Abstract
The regulation of vitamin D3 actions in humans occurs mainly through the Cytochrome P450 24-hydroxylase (CYP24A1) enzyme activity. CYP24A1 hydroxylates both 25-hydroxycholecalciferol (25(OH)D3) and 1,25-dihydroxycholecalciferol (1,25(OH)2D3), which is the first step of vitamin D catabolism. An abnormal status of the upregulation of CYP24A1 occurs in many diseases, including chronic kidney disease (CKD). CYP24A1 upregulation in CKD and diminished activation of vitamin D3 contribute to secondary hyperparathyroidism (SHPT), progressive bone deterioration, and soft tissue and cardiovascular calcification. Previous studies have indicated that CYP24A1 inhibition may be an effective strategy to increase endogenous vitamin D activity and decrease SHPT. This study has designed and synthesized a novel C-24 O-methyloxime analogue of vitamin D3 (VD1-6) to have specific CYP24A1 inhibitory properties. VD1-6 did not bind to the vitamin D receptor (VDR) in concentrations up to 10−7 M, assessed by a VDR binding assay. The absence of VDR binding by VD1-6 was confirmed in human embryonic kidney HEK293T cultures through the lack of CYP24A1 induction. However, in silico docking experiments demonstrated that VD1-6 was predicted to have superior binding to CYP24A1, when compared to that of 1,25(OH)2D3. The inhibition of CYP24A1 by VD1-6 was also evident by the synergistic potentiation of 1,25(OH)2D3-mediated transcription and reduced 1,25(OH)2D3 catabolism over 24 h. A further indication of CYP24A1 inhibition by VD1-6 was the reduced accumulation of the 24,25(OH)D3, the first metabolite of 25(OH)D catabolism by CYP24A1. Our findings suggest the potent CYP24A1 inhibitory properties of VD1-6 and its potential for testing as an alternative therapeutic candidate for treating SHPT.
1. Introduction
The 25-hydroxyvitamin D 24-hydroxylase (CYP24A1) enzyme is a mitochondrial inner membrane cytochrome P450 component that acts naturally to catabolize both 25-hydroxycholecalciferol (25(OH)D3) and 1,25-dihydroxycholecalciferol (1,25(OH)2D3) to control vitamin D3 hormonal actions in different tissues [1]. This catabolism occurs via hydroxylation at either C23 or C24 of the aliphatic side chain of 25(OH)D3 and 1,25(OH)2D3, resulting in less-active catabolites and culminating in the formation of calcitroic acid [2]. CYP24A1 gene expression in healthy individuals is regulated by the effects of 1,25(OH)2D3, parathyroid hormone (PTH), and fibroblast growth factor 23 (FGF23) [3], with 1,25(OH)2D3 being the predominant stimulator of expression [4]. The control of PTH over CYP24A1 expression was found to be tissue-dependent, with PTH increasing CYP24A1 activity in bone cells through enhancing 1,25(OH)2D3 mediated induction [5], while directly suppressing CYP24A1 activity in the kidneys through a non-vitamin D receptor (VDR)-dependent pathway [6]. FGF23, on the other hand, stimulates CYP24A1 expression in kidney cells to lower 1,25(OH)2D3-mediated phosphate absorption [7].
Chronic kidney disease (CKD) is a progressive disease characterized by the gradual deterioration of kidney function. CKD is a growing public health issue, mainly due to aging, with comorbidities such as hypertension and diabetes being major contributors [8,9]. An inevitable event in CKD is the disturbance of the renal vitamin D metabolic pathways, which involve 25-hydroxyvitamin D 1α-hydroxylase (CYP27B1) and CYP24A1. The gradual reduction in renal function translates into a progressive decline in CYP27B1 activity, leading to a decrease in the production of circulating 1,25(OH)2D3 [2]. Additionally, elevated serum FGF23 levels are one of the first indicators of renal disease, which continues to rise as CKD progresses due to hyperphosphatemia resulting from impaired renal phosphate excretion [10,11,12]. Elevated FGF23 suppresses renal CYP27B1 and stimulates renal CYP24A1 expression, contributing to inadequate 1,25(OH)2D3 production [13]. Circulating levels of 25(OH)D3, as a precursor substrate for 1,25(OH)2D3, have also been reported to decline in CKD due to factors including proteinuria [14]. Inappropriately low levels of 1,25(OH)2D3 lead to impaired intestinal calcium absorption and hypocalcaemia, which in turn induce secondary hyperparathyroidism (SHPT) [15,16].
To reduce PTH levels, 1,25(OH)2D3 or VDR activating vitamin D analogues, such as alfacalcidol, maxacalcitol, doxercalciferol, or paricalcitol, are often prescribed for patients with declining renal function [17,18]. However, some factors may limit the usefulness of active vitamin D or vitamin D analogue therapy, such as the resultant hypercalcemia and the inappropriately high levels of CYP24A1 activity, which, besides the deactivation of 1,25(OH)2D3, also have the potential to deactivate vitamin D analogues [19]. Considering the decline in CYP27B1 and elevated CYP24A1 activity during CKD, preventing 1,25(OH)2D3 breakdown to improve the half-life of 1,25(OH)2D3 could offer a strategy that may improve vitamin D activity and possibly improve responsiveness to vitamin D therapies [20,21].
Previously, 1,25(OH)2D3 analogue-derived specific CYP24A1 inhibition was shown to significantly lower PTH levels in normal rats without hypercalcemia [19]. Compounds with dual CYP24A1 inhibition and VDR activation were also demonstrated to ameliorate SHPT in a dose-dependent manner in uremic rats with efficient reduction of plasma PTH at doses that did not elevate serum calcium or phosphate, suggesting potential usefulness in human CKD. However, the therapeutic effect of mixed-mode analogues could not be solely attributed to the prevention of endogenous 1,25(OH)2D3 breakdown [19]. Furthermore, it has been suggested that a targeted CYP24A1 inhibition approach could be of therapeutic usefulness for other conditions associated with CYP24A1 dysregulation, such as some malignancies, psoriasis, and diabetes [19,22,23]. Recently, attention has been brought to side-chain oxime-substituted vitamin D3 analogues as potential bioactive candidates, where the synthesis of three C-24 oxime derivatives has been described without any CYP24A1 modulatory activity evaluation [22].
Among the reported synthetic analogues for calcitriol, different side-chain C-24 and C-25 oxime and oxime ether analogues have been previously described and biologically evaluated [23,24]. Recently, attention has been brought to side-chain oxime-substituted vitamin D3 analogues as potential bioactive candidates, where the synthesis of three C-24 oxime derivatives has been described [22]. However, none of these studies evaluated the CYP24A1 modulatory activity of the oxime analogues.
This study has established a CYP24A1 inhibitor with prospective therapeutic value for conditions showing low vitamin D activity, such as CKD. Here, we report the synthesis of a novel C-24 O-methyl oxime derivative of 1,25(OH)2D3 (VD1-6), together with an evaluation of its VDR binding affinity and its in silico docking evaluation in CYP24A1 compared to 1,25(OH)2D3. We also assess the biological effects of VD1-6 on CYP24A1 inhibition by assessing 1,25(OH)2D3 preservation and the reduction in C-24 hydroxylation of 25(OH)D3 in human embryonic kidney HEK293T cells.
2. Materials and Methods
2.1. Synthesis of VD1-6
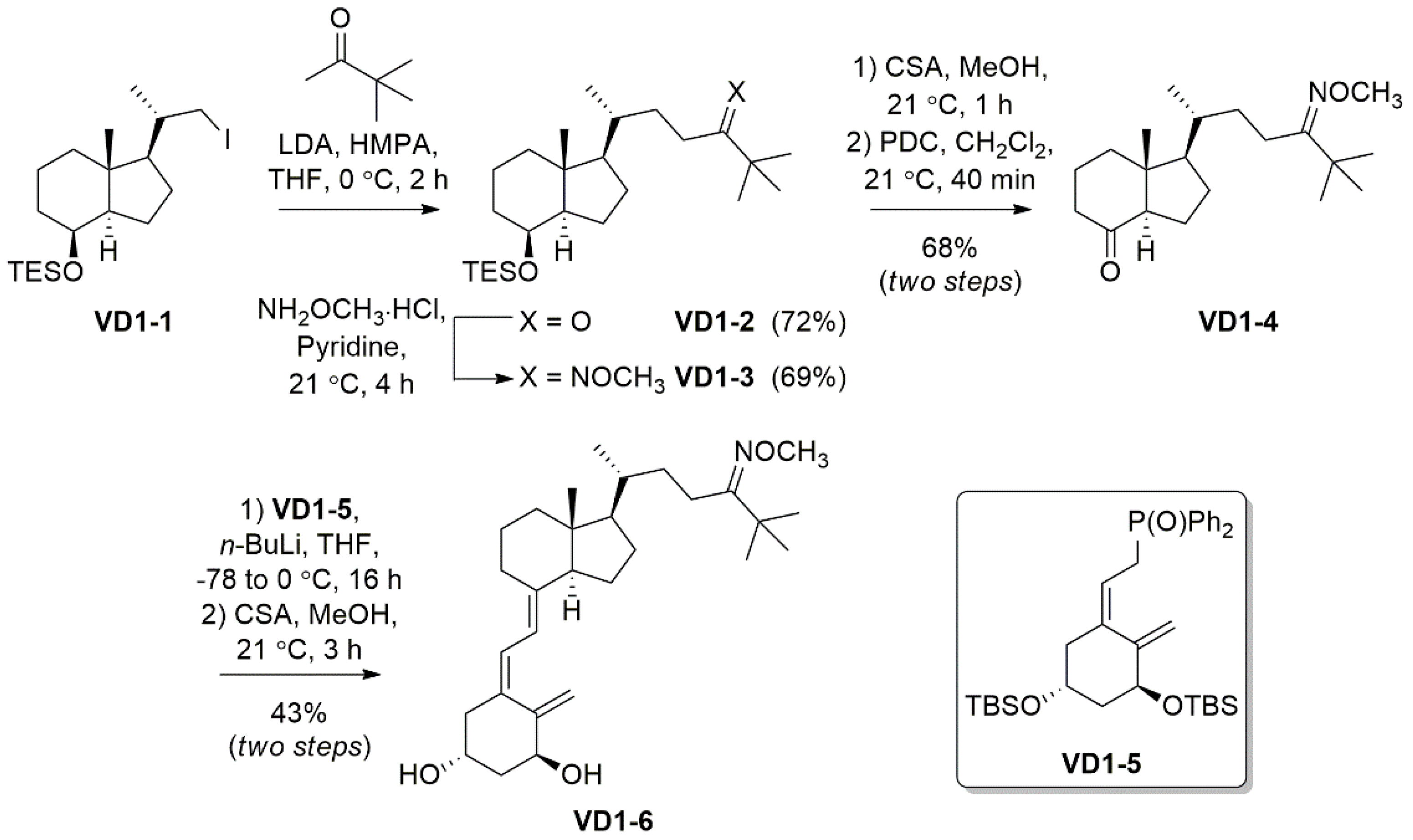
The reagents, chemicals, and apparatus used for the synthesis and purification of VD1-6 are detailed in the Supplementary Section (S.1). The synthetic pathway followed to synthesize VD1-6 is described in Scheme 1. Nuclear magnetic resonance (NMR) and mass spectrometry (MS) spectra are provided in the Supplementary Section for all compounds (Figures S1–S12).
