
The Conspicuous Link between Ear, Brain and Heart–Could Neurotrophin-Treatment of Age-Related Hearing Loss Help Prevent Alzheimer’s Disease and Associated Amyloid Cardiomyopathy?

 ,
, 
 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: Alzheimer’s—A Systemic Disease?
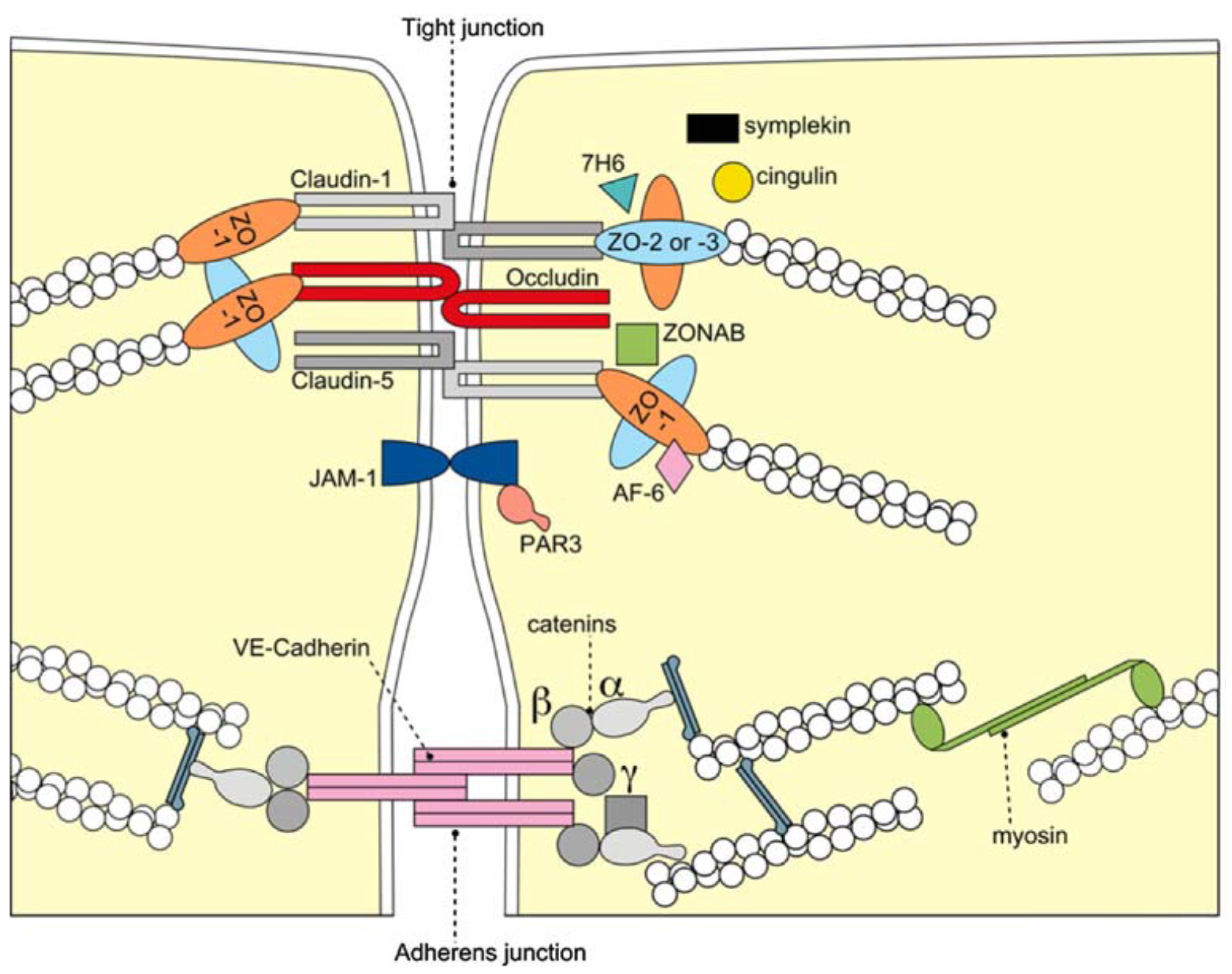
2. The Blood–Brain Barrier
BBB Dysfunction and Vascular Risk Factors for AD
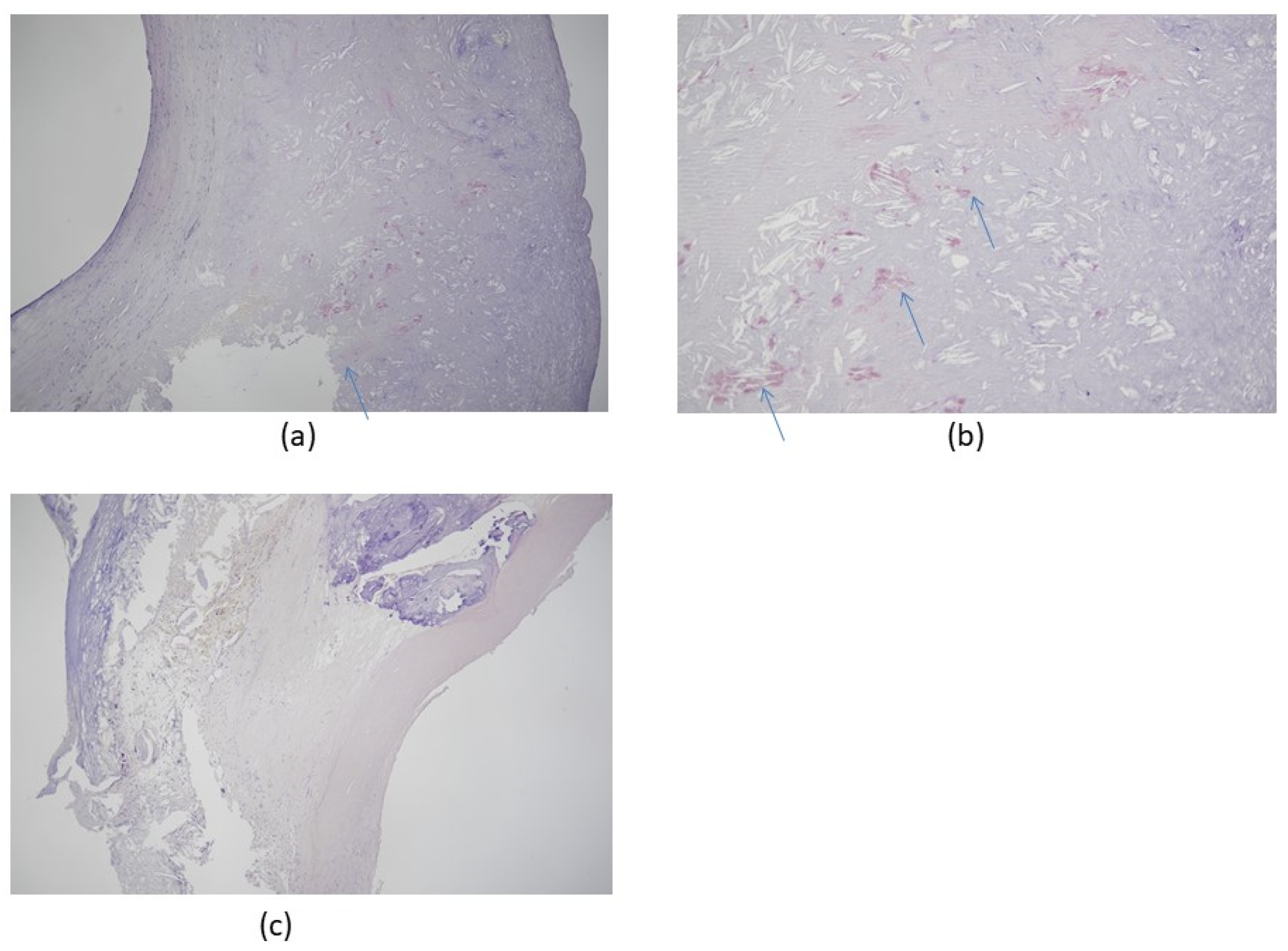
3. Amyloid Cardiomyopathy
4. Current and Novel Treatment Modalities
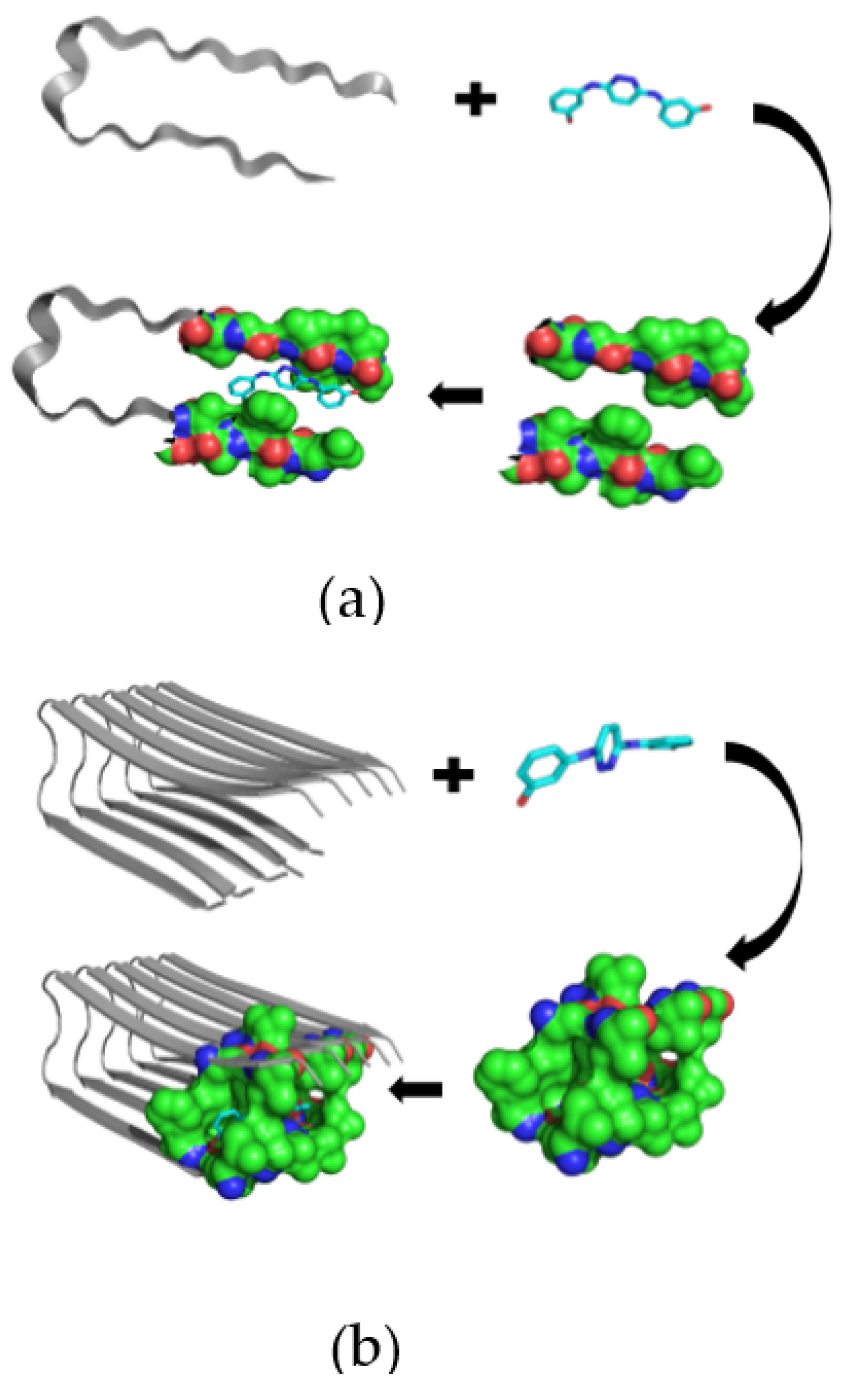
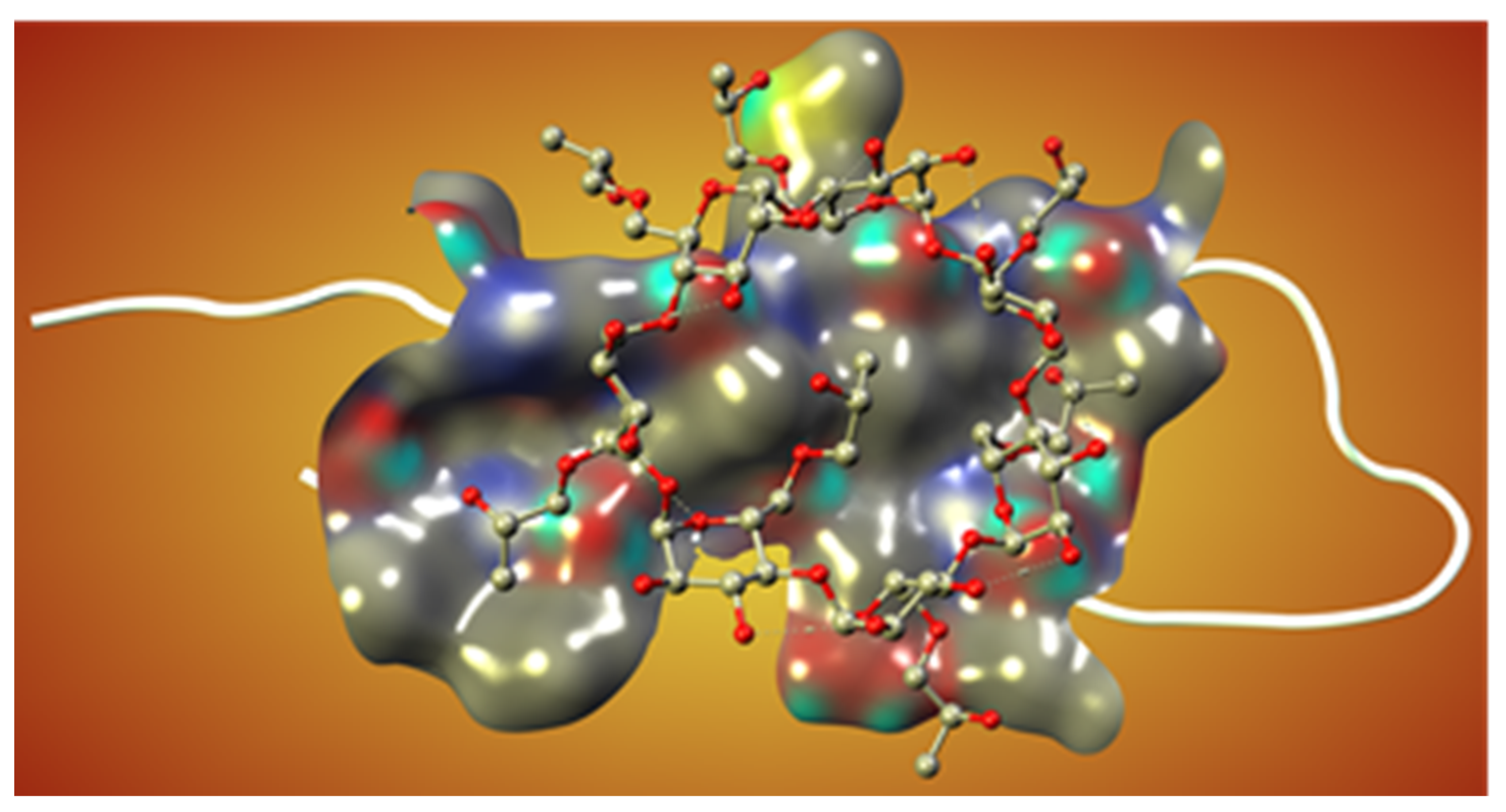
Novel Inhibitors of Amyloid Fibril Formation in AD
5. Age-Related Hearing Loss—A Risk Factor for Dementia and AD?
Neurovascular Dysfunction at the Root of ARHL and AD?
6. Neurotrophic Factors at the Interface of AD Pathophysiology and Neural ARHL
6.1. AD and Neurotrophins
6.2. Neural ARHL and Neurotrophins
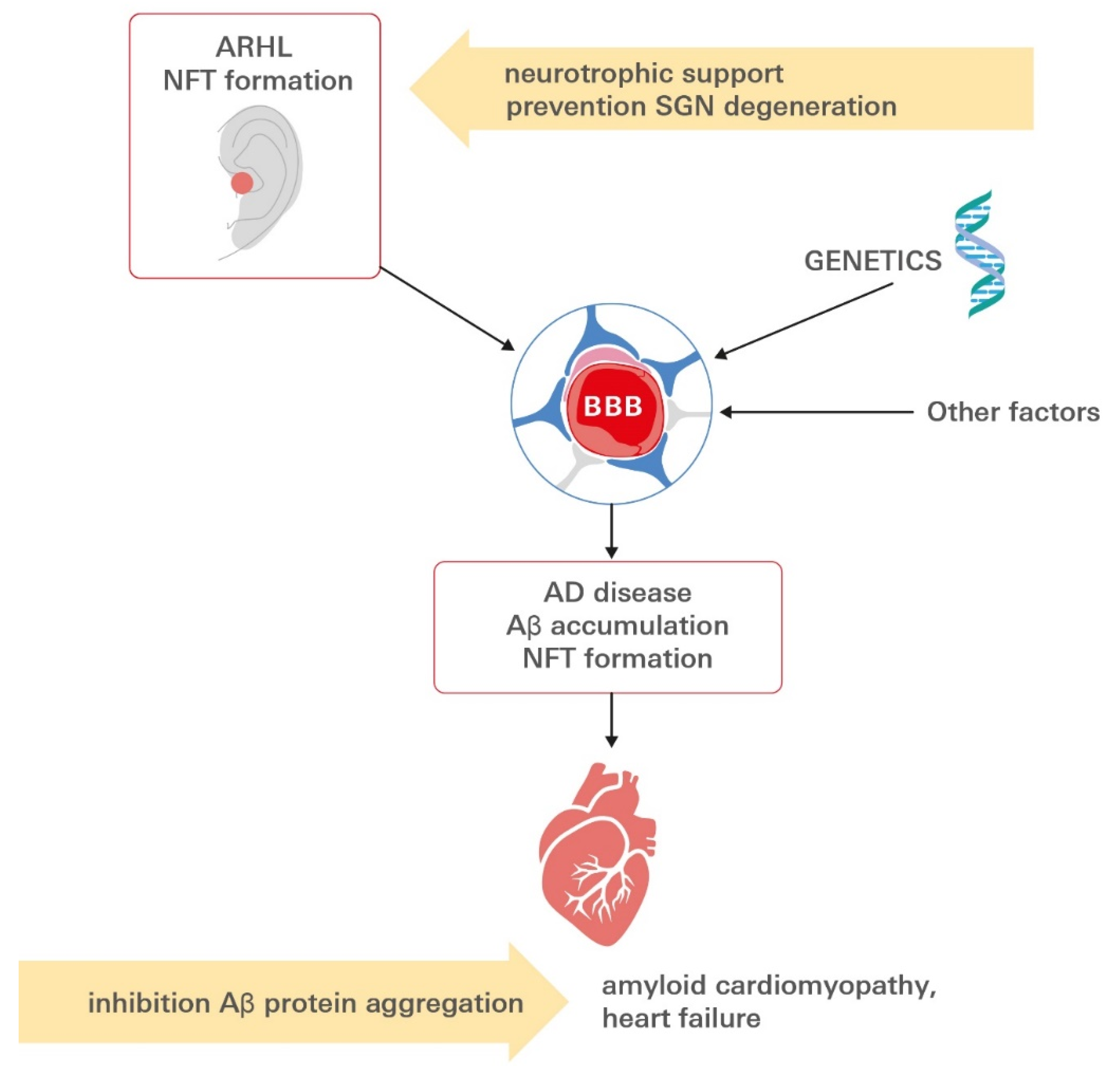
7. Conclusions and Open Questions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular dysfunction and faulty amyloid beta-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452. [Google Scholar] [CrossRef] [PubMed]
- Swanberg, M.M.; Tractenberg, R.E.; Mohs, R.; Thal, L.J.; Cummings, J.L. Executive Dysfunction in Alzheimer Disease. Arch. Neurol. 2004, 61, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.H. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar]
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 1–7. [Google Scholar] [CrossRef]
- Cummings, J.L. Alzheimer’s disease. N. Engl. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113. [Google Scholar] [CrossRef]
- Deane, R.; Yan, S.D.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Takuma, K.; Fang, F.; Zhang, W.; Yan, S.; Fukuzaki, E.; Du, H.; Sosunov, A.; McKhann, G.; Funatsu, Y.; Nakamichi, N.; et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 20021–20026. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Obermuller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010, 330, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Human prion diseases and neurodegeneration. Curr. Top. Microbiol. Immunol. 1996, 207, 1–17. [Google Scholar]
- Zlokovic, B.V. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurother. J. Am. Soc. Exp. Neurother. 2008, 5, 409–414. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. The vascular hypothesis of Alzheimer’s disease: Bench to bedside and beyond. Neuro-Degener. Dis. 2010, 7, 116–121. [Google Scholar] [CrossRef]
- Marchesi, V.T. Alzheimer’s dementia begins as a disease of small blood vessels, damaged by oxidative-induced inflammation and dysregulated amyloid metabolism: Implications for early detection and therapy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208. [Google Scholar] [CrossRef]
- Przybyłowska, M.; Dzierzbicka, K.; Kowalski, S.; Chmielewska, K.; Inkielewicz-Stepniak, I. Therapeutic Potential of Multifunctional Derivatives of Cholinesterase Inhibitors. Curr. Neuropharmacol. 2020, 19, 1–24. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2020, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Salvador, E.; Pastorin, G.; Forster, C. Blood-brain barrier transport studies, aggregation, and molecular dynamics simulation of multiwalled carbon nanotube functionalized with fluorescein isothiocyanate. Int. J. Nanomed. 2015, 10, 1703–1713. [Google Scholar] [CrossRef]
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.; Shusta, E.V.; et al. In vitro models of the blood–brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. Br. J. Pharmacol. 2016, 36, 862–890. [Google Scholar] [CrossRef]
