
Synthesis of C2-Alkoxy-Substituted 19-Nor Vitamin D3 Derivatives: Stereoselectivity and Biological Activity

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. Vitamin D Receptor Binding Analysis
2.1.2. Evaluation of the Differentiation-Inducing Activity by NBT Staining
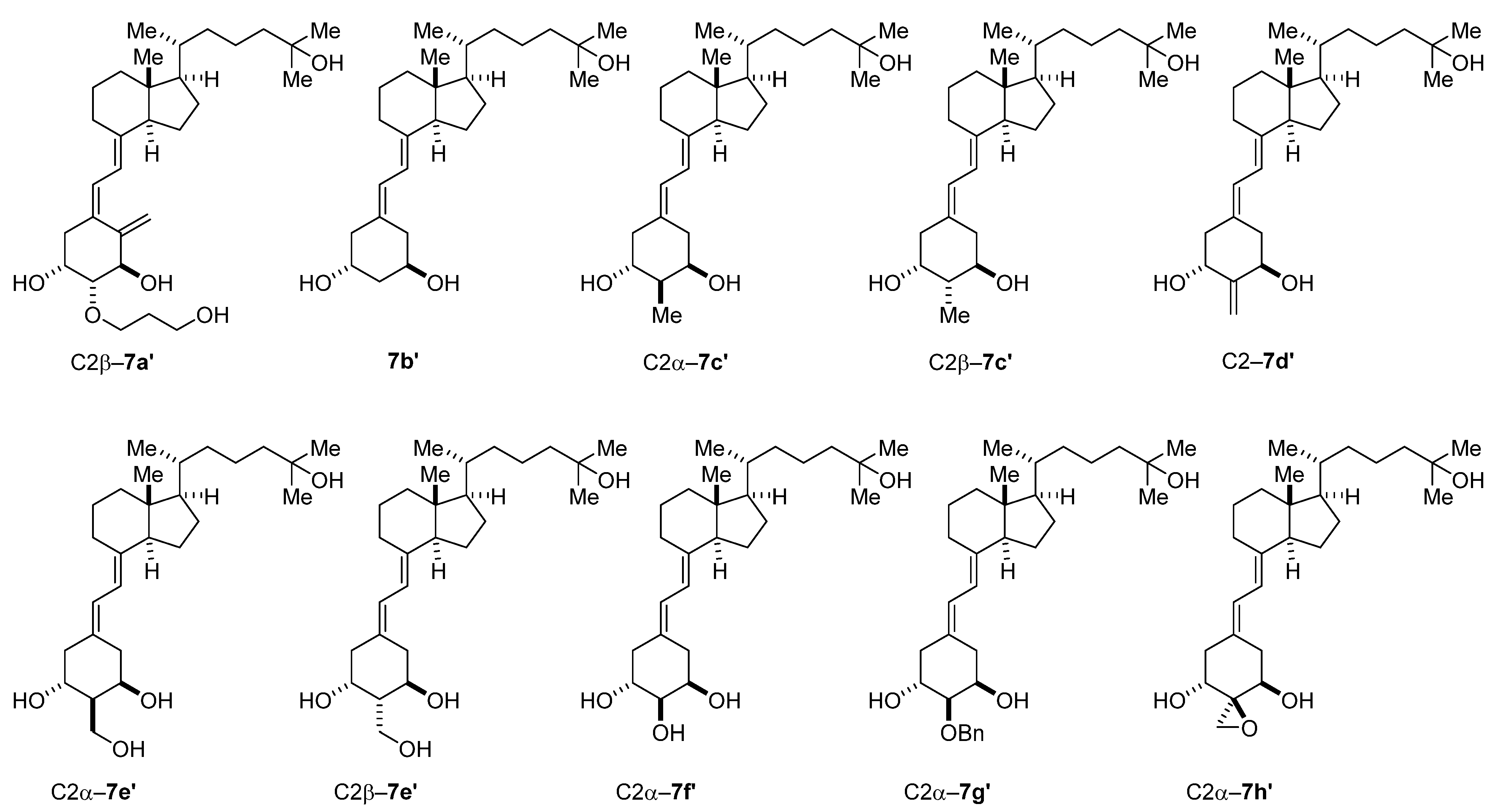
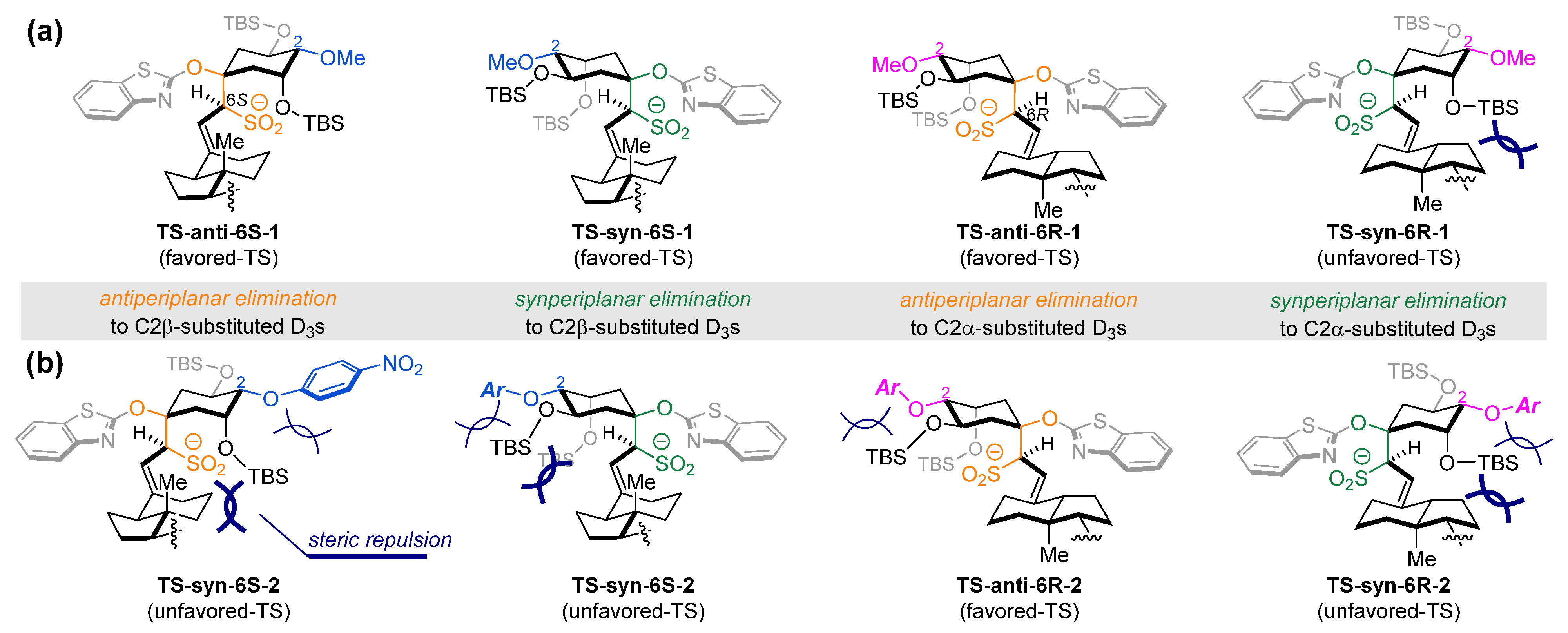
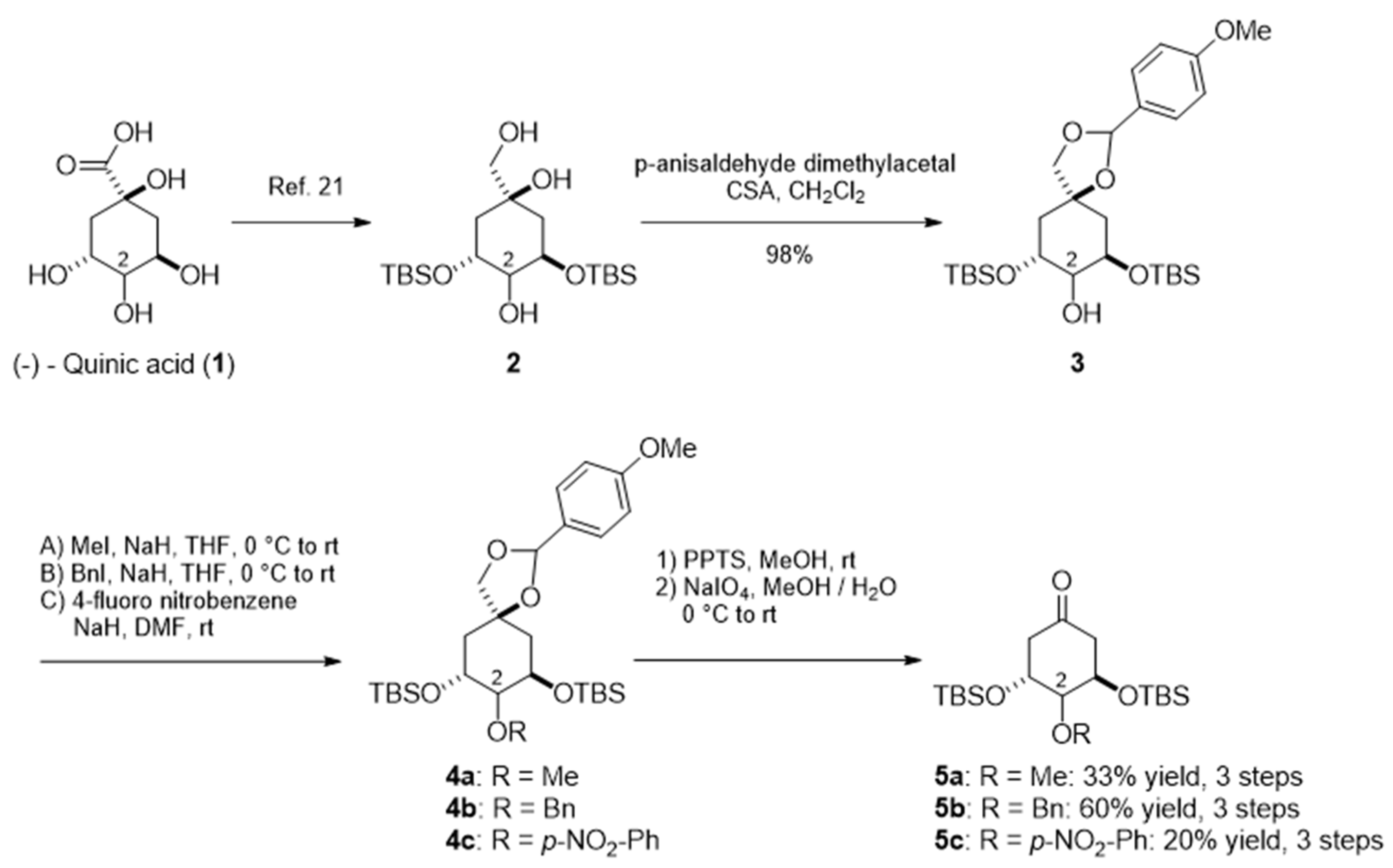
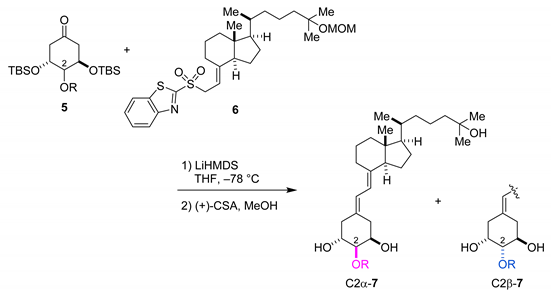
3. Results and Discussion
3.1. Chemistry
3.2. Biological Evaluation of the 19-Nor Type C2 Alkoxy-Substituted D3 Derivatives
3.2.1. Evaluation of the VDR Binding Affinity of 7a–c
3.2.2. Evaluation of the HL-60 Cell Differentiation-Inducing Activity of 7a–c
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References and Notes
- Anthony, W.N.; Helen, L.H.; June, E.B.; Xin, D.S.; Craig, B.; William, H.O. Different shapes of the steroid hormone 1a,25(OH)2-vitamin D3 act as agonists for two different receptors in the vitamin D endocrine system to mediate genomic and rapid responses. Steroids 2001, 66, 147–158. [Google Scholar]
- Anthony, W.N.; James, F.M.; Ronald, J.M.; Henry, G.N.; Vincent, W.; Popják, G. 1,25-Dihydroxycholecalciferol: Identification of the Proposed Active Form of Vitamin D3 in the Intestine. Science 1971, 173, 51–54. [Google Scholar]
- Holick, M.F.; Schnoes, H.K.; Deluca, H.F. Identification of 1,25-dihydroxycholecalciferol, a form of vitamin D3 metabolically active in the intestine. Proc. Nat. Acad. Sci. USA 1971, 68, 803–804. [Google Scholar] [CrossRef] [Green Version]
- Abe, E.; Miyaura, C.; Sakagami, C.; Takeda, H.; Konno, M.; Yamazaki, T.; Suda, T. Differentiation of mouse myeloid leukemia cells induced by 1a,25-dihydroxyvitamin D3. Proc. Nat. Acad. Sci. USA 1981, 78, 4990–4994. [Google Scholar] [CrossRef] [Green Version]
