
The Role of the Key Effector of Necroptotic Cell Death, MLKL, in Mouse Models of Disease


Abstract
1. Introduction
Necroptosis and Disease
2. Mlkl Knock-Out (KO) and Constitutively Active (CA) Mice at Steady State
3. Mlkl−/− Mice in Ischemia and Reperfusion Injury (IRI)
4. Sterile Inflammation
5. Infection: Bacterial
6. Infection: Viral
7. Metabolic Disease
8. Neuromuscular
9. Hematological
10. Cancer and Cancer Treatment
11. Reproductive System
12. Important Experimental Determinants in MLKL-Related Mouse Research
13. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef]
- Weir, A.; Hughes, S.; Rashidi, M.; Hildebrand, J.M.; E Vince, J. Necroptotic movers and shakers: Cell types, inflammatory drivers and diseases. Curr. Opin. Immunol. 2021, 68, 83–97. [Google Scholar] [CrossRef]
- Samson, A.L.; Garnish, S.E.; Hildebrand, J.M.; Murphy, J.M. Location, location, location: A compartmentalized view of TNF-induced necroptotic signaling. Sci. Signal. 2021, 14, eabc6178. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.-G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Newton, K.; Sun, X.; Dixit, V.M. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol. Cell. Biol. 2004, 24, 1464–1469. [Google Scholar] [CrossRef]
- Wu, J.; Huang, Z.; Ren, J.; Zhang, Z.; He, P.; Li, Y.; Ma, J.; Chen, W.; Zhang, Y.; Zhou, X.; et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013, 23, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.S.; Murphy, J.M. Down the rabbit hole: Is necroptosis truly an innate response to infection? Cell. Microbiol. 2017, 19, e12750. [Google Scholar] [CrossRef] [PubMed]
- Petrie, E.J.; Sandow, J.J.; Lehmann, W.I.; Liang, L.-Y.; Coursier, D.; Young, S.N.; Kersten, W.J.; Fitzgibbon, C.; Samson, A.L.; Jacobsen, A.V.; et al. Viral MLKL Homologs Subvert Necroptotic Cell Death by Sequestering Cellular RIPK3. Cell Rep. 2019, 28, 3309–3319. [Google Scholar] [CrossRef] [PubMed]
- Silke, J.; Hartland, E.L. Masters, marionettes and modulators: Intersection of pathogen virulence factors and mammalian death receptor signaling. Curr. Opin. Immunol. 2013, 25, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Hulpiau, P.; Saeys, Y.; Bertrand, M.J.; Vandenabeele, P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol. 2016, 26, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, M.; Deng, Y.; Whitney, P.; Salvamoser, R.; Engel, S.; Kueh, A.J.; Tai, L.; Bachem, A.; Gressier, E.; Geoghegan, N.D.; et al. Flexible Usage and Interconnectivity of Diverse Cell Death Pathways Protect against Intracellular Infection. Immunity 2020, 53, 533–547. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C. Death by inflammation: Drug makers chase the master controller. Nat. Biotechnol. 2019, 37, 111–113. [Google Scholar] [CrossRef]
- GSK. Q3 2019 Results; GlaxoSmithKline: Brentford, UK, 2019; p. 28. [Google Scholar]
- Weisel, K.; Berger, S.; Papp, K.; Maari, C.; Krueger, J.G.; Scott, N.; Tompson, D.; Wang, S.; Simeoni, M.; Bertin, J.; et al. Response to Inhibition of Receptor-Interacting Protein Kinase 1 (RIPK1) in Active Plaque Psoriasis: A Randomized Placebo-Controlled Study. Clin. Pharmacol. Ther. 2020, 108, 808–816. [Google Scholar] [CrossRef]
