
Tissue-Nonspecific Alkaline Phosphatase (TNAP) as the Enzyme Involved in the Degradation of Nucleotide Analogues in the Ligand Docking and Molecular Dynamics Approaches



Abstract
:1. Introduction
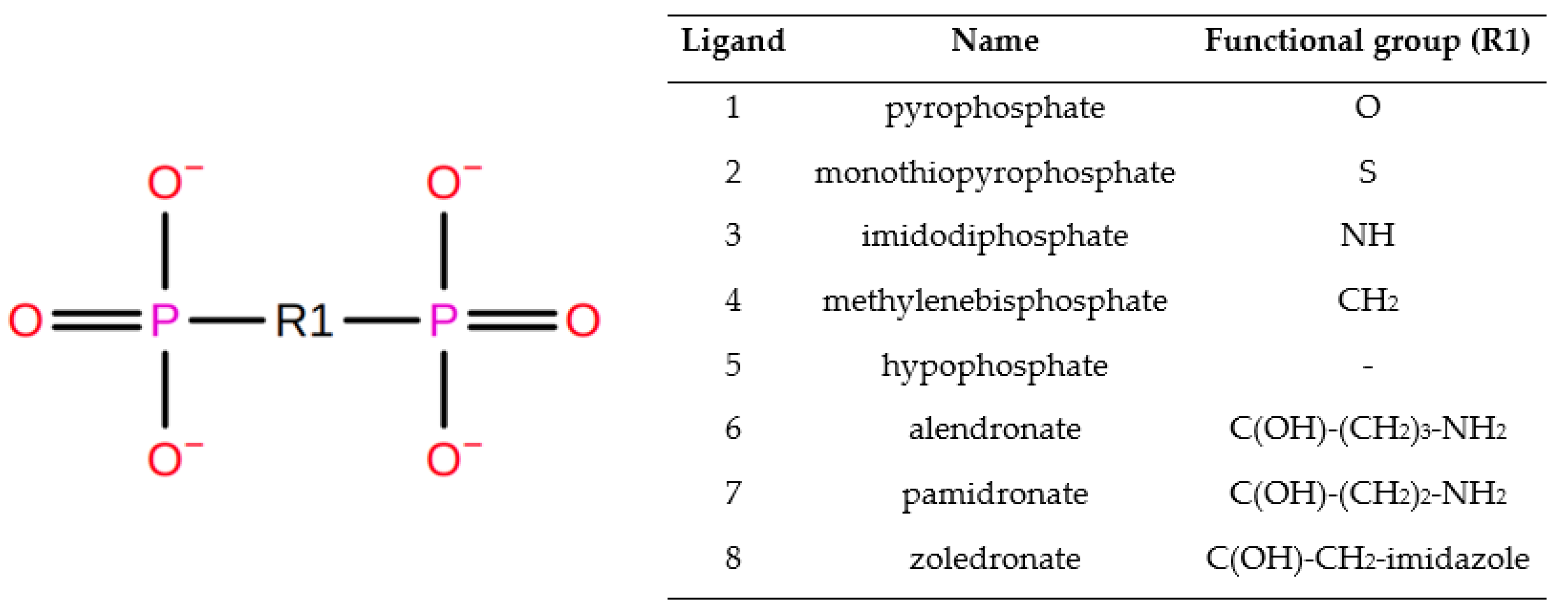
2. Materials and Methods
2.1. The Structure Preparation
2.2. Molecular Docking
2.3. Conventional Molecular Dynamics and Postprocessing
2.4. Thermodynamic Integration
2.5. Calculation Platform
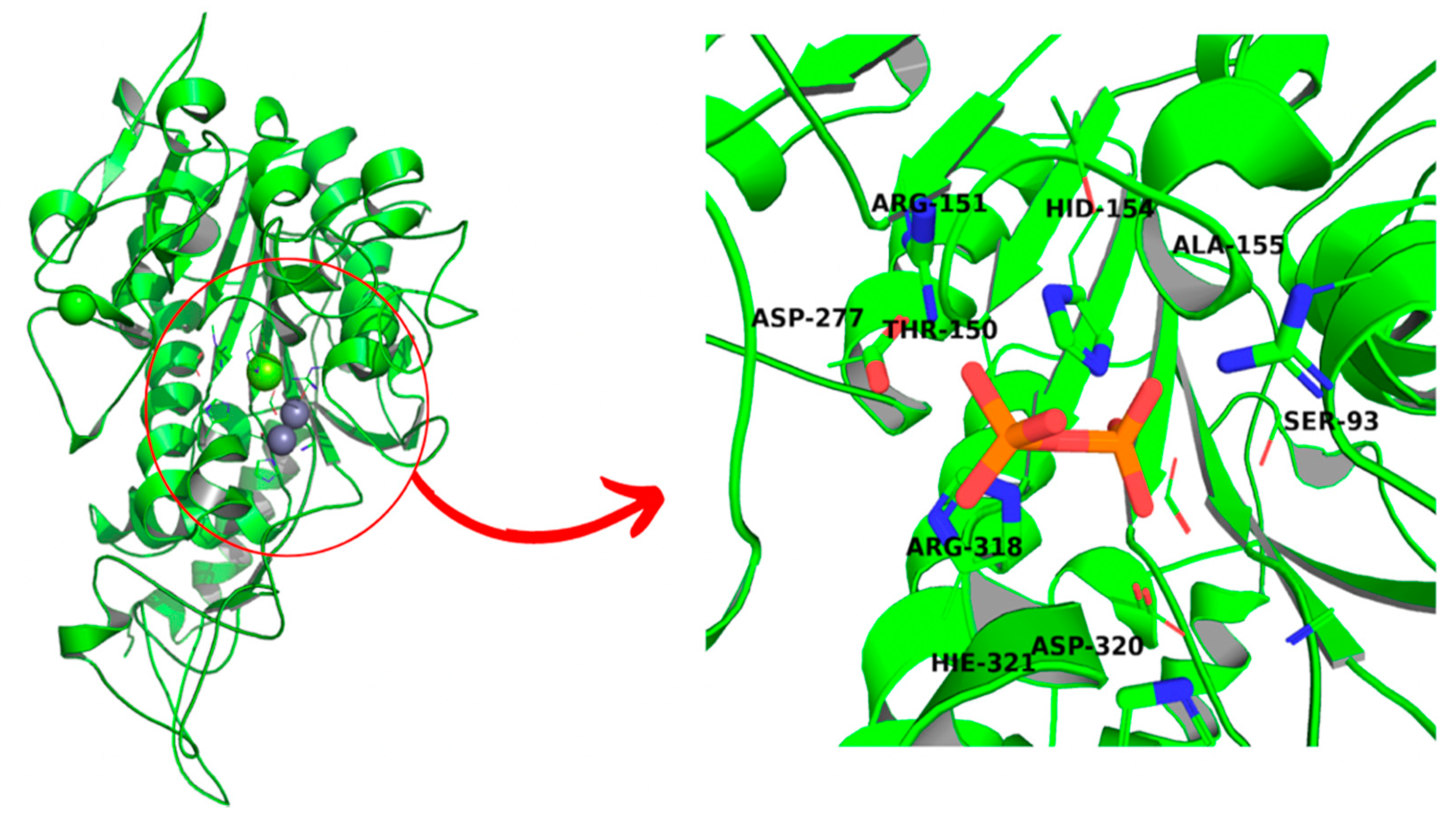
3. Results
3.1. Structure Validation
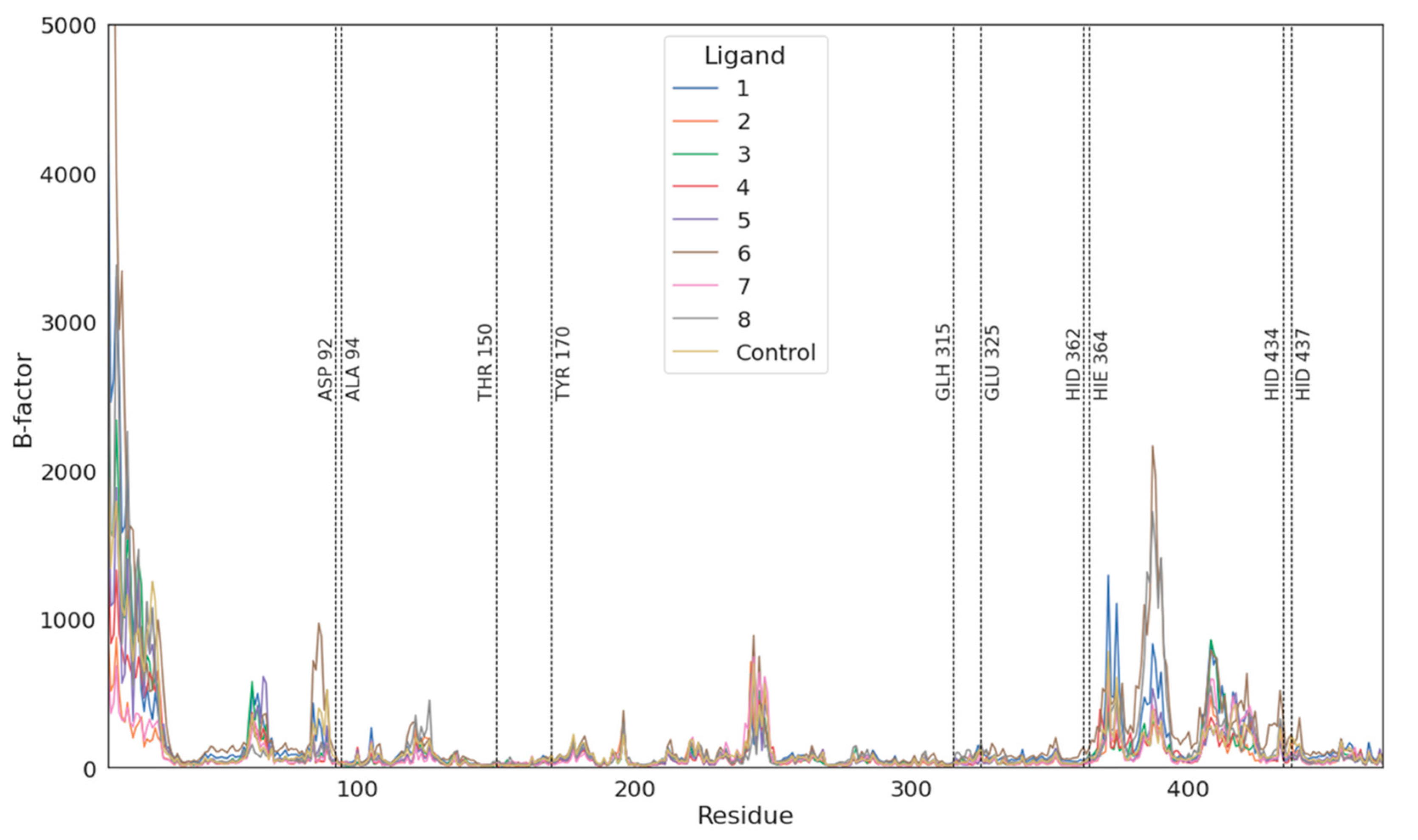
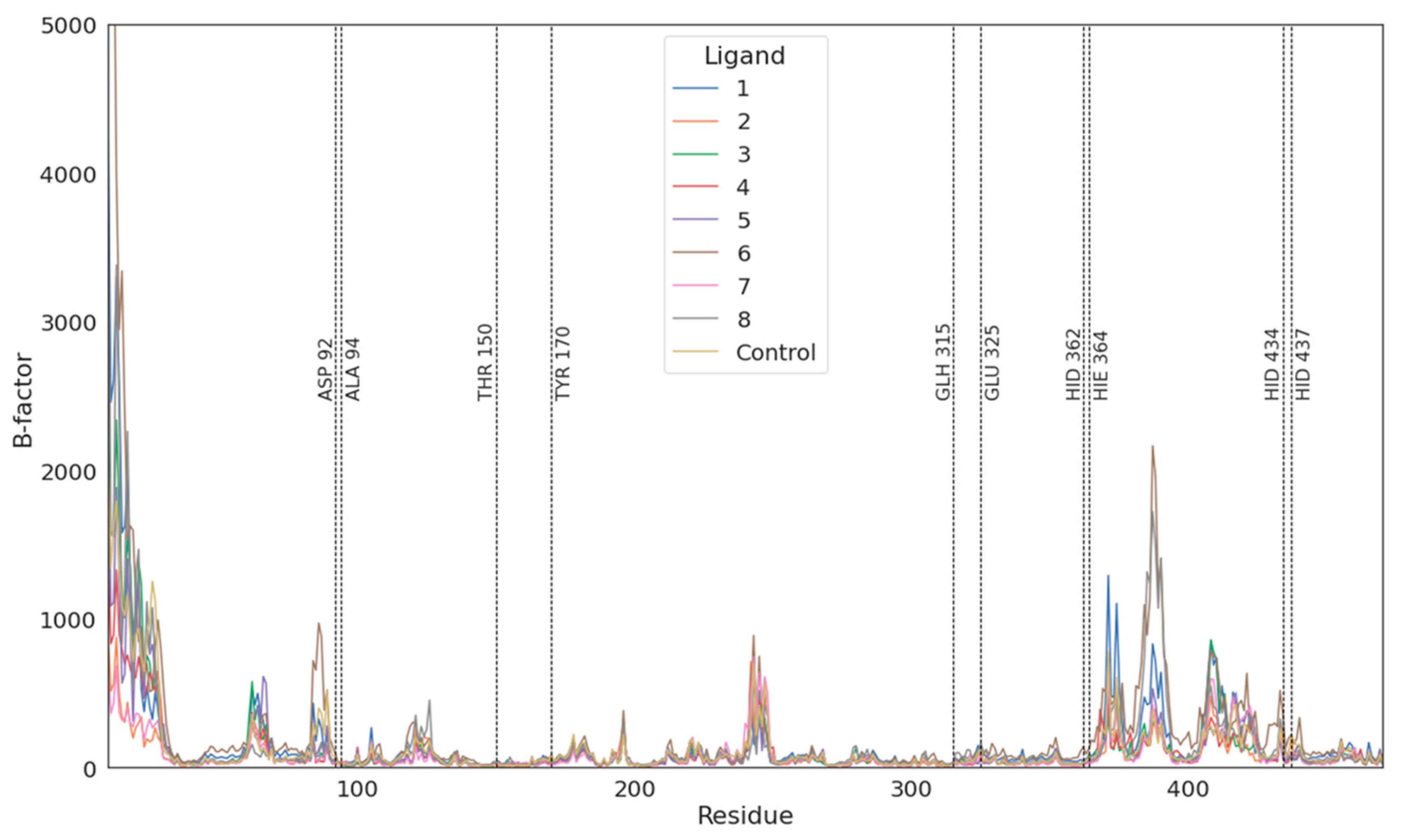
3.2. Molecular Dynamics Simulations Validation
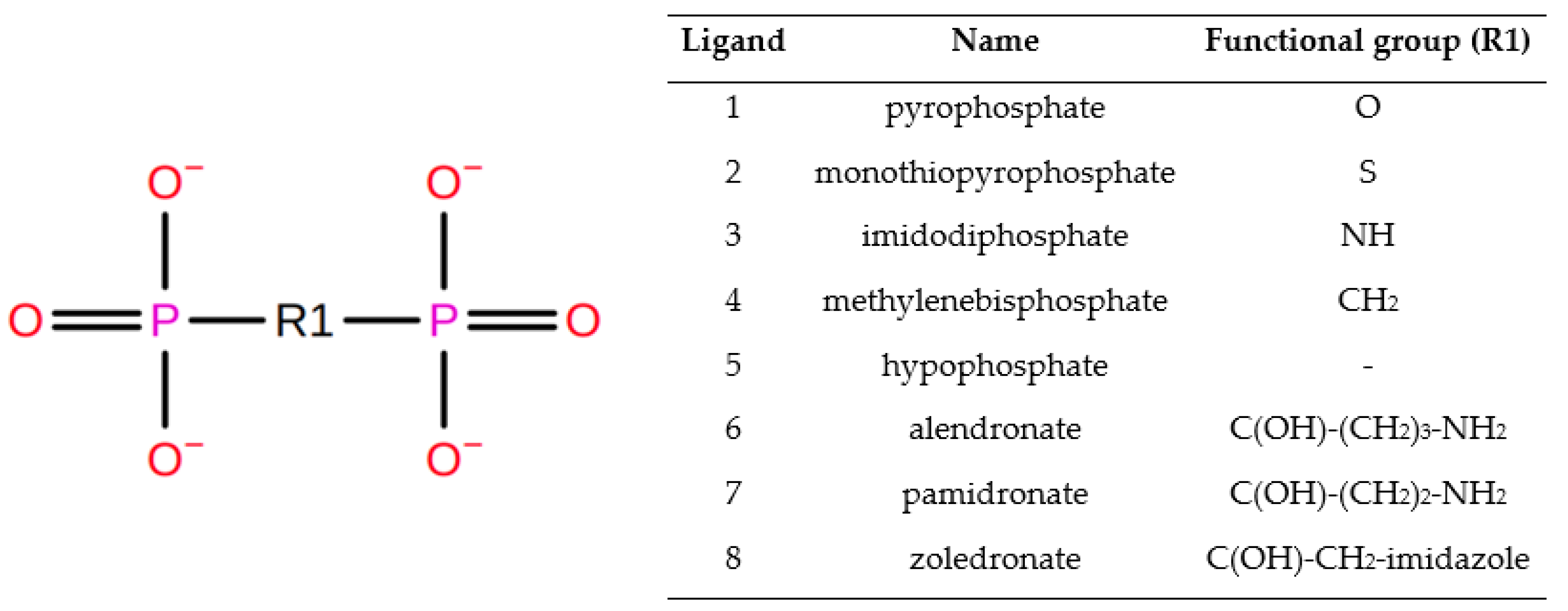
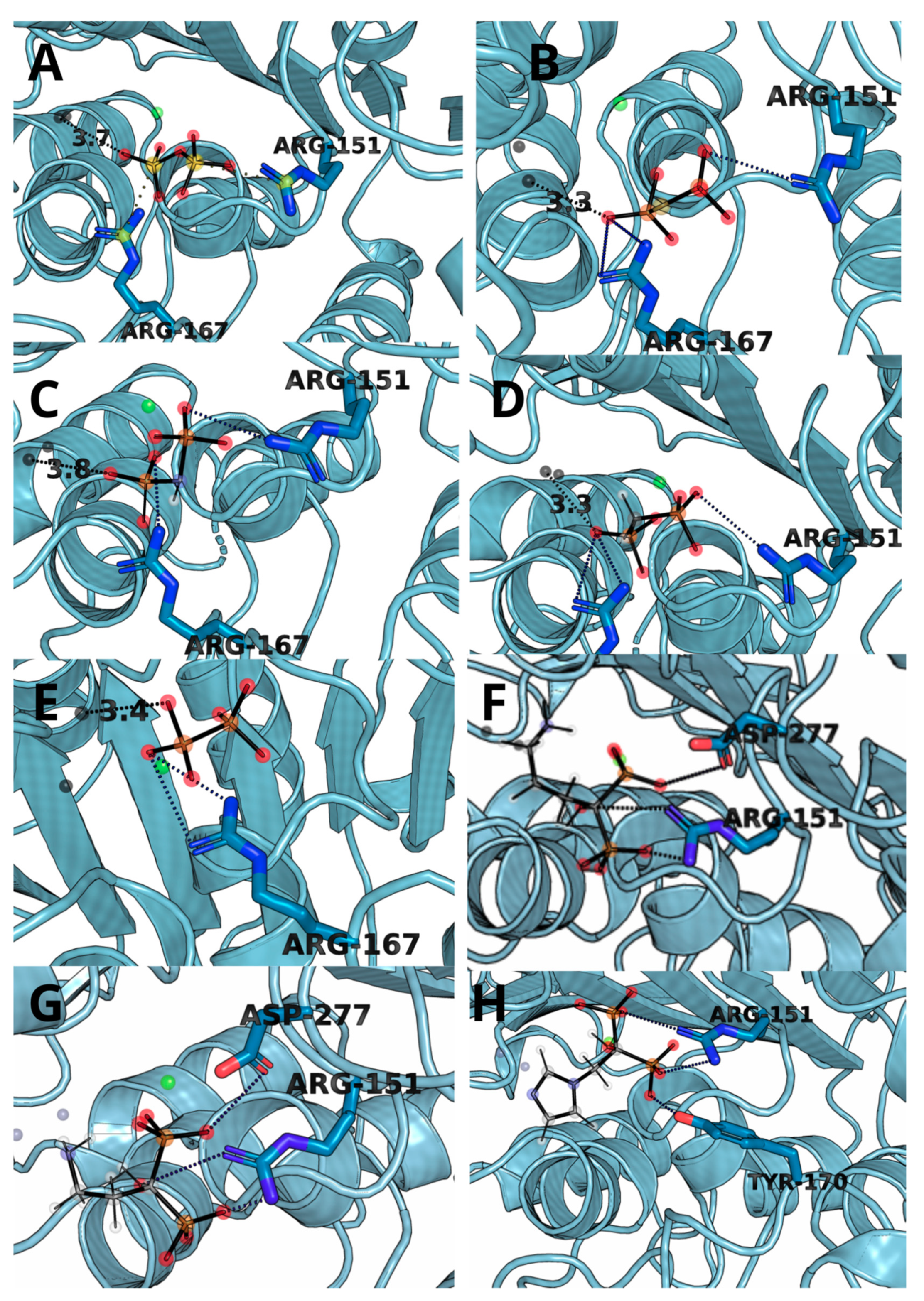
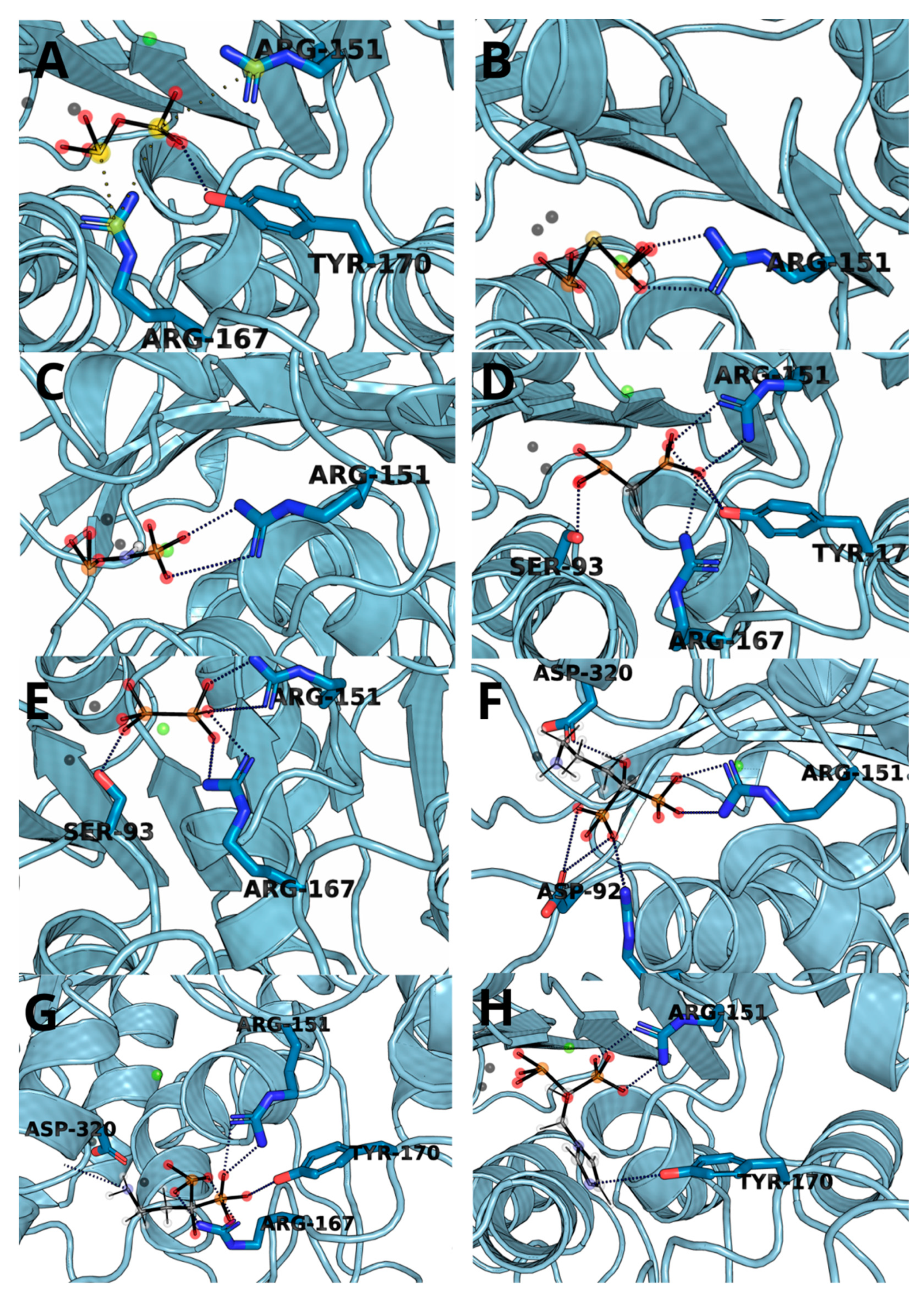
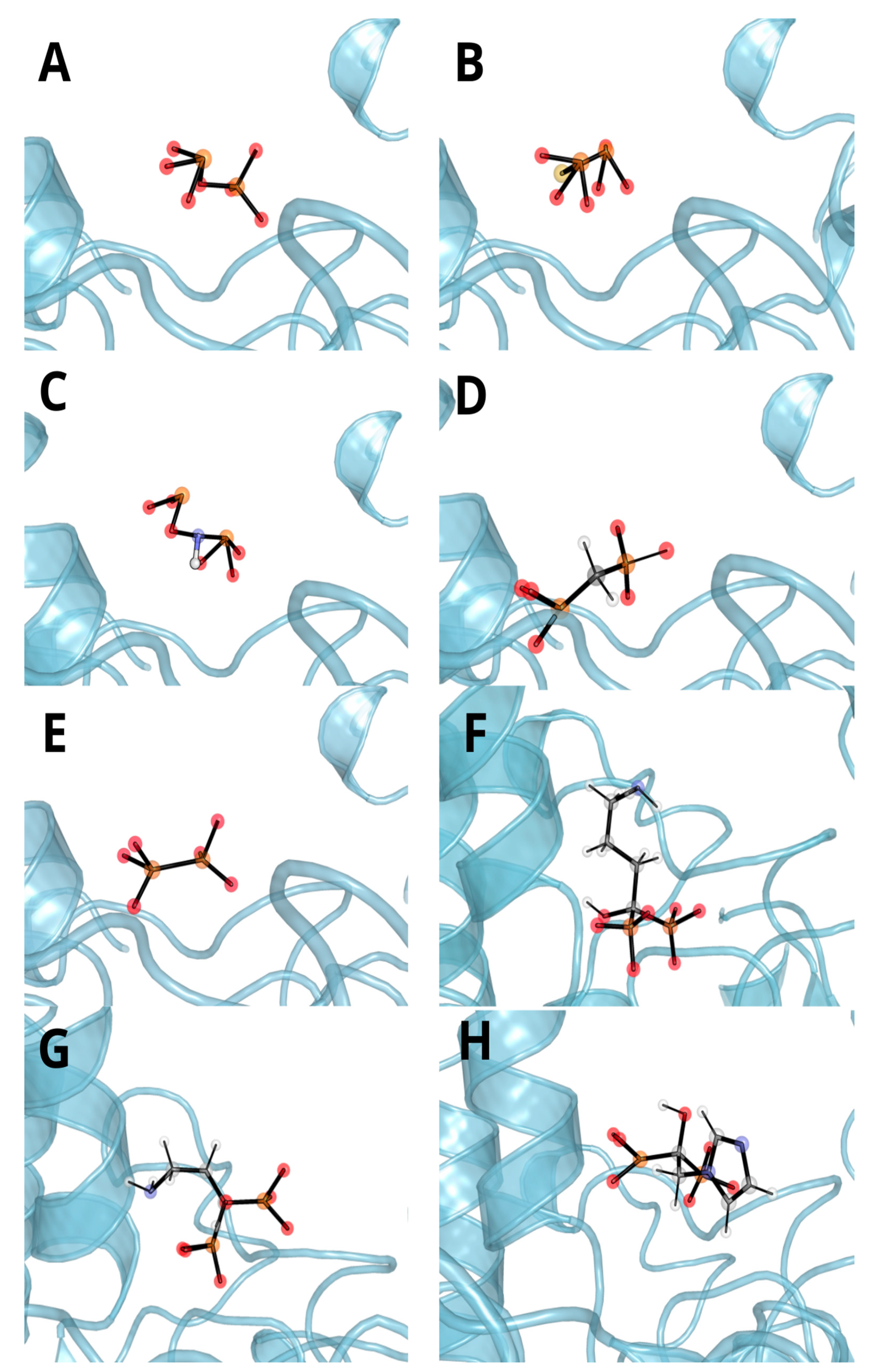
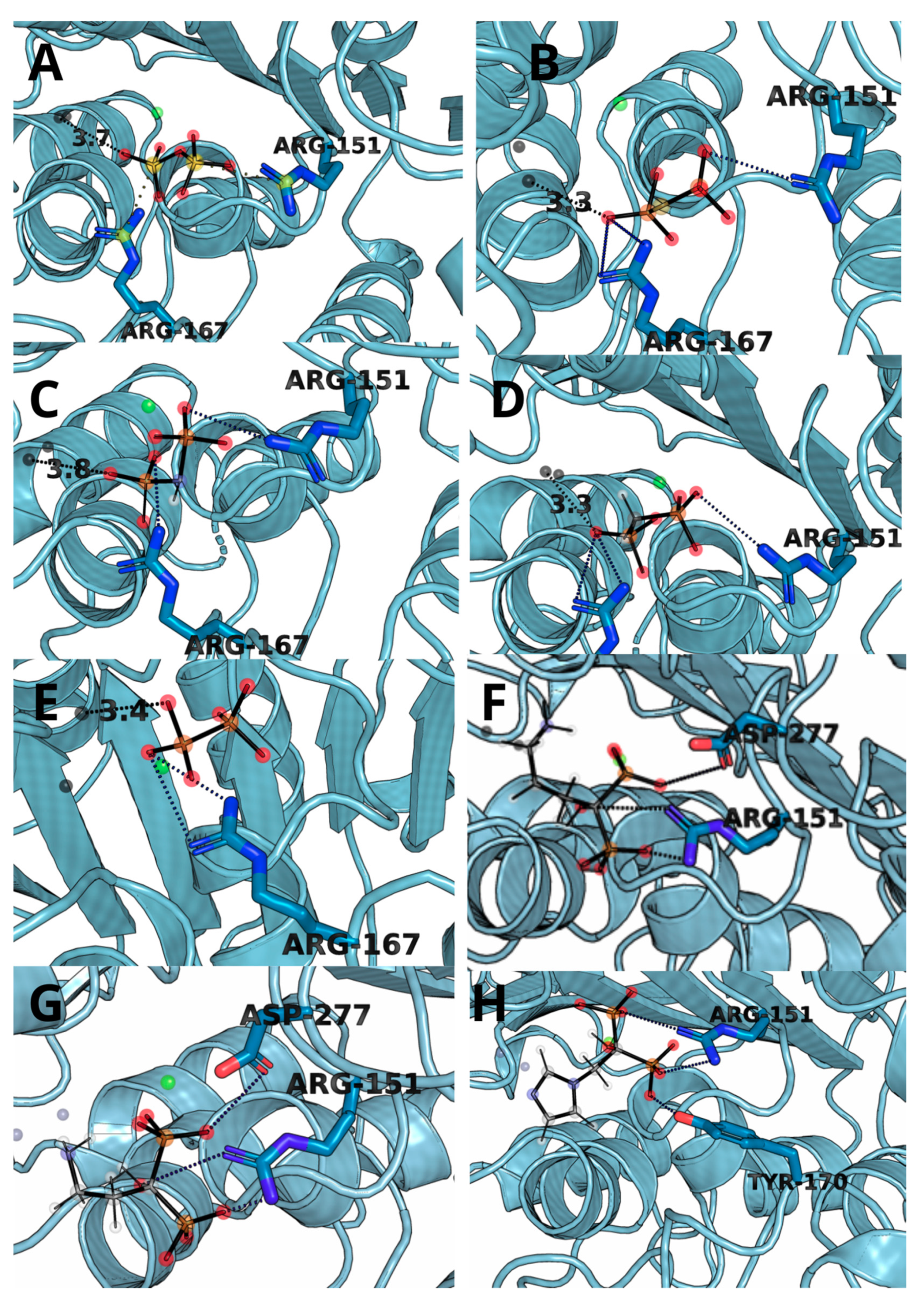
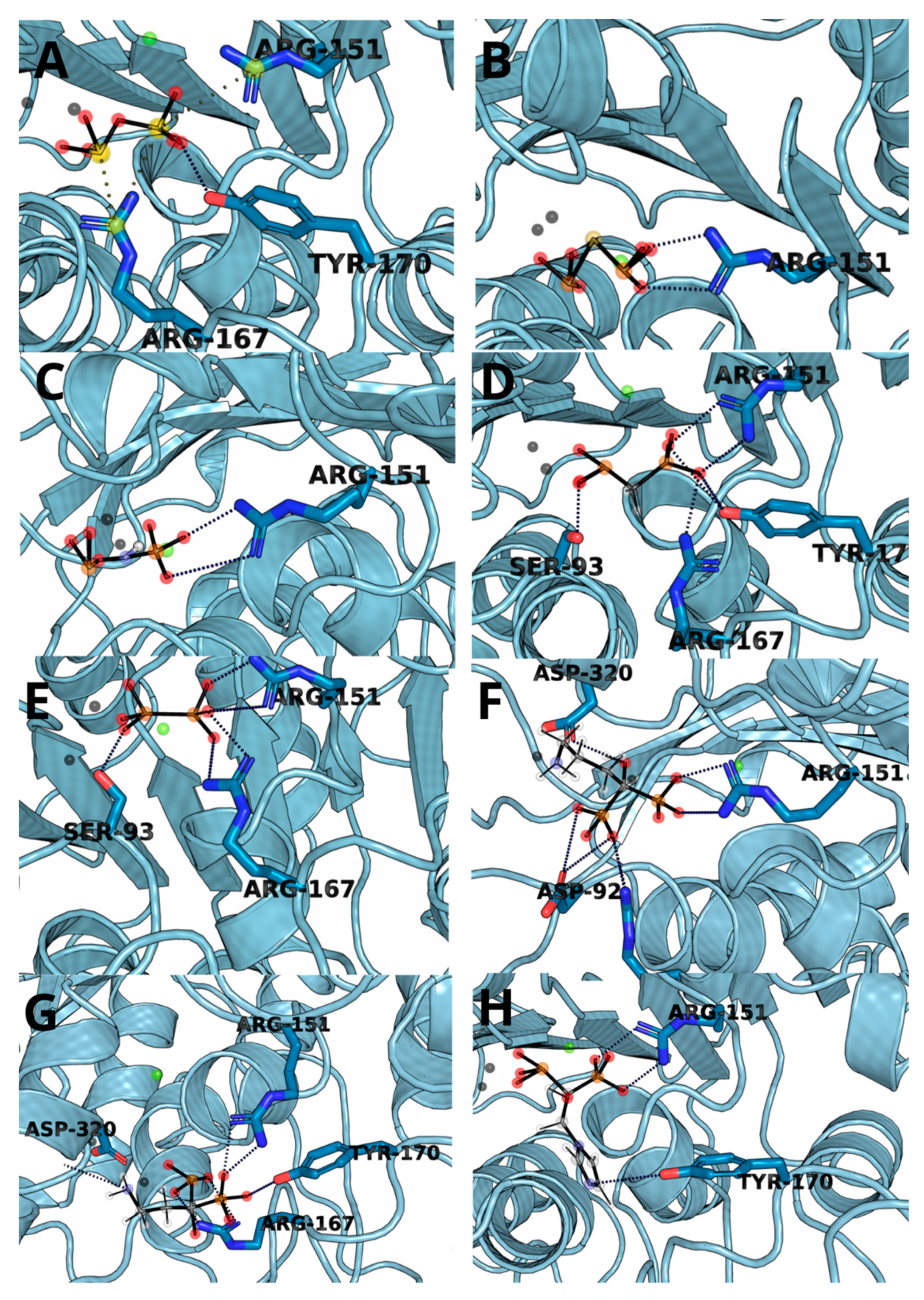
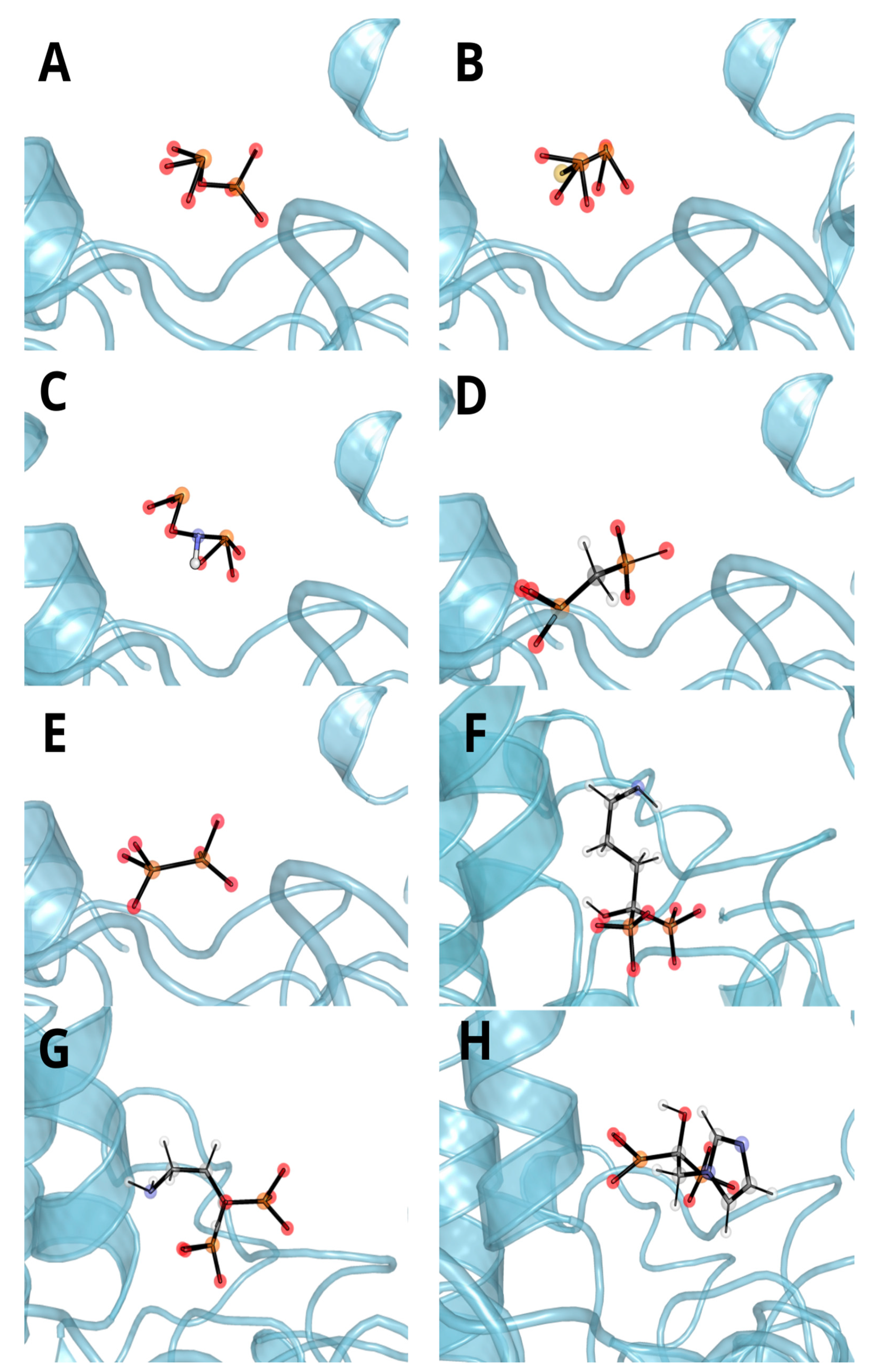