- McConnell, H.; Li, Z.; Woltjer, R.L.; Mishra, A. Astrocyte dysfunction and neurovascular impairment in neurological disorders: Correlation or causation? Neurochem. Int. 2019, 128, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, S.M.; Gurol, E.; Rosand, J.; Smith, E.E. Amyloid Angiopathy-Related Vascular Cognitive Impairment. Stroke 2004, 35, 2616–2619. [Google Scholar] [CrossRef] [PubMed]
- DeSimone, C.V.; Graff-Radford, J.; El-Harasis, M.A.; Rabinstein, A.A.; Asirvatham, S.J.; Holmes, D.R. Cerebral amyloid angiopathy and implications for atrial fibrillation management. Lancet 2017, 390, 9–11. [Google Scholar] [CrossRef]
- Forster, C. Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 2008, 130, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; BeMiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dementia: Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Engelhardt, B.; Ransohoff, R.M. Capture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends Immunol. 2012, 33, 579–589. [Google Scholar] [CrossRef]
- Salvador, E.; Burek, M.; Forster, C.Y. Tight Junctions and the Tumor Microenvironment. Curr. Pathobiol. Rep. 2016, 4, 135–145. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Blecharz, K.; Kahles, T.; Schwarz, T.; Kraft, P.; Göbel, K.; Meuth, S.G.; Burek, M.; Thum, T.; Stoll, G.; et al. Glucocorticoid Insensitivity at the Hypoxic Blood–Brain Barrier Can Be Reversed by Inhibition of the Proteasome. Stroke 2011, 42, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965. [Google Scholar] [CrossRef]
- Hartz, A.M.; Bauer, B.; Soldner, E.L.; Wolf, A.; Boy, S.; Backhaus, R.; Mihaljevic, I.; Bogdahn, U.; Klunemann, H.H.; Schuierer, G.; et al. Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 2012, 43, 514–523. [Google Scholar] [CrossRef]
- Atwood, C.S.; Martins, R.N.; Smith, M.A.; Perry, G. Senile plaque composition and posttranslational modification of amyloid-beta peptide and associated proteins. Peptides 2002, 23, 1343–1350. [Google Scholar] [CrossRef]
- Kumar-Singh, S.; Pirici, D.; McGowan, E.; Serneels, S.; Ceuterick, C.; Hardy, J.; Duff, K.; Dickson, D.; Van Broeckhoven, C. Dense-Core Plaques in Tg2576 and PSAPP Mouse Models of Alzheimer’s Disease Are Centered on Vessel Walls. Am. J. Pathol. 2005, 167, 527–543. [Google Scholar] [CrossRef]
- Hellberg, S.; Silvola, J.M.; Liljenbäck, H.; Kiugel, M.; Eskola, O.; Hakovirta, H.; Hörkkö, S.; Morisson-Iveson, V.; Hirani, E.; Saukko, P.; et al. Amyloid-Targeting PET Tracer [18F]Flutemetamol Accumulates in Atherosclerotic Plaques. Molecules 2019, 24, 1072. [Google Scholar] [CrossRef]
- Van Assema, D.M.; Lubberink, M.; Rizzu, P.; Van Swieten, J.C.; Schuit, R.C.; Eriksson, J.; Scheltens, P.; Koepp, M.; Lammertsma, A.A.; Van Berckel, B.N. Blood–brain barrier P-glycoprotein function in healthy subjects and Alzheimer’s disease patients: Effect of polymorphisms in the ABCB1 gene. EJNMMI Res. 2012, 2, 57. [Google Scholar] [CrossRef]
- Wolf, A.; Bauer, B.; Hartz, A.M. ABC Transporters and the Alzheimer’s Disease Enigma. Front. Psychiatry 2012, 3, 54. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; Rozemuller, A.J.; van Horssen, J.; de Vries, H.E. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimer’s Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Goos, J.D.; Kester, M.I.; Barkhof, F.; Klein, M.; Blankenstein, M.A.; Scheltens, P.; van der Flier, W.M. Patients with Alzheimer disease with multiple microbleeds: Relation with cerebrospinal fluid biomarkers and cognition. Stroke 2009, 40, 3455–3460. [Google Scholar] [CrossRef] [PubMed]
- Lanfranconi, S.; Franco, G.; Borellini, L.; Denaro, F.; Basilico, P.; Parati, E.; Micieli, G.; Bersano, A. Genetics of cerebral hemorrhage and microbleeds. Panminerva Med. 2013, 55, 11–28. [Google Scholar] [PubMed]