- Ni, H.-M.; Chao, X.; Kaseff, J.; Deng, F.; Wang, S.; Shi, Y.-H.; Li, T.; Ding, W.-X.; Jaeschke, H. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)-Mixed Lineage Kinase Domain-Like Protein (MLKL)-Mediated Necroptosis Contributes to Ischemia-Reperfusion Injury of Steatotic Livers. Am. J. Pathol. 2019, 189, 1363–1374. [Google Scholar] [CrossRef]
- Shi, C.; Kim, T.; Steiger, S.; Mulay, S.R.; Klinkhammer, B.M.; Baeuerle, T.; Melica, M.E.; Romagnani, P.; Möckel, D.; Baues, M.; et al. Crystal Clots as Therapeutic Target in Cholesterol Crystal Embolism. Circ. Res. 2020, 126, e37–e52. [Google Scholar] [CrossRef]
- Moerke, C.; Bleibaum, F.; Kunzendorf, U.; Krautwald, S. Combined Knockout of RIPK3 and MLKL Reveals Unexpected Outcome in Tissue Injury and Inflammation. Front. Cell Dev. Biol. 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-McNulty, B.; Carano, R.A.D.; Cao, T.C.; Van Bruggen, N.; Bernstein, L.; et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016, 23, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhao, Y.; Zhang, L.; Fan, H.; Qi, C.; Zhang, K.; Liu, X.; Fei, L.; Chen, S.; Wang, M.; et al. RIPK3/MLKL-Mediated Neuronal Necroptosis Modulates the M1/M2 Polarization of Microglia/Macrophages in the Ischemic Cortex. Cereb Cortex 2018, 28, 2622–2635. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Desai, J.; Kumar, S.V.; Eberhard, J.N.; Thomasova, D.; Romoli, S.; Grigorescu, M.; Kulkarni, O.P.; Popper, B.; Vielhauer, V.; et al. Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat. Commun. 2016, 7, 10274. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015, 26, 2647–2658. [Google Scholar] [CrossRef]
- Günther, C.; Christopher, P.; Kremer, A.E.; Murphy, J.M.; Petrie, E.J.; Amann, K.; Vandenabeele, P.; Linkermann, A.; Poremba, C.; Schleicher, U.; et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J. Clin. Investig. 2016, 126, 4346–4360. [Google Scholar] [CrossRef]
- Hamon, A.; Piquet-Pellorce, C.; Dimanche-Boitrel, M.-T.; Samson, M.; Le Seyec, J. Intrahepatocytic necroptosis is dispensable for hepatocyte death in murine immune-mediated hepatitis. J. Hepatol. 2020, 73, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Dara, L.; Johnson, H.; Suda, J.; Win, S.; Gaarde, W.; Han, D.; Kaplowitz, N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 2015, 62, 1847–1857. [Google Scholar] [CrossRef]
- Schreiber, A.; Rousselle, A.; Becker, J.U.; von Mässenhausen, A.; Linkermann, A.; Kettritz, R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E9618–E9625. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Spall, S.K.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kumari, S.; Kim, C.; Van, T.-M.; Wachsmuth, L.; Polykratis, A.; Pasparakis, M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 2016, 540, 124–128. [Google Scholar] [CrossRef]
- Polykratis, A.; Martens, A.; Eren, R.O.; Shirasaki, Y.; Yamagishi, M.; Yamaguchi, Y.; Uemura, S.; Miura, M.; Holzmann, B.; Kollias, G.; et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 2019, 21, 731–742. [Google Scholar] [CrossRef]
- Schwarzer, R.; Jiao, H.; Wachsmuth, L.; Tresch, A.; Pasparakis, M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity 2020, 52, 978–993. [Google Scholar] [CrossRef]
- Xie, Y.; Zhao, Y.; Shi, L.; Li, W.; Chen, K.; Li, M.; Chen, X.; Zhang, H.; Li, T.; Matsuzawa-Ishimoto, Y.; et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J. Clin. Investig. 2020, 130, 2111–2128. [Google Scholar] [CrossRef]