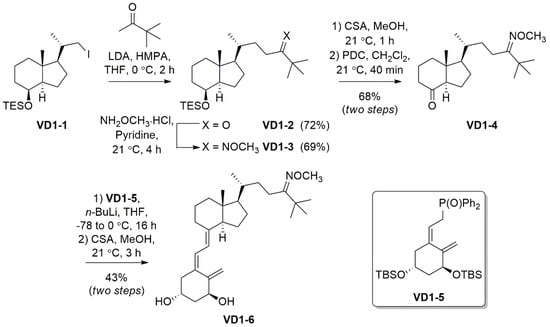
Scheme 1.
Synthesis of the 24-O-methyloxime derivative VD1-6.
2.2. VDR Competitor Binding Assay
The PolarScreen™ VDR Competitor Assay Kit, Red (Life Technologies Australia Pty Ltd., Mulgrave, VIC, Australia) was used for the assay. VDR recombinant human protein (32,100 nM), Fluormone™ VDR Red tracer (100 nM), and dithiothreitol (DTT) solutions provided by the kit manufacturer were thawed for 30 min on ice before use. A sufficient volume of complete VDR Red screening buffer “complete buffer” (5 µL of DTT for each 1 mL of the VDR Red screening buffer) was prepared and kept on ice until use. Primary stock solutions (1 mM) of 1,25(OH)2D3, 25(OH)D3 (Sigma Aldrich, Castle Hill, NSW, Australia), and VD1-6 were prepared separately in DMSO. From these solutions, 15 further 3-fold serial dilutions were prepared for each of the three compounds in DMSO, with the lowest concentration in the series of 0.07 nM. For conducting the assay, each of the 16 DMSO dilutions for each compound was further diluted 50-fold using the complete buffer for the resultant highest and lowest concentrations, being 2 × 104 nM and 0.0014 nM, respectively, and for a post-dilutional DMSO concentration of 2%. To prepare the VDR/Fluormone™ complex, separate 4 nM Fluormone™ and 14.48 nM, VDR full-length protein solutions were prepared by appropriate dilution of the manufacturer’s stock solutions using complete buffer followed by vortex mixing of the Fluormone™ solution for 10 sec, and inversion mixing only of the VDR solution. Sufficient equal volumes of the Fluormone™ and VDR dilutions were combined and mixed by inversion, and the final solution (2 nM Fluormone™/7.24 nM VDR) was kept on ice until use.
In a 384 microwell plate, 10 µL aliquots from each of the 16 (0.0014–2 × 104 nM) serial dilutions for each of the three tested compounds were pipetted into triplicate adjacent wells. To those wells, 10 µL of the (2 nM Fluormone™/7.24 nM VDR) solution was then added for final concentrations of test compounds ranging from 0.0007 to 1 × 104 nM, final Fluormone™ concentration of 1 nM, and a final VDR full-length concentration of 3.62 nM (the manufacturer’s recommended lot-specific concentration). Maximum and minimum polarization controls were similarly prepared for monitoring assay performance by the addition of the Fluormone™/VDR complex to a column of wells with 10 µL blank 2% DMSO in complete buffer and a column of wells with 10 µL 2 × 104 nM 1,25(OH)2D3, respectively. The final DMSO concentration in each well was 1%, matching the manufacturer’s recommended solvent tolerance for the assay. The plate was lid covered and shaken horizontally on an orbital shaker at 60 rpm for 5 min, then incubated for 3 h at room temperature. Fluorescence polarization values (mP) were then measured on the plate reader Cytation™ 5 multi-mode (Biotek, Winooski, VT, USA) equipped with a red fluorescence polarization filter (Excitation/Emission, 530/590 nm) at 25 °C. IC50 was determine by modelling of the mean fluorescence polarization readings versus corresponding concentrations for each compound, as recommended by the kit manufacturer [25]. Briefly, sigmoidal dose-response curve fitting with a variable slope was performed using GraphPad Prism® 8.3.0 for Windows (GraphPad Software, San Diego, CA, USA) with the equation: Y = mP100% + (mP0% − mP100%)/[1 + 10((LogIC50 − X) × Hill Slope)], where: Y = mP, X = Log [inhibitor], mP100% = 100% competition, and mP0% = 0% competition.
2.3. CYP24A1 Molecular Docking Studies
A protein crystal structure for the human CYP24A1 enzyme has not previously been reported. As such, we utilized the crystal structure of a rat mitochondrial cytochrome P450 24A1 co-crystallized with a zwitterionic surfactant, protein data bank (PDB) ID: 3K9V (Resolution = 2.5 Å and receptor pocket-size = 2512 Å) [26]. Ligand structures for docking were acquired by determining the appropriate SMILES string for 1,25(OH)2D3 and VD1-6. Enumeration of 3D conformers was undertaken using OMEGA 3.3.0.3, with the default maximum number of conformers (200) [27,28]. VD1-6 and 1,25(OH)2D3 produced 60 and 62 conformations, respectively. With the selected crystal structure for CYP24A1, binding pockets were derived using Make Receptor 3.3.0.3 [29]. Appropriate protein and co-crystallized ligand sections were chosen, and auto-generated constraints were not included, as per the default settings. No alterations to the site shape potential were made. Docking studies on the receptor were performed using FRED 3.3.0.3 as part of the OE Docking suite [30,31,32]. Docking resolution was set to “High,” and the number of poses was limited to 1. The two ligands docked successfully, and the predicted orientations were examined using VIDA 3.3.0.3 [33]. Protein–ligand interactions were further investigated using the OEChem toolkit (specifically complex2img.py). Generated Chemgauss4 scores ranked the docked 1,25(OH)2D3 and VD1-6 according to their goodness of fit into the receptor pocket and the interactions with surrounding residues.
2.4. HEK293T Cell Culture Experiments
With relevance to kidney physiology, the HEK393T cell line has been sought as a common normal kidney cell line that is used to model renal physiology [34] and is known to express vitamin D responsive genes, in which adequate response to 1,25(OH)2D3 by CYP24A1 induction has been previously demonstrated [35]. HEK293T cells were maintained at 37 °C in a growth media comprised of minimum essential medium alpha (alpha-MEM), 10% v/v fetal bovine serum (FBS), and the standard usage of tissue culture additives, including 25 nM HEPES, 2 mM ʟ-glutamine, and antibiotics: 100 U/mL penicillin G/streptomycin. For experimentation, cells were seeded in 24-well tissue culture plates with 30% confluency and were allowed to attach for 24 h before treatment application.
2.4.1. Assessment of Relevant Genetic Response to VD1-6
Upon cell attachment, culture wells (n = 5) were exposed for 24 h to vehicle control (0.2% v/v ethanol) or treatments of VD1-6 (10−7 M), 1,25(OH)2D3 (10−9 M), or a combination of VD1-6 (10−11 M or 10−10 M) and 1,25(OH)2D3 (10−9 M). The cells from replicate wells for each of the three treatment groups were then treated for RNA extraction using TRIzol® reagent (Thermo-Fisher Scientific, Waltham, MA, USA) at room temperature, following the manufacturer’s protocol [36]. Briefly, after media removal, 200 µL TRIzol® was added to unwashed cells in each well to lyse them. The lysate of each well was then transferred to 1.5 mL Eppendorf tubes, and 40 µL of chloroform was added; then the tube content was mixed by inverting and centrifuged for 15 min at 12,000× g rpm at 4 °C. The upper aqueous layer was transferred to a new tube, to which 100 µL of isopropanol was added, and the tube content was incubated for 10 min, followed by centrifugation for 10 min at 12,000× g rpm at 4 °C; the supernatant was then discarded. For washing, 200 µL of 70% ethanol was added to the RNA pellet, vortexed for 5 s, and centrifuged for 5 min at 7500× g rpm at 4 °C. The supernatant was discarded, and the RNA pellet was air-dried for 10 min. The RNA pellet was resuspended in 25 µL RNase-free water. The tubes were incubated in a 60 °C water bath for 10 min before storage at −80 °C for real-time PCR analysis.