- Brun, A.; Englund, E. A white matter disorder in dementia of the Alzheimer type: A pathoanatomical study. Ann. Neurol. 1986, 19, 253–262. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Yan, S.D.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.N.; Lee, L.; Saxena, R.; Bemis, K.G.; Wang, M.; Theodorakis, J.L.; Vuppalanchi, R.; Alloosh, M.; Sturek, M.; Chalasani, N. Serum proteomic analysis of diet-induced steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Am. J. Physiol. Liver Physiol. 2010, 298, G746–G754. [Google Scholar] [CrossRef]
- Jellinger, K.A. Prevalence and impact of cerebrovascular lesions in Alzheimer and lewy body diseases. Neuro-Degener. Dis. 2010, 7, 112–115. [Google Scholar] [CrossRef]
- Ruitenberg, A.; Heijer, T.D.; Bakker, S.L.M.; Van Swieten, J.C.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Cerebral hypoperfusion and clinical onset of dementia: The Rotterdam study. Ann. Neurol. 2005, 57, 789–794. [Google Scholar] [CrossRef]
- Vermeer, S.E.; Prins, N.D.; Heijer, T.D.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M. Silent Brain Infarcts and the Risk of Dementia and Cognitive Decline. N. Engl. J. Med. 2003, 348, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Snowdon, D.A.; Greiner, L.H.; Mortimer, J.A.; Riley, K.P.; Greiner, P.A.; Markesbery, W.R. Brain Infarction and the Clinical Expression of Alzheimer DiseaseThe Nun Study. JAMA 1997, 277, 813–817. [Google Scholar] [CrossRef]
- Jellinger, K. Cerebellar involvement in progressive supranuclear palsy. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 1104–1105. [Google Scholar] [CrossRef]
- Wendell, C.R.; Waldstein, S.R.; Ferrucci, L.; O’Brien, R.J.; Strait, J.B.; Zonderman, A.B. Carotid atherosclerosis and prospective risk of dementia. Stroke 2012, 43, 3319–3324. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, L.Y.; Venneri, A.; Farkas, E.; Evans, P.C.; Marzo, A.; Frangi, A. Vascular dysfunction in the pathogenesis of Alzheimer’s disease—A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol. Dis. 2015, 82, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef]
- Troncone, L.; Luciani, M.; Coggins, M.; Wilker, E.H.; Ho, C.Y.; Codispoti, K.E.; Frosch, M.P.; Kayed, R.; del Monte, F. Abeta Amyloid Pathology Affects the Hearts of Patients With Alzheimer’s Disease: Mind the Heart. J. Am. Coll. Cardiol. 2016, 68, 2395–2407. [Google Scholar] [CrossRef] [PubMed]
- Petrovitch, H.; White, L.; Izmirilian, G.; Ross, G.; Havlik, R.; Markesbery, W.; Nelson, J.; Davis, D.; Hardman, J.; Foley, D.; et al. Midlife blood pressure and neuritic plaques, neurofibrillary tangles, and brain weight at death: The HAAS☆. Neurobiol. Aging 2000, 21, 57–62. [Google Scholar] [CrossRef]
- Tzourio, C. Hypertension, cognitive decline, and dementia: An epidemiological perspective. Dialogues Clin. Neurosci. 2007, 9, 61–70. [Google Scholar] [PubMed]
- Chakraborty, A.; de Wit, N.M.; van der Flier, W.M.; de Vries, H.E. The blood brain barrier in Alzheimer’s disease. Vasc. Pharmacol. 2017, 89, 12–18. [Google Scholar] [CrossRef]
- Touyz, R.M. Oxidative stress and vascular damage in hypertension. Curr. Hypertens. Rep. 2000, 2, 98–105. [Google Scholar] [CrossRef]
- Roberts, R.O.; Geda, Y.E.; Knopman, D.S.; Cha, R.H.; Boeve, B.F.; Ivnik, R.J.; Pankratz, V.S.; Tangalos, E.G.; Petersen, R.C. Metabolic syndrome, inflammation, and nonamnestic mild cognitive impairment in older persons: A population-based study. Alzheimer Dis. Assoc. Disord. 2010, 24, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Dar, T.; Sheikh, I.; Ganie, S.; Ali, R.; Singh, L.; Gan, S.H.; Kamal, M.A.; Zargar, M. Molecular Linkages Between Diabetes and Alzheimer’s Disease: Current Scenario and Future Prospects. CNS Neurol. Disord. Drug Targets 2014, 13, 290–298. [Google Scholar] [CrossRef]
