 g, 2.59 mmol) in dry acetone (30 mL). The mixture was stirred for 30 min at room temperature, then quenched with 2-propanol, and the solvent was removed in vacuo. The extract was extracted with diethyl ether. The organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1:4) to give 13 (1.148 g, 98%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.36-7.33 (m, 4 H), 7.29-7.27 (m, 1 H), 4.51 (d, J = 12.6 Hz, 1 H), 4.49 (d, J = 12.6 Hz, 1 H), 3.47-3.40 (m, 2 H), 2.70 (t, J = 14.4 Hz, 1 H), 2.33 (td, J = 14.4, 5.4 Hz, 1 H), 2.17-2.14 (m, 1 H), 2.05-2.00 (m, 3 H), 1.88-1.79 (m, 3 H), 1.70-1.08 (m, 19 H), 1.01 (s, 3 H), 0.92 (d, J = 6.6 Hz, 3 H), 0.67 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 213.64, 138.62, 128.31, 127.60, 127.44, 72.81, 70.99, 56.40, 56.12, 44.32, 42.68, 42.35, 40.65, 40.01, 37.21, 36.99, 35.54, 35.48, 34.85, 32.12, 28.22, 26.58, 26.17, 25.74, 24.14, 22.63, 21.15, 18.57, 12.03; HRMS calcd for C31H46NaO2 (M + Na)+ 473.3390, found 473.3379.
Synthesis of compound 14: A mixture of (methoxylmethyl)triphenylphosphonium chloride (18.111 g, 52.8 mmol) and potassium tert-butoxide (6.188 g, 46.7 mmol) in distilled THF (112 mL) was stirred for 45 min at 0 °C under an argon atmosphere. A solution of 13 (6.812 g, 15.1 mmol) in distilled THF (18 mL) was added to it. The resulting mixture was allowed to warm to room temperature, stirred for 2 h, quenched with water, and extracted with ethyl acetate. The organic layer was washed with water and brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (chloroform/n-hexane = 3:2) to give 14 (7.280 g, quant.) as a mixture of geometrical isomers. Each isomer was isolated in a small amount to determine the structure. (E)-14: 1H NMR (600 MHz, CDCl3) δ 7.35-7.33 (m, 4 H), 7.29-7.27 (m, 1 H), 5.76 (s, 1 H), 4.51 (d, J = 12.0 Hz, 1 H), 4.49 (d, J = 12.0 Hz, 1 H), 3.52 (s, 3 H), 3.46-3.40 (m, 2 H), 2.31 (dd, J = 13.8, 3.6 Hz, 1 H), 2.12 (t, J = 13.8 Hz, 1 H), 1.98-1.95 (m, 1 H), 1.86-1.79 (m, 3 H), 1.71-1.66 (m, 2 H), 1.57-0.92 (m, 20 H), 0.91 (s, 3 H), 0.91 (d, J = 5.4 Hz, 3 H), 0.64 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 138.75, 138.63, 128.32, 127.61, 127.43, 118.80, 72.82, 71.07, 59.28, 56.60, 56.23, 43.67, 42.72, 40.24, 40.15, 38.68, 35.76, 35.59, 32.22, 28.29, 26.99, 26.34, 26.28, 25.71, 25.12, 24.24, 23.73, 20.97, 18.61, 12.06; HRMS calcd for C33H50NaO2 (M + Na)+ 501.3707, found 501.3698. (Z)-14: 1H NMR (600 MHz, CDCl3) δ 7.35-7.33 (m, 4 H), 7.29-7.27 (m, 1 H), 5.72 (s, 1 H), 4.51 (d, J = 12.0 Hz, 1 H), 4.49 (d, J = 12.0 Hz, 1 H), 3.52 (s, 3 H), 3.46-3.40 (m, 2 H), 2.46 (d, J = 13.8 Hz, 1 H), 2.39 (t, J = 13.6 Hz, 1 H), 1.98-1.95 (m, 1 H), 1.86-1.79 (m, 3 H), 1.71-1.66 (m, 2 H), 1.57-0.92 (m, 20 H), 0.91 (s, 3 H), 0.91 (d, J = 5.4 Hz, 3 H), 0.64 (s, 3 H). 13C; HRMS calcd for C33H50NaO2 (M + Na)+ 501.3707, found 501.3698.
Synthesis of compound 15: 6 M hydrochloric acid (30 mL) was added to a solution of 14 (6.975 g, 14.6 mmol) in distilled THF (80 mL), and the mixture was stirred for 5 h at room temperature. The reaction mixture was quenched with water, and extracted with ethyl acetate. The organic layer was washed with water and brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column flash chromatography (dichloromethane/n-hexane = 1:1) to give a mixture of 15a and 15b (6.664 g, 98%, 15a:15b = 8:2) as a colorless oil. 15a: 1H NMR (600 MHz, CDCl3) δ 9.64 (d, J =1.8 Hz, 1 H), 7.36-7.33 (m, 4 H), 7.29-7.27 (m, 1 H), 4.51 (d, J = 13.2 Hz, 1 H), 4.49 (d, J = 12.0 Hz, 1 H), 3.46-3.40 (m, 2 H), 2.27 (dtt, J = 12.6, 3.6, 1.8 Hz, 1 H), 1.97-0.97 (m, 28 H), 0.96 (s, 3 H), 0.91 (d, J = 6.6 Hz, 3 H), 0.63 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 205.13, 138.64, 128.31, 127.61, 127.43, 72.80, 71.01, 56.36, 56.11, 51.32, 42.62, 42.56, 40.32, 40.03, 36.06, 35.74, 35.56, 35.10, 32.12, 28.24, 27.16, 26.28, 24.16, 23.81, 20.78, 18.55, 12.00; HRMS calcd for C32H48NaO2 (M + Na)+ 487.3547, found 487.3535. 15b: 1H NMR (600 MHz, CDCl3) δ 9.71 (s, 1 H), 7.36-7.33 (m, 4 H), 7.23-7.27 (m, 1 H), 4.51 (d, J = 12.6 Hz, 1 H), 4.49 (d, J = 12.6 Hz, 1 H), 3.46-3.39 (m, 2 H), 2.45 (br, 1 H), 2.04-0.85 (m, 28 H), 0.91 (d, J = 6.6 Hz, 3 H), 0.87 (s, 3 H), 0.63 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 206.55, 138.64, 128.33, 127.63, 127.46, 72.82, 71.03, 56.56, 56.16, 47.08, 42.68, 40.15, 39.98, 39.60, 35.58, 35.54, 34.90, 33.48, 32.14, 28.26, 26.91, 26.26, 26.07, 25.00, 24.18, 23.85, 20.85, 19.60, 18.58, 12.02; HRMS calcd for C32H48NaO2 (M + Na)+ 487.3547, found 487.3531.