- Dannappel, M.; Vlantis, K.; Kumari, S.; Polykratis, A.; Kim, C.; Wachsmuth, L.; Eftychi, C.; Lin, J.; Corona, T.; Hermance, N.; et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 2014, 513, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Shindo, R.; Ohmuraya, M.; Komazawa-Sakon, S.; Miyake, S.; Deguchi, Y.; Yamazaki, S.; Nishina, T.; Yoshimoto, T.; Kakuta, S.; Koike, M.; et al. Necroptosis of Intestinal Epithelial Cells Induces Type 3 Innate Lymphoid Cell-Dependent Lethal Ileitis. iScience 2019, 15, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Diaz, S.; Preaudet, A.; Samson, A.L.; Nguyen, P.M.; Fung, K.Y.; Garnham, A.L.; Alexander, W.S.; Strasser, A.; Ernst, M.; Putoczki, T.L.; et al. Necroptosis is dispensable for the development of inflammation-associated or sporadic colon cancer in mice. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Devos, M.; Tanghe, G.; Gilbert, B.; Dierick, E.; Verheirstraeten, M.; Nemegeer, J.; De Reuver, R.; Lefebvre, S.; De Munck, J.; Rehwinkel, J.; et al. Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J. Exp. Med. 2020, 217, e20191913. [Google Scholar] [CrossRef]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Rickard, J.A.; Anderton, H.; Etemadi, N.; Nachbur, U.; Darding, M.; Peltzer, N.; Lalaoui, N.; E Lawlor, K.; Vanyai, H.; Hall, C.; et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 2014, 3, e03464. [Google Scholar] [CrossRef]
- Tummers, B.; Mari, L.; Guy, C.S.; Heckmann, B.L.; Rodriguez, D.; Rühl, S.; Moretti, J.; Crawford, J.C.; Fitzgerald, P.; Kanneganti, T.-D.; et al. Caspase-8-Dependent Inflammatory Responses Are Controlled by Its Adaptor, FADD, and Necroptosis. Immunity 2020, 52, 994–1006. [Google Scholar] [CrossRef]
- Hildebrand, J.M.; Kauppi, M.; Majewski, I.J.; Liu, Z.; Cox, A.J.; Miyake, S.; Petrie, E.J.; Silk, M.A.; Li, Z.; Tanzer, M.C.; et al. A missense mutation in the MLKL brace region promotes lethal neonatal inflammation and hematopoietic dysfunction. Nat. Commun. 2020, 11, 3150. [Google Scholar] [CrossRef]
- Kitur, K.; Wachtel, S.; Brown, A.; Wickersham, M.; Paulino, F.; Peñaloza, H.F.; Soong, G.; Bueno, S.M.; Parker, D.; Prince, A. Necroptosis Promotes Staphylococcus aureus Clearance by Inhibiting Excessive Inflammatory Signaling. Cell Rep. 2016, 16, 2219–2230. [Google Scholar] [CrossRef]
- D’Cruz, A.A.; Speir, M.; Bliss-Moreau, M.; Dietrich, S.; Wang, S.; Chen, A.A.; Gavillet, M.; Al-Obeidi, A.; Lawlor, K.E.; Vince, J.E.; et al. The pseudokinase MLKL activates PAD4-dependent NET formation in necroptotic neutrophils. Sci. Signal. 2018, 11, eaao1716. [Google Scholar] [CrossRef]
- Stutz, M.D.; Ojaimi, S.; Allison, C.; Preston, S.; Arandjelovic, P.; Hildebrand, J.M.; Sandow, J.J.; Webb, A.I.; Silke, J.; Alexander, W.S.; et al. Necroptotic signaling is primed in Mycobacterium tuberculosis-infected macrophages, but its pathophysiological consequence in disease is restricted. Cell Death Differ. 2018, 25, 951–965. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Wu, J.; Li, G.; Tao, X.; Lai, K.; Yuan, Y.; Zhang, X.; Zou, Z.; Xu, Y. RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. 2020, 27, 2568–2585. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Patino, E.; Ma, K.C.; Laursen, K.; Finkelsztein, E.J.; Akchurin, O.; Muthukumar, T.; Ryter, S.W.; Gudas, L.; Choi, A.M.K.; et al. RIPK3 promotes sepsis-induced acute kidney injury via mitochondrial dysfunction. JCI Insight 2018, 3, e98411. [Google Scholar] [CrossRef] [PubMed]