- Ott, A.; Stolk, R.; Van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937. [Google Scholar] [CrossRef]
- Muhammad, S.; Bierhaus, A.; Schwaninger, M. Reactive oxygen species in diabetes-induced vascular damage, stroke, and Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2009, 16, 775–785. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064. [Google Scholar] [CrossRef]
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.C.; Killcross, A.S.; Jenkins, T.A. Obesity and cognitive decline: Role of inflammation and vascular changes. Front. Neurosci. 2014, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Wood, W.G.; Eckert, G.P.; Igbavboa, U.; Müller, W.E. Amyloid beta-protein interactions with membranes and cholesterol: Causes or casualties of Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Biomembr. 2003, 1610, 281–290. [Google Scholar] [CrossRef]
- Dempsey, R.J.; Vemuganti, R.; Varghese, T.; Hermann, B.P. A review of carotid atherosclerosis and vascular cognitive decline: A new understanding of the keys to symptomology. Neurosurgery 2010, 67, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Masoudi, F.A.; Rumsfeld, J.S.; Havranek, E.P.; House, J.A.; Peterson, E.D.; Krumholz, H.M.; Spertus, J.A. Age, functional capacity, and health-related quality of life in patients with heart failure. J. Card. Fail. 2004, 10, 368–373. [Google Scholar] [CrossRef]
- Rich, M.W.; Goyal, P.; Forman, D.E. Age and Heart Failure Trials—Lessons from DAPA-HF. J. Card. Fail. 2020, 26, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Ihne, S.; Morbach, C.; Obici, L.; Palladini, G.; Störk, S. Amyloidosis in Heart Failure. Curr. Hear. Fail. Rep. 2019, 16, 285–303. [Google Scholar] [CrossRef]
- Debette, S.; Bauters, C.; Leys, D.; Lamblin, N.; Pasquier, F.; de Groote, P. Prevalence and Determinants of Cognitive Impairment in Chronic Heart Failure Patients. Congest. Hear. Fail. 2007, 13, 205–208. [Google Scholar] [CrossRef]
- Moreira, P.I.; Smith, M.A.; Zhu, X.; Nunomura, A.; Castellani, R.J.; Perry, G. Oxidative Stress and Neurodegeneration. Ann. N. Y. Acad. Sci. 2005, 1043, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.C.; Robinson-Papp, J. Amyloid Neuropathies. Mt. Sinai J. Med. A J. Transl. Pers. Med. 2012, 79, 733–748, . [Google Scholar] [CrossRef] [PubMed]
- Liao, R.; Ward, J. Amyloid Cardiomyopathy. Circ. Res. 2017, 120, 1865–1867, . [Google Scholar] [CrossRef] [PubMed]
- Jensen-Dahm, C.; Waldemar, G.; Jensen, T.S.; Malmqvist, L.; Moeller, M.M.; Andersen, B.B.; Høgh, P.; Ballegaard, M. Autonomic Dysfunction in Patients with Mild to Moderate Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 681–689, . [Google Scholar] [CrossRef]
- Serhiyenko, V.A. Cardiac autonomic neuropathy: Risk factors, diagnosis and treatment. World J. Diabetes 2018, 9, 1–24, . [Google Scholar] [CrossRef] [PubMed]
- Sainz, A.L.; Moral, F.J.D.H.-D.; Dominguez, F.; Restrepo-Cordoba, A.; Amor-Salamanca, A.; Hernandez-Hernandez, A.; Ruiz-Guerrero, L.; Krsnik, I.; Cobo-Marcos, M.; Castro, V.; et al. Prevalence of cardiac amyloidosis among elderly patients with systolic heart failure or conduction disorders. Amyloid 2019, 26, 156–163, . [Google Scholar] [CrossRef]
- Toledo, J.B.; Toledo, E.; Weiner, M.W.; Jack, C.R.; Jagust, W., Jr.; Lee, V.M.; Shaw, L.M.; Trojanowski, J.Q. Cardiovascular risk factors, cortisol, and amyloid-beta deposition in Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2012, 8, 483–489. [Google Scholar]