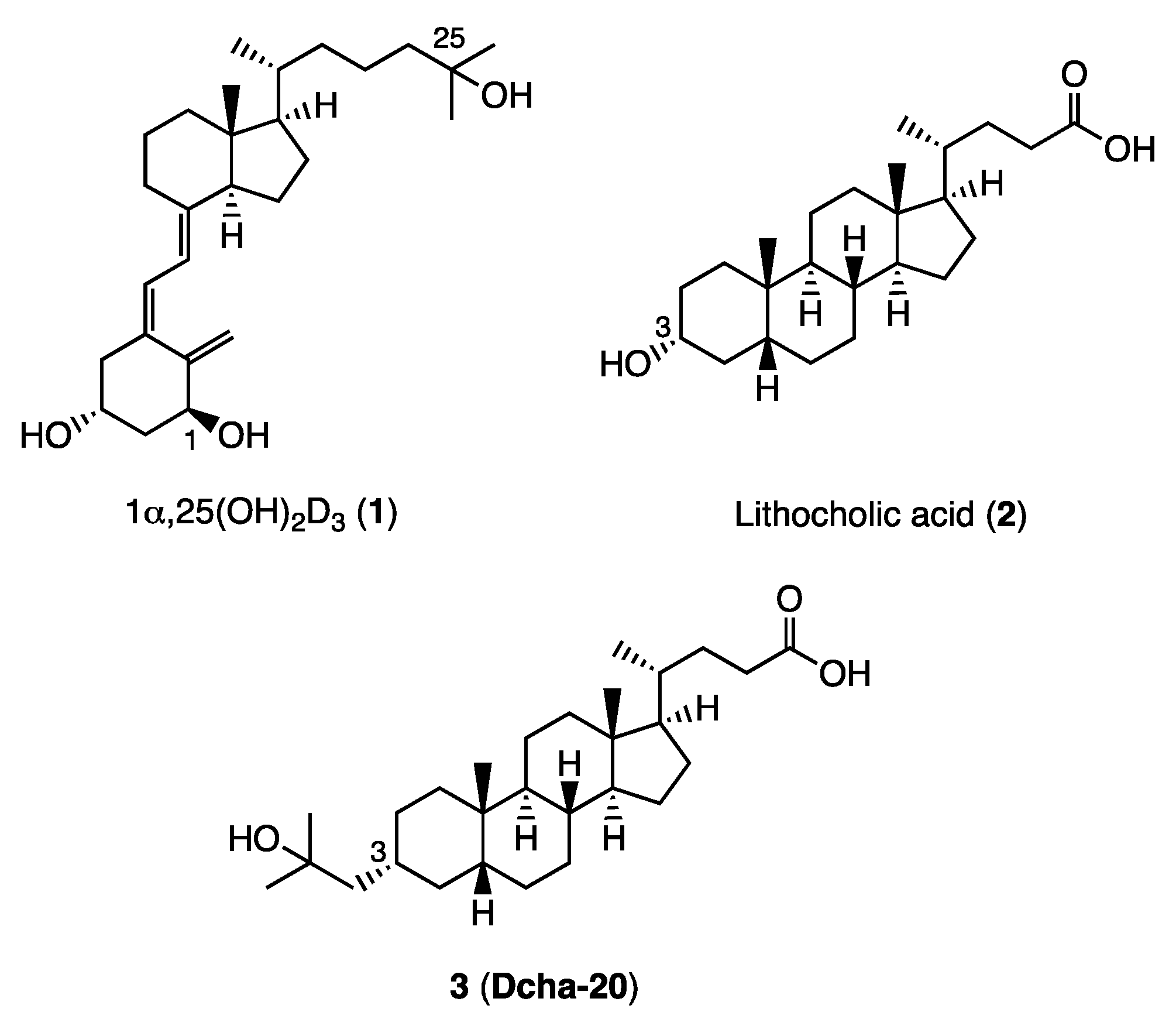
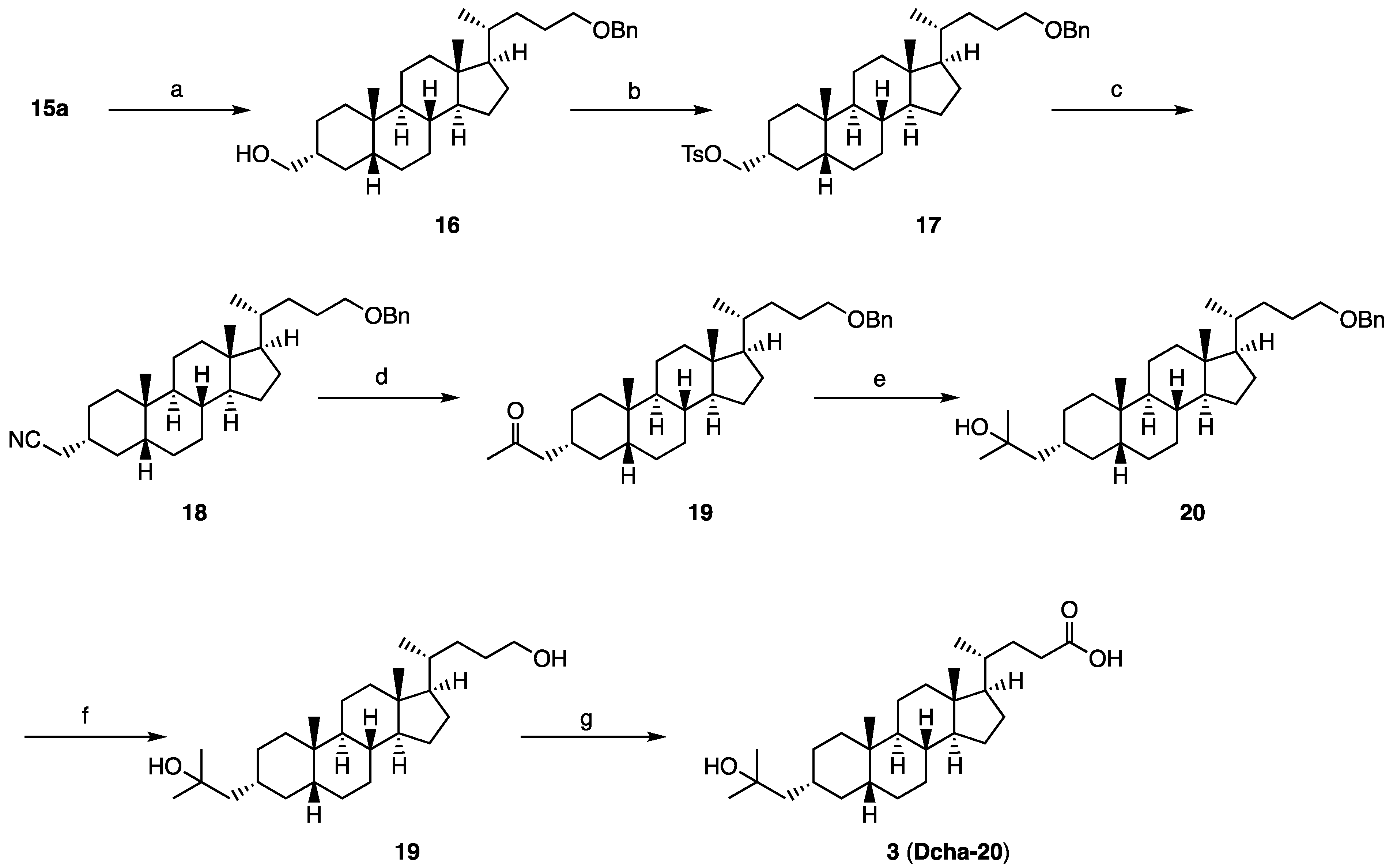
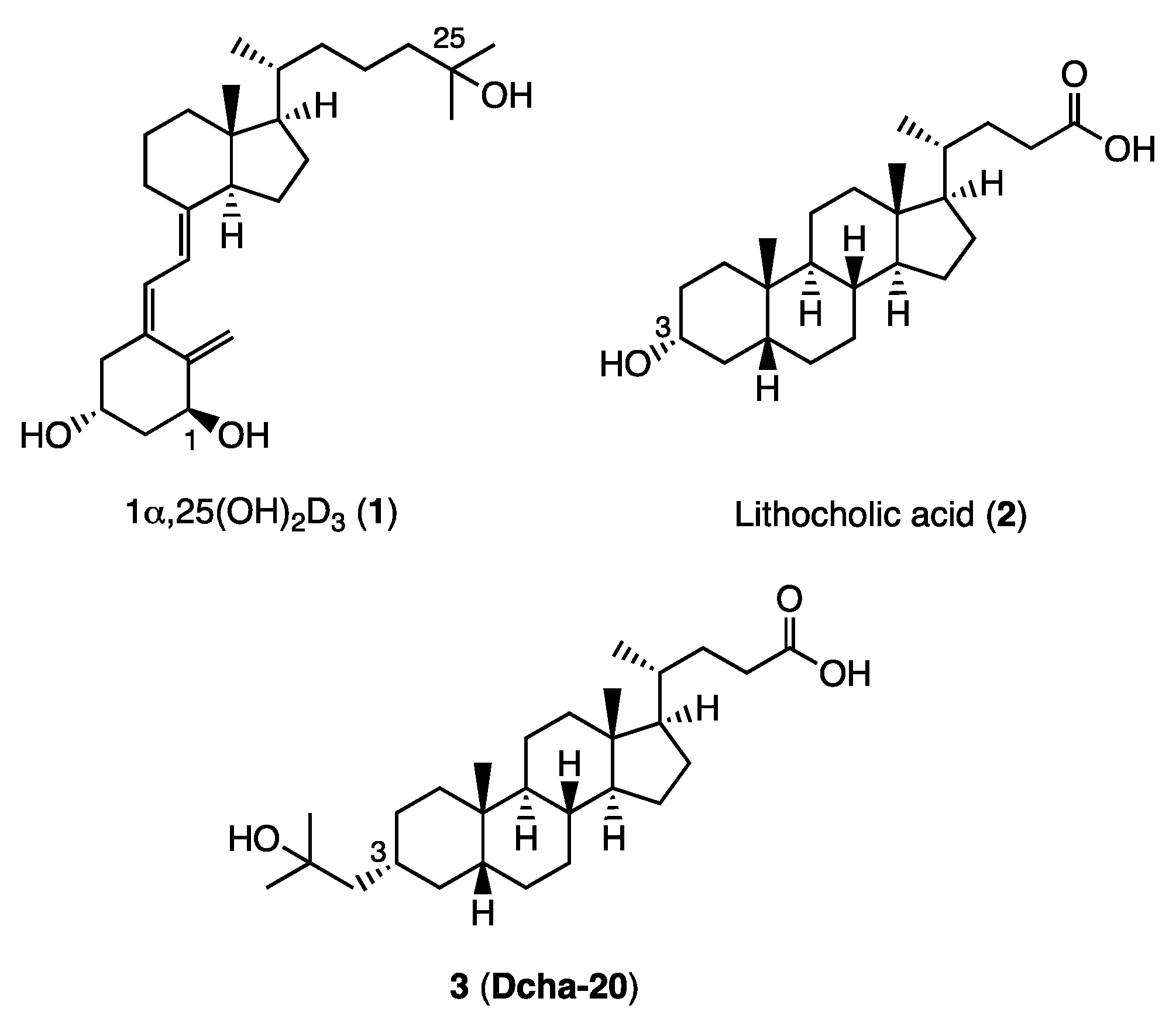
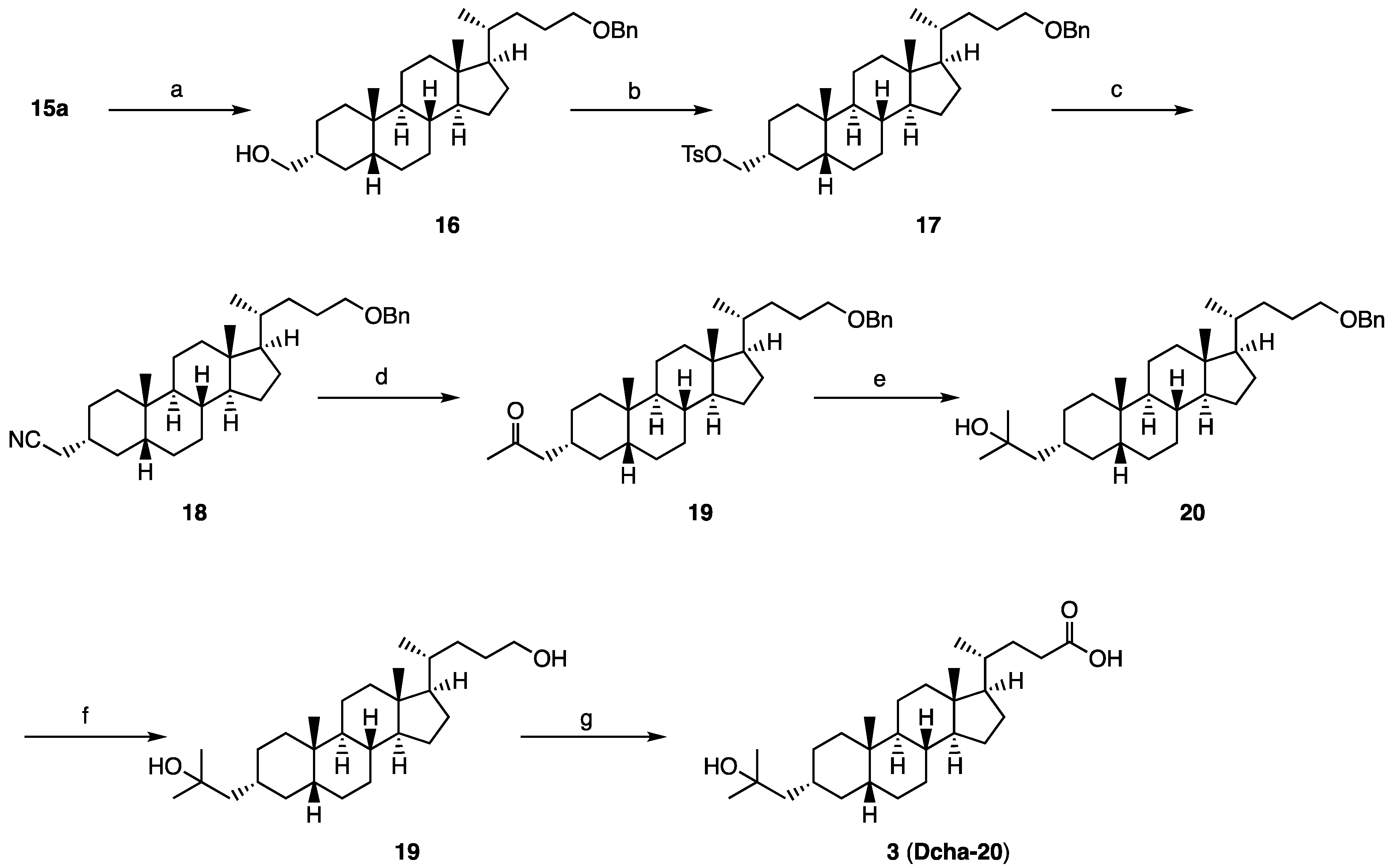
The ratio of 15a in the mixture of the epimers was increased by treatment of the mixture with K2CO3, MeOH, THF (66%), and the isolated 15a was converted to compound 3 (Dcha-20), according to our reported method.