Replicate total RNA samples were analyzed for CYP24A1, CYP27B1, VDR, CALB1, and SLC34A3 mRNA and housekeeping gene, ACTB, by quantitative PCR. A total of 2µg of total RNA was reverse transcribed using the iScript™ cDNA synthesis kit (Bio-Rad, Hercules, CA, USA), following the manufacturer’s protocol [37]. A total of 12.5 ng of cDNA per sample was used for real-time RT-PCR analyses of the target gene mRNA levels using Forget-Me-Not™ EvaGreen® qPCR Master Mix (Biotium, Fremont, CA, USA). Primer sequences for target mRNA were: CYP24A1, F 5′-CCTGCTGCCAGATTCTCTGGAA-3′, R 5′-TTGCCATACTTCTTGTGGTACTCC-3′; CYP27B1, F 5′-CAGACAAAGACATTCATGTGGG-3′, R 5′-GTTGATGCTCCTTTCAGGTAC-3′; VDR, F 5′-CCAGTTCGTGTGAATGATGG-3′, R 5′-GTCGTCCATGGTGAAGGA-3′; CALB1, F 5′-AGAAACTGAGGAGCTTAAGAACT-3′, R 5′-ACTCGGCTAATTTTGTGTCATCA-3′; SLC34A3, F 5′-ACACCTCATCGTGCAGTTGG, R 5′- AGACTGCTGTTAGTGGCGTT-3′; ACTB, F 5′-AAGAGATGGCCACGGCT-3′, R 5′-CAATGATCTTGATCTTCATTGTGC-3′.
2.4.2. Assessment of 1,25(OH)2D3 Preservation by VD1-6
Cultured wells (N = 4) were exposed for 8 and 24 h to a vehicle (0.2% v/v ethanol) and VD1-6 at 10−7 M as controls and to separate treatments of 1,25(OH)2D3 at 5 × 10−10 M, without and with VD1-6, at 10−8 M and 10−7 M. Supernatants from all wells were collected at 8 and 24 h post-treatment and stored with time zero samples at −80 °C for 1,25(OH)2D3 LC-MS/MS analysis.
2.4.3. Assessment of 25(OH)D3 Catabolism Inhibition by VD1-6
A total of 72 h exposure of cultured wells (N = 4) was performed for similar controls, using the procedure detailed in Section 2.4.2, and for 25(OH)D3 at 10−6 M, without and with VD1-6, at 10−11, 10−10, 10−9, 10−8, and 10−7 M, each as a separate treatment. At the end of treatment, supernatants from quadruplicate wells for each group were collected and stored at −80 °C for 24,25(OH)2D3 LC-MS/MS analysis. Cells were then treated for total RNA isolation and mRNA analyses, as described in Section 2.4.1.
2.5. LC-MS/MS Analysis
The 24,25(OH)2D3 concentrations were analyzed in cell culture samples from Section 2.4.3, using DAPTAD derivatization and LC-MS/MS, as previously described [38]. The calibrators were prepared in phosphate-buffered saline (PBS), and the samples were appropriately diluted with PBS to bring them into the linear range of the assay. The same LC-MS/MS method was modified for 1,25(OH)2D3 analysis to enhance the sensitivity by using a higher initial sample volume (200 µL instead of 50 µL) and reconstituting the final derivatized 1,25(OH)2D3 in 20 µL and injecting 15 µL (rather than reconstituting in 25 µL and injecting 5 µL) onto the LC-MS/MS column. Multiple reaction monitoring (MRM) transitions were used for quantifying the 1,25(OH)2D3-DAPTAD derivative, and the derivative of its deuterated isotope (1,25(OH)2D3-d6), as previously reported [39]. Similar twin chromatographic peaks for R and S isomers of the derivatives [39] (Figure S13) were also obtained and used similarly for quantification. The linearity, accuracy, and precision data for 1,25(OH)2D3 LC-MS/MS analysis are provided in Table S1.
2.6. Statistical Analysis
Statistical analysis was performed using one-way analysis of variance (ANOVA) for non-parametric data, along with a secondary Tukey’s multiple comparisons test to determine the difference between treatment groups. A p-value less than 0.05 was considered statistically significant.
3. Results
3.1. Chemistry
VD1-6 was synthesized as outlined in Scheme 1. Iodo VD1-1, which was accessed from vitamin D2 following previously reported methods [40,41], was reacted with an enolate formed in situ from 3,3-dimethyl-2-butanone, to give ketone VD1-2 in 72% yield. The O-methyloxime functionality was installed using methoxyamine hydrochloride in pyridine to give VD1-3 in 69%. Desilylation for 1 h at room temperature using camphor sulphonic acid (CSA) revealed the secondary alcohol, which was then oxidized using pyridinium chromate (PDC) to give ketone VD1-4 in a 68% yield over the two steps. Horner–Wadsworth–Emmons (HWE) chemistry was then employed to react ketone VD1-4 with commercially available phosphonate VD1-5, under basic conditions, to conjugate the two fragments by an olefin linker. Removal of the silyl protecting groups, again using CSA, revealed the anti-planar hydroxy groups in VD1-6 in a 43% yield over two steps.
3.2. Binding of VD1-6 to VDR
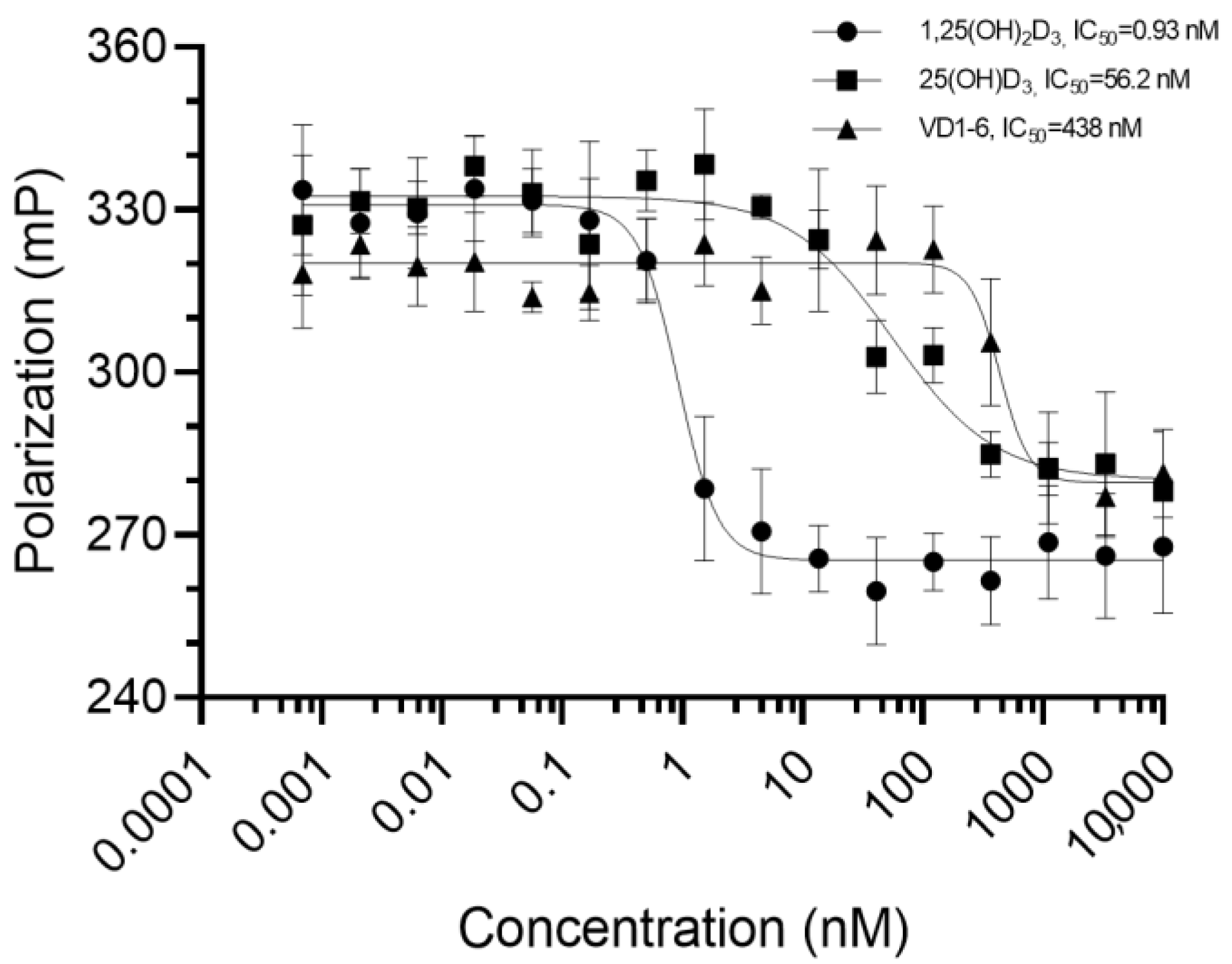
Results of VDR competitor assay experiments for VD1-6, 1,25(OH)2D3, and 25(OH)D3 are represented in Figure 1. Curve fitting (solid connecting lines, Figure 1) using a sigmoidal dose–response (variable slope) curve yielded modelled binding IC50 values of 0.93, 56.2, and 438 nM for 1,25(OH)2D3, 25(OH)D3 and VD1-6, respectively. These concentrations are required to displace half of the high affinity fluorescent tracer from its VDR binding, resulting in a corresponding drop in the mean fluorescence polarization by 50%. This illustrates that VD1-6 has approximately 470-fold (438/0.93) lower binding affinity than 1,25(OH)2D3 to the active site of VDR.