- Sanna, G.D.; Nusdeo, G.; Piras, M.R.; Forteleoni, A.; Murru, M.R.; Saba, P.S.; Dore, S.; Sotgiu, G.; Parodi, G.; Ganau, A. Cardiac Abnormalities in Alzheimer Disease: Clinical Relevance Beyond Pathophysiological Rationale and Instrumental Findings? JACC Heart Fail. 2019, 7, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein Aggregates and Novel Presenilin Gene Variants in Idiopathic Dilated Cardiomyopathy. Circulation 2010, 121, 1216–1226, . [Google Scholar] [CrossRef]
- Subramanian, K.; Gianni, D.; Balla, C.; Assenza, G.E.; Joshi, M.; Semigran, M.J.; Macgillivray, T.E.; van Eyk, J.E.; Agnetti, G.; Paolocci, N.; et al. Cofilin-2 phosphorylation and sequestration in myocardial aggregates: Novel pathogenetic mechanisms for idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1199–1214. [Google Scholar] [CrossRef]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of Presenilin Genes in Dilated Cardiomyopathy and Heart Failure. Am. J. Hum. Genet. 2006, 79, 1030–1039, . [Google Scholar] [CrossRef]
- Backs, D.; Saglam, I.; Löffler, C.; Ihne, S.; Morbach, C.; Brenner, S.; Angermann, C.; Ertl, G.; Frantz, S.; Störk, S.; et al. Prevalence of cardiovascular risk factors and diseases in patients with multiple myeloma undergoing autologous peripheral blood stem cell transplantation. Oncotarget 2019, 10, 3154–3165, . [Google Scholar] [CrossRef] [PubMed]
- CQuarta, C.; Kruger, J.L.; Falk, R.H. Cardiac amyloidosis. Circulation 2012, 126, e178–e182. [Google Scholar] [CrossRef]
- Cordell, C.B.; Borson, S.; Boustani, M.; Chodosh, J.; Reuben, D.; Verghese, J.; Thies, W.; Fried, L.B.; Medicare Detection of Cognitive Impairment Workgroup. Alzheimer’s Association recommendations for operationalizing the detection of cognitive impairment during the Medicare Annual Wellness Visit in a primary care setting. Alzheimer’s Dement. 2013, 9, 141–150, . [Google Scholar] [CrossRef]
- Thies, W.; Bleiler, L. Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013, 9, 208–245. [Google Scholar]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-beta oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef]
- Cieslik, M.; Czapski, G.A.; Wojtowicz, S.; Wieczorek, I.; Wencel, P.L.; Strosznajder, R.P.; Jaber, V.; Lukiw, W.J.; Strosznajder, J.B. Alterations of Transcription of Genes Coding Anti-oxidative and Mitochondria-Related Proteins in Amyloid beta Toxicity: Relevance to Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 1374–1388. [Google Scholar] [CrossRef] [PubMed]
- Nakagami, Y.; Nishimura, S.; Murasugi, T.; Kaneko, I.; Meguro, M.; Marumoto, S.; Kogen, H.; Koyama, K.; Oda, T. A novel beta-sheet breaker, RS-0406, reverses amyloid beta-induced cytotoxicity and impairment of long-term potentiation in vitro. Br. J. Pharmacol. 2002, 137, 676–682. [Google Scholar] [CrossRef]
- Mehrazma, B.; Robinson, M.; Opare, S.K.A.; Petoyan, A.; Lou, J.; Hane, F.T.; Rauk, A.; Leonenko, Z. Pseudo-peptide amyloid-beta blocking inhibitors: Molecular dynamics and single molecule force spectroscopy study. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1707–1718. [Google Scholar] [CrossRef]
- Jha, A.; Kumar, M.G.; Gopi, H.N.; Paknikar, K.M. Inhibition of beta-Amyloid Aggregation through a Designed beta-Hairpin Peptide. Langmuir 2018, 34, 1591–1600. [Google Scholar] [CrossRef]
- Shin, J.; Lee, S.; Cha, M. Neuroprotective effect of single-wall carbon nanotubes with built-in peroxidase-like activity against beta-amyloid-induced neurotoxicity. Medchemcomm 2017, 8, 625–632. [Google Scholar] [CrossRef]
- Shityakov, S.; Sohajda, T.; Puskas, I.; Roewer, N.; Forster, C.; Broscheit, J.A. Ionization states, cellular toxicity and molecular modeling studies of midazolam complexed with trimethyl-beta-cyclodextrin. Molecules 2014, 19, 16861–16876. [Google Scholar] [CrossRef] [PubMed]