Synthesis of compound 20: Acetyl chloride (0.01 mL, 0.140 mmol) was added to a cooled solution of 3 (64 mg, 0.15 mmol) in methanol (7 mL) at 0 °C under an argon atmosphere. The mixture was stirred for 4 h at room temperature, then quenched with water at 0 °C, and the precipitate was collected to obtain 20 (72 mg, quant.) as a colorless solid. 1H NMR (400 MHz, CDCl3) δ 3.66 (s, 3H), 2.39-2.31 (m, 1H), 2.25-2.17 (m, 1H), 1.94 (d, J = 11.4 Hz, 1H), 1.89-1.73 (m, 4H), 1.59-0.90 (m, 25H), 1.22 (s, 6H), 0.91 (d, J = 6.4 Hz, 3H), 0.91 (s, 3H), 0.64 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 174.84, 71.65, 60.39, 56.50, 55.89, 51.50, 51.23, 43.55, 42.70, 40.46, 40.15, 37.43, 35.89, 35.80, 35.34, 34.97, 34.68, 31.01, 30.97, 30.02, 29.97, 28.18, 27.40, 26.45, 24.17, 23.97, 21.07, 20.78, 18.24, 14.17, 12.01; HRMS calcd for C29H50NaO3 (M + Na)+ 469.3652, found 484.3.655.
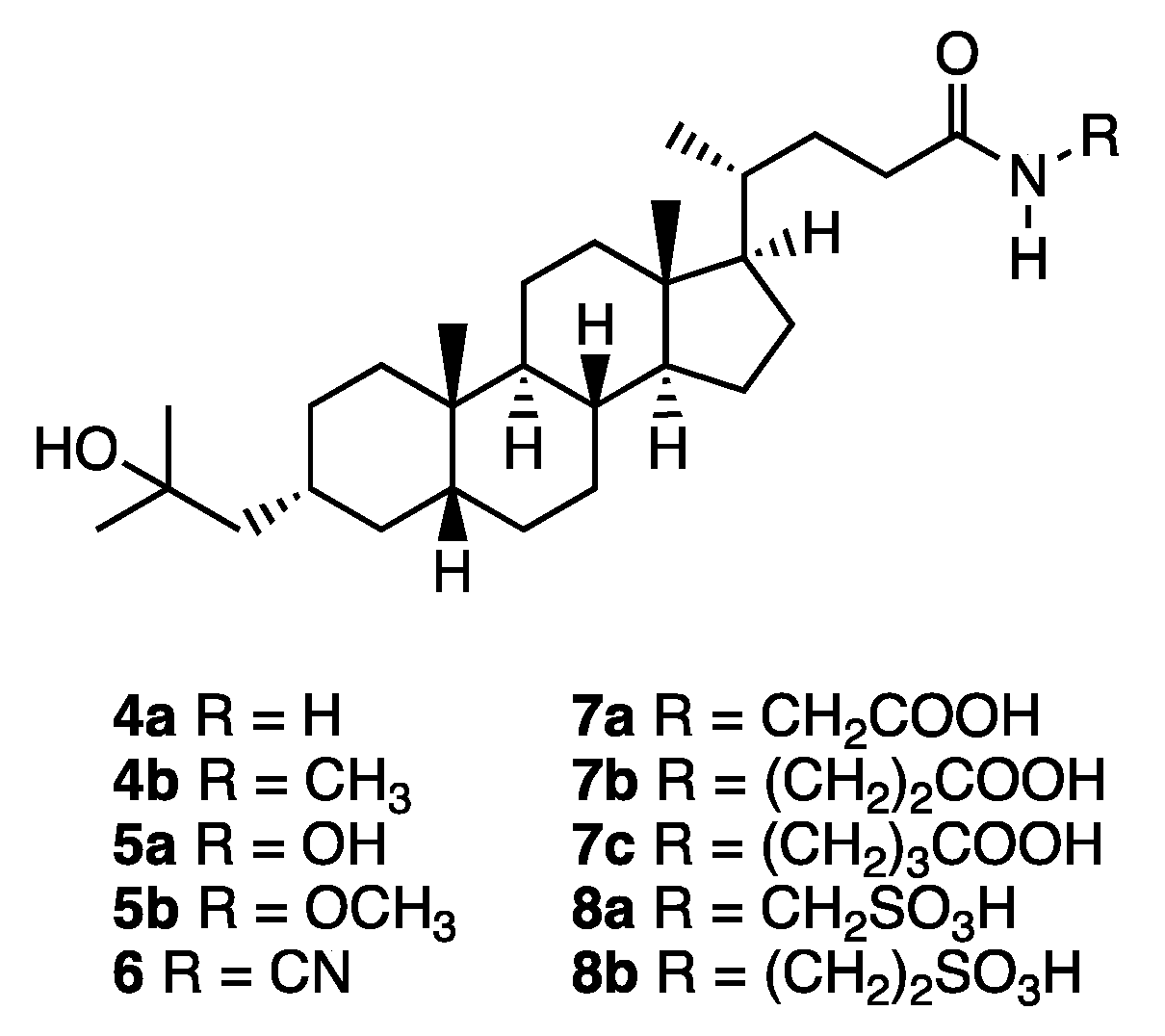
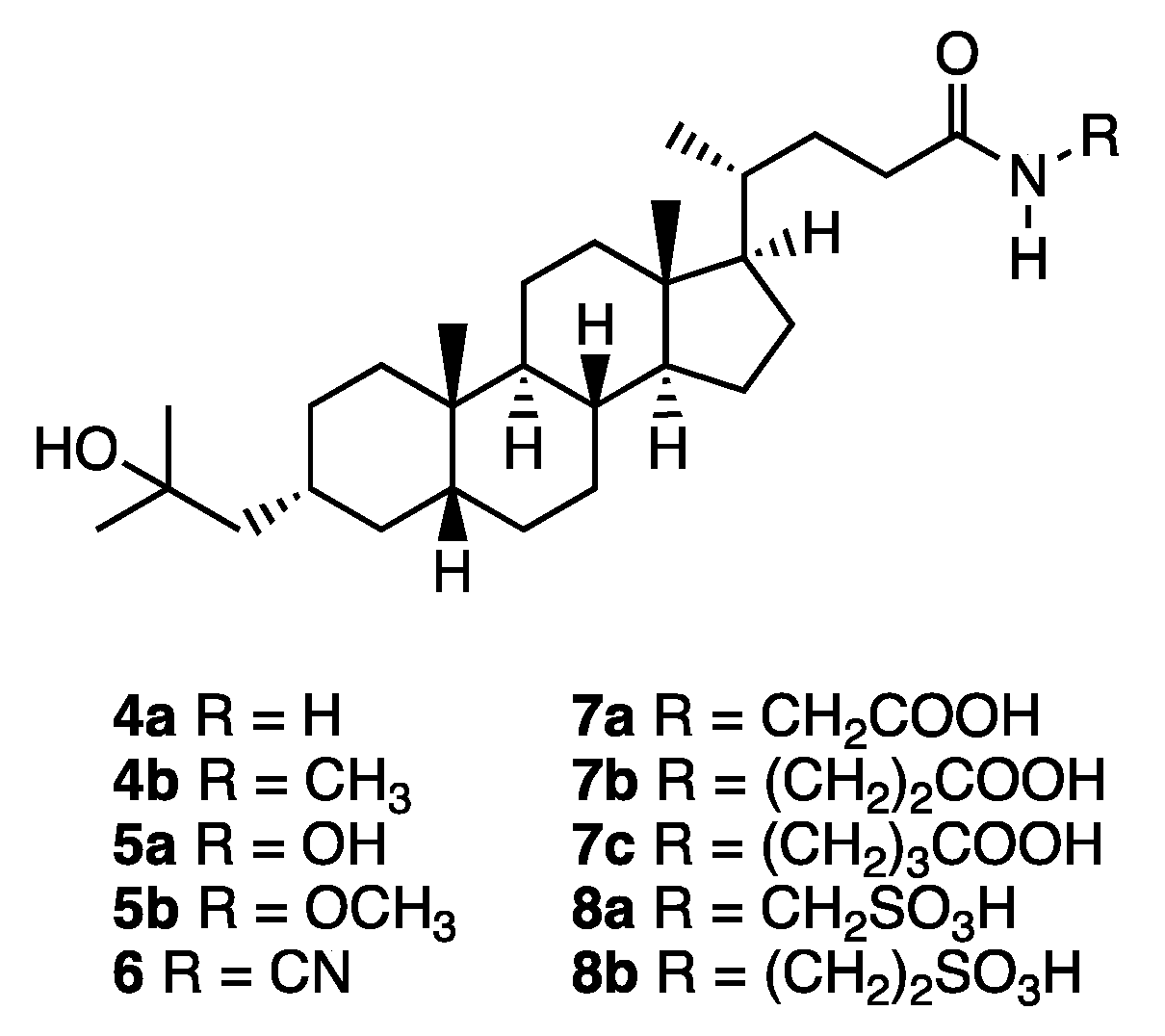
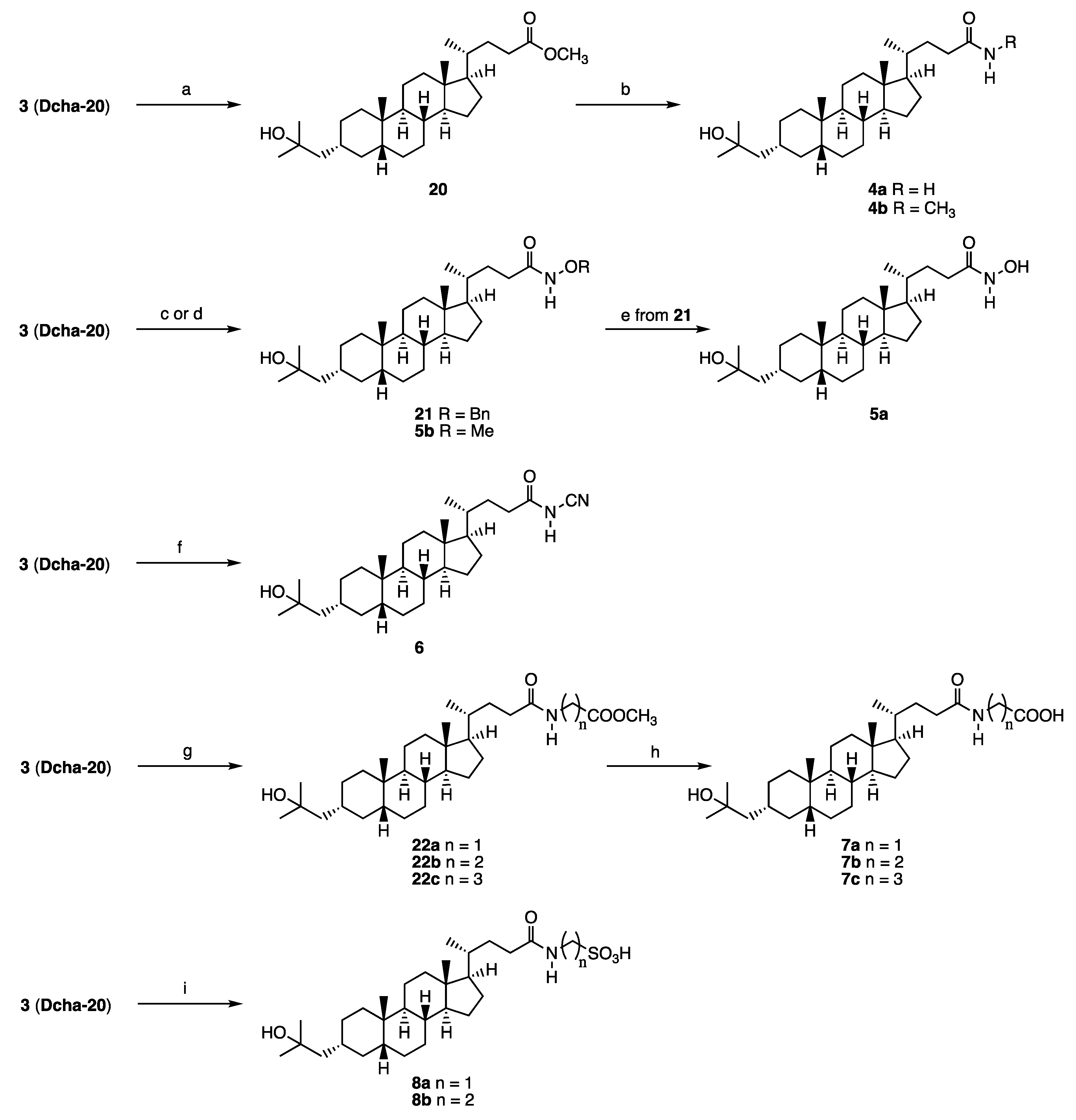
Synthesis of compound 4: A methanolic solution of conc. ammonia (4 mL) was added to 20 (37 mg, 0.084 mmol). The mixture was stirred for 7 d at room temperature, and then for 4 d at 30 °C. The solvent was removed in vacuo, and the residue was purified by GPC (chloroform) to give 4a (18 mg, 51%) as a colorless solid. 1H NMR (400 MHz, CDCl3) δ 5.36 (brs, 1H), 5.22 (brs, 1H), 2.32-2.25 (m, 1H), 2.15-2.07 (m, 1H), 1.95 (d, J = 11.4 Hz, 1H), 1.87-1.73 (m, 4H), 1.59-0.91 (m, 25H), 1.22 (s, 6H), 0.93 (d, J = 6.4 Hz, 3H), 0.91 (s, 3H), 0.64 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 