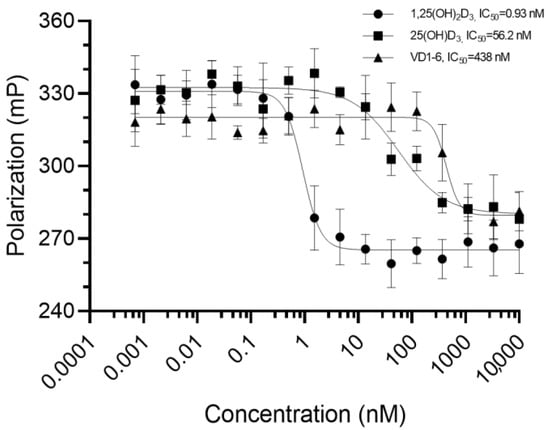
Figure 1.
Mean fluorescence polarization (n = 3) measurements in millipolarization (mP) for the bound 1 nM Fluormone™/3.62 nM VDR full-length protein interacting with each of the 16 tested concentrations (0.0007 to 1 × 104 nM, represented at Log scale in the figure) for each of the three tested compounds. Error bars represent the standard deviation from the mean. Solid lines connecting the points represent the curve fitting, using sigmoidal dose–response modelling with varying slopes.
3.3. Comparative In Silico Docking of 1,25(OH)2D3 and VD1-6 into CYP24A1
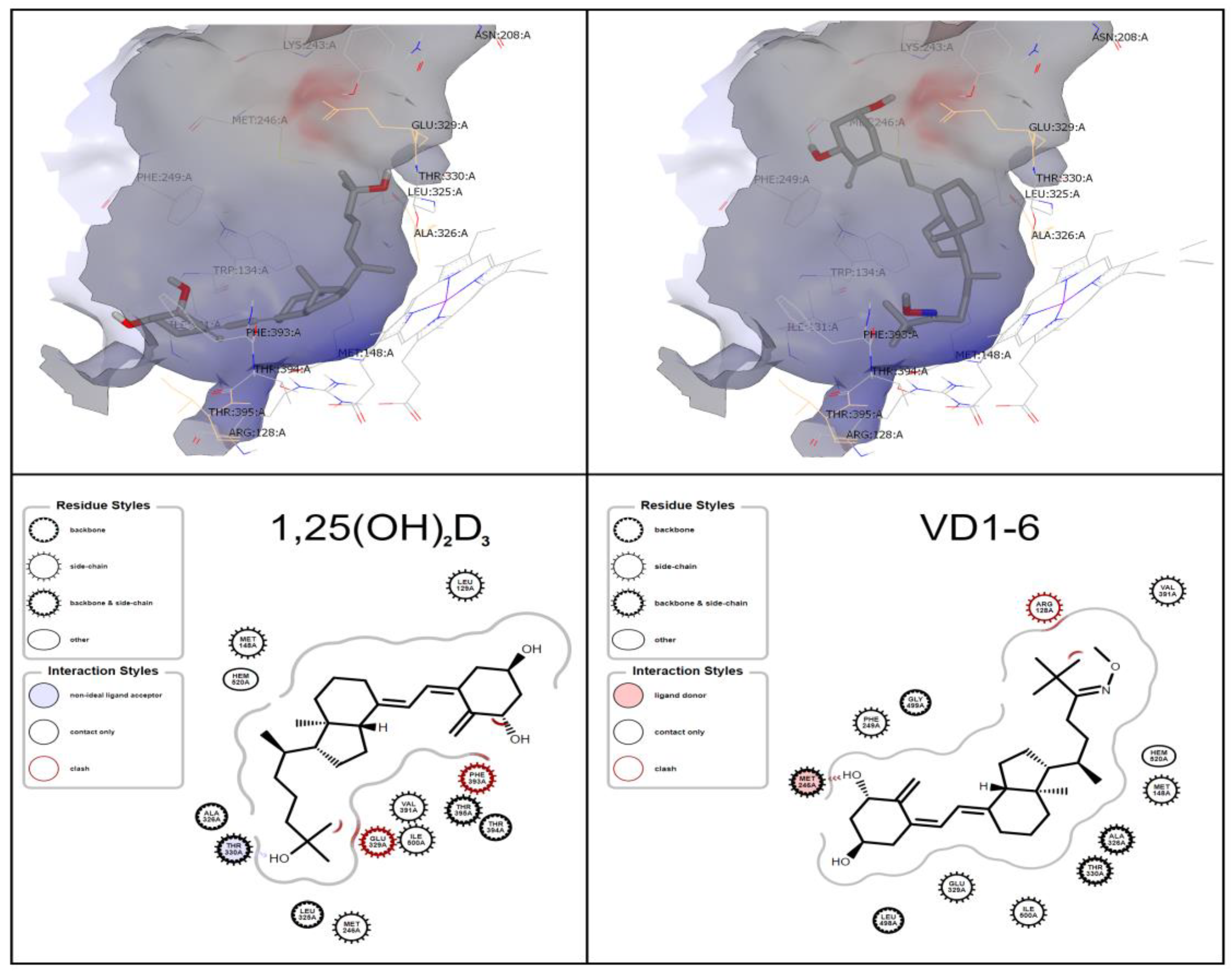
The ligand-binding site demonstrates the prominence of similar amino acid residues to those indicated by Jayaraj et al. [42], i.e., GLU 329 and THR 330 for the CYP24A1 active binding tunnel (Figure 2). Docking studies revealed that 1,25(OH)2D3 is predicted to have one hydrogen bond between its terminal C-25 hydroxyl group and THR 330. At the same time, VD1-6 displays a single hydrogen bond to MET 246 from the C-1 hydroxyl group (Figure 2). The predicted docking pose of 1,25(OH)2D3 is consistent with the results of Annalora et al. [26], where the C-1 and C-3 hydroxyl groups are located in the vicinity of the THR 395 residue, while VD1-6 displayed an inverse orientation to that of 1,25(OH)2D3, as shown in Figure 2. Both molecules have suitable flexibility in their structure and availability of space within the binding pocket to interact with the CYP24A1 heme core at their respective C21-25 ends. Docking scores revealed VD1-6 to rank higher than the endogenous ligand 1,25(OH)2D3 (−12.30 and −11.64 relative Chemgauss4 docking scores) in their docking to the CYP24A1 active binding site, which indicates a higher relative predicted binding affinity of VD1-6 to CYP24A1.
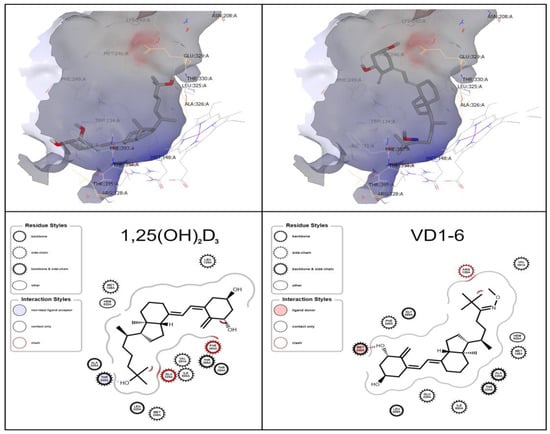
Figure 2.
OEDocking simulated orientation of 1,25(OH)2D3 (left) and VD1-6 (right) in the CYP24Al active site. Depicted as a 3D binding pocket view, using VIDA (upper panels), and ligand-interaction diagram (lower panels), using the OEChem toolkit.