- Shityakov, S.; Broscheit, J.; Forster, C. Alpha-Cyclodextrin dimer complexes of dopamine and levodopa derivatives to assess drug delivery to the central nervous system: ADME and molecular docking studies. Int. J. Nanomed. 2012, 7, 3211–3219. [Google Scholar] [CrossRef]
- Yao, J.; Ho, D.; Calingasan, N.Y.; Pipalia, N.H.; Lin, M.T.; Beal, M.F. Neuroprotection by cyclodextrin in cell and mouse models of Alzheimer disease. J. Exp. Med. 2012, 209, 2501–2513, . [Google Scholar] [CrossRef]
- Stefanescu, R.; Stanciu, G.D.; Luca, A.; Caba, I.C.; Tamba, B.I.; Mihai, C.T. Contributions of Mass Spectrometry to the Identification of Low Molecular Weight Molecules Able to Reduce the Toxicity of Amyloid-beta Peptide to Cell Cultures and Transgenic Mouse Models of Alzheimer’s Disease. Molecules 2019, 24, 1167. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Cuttler, J.M.; Abdellah, E.; Goldberg, Y.; Al-Shamaa, S.; Symons, S.P.; Black, S.E.; Freedman, M. Low Doses of Ionizing Radiation as a Treatment for Alzheimer’s Disease: A Pilot Study. J. Alzheimer’s Dis. 2021, 80, 1119–1128, . [Google Scholar] [CrossRef]
- Loughrey, D.G.; Kelly, M.E.; Kelley, G.A.; Brennan, S.; Lawlor, B.A. Association of Age-Related Hearing Loss With Cognitive Function, Cognitive Impairment, and Dementia. JAMA Otolaryngol. Neck Surg. 2018, 144, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Lin, P.-J.; Yang, Z.; Fillit, H.M.; Cohen, J.T.; Neumann, P.J. Unintended Benefits: The Potential Economic Impact Of Addressing Risk Factors To Prevent Alzheimer’s Disease. Health Aff. 2014, 33, 547–554. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef]
- Edwards, G., III; Gamez, N.; Escobedo, G., Jr.; Calderon, O.; Moreno-Gonzalez, I. Modifiable Risk Factors for Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Sardone, R.; Battista, P.; Piccininni, M.; Dibello, V.; La Montagna, M.; Stallone, R.; Venezia, P.; Liguori, A.; et al. Sensorial frailty: Age-related hearing loss and the risk of cognitive impairment and dementia in later life. Ther. Adv. Chronic Dis. 2019, 10, 2040622318811000. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.Z.; Liberman, L.D.; Bennett, K.; de Gruttola, V.; O’Malley, J.T.; Liberman, M.C. Primary Neural Degeneration in the Human Cochlea: Evidence for Hidden Hearing Loss in the Aging Ear. Neuroscience 2019, 407, 8–20. [Google Scholar] [CrossRef]
- Bao, J.; Ohlemiller, K.K. Age-related loss of spiral ganglion neurons. Hear. Res. 2010, 264, 93–97. [Google Scholar] [CrossRef]
- Förster, C.Y.; Scheper, V.; Lenarz, T. Hearing loss and strial microvascular pathology towards unravelling the functional contribution of the bloodlabyrinth.barrier. Otorhinolaryngol. Head. Neck. Surg. 2019. [Google Scholar] [CrossRef]
- Nyberg, S.; Abbott, N.J.; Shi, X.; Steyger, P.S.; Dabdoub, A. Delivery of therapeutics to the inner ear: The challenge of the blood-labyrinth barrier. Sci. Transl. Med. 2019, 11, eaao0935. [Google Scholar] [CrossRef]
- Sinha, U.K.; Hollen, K.M.; Rodriguez, R.; Miller, C.A. Auditory system degeneration in Alzheimer’s disease. Neurology 1993, 43, 779. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, C.; Lin, S.; Wang, Y.; Seicol, B.J.; Ariss, R.W.; Xie, R. Biased auditory nerve central synaptopathy is associated with age-related hearing loss. J. Physiol. 2021, 599, 1833–1854. [Google Scholar] [CrossRef] [PubMed]
- Keithley, E.M.; Croskrey, K.L. Spiral ganglion cell endings in the cochlear nucleus of young and old rats. Hear. Res. 1990, 49, 169–177. [Google Scholar] [CrossRef]
- Schuknecht, H.F. The Pathology of Several Disorders of the Inner Ear Which Cause Vertigo. South. Med. J. 1964, 57, 1161–1167. [Google Scholar] [CrossRef]