175.82, 71.65, 56.50, 55.93, 51.22, 43.54, 42.72, 40.46, 40.17, 37.42, 35.89, 35.80, 35.45, 34.97, 34.67, 32.75, 31.59, 30.03, 29.96, 29.76, 28.25, 27.39, 26.45, 24.17, 23.96, 20.78, 18.33, 12.02; HRMS calcd for C28H49NaO2 (M+Na)+ 454.3656, found 454.3649.
Compound 4b was synthesized similarly. 4b: 1H NMR (400 MHz, CDCl3) δ 5.38 (brs, 1H), 2.80 (d, J = 5.0 Hz, 3H), 2.23-2.19 (m, 1H), 2.08-2.02 (m, 1H), 1.94 (d, J = 11.4 Hz, 1H), 1.85-1.73 (m, 4H), 1.49-0.90 (m, 25H), 1.22 (s, 6H), 0.91 (d, J = 6.4 Hz, 3H), 0.91 (s, 3H), 0.63 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 174.19, 71.66, 56.51, 55.97, 51.23, 43.55, 42.70, 40.47, 40.17, 37.43, 35.89, 35.80, 35.52, 34.97, 34.68, 33.55, 31.82, 30.03, 29.96, 29.77, 28.24, 27.40, 26.45, 26.30, 24.17, 23.97, 20.78, 18.35, 12.02; HRMS calcd for C29H51NaO2 (M + Na)+ 468.3796, found 468.3805.
Synthesis of compound 21: O-Benzylhydroxylamine hydrochloride (24 mg, 0.15 mmol), N,N-diisopropylethylamine (20 mg, 0.16 mmol) and 1-hydroxybenzotriazole (17 mg, 0.13 mmol) were successively added to a solution of 3 (49 mg, 0.11 mmol) in dry dichloromethane (10 mL) at room temperature. After 10 min, N,N’-dicyclohexylcarbodiimide (29 mg, 0.14 mmol) was added to it. The mixture was stirred for 23 h at room temperature, and then filtered. The filtrate was washed with 5% hydrochloric acid, and brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 1:2) to give 21 (68 mg, quant.) as a colorless solid. 