3.3. Molecular Docking and Dynamic’s Results
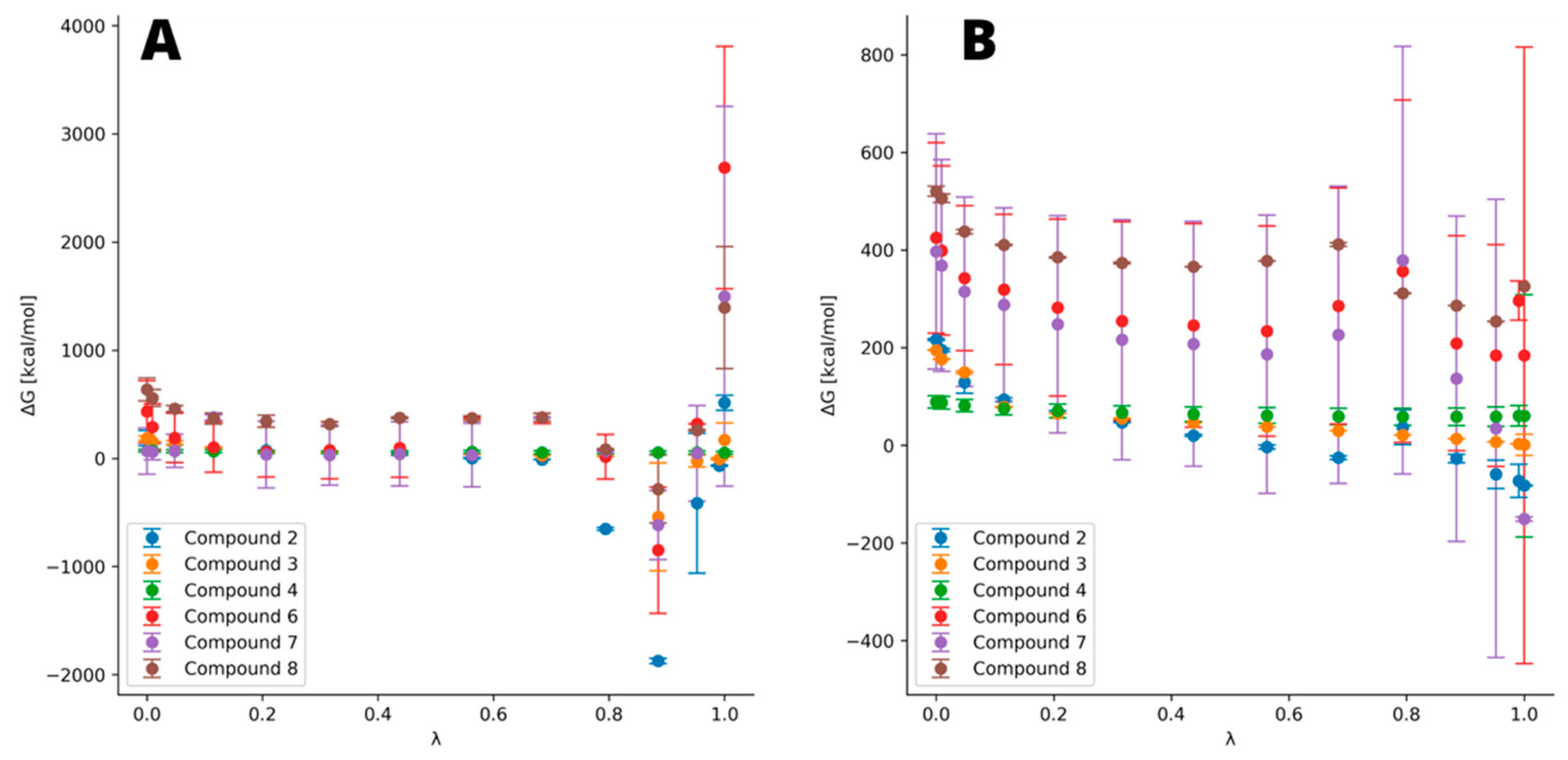
3.4. Thermodynamic Integration
3.5. Molecular Dynamics and MM/GBSA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sun, Y.Z.; Rahbani, J.F.; Jedrychowski, M.P.; Riley, C.L.; Vidoni, S.; Bogoslavski, D.; Hu, B.; Dumesic, P.A.; Zeng, X.; Wang, A.B.; et al. Mitochondrial TNAP controls thermogenesis by hydrolysis of phosphocreatine. Nature 2021, 593, 580–585. [Google Scholar] [CrossRef]
- Hoshi, K.; Amizuka, N.; Oda, K.; Ikehara, Y.; Ozawa, H. Immunolocalization of tissue non-specific alkaline phosphatase in mice. Histochem. Cell Biol. 1997, 107, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Nwafor, D.C.; Chakraborty, S.; Brichacek, A.L.; Jun, S.; Gambill, C.A.; Wang, W.; Engler-Chiurazzi, E.B.; Dakhlallah, D.; Pinkerton, A.B.; Millán, J.L.; et al. Loss of tissue-nonspecific alkaline phosphatase (TNAP) enzyme activity in cerebral microvessels is coupled to persistent neuroinflammation and behavioral deficits in late sepsis. Brain Behav. Immun. 