- Okano, T.; Tsugawa, N.; Masuda, S.; Takeuchi, A.; Kobayashi, T.; Takita, Y.; Nishii, Y. Regulatory activities of 2b-(3-hydroxypropoxy)-1a,25-dihydroxyvitamin D3, a novel synthetic vitamin D3 derivative, on calcium metabolism. Biochem. Biophys. Res. Commun. 1989, 163, 1444–1449. [Google Scholar] [CrossRef]
- Tsurukami, H.; Nakamura, T.; Suzuki, K.; Sato, K.; Higuchi, Y.; Nishii, Y. A novel synthetic vitamin D analogue, 2b-(3-hydroxypropoxy)1a, 25-dihydroxyvitamin D3 (ED-71), increases bone mass by stimulating the bone formation in normal and ovariectomized rats. Calcif. Tissue Int. 1994, 54, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Perlman, K.L.; Sicinski, R.R.; Schnoes, H.K.; DeLuca, H.F. 1a,25- Dihydroxy-19-nor-vitaminD3, a novel vitamin D-related compound with potential therapeutic activity. Tetrahedron Lett. 1990, 31, 1823–1824. [Google Scholar] [CrossRef]
- Sicinski, R.R.; Prahl, J.M.; Smith, C.M.; DeLuca, H.F. New 1a,25-dihydroxy-19-norvitamin D3 compounds of high biological activity: Synthesis and biological evaluation of 2-hydroxymethyl, 2-methyl, and 2-methylene analogues. J. Med. Chem. 1998, 41, 4662–4674. [Google Scholar] [CrossRef]
- Abella, L.S.; Fernández, S.; Verstuyf, A.; Verlinden, L.; Gotor, V.; Ferrero, M. Synthesis, conformational analysis, and biological evaluation of 19-nor-vitamin D3 analogues with A-ring modifications. J. Med. Chem. 2009, 52, 6158–6162. [Google Scholar] [CrossRef]
- Sicinski, R.R.; Perlman, K.L.; DeLuca, H.F. Synthesis and biological activity of 2-hydroxy and 2-alkoxy analogs of 1a,25-dihydroxy-19-norvitaminD3. J. Med. Chem. 1994, 37, 3730–3738. [Google Scholar] [CrossRef]