1H NMR (600 MHz, CDCl3) δ 7.85 (br, 1 H), 7.41-7.39 (m, 5 H), 4.92 (br, 2 H), 1.94-0.92 (m, 31 H), 1.22 (s, 6 H), 0.91 (s, 3 H), 0.88 (d, J = 4.8 Hz, 3 H), 0.62 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 171.44, 137.83, 129.24, 128.59, 128.18, 78.03, 71.66, 56.48, 55.88, 51.21, 43.53, 42.69, 40.45, 40.15, 37.41, 35.88, 35.78, 35.41, 34.95, 34.66, 31.43, 30.02, 29.93, 29.75, 29.66, 28.19, 27.38, 26.43, 24.15, 23.95, 20.76, 18.26, 12.02; HRMS calcd for C35H56NO3 (M + H)+ 538.4255, found 538.4241.
Synthesis of compound 5b: Compound 3 (20 mg, 0.047 mmol) in DMF (0.5 mL) was added to a solution of triethylamine (12 mg, 0.12 mmol) in DMF (0.5 mL). The mixture was stirred at 0 °C for 10 min, and then ethyl chloroformate (6 mg, 0.055 mmol) in DMF (0.5 mL) was added to it. The resulting mixture was stirred at 0 °C for 45 min, and a mixture of methoxyamine hydrochloride (4 mg, 0.052 mmol) and triethylamine (12 mg, 0.12 mmol) in DMF (1.0 mL) was added to it. Stirring was continued at room temperature for 4 h, then the solvent was removed in vacuo, and the residue was extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by GPC (chloroform) to give 5b (16 mg, 73%) as a colorless solid. 1H NMR (400 MHz, CDCl3) δ 7.98 (brs, 1H), 3.76 (s, 3H), 2.17 (brs, 1H), 1.94 (d, J = 10.5, 1H), 1.87-1.73 (m, 4H), 1.59-0.91 (m, H), 1.22 (s, 6H), 0.92 (d, J = 8.2 Hz, 3H), 0.91 (s, 3H), 0.64 (s, 3H) ; 13C NMR (150MHz, CDCl3) δ 171.64, 71.73, 64.56, 56.56, 55.95, 51.28, 43.60, 42.78, 40.52, 40.22, 37.48, 35.95, 35.85, 35.51, 35.03, 34.73, 31.45, 30.25, 30.09, 30.01, 29.83, 28.28, 27.45, 26.50, 24.22, 24.02, 20.84, 18.38, 12.08; HRMS calcd for C29H51NaO3 (M + Na)+ 484.3761, found 484.3762.