2020, 84, 115–131. [Google Scholar] [CrossRef]
- Shimada, B.K.; Pomozi, V.; Zoll, J.; Kuo, S.R.; Martin, L.; Le Saux, O. ABCC6, Pyrophosphate and Ectopic Calcification: Therapeutic Solutions. Int. J. Mol. Sci. 2021, 22, 4555. [Google Scholar] [CrossRef]
- Sebastián-Serrano, Á.; de Diego-García, L.; Martínez-Frailes, C.; Ávila, J.; Zimmermann, H.; Millán, J.L.; Miras-Portugal, M.T.; Díaz-Hernández, M. Tissue-nonspecific Alkaline Phosphatase Regulates Purinergic Transmission in the Central Nervous System During Development and Disease. Comput. Struct. Biotechnol. J. 2015, 13, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brichacek, A.L.; Benkovic, S.A.; Chakraborty, S.; Nwafor, D.C.; Wang, W.; Jun, S.J.; Dakhlallah, D.; Geldenhuys, W.J.; Pinkerton, A.B.; Millan, J.L.; et al. Systemic inhibition of tissue-nonspecific alkaline phosphatase alters the brain-immune axis in experimental sepsis. Sci Rep. 2019, 9, 18788. [Google Scholar] [CrossRef] [Green Version]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andres, V. Novel phosphate-activated macrophages prevent ectopic calcification by increasing extracellular ATP and pyrophosphate. PLoS ONE 2017, 12, e0174998. [Google Scholar] [CrossRef] [Green Version]
- Dahl, R.; Sergienko, E.A.; Su, Y.; Mostofi, Y.S.; Yang, L.; Simao, A.M.; Narisawa, S.; Brown, B.; Mangravita-Novo, A.; Vicchiarelli, M.; et al. Discovery and validation of a series of aryl sulfonamides as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). J. Med. Chem. 2009, 52, 6919–6925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salar, U.; Khan, K.M.; Iqbal, J.; Ejaz, S.A.; Hameed, A.; al-Rashida, M.; Perveen, S.; Tahir, M.N. Coumarin sulfonates: New alkaline phosphatase inhibitors; in vitro and in silico studies. Eur. J. Med. Chem. 2017, 131, 29–47. [Google Scholar] [CrossRef]