- Fernández, S.; Ferrero, M. Strategies for the Synthesis of 19-nor-Vitamin D Analogs. Pharmaceuticals 2020, 13, 159. [Google Scholar] [CrossRef]
- Saito, N.; Honzawa, S.; Kittaka, A. Recent results on A-ring modification of 1a,25-dihydroxyvitamin D3: Design and synthesis of VDR-agonists and antagonists with high biological activity. Curr. Top. Med. Chem. 2006, 6, 1273–1288. [Google Scholar] [CrossRef]
- Glebocka, A.; Chiellini, G. A-ring analogs of 1,25-dihydroxyvitamin D3. Arch. Biochem. Biophys. 2012, 523, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, S.; Chodyński, M.; Corcoran, A.; Marcinkowska, E.; Brown, G.; Kutner, A. Double point modified analogs of vitamin D as potent activators of vitamin D receptor. Curr. Pharm. Des. 2015, 21, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Iwasaki, Y.; Shimazaki, M.; Amano, Y.; Yamamoto, K.; Reischl, W.; Yamada, S. New derivatives of 1α,25-dihydroxy-19-norvitamin D3 with two substituents at C-2: Synthesis and biological activity. Bioorg. Med. Chem. Lett. 2005, 15, 1451–1455. [Google Scholar] [CrossRef]
- In 2009, Abella et al. synthesized 19-norvitamin D3 with OTBS and OMs groups at C2 using the Julia-type coupling reaction resulting in diastereomeric ratios of 2a:2b = 4:1 and 3:2, respectively [ref. 9].
- Perlman, K.L.; Swenson, R.E.; Paaren, H.E.; Schnoes, H.K.; DeLuca, H.F. Novel synthesis of 19-nor-vitamin D compounds. Tetrahedron Lett. 1991, 32, 7663–7666. [Google Scholar] [CrossRef]
- Under the high concentration conditions, some protective groups in the substrate fall off.