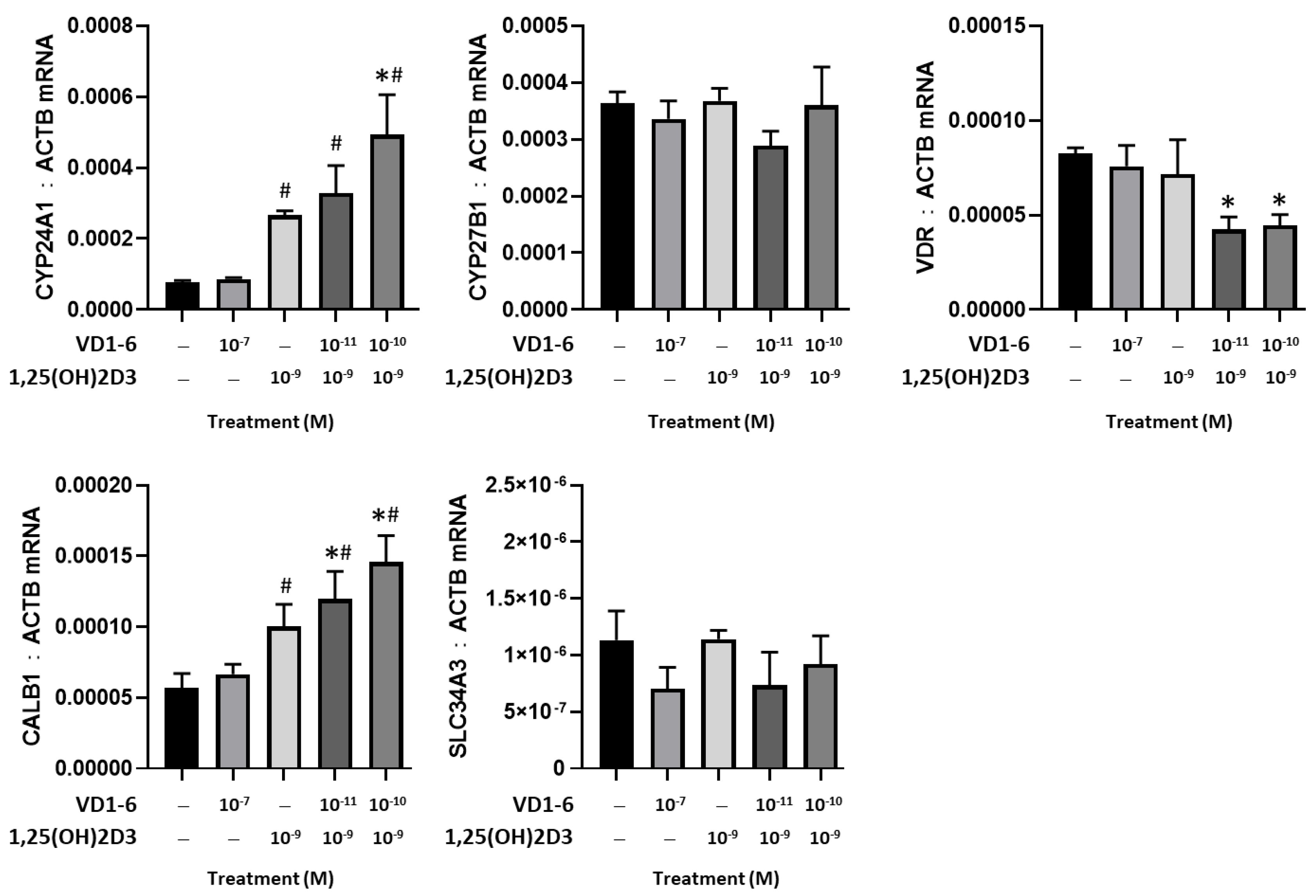
3.4. Effect of VD1-6 on CYP24A1, CYP27B1, and VDR Genes
A comparative study of the effects of VD1-6 versus 1,25(OH)2D3, alone and in combination, on the mRNA levels of CYP24A1, CYP27B1, VDR, CALB1, and SLC34A3 in HEK293T cells at 24 h is illustrated in Figure 3. At 10−7 M, VD1-6 did not affect CYP24A1 mRNA levels, compared to vehicle control. However, 1,25(OH)2D3 at 10−9 M concentration was associated with a significant (p < 0.05) elevation in CYP24A1 and CalB1 mRNA levels. The combination of VD1-6 (10−10 M) and 1,25(OH)2D3 (10−9 M) resulted in a 2-fold increase in CYP24A1 mRNA (p < 0.05) and a 1.3-fold increase in CALB1 mRNA, when compared to levels with 1,25(OH)2D3 treatment alone. In contrast, mRNA levels for CYP27B1, VDR, and SLC34A3 were not elevated by 1,25(OH)2D3, with or without VD1-6. Furthermore, 1,25(OH)2D3, in combination with VD1-6 at 10−11 M and 10−10 M, decreased VDR mRNA levels when compared to vehicle control levels.
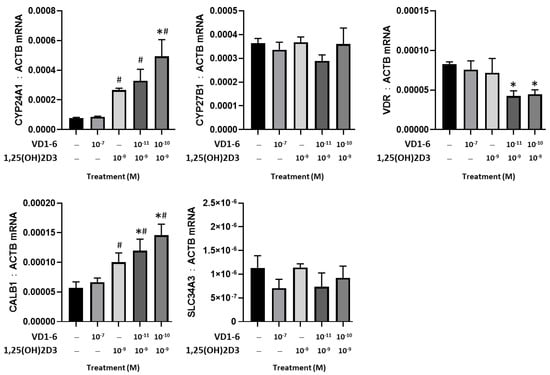
Figure 3.
Effect of VD1-6 (10−11–10−7 M), 1,25(OH)2D3 (10−9 M), and in combination on the mRNA levels of CYP24A1, CYP27B1, and VDR, CALB1, and SLC34A3 in HEK293T cell culture at 24 h. * p < 0.05 vs. vehicle control; # p < 0.05 vs. 1,25(OH)2D3 (10−7 M) treatment. Values are represented as mean ± SD.
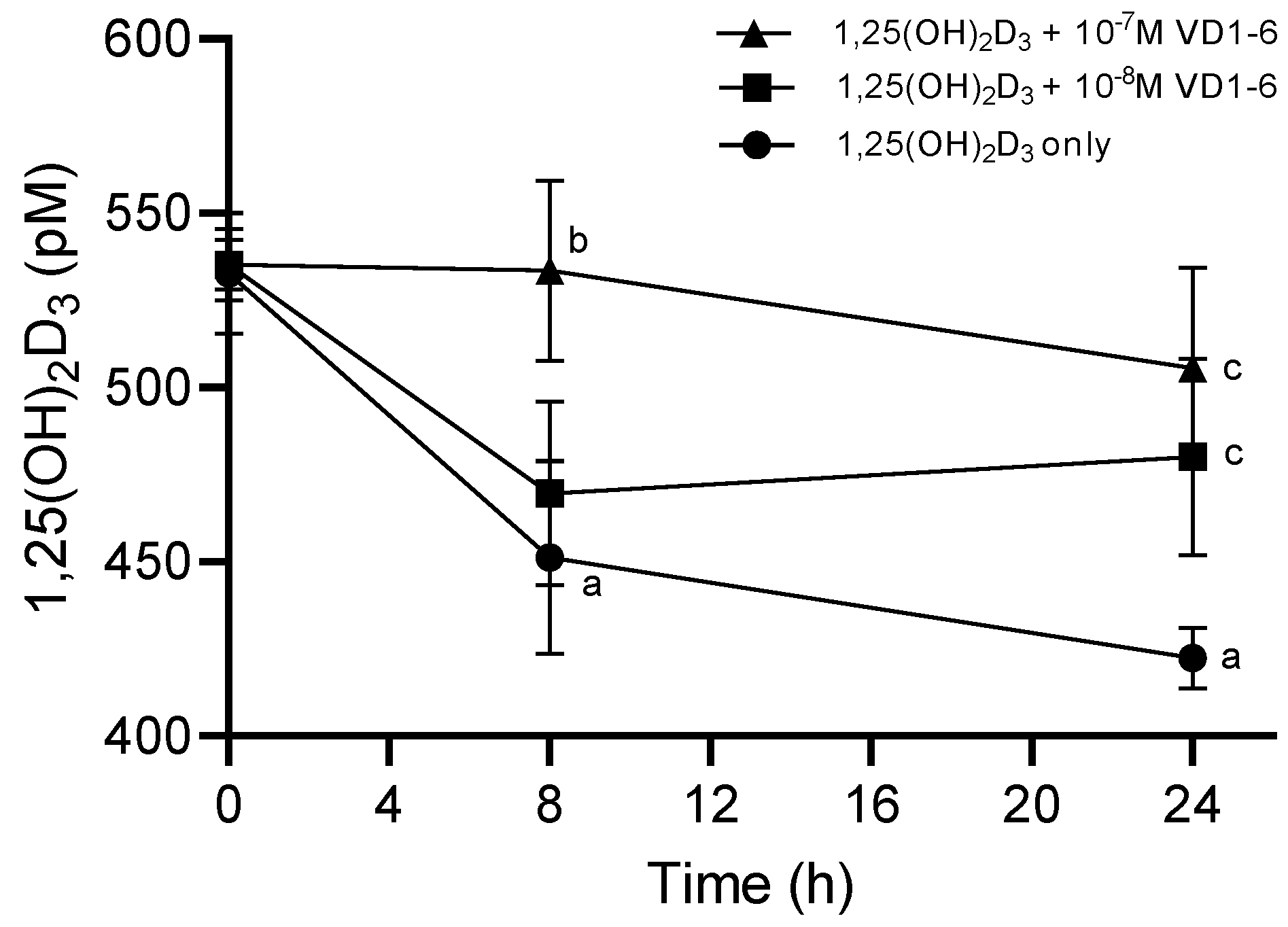
3.5. Effect of VD1-6 on Preserving 1,25(OH)2D3 in HEK293T Cell Culture
Results of LC-MS/MS analysis of the concentration of the added 1,25(OH)2D3 to HEK293T cell culture, in the absence and presence of VD1-6, at concentrations of 10−8 and 10−7 M at time zero and 8 h and 24 h post-treatment, are illustrated in Figure 4. In the absence of VD1-6, 1,25(OH)2D3 concentrations exhibited a rapid mean decline of 83 pM (p < 0.05) over 8 h to reach a mean concentration of 451 ± 27.7 pM. This decline rate was found to slow beyond 8 h (i.e., 1st order rate) with a further, but non-significant mean decline of 29 pM over the following 16 h. This resulted in a mean of 1,25(OH)2D3 levels of 422 ± 11.9 pM at 24 h, which was significantly different from the time zero mean levels (p < 0.05) (Figure 4).
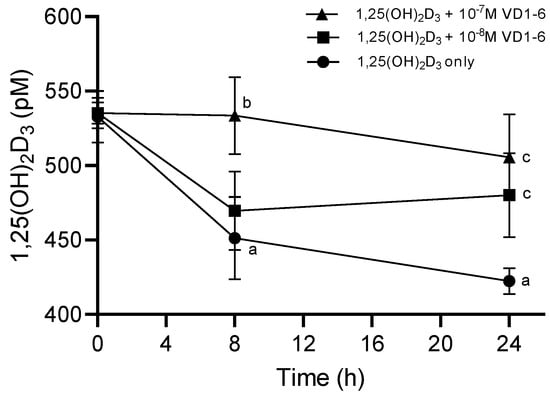
Figure 4.
Levels of 1,25(OH)2D3 in HEK293T cell culture at 0, 8, and 24 h in the absence and presence of VD1-6 at 10−8 M and 10−7 M. Mean ± S.D, n = 4/group; a p < 0.05 vs 0 h; b p < 0.05 vs. 1,25(OH)2D3 alone at 8 h; c p < 0.05 vs. 1,25(OH)2D3 alone at 24 h.