- Riegler, A.N.; Brissac, T.; Gonzalez-Juarbe, N.; Orihuela, C.J. Necroptotic Cell Death Promotes Adaptive Immunity Against Colonizing Pneumococci. Front. Immunol. 2019, 10, 615. [Google Scholar] [CrossRef]
- González-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015, 11, e1005337. [Google Scholar] [CrossRef] [PubMed]
- Sai, K.; Parsons, C.; House, J.S.; Kathariou, S.; Ninomiya-Tsuji, J. Necroptosis mediators RIPK3 and MLKL suppress intracellular Listeria replication independently of host cell killing. J. Cell Biol. 2019, 218, 1994–2005. [Google Scholar] [CrossRef]
- Yu, S.-X.; Chen, W.; Liu, Z.-Z.; Zhou, F.-H.; Yan, S.-Q.; Hu, G.-Q.; Qin, X.-X.; Zhang, J.; Ma, K.; Du, C.-T.; et al. Non-Hematopoietic MLKL Protects Against Salmonella Mucosal Infection by Enhancing Inflammasome Activation. Front. Immunol. 2018, 9, 119. [Google Scholar] [CrossRef]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H.; Ingram, J.P.; Rodriguez, D.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef]
- Shubina, M.; Tummers, B.; Boyd, D.F.; Zhang, T.; Yin, C.; Gautam, A.; Guo, X.-Z.J.; Rodriguez, D.A.; Kaiser, W.J.; Vogel, P.; et al. Necroptosis restricts influenza A virus as a stand-alone cell death mechanism. J. Exp. Med. 2020, 217, e20191259. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Platt, M.P.; Gilley, R.P.; Brown, D.; Dube, P.H.; Yu, Y.; Gonzalez-Juarbe, N. Influenza Causes MLKL-Driven Cardiac Proteome Remodeling During Convalescence. Circ. Res. 2021. [Google Scholar] [CrossRef]
- Gonzalez-Juarbe, N.; Riegler, A.N.; Jureka, A.S.; Gilley, R.P.; Brand, J.D.; Trombley, J.E.; Scott, N.R.; Platt, M.P.; Dube, P.H.; Petit, C.M.; et al. Influenza-Induced Oxidative Stress Sensitizes Lung Cells to Bacterial-Toxin-Mediated Necroptosis. Cell Rep. 2020, 32, 108062. [Google Scholar] [CrossRef]
- Daniels, B.P.; Snyder, A.G.; Olsen, T.M.; Orozco, S.L.; Oguin, T.H.; Tait, S.; Martinez, J.; Gale, M.; Loo, Y.-M.; Oberst, A. RIPK3 Restricts Viral Pathogenesis via Cell Death-Independent Neuroinflammation. Cell 2017, 169, 301–313. [Google Scholar] [CrossRef]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Kok, C.Y.-Y.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef]
- Saeed, W.K.; Jun, D.W.; Jang, K.; Oh, J.H.; Chae, Y.J.; Lee, J.S.; Koh, D.H.; Kang, H.T. Decrease in fat de novo synthesis and chemokine ligand expression in non-alcoholic fatty liver disease caused by inhibition of mixed lineage kinase domain-like pseudokinase. J. Gastroenterol. Hepatol. 2019, 34, 2206–2218. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Poulsen, K.L.; Sanz-Garcia, C.; Huang, E.; McMullen, M.R.; Roychowdhury, S.; Dasarathy, S.; Nagy, L.E. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J. Hepatol. 2020, 73, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Wu, X.; Fan, X.; Huang, E.; Sanz-Garcia, C.; Ross, C.K.C.-D.; Roychowdhury, S.; Bellar, A.; McMullen, M.R.; Dasarathy, J.; et al. Differential role of MLKL in alcohol-associated and non-alcohol-associated fatty liver diseases in mice and humans. JCI Insight 2021, 6, e140180. [Google Scholar] [CrossRef]