- Hoang, C.T.; Bouillère, F.; Johannesen, S.; Zulauf, A.; Panel, C.; Pouilhès, A.; Gori, D.; Alezra, V.; Kouklovsky, C. Amino acid homologation by the Blaise reaction: A new entry into nitrogen heterocycles. J. Org. Chem. 2009, 74, 4177–4187. [Google Scholar] [CrossRef] [PubMed]
- Purification of C2α-7b was quite difficult even with the HPLC. However, we believe that a quite small amount of the impurities would not significantly affect the results of the biological assays.
- Collins, S.J.; Ruscetti, F.W.; Gallagher, R.E.; Gallo, R.C. Normal functional characteristics of cultured human promyelocytic leukemia cells (HL-60) after induction of differentiation by dimethylsulfoxide. J. Exp. Med. 1979, 149, 969–974. [Google Scholar] [CrossRef]
- Ostrem, V.K.; Lau, W.F.; Lee, S.H.; Perlman, K.; Prahl, J.; Schnoes, H.K.; DeLuca, H.F. Induction of monocytic differentiation of HL-60 cells by 1,25-dihydroxyvitamin D analogs. J. Biol. Chem. 1987, 262, 14164–14171. [Google Scholar] [CrossRef]
- Yoshimoto, N.; Inaba, Y.; Yamada, S.; Makishima, M.; Shimizu, M.; Yamamoto, K. 2-Methylene 19-nor-25-dehydro-1α-hydroxyvitamin D3 26,23-lactones: Synthesis, biological activities and molecular basis of passive antagonism. Bioorg. Med. Chem. 2008, 16, 457–473. [Google Scholar] [CrossRef]
- Ono, K.; Yoshida, A.; Saito, N.; Fujishima, T.; Honzawa, S.; Suhara, Y.; Kishimoto, S.; Sugiura, T.; Waku, K.; Takayama, H.; et al. Efficient synthesis of 2-modified 1α,25-dihydroxy-19-norvitamin D3 with Julia olefination: High potency in induction of differentiation on HL-60 cells. J. Org. Chem. 2003, 68, 7407–7415. [Google Scholar] [CrossRef]
- The stereoselectivity was determined by 1H NMR with diastereomer mixtures 7.