The inclusion of VD1-6 resulted in the dose-dependent preservation of the added 1,25(OH)2D3 levels. VD1-6 at 10−8 M did not exert a significant inhibition of catabolism at 8 h in terms of mean 1,25(OH)2D3 levels, compared to levels in the absence of VD1-6 at a similar time. However, with that VD1-6 concentration, the level of catabolism inhibition seen at 8 h seemed to be sustained up to 24 h, which resulted in a mean 1,25(OH)2D3 concentration of 480 ± 28.3 pM, which was significantly different (p < 0.05) when compared to that at 24 h without VD1-6 (Figure 4). With a higher VD1-6 concentration though, i.e., 10−7 M, significant catabolism inhibition was evident by 8 h, with mean 1,25(OH)2D3 concentrations of 534 ± 25.9 pM, which was significantly different (p < 0.05) from the mean concentration in the absence of VD1-6 at 8 h and non-significantly different from the mean concentrations at time zero (Figure 4). This inhibition of the catabolism effect persisted up to 24 h (i.e., non-significantly different from that at 8 h and zero time points), with a mean 1,25(OH)2D3 concentration of 506 ± 28.9 pM, which was significantly different (p < 0.05) from mean concentrations at 24 h in the absence of VD1-6 (Figure 4). It is of note that at 24 h, mean 1,25(OH)2D3 concentrations with both 10−8 M and 10−7 M VD1-6 were not significantly different, possibly indicating the comparable longer-term preservation effects of these two concentrations.
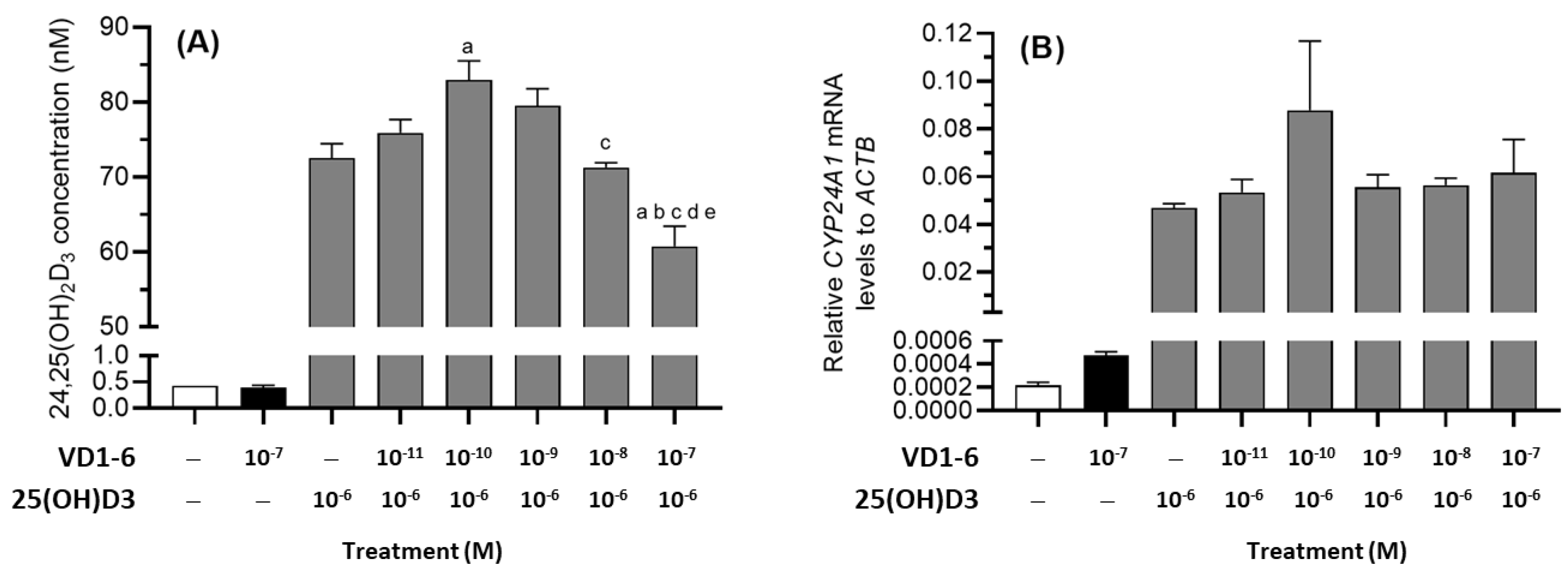
3.6. Effect of VD1-6 on 24,25(OH)2D3 Production in HEK293T Cell Culture
To assess the effects of VD1-6 on 25(OH)D3 catabolism, HEK293T cells were treated with 25(OH)D3 (10−6 M) over 72 h, and media levels of 24,25(OH)2D3 and mRNA levels for CYP24A1 were measured in the absence or presence of VD1-6 (10−11–10−7 M) (Figure 5A,B). In the presence of 25(OH)D3 (10−6 M), HEK293T cells produced 24,25(OH)2D3, which was approximately 170-fold higher than the levels in the vehicle-treated cells (Figure 5A). The inclusion of VD1-6 resulted in a biphasic concentration-dependent effect on the mean accumulated 24,25(OH)2D3 concentrations. At 10−10 M VD1-6, a 14% increase in mean 24,25(OH)2D3 concentration was observed when compared to levels with 25(OH)D3 treatment alone (p < 0.05) (Figure 5A). However, at 10−7 M VD1-6, a marked decrease in 24,25(OH)2D3 levels was seen when compared to all other treatment concentrations (Figure 5A). Although 25(OH)D3 treatment increased CYP24A1 mRNA levels in HEK293T cells, VD1-6 inclusion did not elevate CYP24A1 mRNA levels (Figure 5B). While the elevated 24,25(OH)2D3 levels at 10−10 M VD1-6 appeared to correlate with elevated CYP24A1 mRNA levels, this was not statistically significant. The decline in 24,25(OH)2D3 levels in 25(OH)D3 treatments with 10−7 M VD1-6 did not correspond to a change in CYP24A1 mRNA levels when compared to the mRNA levels in 25(OH)D3 only treatments, suggesting that the 24,25(OH)2D3 decline was not due to reduced CYP24A1 expression.
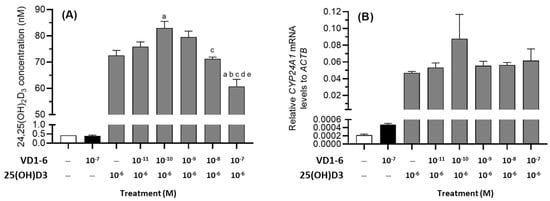
Figure 5.
24,25(OH)2D3 levels (A) and relative CYP24A1 mRNA levels (B) in HEK293T cell culture at 72 h in the absence and presence of VD1-6 (10−11–10−7 M). Mean ± S.D, n = 4/group; a p < 0.05 vs. 25(OH)D3 only treatment; b p < 0.05 vs. 25(OH)D3 + 10−11 M VD1-6 treatment; c p < 0.05 vs. 25(OH)D3 + 10−10 M VD1-6 treatment; d p < 0.05 vs. 25(OH)D3 + 10−9 M VD1-6 treatment; e p < 0.05 vs. 25(OH)D3 + 10−8 M VD1-6 treatment.
4. Discussion
In this study, we have synthesized and evaluated a novel C-24 O-methyl oxime derivative of vitamin D3 for its use as a CYP24A1 inhibitor. Since it was previously shown that the absence of the terminal hydroxyl group of the 1,25(OH)2D3 side chain is not an indication that VDR binding would be abolished, it was essential to establish whether VD1-6 has an affinity for VDR binding at concentrations used in the biological assessment. We have conducted an in vitro competition VDR binding assay based on the variable displacement of Fluormone™, a high-affinity VDR-binding fluorescent tracer [43]. This assay demonstrated that VD1-6 has a 470-fold lower affinity to the VDR binding pocket than 1,25(OH)2D3. Furthermore, VD1-6 binding to VDR was only evident at concentrations greater than 10−7 M (Figure 1). The 25(OH)D3, known to bind VDR with markedly less affinity than 1,25(OH)2D3 [44], had an 8-fold higher affinity for VDR than VD1-6.
The computer-aided simulated measurement of the change of molecular free energy that occurs due to binding (relative binding free energy) is considered a potentially reliable measure for predicting binding affinities of small ligand molecules to protein targets [45,46]. In silico docking of VD1-6 into the CYP24A1 binding pocket was conducted as a predictive assessment of CYP24A1 binding site affinity compared to 1,25(OH)2D3. The lack of a crystal structure of the human CYP24A1 in the Protein Data Bank has led us to utilize the available rat CYP24A1 protein structure for our docking studies. The reported similarity between human and rat CYP24A1 is 85%, with the rat variant matching 11 of 13 key amino acids for substrate binding and catalysis [26,42]. Simulated docking into the active binding pocket of CYP24A1 has shown that the binding of VD1-6 involves a change in the hydrogen bonding pattern relative to 1,25(OH)2D3. This may be attributed to the lack of a terminal hydroxyl moiety at the C-25 end of VD1-6, which may also explain the predicted inversion of orientation inside the binding pocket, as seen in Figure 2. The docking score of VD1-6 was superior to that of 1,25(OH)2D3, reflected by the lower relative binding free energy of −12.30 compared to −11.64 of 1,25(OH)2D3, which indicates a higher simulated binding affinity of VD1-6 to CYP24A1.