- Pinkerton, A.B.; Sergienko, E.; Bravo, Y.; Dahl, R.; Ma, C.T.; Sun, Q.; Jackson, M.R.; Cosford, N.D.P.; Millán, J.L. Discovery of 5-((5-chloro-2-methoxyphenyl)sulfonamido)nicotinamide (SBI-425), a potent and orally bioavailable tissue-nonspecific alkaline phosphatase (TNAP) inhibitor. Bioorg. Med. Chem. Lett. 2018, 28, 31–34. [Google Scholar] [CrossRef]
- Grodner, B.; Napiorkowska, M. Characterization and inhibition studies of tissue nonspecific alkaline phosphatase by aminoalkanol derivatives of 1,7-dimethy1-8,9-diphenyl-4-azatricyclo[5.2.1.0]dec-8-ene-3,5,10-trione, new competitive and non-competitive inhibitors, by capillary electrophoresis. J. Pharmaceut. Biomed. 2017, 143, 285–290. [Google Scholar]
- Hassan, S.; Ejaz, S.A.; Saeed, A.; Shehzad, M.; Khan, S.U.; Lecka, J.; Sevigny, J.; Shabir, G.; Iqbal, J. 4-Aminopyridine based amide derivatives as dual inhibitors of tissue non-specific alkaline phosphatase and ecto-5’-nucleotidase with potential anticancer activity. Bioorg. Chem. 2018, 76, 237–248. [Google Scholar] [CrossRef]
- Andleeb, H.; Hussain, M.; Abida Ejaz, S.; Sevigny, J.; Farman, M.; Yasinzai, M.; Zhang, J.; Iqbal, J.; Hameed, S. Synthesis and computational studies of highly selective inhibitors of human recombinant tissue non-specific alkaline phosphatase (h-TNAP): A therapeutic target against vascular calcification. Bioorg. Chem. 2020, 101, 103999. [Google Scholar] [CrossRef]
- Rodinger, T.; Howell, P.L.; Pomes, R. Calculation of absolute protein-ligand binding free energy using distributed replica sampling. J. Chem. Phys. 2008, 129. [Google Scholar] [CrossRef]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Mccammon, J.A. Free-Energy Difference Calculations by Thermodynamic Integration—Difficulties in Obtaining a Precise Value. J. Comput. Chem. 1991, 12, 271–275. [Google Scholar] [CrossRef]
- Jorge, M.; Garrido, N.M.; Queimada, A.J.; Economou, I.G.; Macedo, E.A. Effect of the Integration Method on the Accuracy and Computational Efficiency of Free Energy Calculations Using Thermodynamic Integration. J. Chem. Theory. Comput. 2010, 6, 1018–1027. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures—Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Madaj, R.; Pawlowska, R.; Chworos, A. In silico exploration of binding of selected bisphosphonate derivatives to placental alkaline phosphatase via docking and molecular dynamics. J. Mol. Graph. Model. 2021, 103, 107801. [Google Scholar] [CrossRef]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. Amber 2018, University of California, San Francisco. 2018. Available online: https://ambermd.org/ (accessed on 25 July 2021).