- Sicinski, R.R.; Rotkiewicz, P.; Kolinski, A.; Sicinska, W.; Prahl, J.M.; Smith, C.M.; DeLuca, H.F. 2-Ethyl and 2-Ethylidene Analogues of 1α,25-Dihydroxy-19-norvitamin D3: Synthesis, Conformational Analysis, Biological Activities, and Docking to the Modeled rVDR Ligand Binding Domain. J. Med. Chem. 2002, 45, 3366–3380. [Google Scholar] [CrossRef] [PubMed]
- Ibe, K.; Yamada, T.; Okamoto, S. Synthesis and vitamin D receptor affinity of 16-oxa vitamin D3 analogues. Org. Biomol. Chem. 2019, 17, 10188–10200. [Google Scholar] [CrossRef] [PubMed]
- Rochel, N.; Wurtz, J.M.; Mitschler, A.; Klaholz, B.; Moras, D. The Crystal Structure of the Nuclear Receptor for Vitamin D Bound to Its Natural Ligand. Mol. Cell 2000, 5, 173–179. [Google Scholar] [CrossRef]
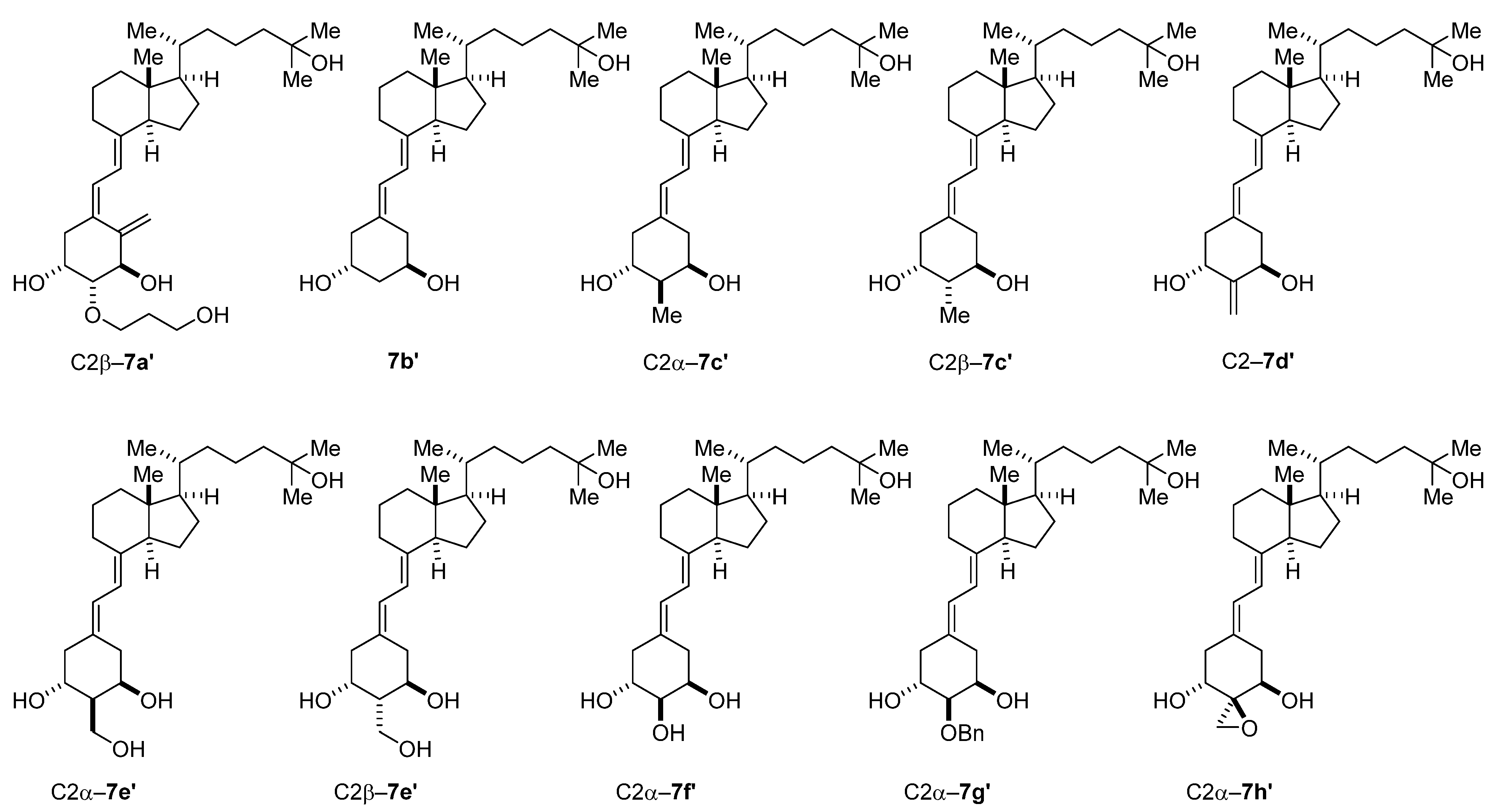
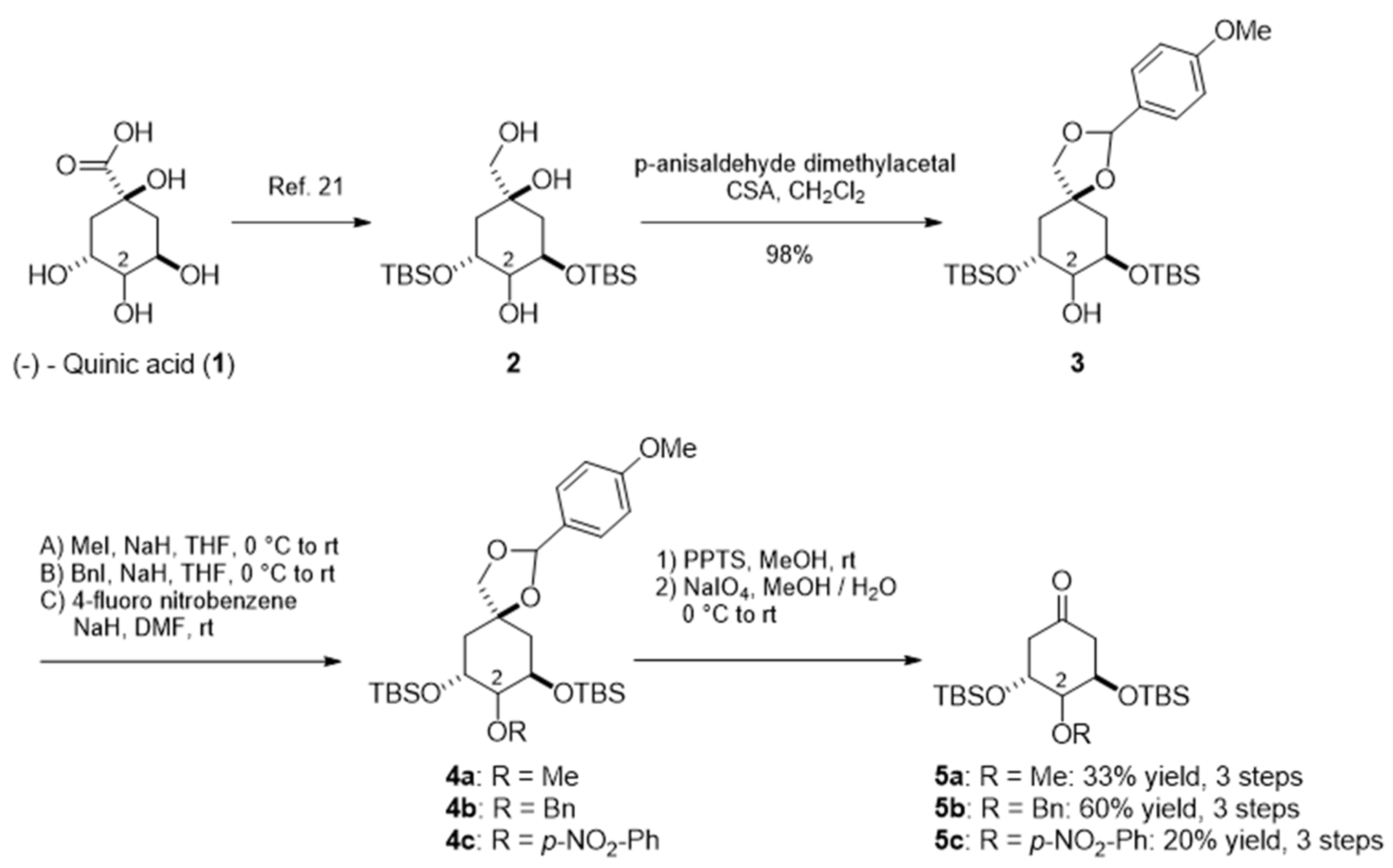
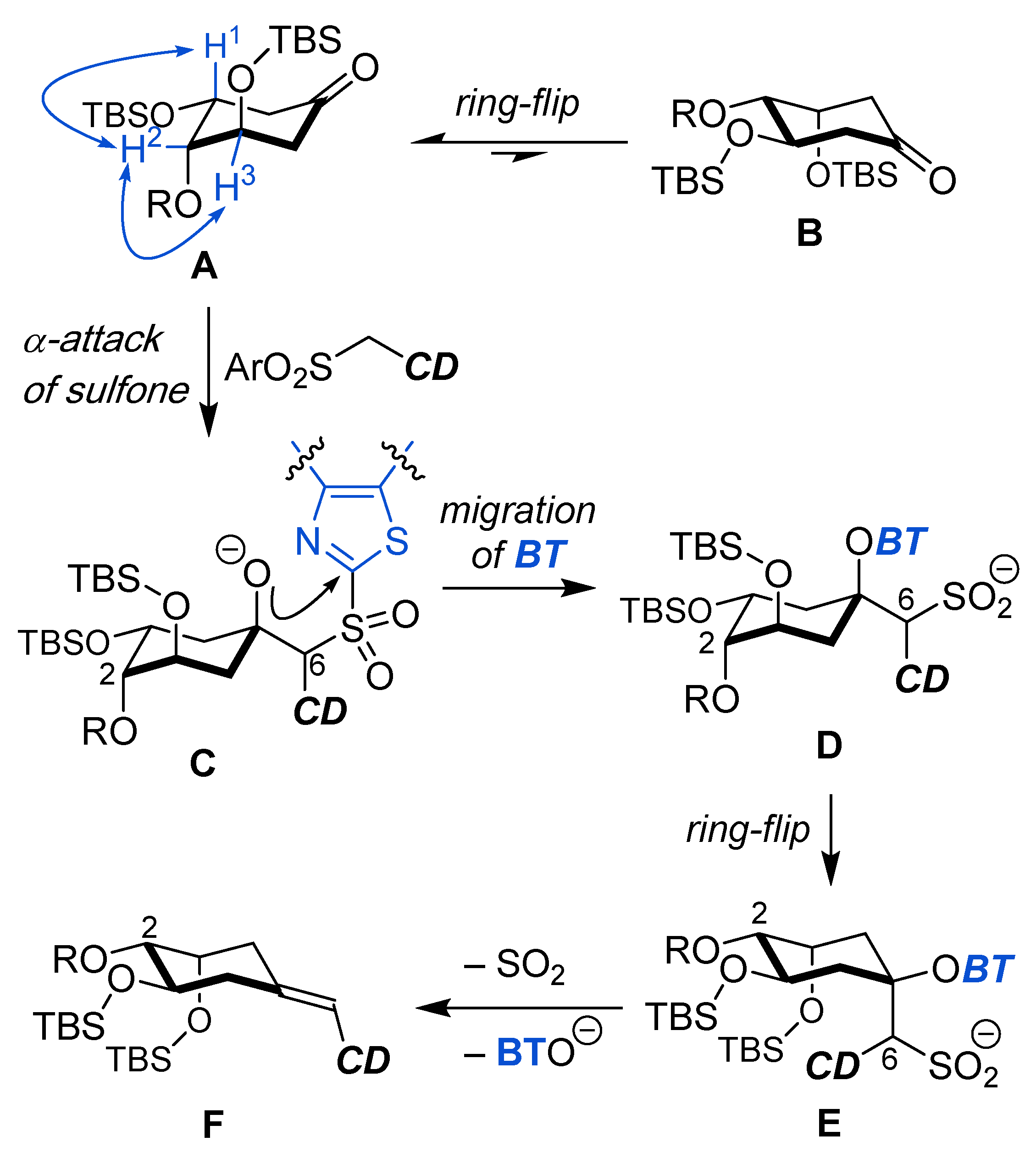
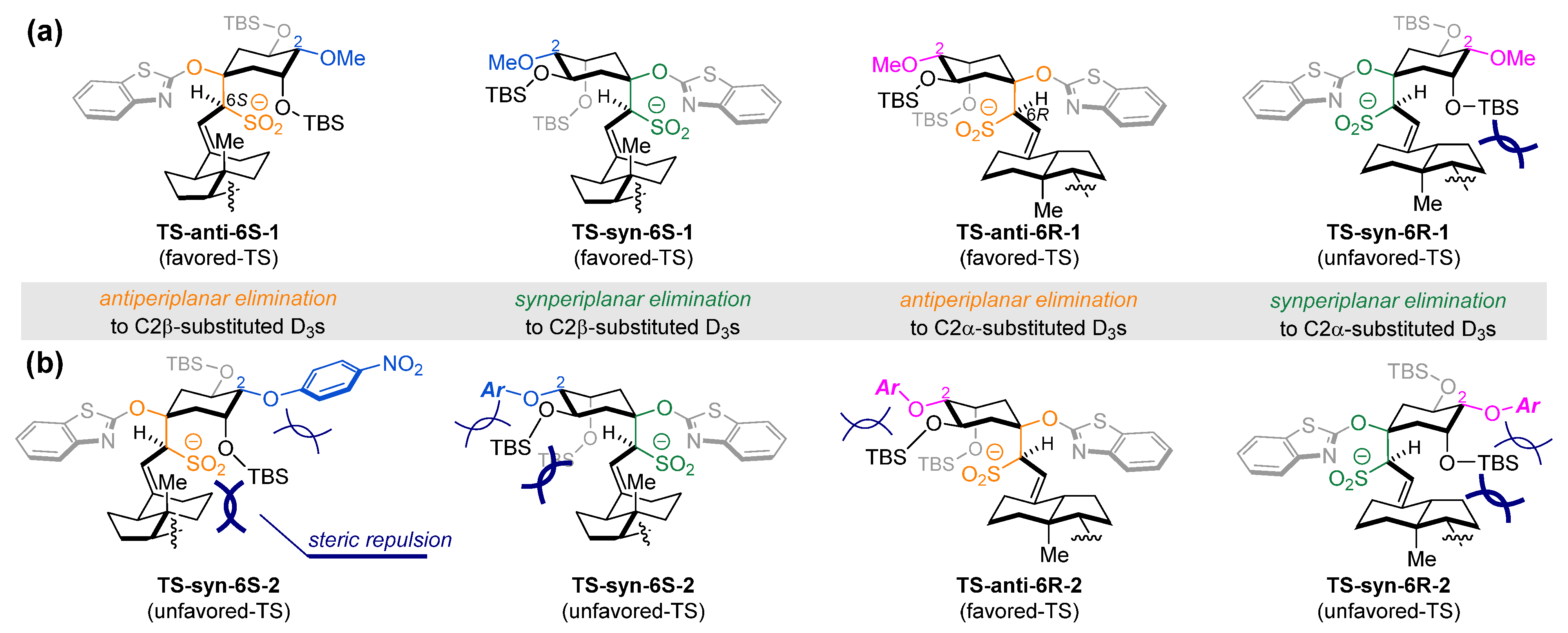

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coupling Product 7 | ||||
|---|---|---|---|---|
| Entry | Ketone 5 | Yield [%] 1 | Ratio [C2α/C2β] 2 | |
| 1 | 5a (R = Me) | 7a | 31 | 1:3.5 |
| 2 | 5b (R = Bn) | 7b | 27 | 1:1 |
| 3 | 5c (R = p-NO2-PhO) | 7c | 62 | 1:0 |
| VDR Binding | HL-60 Differentiation | ||
|---|---|---|---|
| Compound | Activity Ratio 1 | EC50 (M) | Activity Ratio 1 |
| 1,25D3 | 100.0 | 1.01 × 10−8 | 100 |
| 2α-OMe (C2α-7a) | 100.8 | 3.80 × 10−10 | 2655 |