- Xu, H.; Du, X.; Liu, G.; Huang, S.; Du, W.; Zou, S.; Tang, D.; Fan, C.; Xie, Y.; Wei, Y.; et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol. Metab. 2019, 23, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Robichaud, S.; Nguyen, M.-A.; Geoffrion, M.; Wyatt, H.; Cottee, M.L.; Dennison, T.; Pietrangelo, A.; Lee, R.; Lagace, T.A.; et al. Loss of MLKL (Mixed Lineage Kinase Domain-Like Protein) Decreases Necrotic Core but Increases Macrophage Lipid Accumulation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Brumatti, G.; Ma, C.; Lalaoui, N.; Nguyen, N.-Y.; Navarro, M.; Tanzer, M.C.; Richmond, J.; Ghisi, M.; Salmon, J.M.; Silke, N.; et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Transl. Med. 2016, 8, 339ra69. [Google Scholar] [CrossRef]
- Zhao, Q.; Yu, X.; Li, M.; Liu, Y.; Han, Y.; Zhang, X.; Li, X.M.; Wu, X.; Qin, J.; Fang, J.; et al. MLKL attenuates colon inflammation and colitis-tumorigenesis via suppression of inflammatory responses. Cancer Lett. 2019, 459, 100–111. [Google Scholar] [CrossRef]
- Zhao, Q.; Cheng, X.; Guo, J.; Bi, Y.; Kuang, L.; Ren, J.; Zhong, J.; Pan, L.; Zhang, X.; Guo, Y.; et al. MLKL inhibits intestinal tumorigenesis by suppressing STAT3 signaling pathway. Int. J. Biol. Sci. 2021, 17, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fan, C.; Zhang, H.; Zhao, Q.; Liu, Y.; Xu, C.; Xie, Q.; Wu, X.; Yu, X.; Zhang, J.; et al. MLKL and FADD Are Critical for Suppressing Progressive Lymphoproliferative Disease and Activating the NLRP3 Inflammasome. Cell Rep. 2016, 16, 3247–3259. [Google Scholar] [CrossRef]
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.; O’Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Perera, N.D.; Chiam, M.D.F.; Cuic, B.; Wanniarachchillage, N.; Tomas, D.; Samson, A.L.; Cawthorne, W.; Valor, E.N.; Murphy, J.M.; et al. Necroptosis is dispensable for motor neuron degeneration in a mouse model of ALS. Cell Death Differ. 2020, 27, 1728–1739. [Google Scholar] [CrossRef]
- Ying, Z.; Pan, C.; Shao, T.; Liu, L.; Li, L.; Guo, D.; Zhang, S.; Yuan, T.; Cao, R.; Jiang, Z.; et al. Mixed Lineage Kinase Domain-like Protein MLKL Breaks Down Myelin following Nerve Injury. Mol. Cell 2018, 72, 457–468. [Google Scholar] [CrossRef]
- Zhang, S.; Su, Y.; Ying, Z.; Guo, D.; Pan, C.; Guo, J.; Zou, Z.; Wang, L.; Zhang, Z.; Jiang, Z.; et al. RIP1 kinase inhibitor halts the progression of an immune-induced demyelination disease at the stage of monocyte elevation. Proc. Natl. Acad. Sci. USA 2019, 116, 5675–5680. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.-S.; Chen, P.; Wang, W.-X.; Lin, C.-C.; Zhou, Y.; Yu, L.-H.; Lin, Y.-X.; Xu, Y.-F.; Kang, D.-Z. RIP1/RIP3/MLKL mediates dopaminergic neuron necroptosis in a mouse model of Parkinson disease. Lab. Investig. 2020, 100, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhang, W.; Cai, G.; Ding, Y.; Wei, C.; Li, S.; Yang, Y.; Qin, J.; Liu, D.; Zhang, H.; et al. Myofiber necroptosis promotes muscle stem cell proliferation via releasing Tenascin-C during regeneration. Cell Res. 2020, 30, 1063–1077. [Google Scholar]