It is noteworthy that for 1,25(OH)2D3 to be hydroxylated at either C-23 or C-24 in the active site of CYP24A1, the terminal side chain (C-21 to C-25) needs to be positioned towards the heme moiety of the enzyme [47]. Our simulated poses for 1,25(OH)2D3 and VD1-6 appear consistent with this concept (Figure 2), with aliphatic side chains pitched toward the heme moiety, with appropriate flexibility. From a metabolism perspective, the C-24 O-methyl oxime moiety of VD1-6 would block C-24 hydroxylation, making it only possible to proceed down the C-23 pathway. On the other hand, the endogenous ligand 1,25(OH)2D3 can proceed through both the C-23 and C-24 catabolic routes. The greater potential for the interaction of VD1-6 with CYP24A1, besides its occupied C-24, suggests that VD1-6 is a more efficient CYP24A1 binder, with partial catabolic immunity, i.e., a likely inhibitor of vitamin D catabolism, with a presumably longer half-life than the endogenous ligand.
The combined in vitro cell-free VDR binding assay and in silico docking results for VD1-6, in comparison to the endogenous ligand, provide a clear preliminary indication that VD1-6 has the potential to be an inhibitor of CYP24A1, without significant VDR binding, at least up to concentrations of 10−7 M. Consistent with this, VD1-6 alone at 10−7 M did not induce CYP24A1 mRNA, unlike 1,25(OH)2D3, indicating the absence of VDR binding by VD1-6 (Figure 3). However, the inclusion of VD1-6, at lower concentrations, with 1,25(OH)2D3 treatment increased mRNA levels for CYP24A1 and CALB1 when compared to the effects of 1,25(OH)2D3 treatment alone. This suggests that VD1-6 potentiated the transcriptional effects of 1,25(OH)2D3, likely through binding to and inhibiting CYP24A1, increasing the half-life of 1,25(OH)2D3. The gene CALB1 encodes for Calbindin-D28K, and like CYP24A1, is directly induced by 1,25(OH)2D3 [48]. In contrast, VD1-6 did not impact CYP27B1 mRNA levels, suggesting that the effects of VD1-6 did not alter 1,25(OH)2D3 synthesis. VDR mRNA levels declined by 2-fold with the addition of VD1-6 and 1,25(OH)2D3. While the gene for VDR is vitamin D-responsive, previous studies have also demonstrated that 1,25(OH)2D3 binding VDR can increase the half-life of VDR protein without inducing VDR expression [49]. Further studies would be required to establish whether an increased half-life of 1,25(OH)2D3 due to VD1-6 treatment results in an increased VDR half-life. The 1,25(OH)2D3 mRNA levels were also unchanged due to VD1-6 treatment. SCL34A3, encoding for proximal tubular sodium-phosphate cotransporter, NaPi-IIc, has been shown to be stimulated by 1,25(OH)2D3 [50]. However, the effects of 1,25(OH)2D3 on NaPi-IIc have also been shown to be secondary to the regulation by the phosphaturic hormone, FGF23 [50]. While CALB1 mRNA levels were elevated in the presence of 1,25(OH)2D3 and VD1-6, it is worth noting that the elevation of CYP24A1 mRNA may contribute to the effects of 1,25(OH)2D3 and VD1-6 on gene expression in this experiment by responding to an increased half-life of 1,25(OH)2D3 by increasing its catabolic activity, potentially contributing to an absence of mRNA increases for VDR and SCL34A3.
To establish whether reduced 1,25(OH)2D3 catabolism occurred with VD1-6 treatment, we tested the preservation of 1,25(OH)2D3 in HEK293T cell cultures up to 24 h (Figure 4). While 1,25(OH)2D3 levels declined by 16% at 8 h and 22% at 24 h, VD1-6 at 10−7 M prevented this decline (Figure 4). While VD1-6 at 10−8 M was less protective of 1,25(OH)2D3 levels at 8 h, at 24 h, protection was comparable to that of 10−7 M VD1-6 (Figure 4). VD1-6 at 10−7 M was also capable of inhibiting 25(OH)D3 catabolism, as measured by the reduced formation of 24,25(OH)2D3, the first product of 25(OH)D3 catabolism (Figure 5A), most likely through significant competition and occupation of the CYP24A1 active site. Interestingly, the elevation in 24,25(OH)2D3 levels with VD1-6 at 10−10 M may have occurred due to counter-elevation of CYP24A1 activity by the preservation of 1,25(OH)2D3 (Figure 5A,B), given that VD1-6 combined with 1,25(OH)2D3 enhanced CYP24A1 mRNA expression (Figure 3).
5. Conclusions
Herein, we have successfully synthesized a novel C-24 O-methyloxime analogue of vitamin D3 and evaluated its VDR and CYP24A1 interactions and its CYP24A1 inhibitory activity. VD1-6 has negligible VDR binding activity and is predicted to bind, with high affinity, to the CYP24A1 active pocket, thus blocking CYP24A1 catabolic activity. Thus, our findings indicate that this compound is a novel pure inhibitor of CYP24A1. Future work will investigate the capacity for VD1-6 to inhibit CYP24A1 in diseases marked by low vitamin D activity and excessive vitamin D catabolism.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/biom12070960/s1, Figure S1: 1H NMR spectrum of VD1-2 in CDCl3; Figure S2: 13C NMR spectrum of VD1-2 in CDCl3; Figure S3: ESI MS spectrum of VD1-2; Figure S4: 1H NMR spectrum of VD1-3 in CDCl3; Figure S5: 13C NMR spectrum of VD1-3 in CDCl3; Figure S6: ESI MS spectrum of VD1-3; Figure S7: 1H NMR spectrum of VD1-4 in CDCl3; Figure S8: 13C NMR spectrum of VD1-4 in CDCl3; Figure S9: ESI MS spectrum of VD1-4; Figure S10: 1H NMR spectrum of VD1-6 in CDCl3; Figure S11: 13C NMR spectrum of VD1-6 in CDCl3; Figure S12: ESI MS spectrum of VD1-6; Figure S13: Chromatograms of 1,25(OH)2D3-DAPTAD samples; Table S1: Reproducibility of 1,25(OH)2D3 LC-MS/MS analysis assays.
Author Contributions
Conceptualization, P.H.A. and Z.L.; methodology, A.K.A., Y.C., C.S., D.Y., E.S.P., M.J.S., G.J.A., A.M.S., Z.L. and P.H.A.; software, A.K.A., E.S.P. and M.J.S.; validation, A.K.A., Y.C., C.S., D.Y., E.S.P., S.M.H. and M.J.S.; formal analysis, A.K.A. and E.S.P.; investigation, A.K.A., Y.C., C.S., D.Y., E.S.P., K.R.B., R.K.S., J.K.H. and R.P.; resources, M.J.S., G.J.A., M.D.W., Z.L. and P.H.A.; data curation, A.K.A., E.S.P. and P.H.A.; writing—original draft preparation, A.K.A.; writing—review and editing, Y.C., C.S., E.S.P., S.M.H., M.J.S., G.J.A., M.D.W., A.M.S., Z.L. and P.H.A.; visualization, A.K.A., Y.C., E.S.P. and S.M.H.; supervision, M.D.W., A.M.S., Z.L. and P.H.A.; project administration, P.H.A.; funding acquisition, P.H.A. All authors have read and agreed to the published version of the manuscript.
Funding
This project was funded by the National Health and Medical Research Council of Australia (GNT1165107) and support from the China-Australia Centre for Health Science Research.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available, as part of the data will be used for further studies.