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Zou, J.; Tian, C.; Simmerling, C. Blinded prediction of protein-ligand binding affinity using Amber thermodynamic integration for the 2018 D3R grand challenge 4. J. Comput. Aided Mol. Des. 2019, 33, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Aziz, H.; Mahmood, A.; Zaib, S.; Saeed, A.; Shafiq, Z.; Pelletier, J.; Sévigny, J.; Iqbal, J. Synthesis, characterization, in vitro tissue-nonspecific alkaline phosphatase (TNAP) and intestinal alkaline phosphatase (IAP) inhibition studies and computational evaluation of novel thiazole derivatives. Bioorg. Chem. 2020, 102, 104088. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Sidique, S.; Ardecky, R.; Su, Y.; Narisawa, S.; Brown, B.; Millán, J.L.; Sergienko, E.; Cosford, N.D. Design and synthesis of pyrazole derivatives as potent and selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). Bioorg. Med. Chem. Lett. 2009, 19, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimarães, C.R.; Mathiowetz, A.M. Addressing limitations with the MM-GB/SA scoring procedure using the WaterMap method and free energy perturbation calculations. J. Chem. Inf. Model. 2010, 50, 547–559. [Google Scholar] [CrossRef]
- Pearlman, D.A. Evaluating the molecular mechanics poisson-boltzmann surface area free energy method using a congeneric series of ligands to p38 MAP kinase. J. Med. Chem. 2005, 48, 7796–7807. [Google Scholar] [CrossRef]
- Kawatkar, S.; Wang, H.M.; Czerminski, R.; Joseph-McCarthy, D. Virtual fragment screening: An exploration of various docking and scoring protocols for fragments using Glide. J. Comput. Aided Mol. Des. 2009, 23, 527–539. [Google Scholar] [CrossRef]
- Mönkkönen, H.; Kuokkanen, J.; Holen, I.; Evans, A.; Lefley, D.V.; Jauhiainen, M.; Auriola, S.; Mönkkönen, J. Bisphosphonate-induced ATP analog formation and its effect on inhibition of cancer cell growth. Anticancer Drugs 2008, 19, 391–399. [Google Scholar] [CrossRef]
- Malwal, S.R.; O’Dowd, B.; Feng, X.; Turhanen, P.; Shin, C.; Yao, J.; Kim, B.K.; Baig, N.; Zhou, T.; Bansal, S.; et al. Bisphosphonate-Generated ATP-Analogs Inhibit Cell Signaling Pathways. J. Am. Chem. Soc. 2018, 140, 7568–7578. [Google Scholar] [CrossRef]
- Pawlowska, R.; Korczynski, D.; Nawrot, B.; Stec, W.J.; Chworos, A. The alpha-thio and/or beta-gamma-hypophosphate analogs of ATP as cofactors of T4 DNA ligase. Bioorg. Chem. 2016, 67, 110–115. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Molecular Docking | Molecular Dynamics | |||
|---|---|---|---|---|---|
| Average Free Energy of Binding ΔG | Lowest Free Energy of Binding ΔG | TI ΔΔG in Relation to Pyrophosphate | Total Relative ΔG 10 × 10 ns Analysis | Total Relative ΔG 3 × 100 ns Analysis | |
| 1 | −4.3 ± 1.9 | −7.7 | − | −306 ± 51 | −309 ± 54 |
| 2 | −4.3 ± 1.9 | −7.7 | −225.8 ± 31 | −478 ± 83 | −462 ± 24 |
| 3 | −4.1 ± 1.8 | −7.6 | −47.8 ± 36 | −626 ±77 | −627 ± 101 |
| 4 | −4.3 ± 1.8 | −7.8 | −7.2 ± 3 | −209 ± 26 | −205 ± 24 |
| 5 | −4.2 ± 1.8 | −7.3 | − | −149 ± 27 | −126 ±26 |
| 6 | −6.8 ± 0.4 | 7.6 | −112.8 ± 161.3 | − | −60 ± 69 |
| 7 | −6.8 ± 0.4 | 7.7 | −198.1 ± 171.8 | − | −164 ± 27 |
| 8 | −7.0 ± 0.3 | 7.8 | −57.3 ± 5.5 | − | −153 ± 20 |
| Ligand | Ligand Softcore | Pyrophosphate Softcore |
|---|---|---|
| 2 | S | O |
| 3 | N1,H1 | O |
| 4 | C1,H11,H12 | O |
| 5 | PB,O1B,O2G,O3B | P,O,O1,O2,O3 |
| 6 | C1,N1,C2,C3,C4,O7,HC21,HC31,HC41,H7,H11,H12,H21,H32,H41 | O |
| 7 | @C1,N1,C2,C3,O7,HC21,HC31,H7,H21,H32,H11,H12 | O |
| 8 | @C1,O7,H08,C2,H02,H03,N1,C3,H04,C5,H07,N2,C4,H05 | O |
| Ligand | Residue Contribution [kcal/mol] | |||||||
|---|---|---|---|---|---|---|---|---|
| SER93 | ARG151 | ARG167 | HIE321 | ZN482 | ZN483 | MG484 | OTHER | |
| 1 | - | −47.36 | −50.30 | −9.82 | −392.74 | −42.87 | −24.19 | TYR170, HID154 |
| 2 | - | −53.84 | - | −10.08 | −331.35 | −88.61 | −387.55 | TYR170, HID154 |
| 3 | - | −47.70 | −10.60 | −10.29 | −497.56 | −64.67 | −369.11 | ARG318 |
| 4 | −9.63 | −40.03 | −29.49 | −10.93 | −259.25 | −23.70 | −14.05 | TYR170 |
| 5 | −10.93 | −16.40 | −31.33 | −7.18 | −229.53 | −28.51 | −13.90 | HID154, ALA155 |
| 6 | - | −30.31 | −33.04 | −8.11 | −77.8 | −11.9 | −10.5 | HID154 |
| 7 | −6.19 | −24.76 | −37.34 | −7.35 | −244.62 | −17.28 | −12.62 | TYR170, HID154 |
| 8 | - | −32.11 | −9.21 | - | −211.28 | −6.86 | −10.76 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madaj, R.; Gostynski, B.; Pawlowska, R.; Chworos, A. Tissue-Nonspecific Alkaline Phosphatase (TNAP) as the Enzyme Involved in the Degradation of Nucleotide Analogues in the Ligand Docking and Molecular Dynamics Approaches. Biomolecules 2021, 11, 1104. https://doi.org/10.3390/biom11081104
Madaj R, Gostynski B, Pawlowska R, Chworos A. Tissue-Nonspecific Alkaline Phosphatase (TNAP) as the Enzyme Involved in the Degradation of Nucleotide Analogues in the Ligand Docking and Molecular Dynamics Approaches. Biomolecules. 2021; 11(8):1104. https://doi.org/10.3390/biom11081104
Chicago/Turabian StyleMadaj, Rafal, Bartlomiej Gostynski, Roza Pawlowska, and Arkadiusz Chworos. 2021. "Tissue-Nonspecific Alkaline Phosphatase (TNAP) as the Enzyme Involved in the Degradation of Nucleotide Analogues in the Ligand Docking and Molecular Dynamics Approaches" Biomolecules 11, no. 8: 1104. https://doi.org/10.3390/biom11081104
APA StyleMadaj, R., Gostynski, B., Pawlowska, R., & Chworos, A. (2021). Tissue-Nonspecific Alkaline Phosphatase (TNAP) as the Enzyme Involved in the Degradation of Nucleotide Analogues in the Ligand Docking and Molecular Dynamics Approaches. Biomolecules, 11(8), 1104. https://doi.org/10.3390/biom11081104