| 2β-OMe (C2β-7a) | 47.5 | 7.13 × 10−9 | 141 |
| 2α-OBn (C2α-7b) | 2.5 | 3.34 × 10−8 | 30 |
| 2β-OBn (C2β-7b) | 0.4 | 1.34 × 10−7 | 8 |
| 2α-p-NO2-PhO (C2α-7c) | 0 | 4.83 × 10−7 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mizumoto, Y.; Sakamoto, R.; Nagata, A.; Sakane, S.; Kittaka, A.; Odagi, M.; Tera, M.; Nagasawa, K. Synthesis of C2-Alkoxy-Substituted 19-Nor Vitamin D3 Derivatives: Stereoselectivity and Biological Activity. Biomolecules 2022, 12, 69. https://doi.org/10.3390/biom12010069
Mizumoto Y, Sakamoto R, Nagata A, Sakane S, Kittaka A, Odagi M, Tera M, Nagasawa K. Synthesis of C2-Alkoxy-Substituted 19-Nor Vitamin D3 Derivatives: Stereoselectivity and Biological Activity. Biomolecules. 2022; 12(1):69. https://doi.org/10.3390/biom12010069
Chicago/Turabian StyleMizumoto, Yuka, Ryota Sakamoto, Akiko Nagata, Suzuka Sakane, Atsushi Kittaka, Minami Odagi, Masayuki Tera, and Kazuo Nagasawa. 2022. "Synthesis of C2-Alkoxy-Substituted 19-Nor Vitamin D3 Derivatives: Stereoselectivity and Biological Activity" Biomolecules 12, no. 1: 69. https://doi.org/10.3390/biom12010069
APA StyleMizumoto, Y., Sakamoto, R., Nagata, A., Sakane, S., Kittaka, A., Odagi, M., Tera, M., & Nagasawa, K. (2022). Synthesis of C2-Alkoxy-Substituted 19-Nor Vitamin D3 Derivatives: Stereoselectivity and Biological Activity. Biomolecules, 12(1), 69. https://doi.org/10.3390/biom12010069