- Nakazawa, D.; Desai, J.; Steiger, S.; Müller, S.; Devarapu, S.K.; Mulay, S.R.; Iwakura, T.; Anders, H.-J. Activated platelets induce MLKL-driven neutrophil necroptosis and release of neutrophil extracellular traps in venous thrombosis. Cell Death Discov. 2018, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Moujalled, D.; Gangatirkar, P.; Kauppi, M.; Corbin, J.; Lebois, M.; Murphy, J.M.; Lalaoui, N.; Hildebrand, J.M.; Silke, J.; Alexander, W.S.; et al. The necroptotic cell death pathway operates in megakaryocytes, but not in platelet synthesis. Cell Death Dis. 2021, 12, 133. [Google Scholar] [CrossRef]
- Li, D.; Meng, L.; Xu, T.; Su, Y.; Liu, X.; Zhang, Z.; Wang, X. RIPK1-RIPK3-MLKL-dependent necrosis promotes the aging of mouse male reproductive system. eLife 2017, 6, e27692. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Kwon, Y.C.; Park, S.; Zhang, H.; Corr, N.; Ljumanovic, N.; Adedeji, A.O.; Varfolomeev, E.; Goncharov, T.; Preston, J.; et al. RIP1 kinase activity is critical for skin inflammation but not for viral propagation. J. Leukoc. Biol. 2020, 107, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Siempos, I.I.; Ma, K.C.; Imamura, M.; Baron, R.M.; Fredenburgh, L.E.; Huh, J.-W.; Moon, J.-S.; Finkelsztein, E.J.; Jones, D.S.; Lizardi, M.T.; et al. RIPK3 mediates pathogenesis of experimental ventilator-induced lung injury. JCI Insight 2018, 3, e97102. [Google Scholar] [CrossRef]
- Imamura, M.; Moon, J.-S.; Chung, K.-P.; Nakahira, K.; Muthukumar, T.; Shingarev, R.; Ryter, S.W.; Choi, A.M.; Choi, M.E. RIPK3 promotes kidney fibrosis via AKT-dependent ATP citrate lyase. JCI Insight 2018, 3, e94979. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Yan, R.; Tian, J.; Zhang, Y.; Zhang, J.; Chen, M.; Cui, Q.; Zhao, L.; Hu, R.; et al. Receptor-interacting protein kinase 3 promotes platelet activation and thrombosis. Proc. Natl. Acad. Sci. USA 2017, 114, 2964–2969. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Dewitz, C.; Schmitz, J.; Schröder, A.S.; Bräsen, J.H.; Stockwell, B.R.; Murphy, J.M.; Kunzendorf, U.; Krautwald, S. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell. Mol. Life Sci. 2017, 74, 3631–3645. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Ito, Y.; Wijeweera, J.; Liu, J.; Malle, E.; Farhood, A.; McCuskey, R.S.; Jaeschke, H. Reduced inflammatory response and increased microcirculatory disturbances during hepatic ischemia-reperfusion injury in steatotic livers of ob/ob mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1385–G1395. [Google Scholar] [CrossRef]
- Pierotti, C.L.; Tanzer, M.C.; Jacobsen, A.V.; Hildebrand, J.M.; Garnier, J.-M.; Sharma, P.; Lucet, I.S.; Cowan, A.D.; Kersten, W.J.A.; Luo, M.-X.; et al. Potent Inhibition of Necroptosis by Simultaneously Targeting Multiple Effectors of the Pathway. ACS Chem. Biol. 2020, 15, 2702–2713. [Google Scholar] [CrossRef]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra40. [Google Scholar] [CrossRef] [PubMed]