Synthesis of compound 5a: Palladium hydroxide (13 mg) was added to a solution of 21 (68 mg, 0.11 mmol) in dry methanol (15 mL). The mixture was stirred for 24 h at room temperature under a hydrogen atmosphere, then filtered, and the filtrate was concentrated. The residue was purified by silica gel column chromatography (ethyl acetate/n-hexane = 2:1, ethyl acetate, then, ethyl acetate/methanol = 20:1) to give 5a (31 mg, 55%) as a colorless solid. 1H NMR (600 MHz, CDCl3) δ 5.40 (br, 1 H), 5.33 (br, 1 H), 2.28 (ddd, J = 15.6, 10.8, 4.2 Hz 1 H), 2.11 (ddd, J = 16.8, 10.8, 6.0 Hz 1 H), 1.95-1.92 (m, 1 H), 1.85-0.94 (m, 28 H), 1.22 (s, 6 H), 0.92 (d, J = 6.6 Hz, 3 H), 0.90 (s, 3 H), 0.63 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 176.04, 71.69, 56.52, 55.94, 51.23, 43.56, 42.73, 40.47, 40.18, 37.44, 35.90, 35.81, 35.46, 34.98, 34.69, 32.77, 31.61, 30.04, 29.97, 29.78, 28.27, 27.41, 26.46, 24.18, 23.98, 20.80, 18.35, 12.04; Anal. calcd for C28H49NO3: C, 75.12; H, 11.03; N, 3.15, found: C, 76.35; H, 11.04; N, 3.20.
Synthesis of compound 6: 4-Dimethylaminopyridine (23 mg, 0.19 mmol), cyanamide (19 mg, 0.45 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (42 mg, 0.22 mmol) and N,N-diisopropylethylamine (39 mg, 0.30 mmol) were successively added to a solution of 3 (59 mg, 0.14 mmol) in dry dichloromethane (5 mL). The mixture was stirred for 18 h at room temperature under an argon atmosphere, then diluted with dichloromethane, washed with 2 M hydrochloric acid and brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel flash column chromatography (chloroform/methanol = 10:1) to give 6 (5 mg, 89%) as a colorless oil. 1H NMR (600 MHz, pyridine-d5) δ 2.65 (br, 1 H), 2.52 (br, 1 H), 2.01-1.95 (m, 1 H), 1.86-1.72 (m, 6 H), 1.65-0.85 (m, 22 H), 1.44 (s, 6 H), 0.92 (s, 3 H), 0.88 (d, J = 6.0 Hz, 3 H), 0.56 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 174.15, 108.26, 71.57, 56.39, 55.76, 51.03, 43.46, 42.64, 40.36, 40.05, 37.34, 35.82, 35.70, 35.21, 34.85, 34.57, 32.21, 30.49, 29.68, 29.62, 29.51, 28.08, 27.30, 26.35, 24.07, 23.85, 20.68, 18.09, 11.90; HRMS calcd for C29H47N2O2 (M-H)- 455.3643, found 455.3627.
Synthesis of compound 22: L-Glycine methyl ester hydrochloride (8 mg, 0.06 mmol) and N-methylmorpholine (13 mg, 0.12 mmol) were added to a solution of 3 (20 mg, 0.047 mmol) in dry dichloromethane (8 mL). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (12 mg, 0.06 mmol) was added to the mixture under an argon atmosphere. The resulting mixture was stirred for 24 h at room temperature, then diluted with dichloromethane, washed with 2 M hydrochloric acid, and brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel column chromatography (dichloromethane/methanol = 19:1) to give 22a (19 mg, 83%) as a colorless solid. 1H NMR (600 MHz, CDCl3) δ 5.91 (br, 1 H), 4.05 (d, J = 5.4 Hz, 2 H), 3.77 (s, 3 H), 2.30 (ddd, J = 15.6, 10.8, 5.4 Hz 1 H), 2.13 (ddd, J = 15.0, 10.2, 6.0 Hz 1 H), 1.96-1.94 (m, 1 H), 1.87-1.73 (m, 5 H), 1.56-0.96 (m, 23 H), 1.22 (s, 6 H), 0.92 (d, J = 6.6 Hz, 3 H), 0.91 (s, 3 H), 0.63 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 173.58, 170.52, 71.55, 56.40, 55.85, 52.19, 51.13, 43.45, 42.61, 41.07, 40.36, 40.07, 37.33, 35.79, 35.70, 35.36, 34.87, 34.58, 33.14, 31.46, 29.93, 29.86, 29.67, 28.13, 27.30, 26.35, 24.07, 23.87, 20.68, 18.23, 11.92; HRMS calcd for C31H53NNaO4 (M + Na)+ 526.3867, found 526.3860.
Compounds 22b and 22c were synthesized similarly. 22b: 1H NMR (600 MHz, CDCl3) δ 6.01 (t, J = 6.6 Hz, 1 H), 3.70 (s, 3 H), 3.51 (q, J = 6.0 Hz, 2 H), 2.54 (t, J = 6.0 Hz, 2 H), 2.24-2.18 (m, 1 H), 2.07-2.01 (m, 1 H), 1.95-1.92 (m, 1 H), 1.87-1.73 (m, 4 H), 1.56-0.96 (m, 24 H), 1.22 (s, 6 H), 0.91 (s, 3 H), 0.90 (d, J = 6.0 Hz, 3 H), 0.63 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 173.64, 173.32, 71.67, 56.50, 55.96, 51.83, 51.22, 43.55, 42.70, 40.46, 40.17, 37.43, 35.89, 35.80, 35.47, 34.97, 34.67, 33.81, 33.62, 31.70, 30.02, 29.95, 29.77, 28.24, 27.40, 26.45, 24.17, 23.96, 20.78, 18.33, 12.01; HRMS calcd for C32H55NNaO4 (M + Na)+ 540.4023, found 540.4033. 22c: 1H NMR (600 MHz, CDCl3) δ 5.63 (br, 1 H), 3.68 (s, 3 H), 3.29 (q, J = 7.2 Hz, 2 H), 2.37 (t, J = 7.2 Hz, 2 H), 2.22 (ddd, J = 15.6, 10.2, 4.2 Hz, 1 H), 2.04 (ddd, J = 15.6, 10.2, 6.0 Hz, 1 H), 1.94-1.02 (m, 1 H), 1.84 (q, J = 7.2 Hz, 2 H), 1.77-1.73 (m, 3 H), 1.57-0.93 (m, 25 H), 1.22 (s, 6 H), 0.91 (d, J = 6.6 Hz, 3 H), 0.91 (s, 3 H), 0.63 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 173.98, 173.80, 71.66, 56.49, 55.95, 51.75, 51.21, 43.54, 42.69, 40.45, 40.16, 38.92, 37.42, 35.88, 35.79, 35.48, 34.96, 34.66, 33.61, 31.77, 31.46, 30.01, 29.94, 29.76, 28.24, 27.39, 26.44, 24.54, 24.16, 23.95, 20.77, 18.34, 12.00; HRMS calcd for C33H57NNaO4 (M + Na)+ 554. 4180, found 554.4179.