- Ernfors, P.; Kucera, J.; Lee, K.F.; Loring, J.; Jaenisch, R. Studies on the physiological role of brain-derived neurotrophic factor and neurotrophin-3 in knockout mice. Int. J. Dev. Biol. 1995, 39, 799–807. [Google Scholar]
- Leake, P.A.; Rebscher, S.J.; Dore‘, C.; Akil, O. AAV-Mediated Neurotrophin Gene Therapy Promotes Improved Survival of Cochlear Spiral Ganglion Neurons in Neonatally Deafened Cats: Comparison of AAV2-hBDNF and AAV5-hGDNF. J. Assoc. Res. Otolaryngol. 2019, 20, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Pinton, S.; Sampaio, T.B.; Savall, A.S.; Gutierrez, M.E.Z. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: Implications for pathogenesis and therapy. Neural Regen. Res. 2017, 12, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Rüttiger, L.; Panford-Walsh, R.; Schimmang, T.; Tan, J.; Zimmermann, U.; Rohbock, K.; Köpschall, I.; Limberger, A.; Müller, M.; Fraenzer, J.-T.; et al. BDNF mRNA expression and protein localization are changed in age-related hearing loss. Neurobiol. Aging 2007, 28, 586–601. [Google Scholar] [CrossRef]
- Leake, P.A.; Akil, O.; Lang, H. Neurotrophin gene therapy to promote survival of spiral ganglion neurons after deafness. Hear. Res. 2020, 394, 107955. [Google Scholar] [CrossRef]
- Ibanez, C.F.; Andressoo, J.O. Biology of GDNF and its receptors—Relevance for disorders of the central nervous system. Neurobiol. Dis. 2017, 97, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, M. Recent advances in understanding neurotrophin signaling. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Altschuler, R.A.; Cho, Y.; Ylikoski, J.; Pirvola, U.; Magal, E.; Miller, J.M. Rescue and Regrowth of Sensory Nerves Following Deafferentation by Neurotrophic Factors. Ann. N.Y. Acad. Sci. 1999, 884, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.; Mufson, E.; Weingartner, J.; Skau, K.; Crutcher, K. Nerve growth factor in Alzheimer’s disease: Increased levels throughout the brain coupled with declines in nucleus basalis. J. Neurosci. 1995, 15, 6213–6221. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Kim, J.H. Brain-derived neurotrophic factor exerts neuroprotective actions against amyloid beta-induced apoptosis in neuroblastoma cells. Exp. Ther. Med. 2014, 8, 1891–1895. [Google Scholar] [CrossRef] [PubMed]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Garcez, M.; Zugno, A.I. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 2015, 6, 331–341. [Google Scholar] [CrossRef]
- Straten, G.; Saur, R.; Laske, C.; Gasser, T.; Annas, P.; Basun, H.; Leyhe, T. Influence of Lithium Treatment on GDNF Serum and CSF Concentrations in Patients with Early Alzheimers Disease. Curr. Alzheimer Res. 2011, 8, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Marksteiner, J.; Kemmler, G.; Weiss, E.M.; Knaus, G.; Ullrich, C.; Mechtcheriakov, S.; Oberbauer, H.; Auffinger, S.; Hinterhölzl, J.; Hinterhuber, H.; et al. Five out of 16 plasma signaling proteins are enhanced in plasma of patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2011, 32, 539–540. [Google Scholar] [CrossRef]
- Faria, M.C.; Gonçalves, G.S.; Rocha, N.P.; de Moraes, E.N.; Bicalho, M.A.; Cintra, M.T.G.; de Paula, J.; De Miranda, L.F.J.R.; Ferreira, A.C.D.S.; Teixeira, A.L.; et al. Increased plasma levels of BDNF and inflammatory markers in Alzheimer’s disease. J. Psychiatr. Res. 2014, 53, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Sharif, M.; Noroozian, M.; Hashemian, F. Do serum GDNF levels correlate with severity of Alzheimer’s disease? Neurol. Sci. 2020. [Google Scholar] [CrossRef]