Acknowledgments
We would like to thank OpenEye Scientific Software for the use of a no-cost academic license. This research was supported by an Australian Government Research Training Program (RTP) Scholarship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anderson, P.H. Vitamin D activity and metabolism in bone. Curr. Osteoporos. Rep. 2017, 15, 443–449. [Google Scholar] [CrossRef] [PubMed]
- St-Arnaud, R.; Jones, G. CYP24A1: Structure, function, and physiological role. In Vitamin D; Elsevier: Amsterdam, The Netherlands, 2018; pp. 81–95. [Google Scholar]
- Jones, G.; Prosser, D.E.; Kaufmann, M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch. Biochem. Biophys. 2012, 523, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Zierold, C.; Reinholz, G.G.; Mings, J.A.; Prahl, J.M.; DeLuca, H.F. Regulation of the porcine 1,25-dihydroxyvitamin D3-24-hydroxylase (CYP24) by 1,25-dihydroxyvitamin D3 and parathyroid hormone in AOK-B50 cells. Arch. Biochem. Biophys. 2000, 381, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Armbrecht, H.; Hodam, T.; Boltz, M.; Partridge, N.; Brown, A.; Kumar, V. Induction of the vitamin D 24-hydroxylase (CYP24) by 1,25-dihydroxyvitamin D3 is regulated by parathyroid hormone in UMR106 osteoblastic cells. Endocrinology 1998, 139, 3375–3381. [Google Scholar] [CrossRef] [PubMed]
- Shinki, T.; Jin, C.H.; Nishimura, A.; Nagai, Y.; Ohyama, Y.; Noshiro, M.; Okuda, K.; Suda, T. Parathyroid hormone inhibits 25-hydroxyvitamin D3-24-hydroxylase mRNA expression stimulated by 1 alpha, 25-dihydroxyvitamin D3 in rat kidney but not in intestine. J. Biol. Chem. 1992, 267, 13757–13762. [Google Scholar] [CrossRef]
- Anderson, P.H.; Atkins, G.J.; Morris, H.A.; Findlay, D.M. Vitamin D Activities in Osteocytes. In Vitamin D; Elsevier: Amsterdam, The Netherlands, 2018; pp. 319–327. [Google Scholar]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic kidney disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Patel, U.D.; Young, E.W.; Ojo, A.O.; Hayward, R.A. CKD progression and mortality among older patients with diabetes. Am. J. Kidney Dis. 2005, 46, 406–414. [Google Scholar] [CrossRef]
- Goltzman, D. Parathyroid Hormone: Its Role in Calcium and Phosphate Homeostasis. In The Physiological Basis of Metabolic Bone Disease; Nordin, B.E.C., Morris, H.A., Anderson, P., Eds.; CRC Press: Boca Raton, FL, USA, 2014; pp. 103–136. [Google Scholar]
- Perwad, F.; Portale, A.A. Phosphate Homeostasis and Metabolic Bone Disease. In The Physiological Basis of Metabolic Bone Disease; CRC Press: Boca Raton, FL, USA, 2014; pp. 137–165. [Google Scholar]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Helvig, C.F.; Cuerrier, D.; Hosfield, C.M.; Ireland, B.; Kharebov, A.Z.; Kim, J.W.; Ramjit, N.J.; Ryder, K.; Tabash, S.P.; Herzenberg, A.M. Dysregulation of renal vitamin D metabolism in the uremic rat. Kidney Int. 2010, 78, 463–472. [Google Scholar] [CrossRef] [Green Version]
- Caravaca-Fontán, F.; Gonzales-Candia, B.; Luna, E.; Caravaca, F. Relative importance of the determinants of serum levels of 25-hydroxy vitamin D in patients with chronic kidney disease. Nefrología 2016, 36, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Elder, G. Pathophysiology and recent advances in the management of renal osteodystrophy. J. Bone Mineral. Res. 2002, 17, 2094–2105. [Google Scholar] [CrossRef]
- Martinez, I.; Saracho, R.; Montenegro, J.; Llach, F. The importance of dietary calcium and phosphorous in the secondary hyperparathyroidism of patients with early renal failure. Am. J. Kidney Dis. 1997, 29, 496–502. [Google Scholar] [CrossRef]
- Drüeke, T.B. Which vitamin D derivative to prescribe for renal patients. Curr. Opin. Nephrol. Hypertens. 2005, 14, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Frazão, J.M.; Elangovan, L.; Maung, H.M.; Chesney, R.W.; Acchiardo, S.R.; Bower, J.D.; Kelley, B.J.; Rodriguez, H.J.; Norris, K.C.; Robertson, J.A. Intermittent doxercalciferol (1α-hydroxyvitamin D2) therapy for secondary hyperparathyroidism. Am. J. Kidney Dis. 2000, 36, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Posner, G.H.; Helvig, C.; Cuerrier, D.; Collop, D.; Kharebov, A.; Ryder, K.; Epps, T.; Petkovich, M. Vitamin D analogues targeting CYP24 in chronic kidney disease. J. Steroid Biochem. Mol. Biol. 2010, 121, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Byford, V.; Arabian, A.; Sakai, Y.; Demay, M.B.; St-Arnaud, R.; Jones, G. Altered pharmacokinetics of 1α,25-dihydroxyvitamin D3 and 25-hydroxyvitamin D3 in the blood and tissues of the 25-hydroxyvitamin D-24-hydroxylase (Cyp24a1) null mouse. Endocrinology 2005, 146, 825–834. [Google Scholar] [CrossRef] [Green Version]
- St-Arnaud, R.; Arabian, A.; Travers, R.; Barletta, F.; Raval-Pandya, M.; Chapin, K.; Depovere, J.; Mathieu, C.; Christakos, S.; Demay, M.B. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1, 25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology 2000, 141, 2658–2666. [Google Scholar] [CrossRef]
- Li, H.; Fang, Z.; Dai, H.; Zhang, H.; Liu, Y. Efficient synthesis of novel oxime analogues of the hormone 1α,25-dihydroxyvitamin D3. J. Chem. Res. 2015, 39, 368–372. [Google Scholar] [CrossRef]
- Kabakoff, B.; Schnoes, H.K.; De Luca, H.F. In vitro inhibitor studies of vitamin D 25-hydroxylase in rat liver microsomes. Arch. Biochem. Biophys. 1983, 221, 38–45. [Google Scholar] [CrossRef]
- Posner, G.H.; Halford, B.A.; Peleg, S.; Dolan, P.; Kensler, T.W. Conceptually new low-calcemic oxime analogues of the hormone 1α,25-dihydroxyvitamin D3: Synthesis and biological testing. J. Med. Chem. 2002, 45, 1723–1730. [Google Scholar] [CrossRef]
- POLARSCREEN™ VITAMIN D RECEPTOR COMPETITOR ASSAY, RED. Available online: http://tools.thermofisher.com/content/sfs/manuals/polarscreen_vdr_red_man.pdf (accessed on 8 July 2022).
- Annalora, A.J.; Goodin, D.B.; Hong, W.-X.; Zhang, Q.; Johnson, E.F.; Stout, C.D. Crystal structure of CYP24A1, a mitochondrial cytochrome P450 involved in vitamin D metabolism. J. Mol. Biol. 2010, 396, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer Generation with OMEGA: Algorithm and Validation Using High Quality Structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Modeling 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- OMEGA, version 3.3.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2019.
- Make Receptor, version 3.3.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2019.
- FRED, version 3.3.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2019.
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput.-Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- VIDA, version 3.3.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2019.
- Brodaczewska, K.K.; Szczylik, C.; Fiedorowicz, M.; Porta, C.; Czarnecka, A.M. Choosing the right cell line for renal cell cancer research. Mol. Cancer 2016, 15, 83. [Google Scholar] [CrossRef] [Green Version]
- Simner, C.L.; Ashley, B.; Cooper, C.; Harvey, N.C.; Lewis, R.M.; Cleal, J.K. Investigating a suitable model for the study of vitamin D mediated regulation of human placental gene expression. J. Steroid Biochem. Mol. Biol. 2020, 199, 105576. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific. TRIzol Reagent User Guide. Available online: https://tools.thermofisher.com/content/sfs/manuals/trizol_reagent.pdf (accessed on 8 July 2022).
- Bio-Rad. iScript™ cDNA Synthesis Kit. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/4106228.pdf (accessed on 8 July 2022).
- Alshabrawy, A.K.; Bergamin, A.; Sharma, D.K.; Hickey, S.M.; Brooks, D.A.; O’Loughlin, P.; Wiese, M.D.; Anderson, P.H. LC-MS/MS analysis of vitamin D3 metabolites in human serum using a salting-out based liquid-liquid extraction and DAPTAD derivatization. J. Chromatogr. B 2021, 1173, 122654. [Google Scholar] [CrossRef]
- Ishige, T.; Satoh, M.; Ogawa, S.; Nishimura, M.; Matsushita, K.; Higashi, T.; Nomura, F. Improved sensitivity of serum/plasma 1α, 25-dihydroxyvitamin D quantification by DAPTAD derivatization. Clin. Chim. Acta 2017, 473, 173–179. [Google Scholar] [CrossRef]
- Posner, G.H.; Lee, J.K.; White, M.C.; Hutchings, R.H.; Dai, H.; Kachinski, J.L.; Dolan, P.; Kensler, T.W. Antiproliferative hybrid analogs of the hormone 1α, 25-dihydroxyvitamin D3: Design, synthesis, and preliminary biological evaluation. J. Org. Chem. 1997, 62, 3299–3314. [Google Scholar] [CrossRef]
- Kahraman, M.; Sinishtaj, S.; Dolan, P.M.; Kensler, T.W.; Peleg, S.; Saha, U.; Chuang, S.S.; Bernstein, G.; Korczak, B.; Posner, G.H. Potent, selective and low-calcemic inhibitors of CYP24 hydroxylase: 24-sulfoximine analogues of the hormone 1α, 25-dihydroxyvitamin D3. J. Med. Chem. 2004, 47, 6854–6863. [Google Scholar] [CrossRef]
- Jayaraj, J.M.; Krishnasamy, G.; Lee, J.K.; Muthusamy, K. In silico identification and screening of CYP24A1 inhibitors: 3D QSAR pharmacophore mapping and molecular dynamics analysis. J. Biomol. Struct. Dyn. 2019, 37, 1700–1714. [Google Scholar] [CrossRef] [PubMed]
- Moerke, N.J. Fluorescence polarization (FP) assays for monitoring peptide-protein or nucleic acid-protein binding. Curr. Protoc. Chem. Biol. 2009, 1, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Okamura, W.H.; Norman, A.W. Structure-function relationships in the vitamin D endocrine system. Endocr. Rev. 1995, 16, 200–257. [Google Scholar] [PubMed]
- Cournia, Z.; Allen, B.; Sherman, W. Relative binding free energy calculations in drug discovery: Recent advances and practical considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Bhati, A.P.; Zasada, S.J.; Coveney, P.V. Rapid, accurate, precise and reproducible ligand–protein binding free energy prediction. Interface Focus 2020, 10, 20200007. [Google Scholar] [CrossRef] [PubMed]
- Schuster, I. Cytochromes P450 are essential players in the vitamin D signaling system. Biochim. Biophys. Acta 2011, 1814, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Lee, S.; Stolz, R.; Gabrielides, C.; Pansini-Porta, A.; Bruns, M.E.; Bruns, D.E.; Miffin, T.E.; Pike, J.W.; Christakos, S. Effect of hormones and development on the expression of the rat 1,25-dihydroxyvitamin D3 receptor gene. Comparison with calbindin gene expression. J. Biol. Chem. 1989, 264, 17454–17461. [Google Scholar] [CrossRef]
- Healy, K.D.; Frahm, M.A.; DeLuca, H.F. 1,25-Dihydroxyvitamin D3 up-regulates the renal vitamin D receptor through indirect gene activation and receptor stabilization. Arch. Biochem. Biophys. 2005, 433, 466–473. [Google Scholar] [CrossRef]
- Kido, S.; Kaneko, I.; Tatsumi, S.; Segawa, H.; Miyamoto, K. Vitamin D and type II sodium-dependent phosphate cotransporters. Contrib. Nephrol. 2013, 180, 86–97. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).