Synthesis of compound 7: 15% w/v aqueous sodium hydroxide (1 mL) was added to a solution of 22a (8 mg, 0.035 mmol) in ethanol (5 mL), and the mixture was stirred for 3 h at room temperature. Ethanol was removed in vacuo, and the solution was acidified with conc. hydrochloric acid until a precipitate was formed. This was collected and washed with water to give 7a (13 mg, 77%) as a colorless solid. 1H NMR (600 MHz, CD3OD) δ 3.86 (s, 2 H), 2.30 (ddd, J = 13.8, 10.2, 5.4 Hz, 1 H), 2.15 (ddd, J = 13.8, 9.6, 6.6 Hz, 1 H), 2.00-1.98 (m, 1 H), 1.91-1.85 (m, 2 H), 1.82-1.76 (m, 2 H), 1.60-0.98 (m, 24 H), 1.17 (s, 6 H), 0.96 (d, J = 6.0 Hz, 3 H), 0.93 (s, 3 H), 0.68 (s, 3 H); 13C NMR (150 MHz, CD3OD) δ 177.10, 173.50, 72.01, 57.95, 57.46, 52.08, 49.56, 45.15, 43.91, 42.03, 41.90, 41.57, 38.69, 37.27, 36.83, 36.22, 35.82, 33.81, 33.08, 31.00, 29.93, 29.91, 29.25, 28.63, 27.75, 25.30, 24.54, 21.94, 18.83, 12.47; HRMS calcd for C30H51NNaO4 (M + Na)+ 512.3710, found 512.3707.
Compounds 7b and 7c were synthesized similarly. 7b: 1H NMR (600 MHz, CD3OD) δ 3.39 (t, J = 6.6 Hz, 2 H), 2.48 (t, J = 6.6 Hz, 2 H), 2.21-2.19 (m, 1 H), 2.10-2.05 (m, 1 H), 2.00-1.98 (m, 1 H), 1.91-1.87 (m, 2 H), 1.78-1.73 (m, 2 H), 1.59-1.49 (m, 4 H), 1.40-0.96 (m, 26 H), 0.94 (d, J = 7.2 Hz, 3 H), 0.93 (s, 3 H), 0.67 (s, 3 H); 13C NMR (150 MHz, CD3OD) δ 176.87, 175.48, 72.00, 57.94, 57.43, 52.08, 45.14, 43.90, 41.89, 41.55, 38.68, 37.26, 36.85, 36.42, 36.21, 35.81, 34.77, 34.00, 33.28, 30.99, 29.92, 29.90, 29.28, 28.62, 27.74, 25.28, 24.53, 21.93, 18.81, 12.45; HRMS calcd for C31H53NNaO4 (M + Na)+ 526.3867, found 526.3862. 7c: 1H NMR (600 MHz, CDCl3) δ 5.63 (br, 1 H), 3.68 (s, 3 H), 3.29 (q, J = 7.2 Hz, 2 H), 2.37 (t, J = 7.2 Hz, 2 H), 2.22 (ddd, J = 15.6, 10.2, 4.2 Hz, 1 H), 2.04 (ddd, J = 15.6, 10.2, 6.0 Hz, 1 H), 1.94-1.02 (m, 1 H), 1.84 (q, J = 7.2 Hz, 2 H), 1.77-1.73 (m, 3 H), 1.57-0.93 (m, 25 H), 1.22 (s, 6 H), 0.91 (d, J = 6.6 Hz, 3 H), 0.91 (s, 3 H), 0.63 (s, 3 H); 13C NMR (150 MHz, CDCl3) δ 173.98, 173.80, 71.66, 56.49, 55.95, 51.75, 51.21, 43.54, 42.69, 40.45, 40.16, 38.92, 37.42, 35.88, 35.79, 35.48, 34.96, 34.66, 33.61, 31.77, 31.46, 30.01, 29.94, 29.76, 28.24, 27.39, 26.44, 24.54, 24.16, 23.95, 20.77, 18.34, 12.00; HRMS calcd for C33H57NNaO4 (M + Na)+ 554. 4180, found 554.4179.
Synthesis of compound 8: 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (123 mg, 0.44 mmol) and triethylamine (437 mg, 4.32 mmol) were added to a solution of 3 (75 mg, 0.17 mmol) in dry N,N-dimethylformamide (8 mL). The mixture was stirred for 10 min at room temperature under an argon atmosphere, and aminomethanesulfuric acid (132.2 mg, 1.190 mmol) was added to it. The mixture was stirred for 20 h at room temperature, then filtered, and the solvent was removed in vacuo. The residue was extracted with ethyl acetate and water. The water layer was cooled to 0 °C, and conc. hydrochloric acid (5.0 mL) was added. The resulting precipitate was collected, and washed with water to give 8a (67 mg, 74%) as a pale yellow solid. 1H NMR (600 MHz, CD3OD) δ 4.16 (q, J = 13.2 Hz, 2 H), 2.18 (br, 1 H), 2.02 (br, 1 H), 1.85 (d, J = 11.4 Hz, 1 H), 1.75 (m, 2 H), 1.63 (d, J = 13.8 Hz, 2 H), 1.44-0.84 (m, 24 H), 1.03 (s, 6 H), 0.81 (d, J = 6.0 Hz, 3 H), 0.79 (s, 3 H), 0.53 (s, 3 H); HRMS calcd for C29H50NO5S (M-H)- 524.3415, found 524.3404.
Compound 8b was synthesized similarly. 8b: 1H NMR (600 MHz, Pyrdine-d5) δ 4.23 (t, J = 5.4 Hz, 2 H), 3.48 (t, J = 5.4 Hz, 2 H), 2.40 (ddd, J = 14.4, 10.2, 4.2 Hz, 1 H), 2.25 (ddd, J = 16.2, 10.2, 6.0 Hz, 1 H), 2.00-1.98 (m, 1 H), 1.82-0.96 (m, 28 H), 1.42 (s, 6 H), 0.90 (s, 3 H), 0.85 (d, J = 6.0 Hz, 3 H), 0.53 (s, 3 H); 13C NMR (150 MHz, Pyrdine-d5) δ 174.07, 70.15, 56.46, 56.19, 52.01, 51.70, 44.00, 42.79, 40.71, 40.33, 37.92, 36.52, 36.41, 35.96, 35.73, 35.45, 34.90, 33.78, 32.26, 30.78, 30.74, 30.33, 28.35, 27.80, 26.75, 24.36, 24.24, 21.08, 18.55, 12.19; HRMS calcd for C30H52NO5S (M-H)- 538.3572, found 528.3576.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}