- Von Bartheld, C.; Patterson, S.; Heuer, J.; Wheeler, E.; Bothwell, M.; Rubel, E. Expression of nerve growth factor (NGF) receptors in the developing inner ear of chick and rat. Development 1991, 113, 455–470. [Google Scholar] [CrossRef]
- Dai, C.F.; Steyger, P.; Wang, Z.; Vass, Z.; Nuttall, A. Expression of Trk A receptors in the mammalian inner ear. Hear. Res. 2004, 187, 1–11. [Google Scholar] [CrossRef]
- Gao, L.; Ge, R.; Xie, G.; Yao, D.; Li, P.; Wang, O.; Ma, X.; Han, F. Hearing Improvement in A/J Mice via the Mouse Nerve Growth Factor. Clin. Exp. Otorhinolaryngol. 2017, 10, 303–308. [Google Scholar] [CrossRef]
- Chacko, L.J.; Blumer, M.J.F.; Pechriggl, E.; Rask-Andersen, H.; Dietl, W.; Haim, A.; Fritsch, H.; Glueckert, R.; Dudas, J.; Schrott-Fischer, A. Role of BDNF and neurotrophic receptors in human inner ear development. Cell Tissue Res. 2017, 370, 347–363. [Google Scholar] [CrossRef]
- Medd, A.M.; Bianchi, L.M. Analysis of BDNF production in the aging gerbil cochlea. Exp. Neurol. 2000, 162, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Szobota, S.; Mathur, P.D.; Siegel, S.; Black, K.; Saragovi, H.U.; Foster, A.C. BDNF, NT-3 and Trk receptor agonist monoclonal antibodies promote neuron survival, neurite extension, and synapse restoration in rat cochlea ex vivo models relevant for hidden hearing loss. PLoS ONE 2019, 14, e0224022. [Google Scholar] [CrossRef] [PubMed]
- Wefstaedt, P.; Scheper, V.; Lenarz, T.; Stöver, T. Brain-derived neurotrophic factor/glial cell line-derived neurotrophic factor survival effects on auditory neurons are not limited by dexamethasone. NeuroReport 2005, 16, 2011–2014. [Google Scholar] [CrossRef] [PubMed]
- Schwieger, J.; Warnecke, A.; Lenarz, T.; Esser, K.-H.; Scheper, V. Neuronal Survival, Morphology and Outgrowth of Spiral Ganglion Neurons Using a Defined Growth Factor Combination. PLoS ONE 2015, 10, e0133680. [Google Scholar] [CrossRef]
- Yurek, D.M.; Fletcher-Turner, A. Differential expression of GDNF, BDNF, and NT-3 in the aging nigrostriatal system following a neurotoxic lesion. Brain Res. 2001, 891, 228–235. [Google Scholar] [CrossRef]
- Yagi, M.; Kanzaki, S.; Kawamoto, K.; Shin, B.; Shah, P.P.; Magal, E.; Sheng, J.; Raphael, Y. Spiral Ganglion Neurons Are Protected from Degeneration by GDNF Gene Therapy. J. Assoc. Res. Otolaryngol. 2000, 1, 315–325. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shityakov, S.; Hayashi, K.; Störk, S.; Scheper, V.; Lenarz, T.; Förster, C.Y. The Conspicuous Link between Ear, Brain and Heart–Could Neurotrophin-Treatment of Age-Related Hearing Loss Help Prevent Alzheimer’s Disease and Associated Amyloid Cardiomyopathy? Biomolecules 2021, 11, 900. https://doi.org/10.3390/biom11060900
Shityakov S, Hayashi K, Störk S, Scheper V, Lenarz T, Förster CY. The Conspicuous Link between Ear, Brain and Heart–Could Neurotrophin-Treatment of Age-Related Hearing Loss Help Prevent Alzheimer’s Disease and Associated Amyloid Cardiomyopathy? Biomolecules. 2021; 11(6):900. https://doi.org/10.3390/biom11060900
Chicago/Turabian StyleShityakov, Sergey, Kentaro Hayashi, Stefan Störk, Verena Scheper, Thomas Lenarz, and Carola Y. Förster. 2021. "The Conspicuous Link between Ear, Brain and Heart–Could Neurotrophin-Treatment of Age-Related Hearing Loss Help Prevent Alzheimer’s Disease and Associated Amyloid Cardiomyopathy?" Biomolecules 11, no. 6: 900. https://doi.org/10.3390/biom11060900
APA StyleShityakov, S., Hayashi, K., Störk, S., Scheper, V., Lenarz, T., & Förster, C. Y. (2021). The Conspicuous Link between Ear, Brain and Heart–Could Neurotrophin-Treatment of Age-Related Hearing Loss Help Prevent Alzheimer’s Disease and Associated Amyloid Cardiomyopathy? Biomolecules, 11(6), 900. https://doi.org/10.3390/biom11060900