- Majdi, A.; Aoudjehane, L.; Ratziu, V.; Islam, T.; Afonso, M.B.; Conti, F.; Mestiri, T.; Lagouge, M.; Foufelle, F.; Ballenghien, F.; et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J. Hepatol. 2020, 72, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Turner, A.W.; Duchez, A.-C.; Soubeyrand, S.; Rasheed, A.; Smyth, D.; Cook, D.P.; Nikpay, M.; Kandiah, J.W.; Pan, C.; et al. RIPK1 gene variants associate with obesity in humans and can be therapeutically silenced to reduce obesity in mice. Nat. Metab. 2020, 2, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Ofengeim, D.; Najafov, A.; Das, S.; Saberi, S.; Li, Y.; Hitomi, J.; Zhu, H.; Chen, H.; Mayo, L.; et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 2016, 353, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Chevin, M.; Sébire, G. Necroptosis in ALS: A hot topic in-progress. Cell Death Discov. 2021, 7, 79. [Google Scholar] [CrossRef]
- Tanzer, M.C.; Tripaydonis, A.; Webb, A.I.; Young, S.N.; Varghese, L.N.; Hall, C.; Alexander, W.S.; Hildebrand, J.M.; Silke, J.; Murphy, J.M. Necroptosis signalling is tuned by phosphorylation of MLKL residues outside the pseudokinase domain activation loop. Biochem. J. 2015, 471, 255–265. [Google Scholar] [CrossRef]
- Hoglen, N.C.; Chen, L.-S.; Fisher, C.D.; Hirakawa, B.P.; Groessl, T.; Contreras, P.C. Characterization of IDN-6556 (3-[2-(2-tert-butyl-phenylaminooxalyl)-amino]-propionylamino]-4-oxo-5-(2,3,5,6-tetrafluoro-phenoxy)-pentanoic acid): A liver-targeted caspase inhibitor. J. Pharmacol. Exp. Ther. 2004, 309, 634–640. [Google Scholar] [CrossRef]
- Li, D.; Ai, Y.; Guo, J.; Dong, B.; Li, L.; Cai, G.; Chen, S.; Xu, D.; Wang, F.; Wang, X. Casein kinase 1G2 suppresses necroptosis-promoted testis aging by inhibiting receptor-interacting kinase 3. eLife 2020, 9, e61564. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2015, 2, e975093. [Google Scholar] [CrossRef]
- Robertson, S.J.; Lemire, P.; Maughan, H.; Goethel, A.; Turpin, W.; Bedrani, L.; Guttman, D.S.; Croitoru, K.; Girardin, S.E.; Philpott, D.J. Comparison of Co-housing and Littermate Methods for Microbiota Standardization in Mouse Models. Cell Rep. 2019, 27, 1910–1919. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef]
- Kadioglu, A.; Cuppone, A.M.; Trappetti, C.; List, T.; Spreafico, A.; Pozzi, G.; Andrew, P.W.; Oggioni, M.R. Sex-based differences in susceptibility to respiratory and systemic pneumococcal disease in mice. J. Infect. Dis. 2011, 204, 1971–1979. [Google Scholar] [CrossRef]
- Petrie, E.J.; Sandow, J.J.; Jacobsen, A.V.; Smith, B.J.; Griffin, M.D.W.; Lucet, I.S.; Dai, W.; Young, S.N.; Tanzer, M.C.; Wardak, A.; et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 2018, 9, 2422. [Google Scholar] [CrossRef]
- Davies, K.A.; FitzGibbon, C.; Young, S.N.; Garnish, S.E.; Yeung, W.; Coursier, D.; Birkinshaw, R.W.; Sandow, J.J.; Lehmann, W.I.L.; Liang, L.-Y.; et al. Distinct pseudokinase domain conformations underlie divergent activation mechanisms among vertebrate MLKL orthologues. Nat. Commun. 2020, 11, 3060. [Google Scholar] [CrossRef] [PubMed]
- Tanzer, M.C.; Matti, I.; Hildebrand, J.M.; Young, S.N.; Wardak, A.; Tripaydonis, A.; Petrie, E.J.; Mildenhall, A.L.; Vaux, D.L.; E Vince, J.; et al. Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ. 2016, 23, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Hofmans, S.; Declercq, W.; Augustyns, K.; Vandenabeele, P. Inhibitors Targeting RIPK1/RIPK3: Old and New Drugs. Trends Pharmacol. Sci. 2020, 41, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Weisel, K.; Berger, S.; Thorn, K.; Taylor, P.C.; Peterfy, C.; Siddall, H.; Tompson, D.; Wang, S.; Quattrocchi, E.; Burriss, S.W.; et al. A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res. Ther. 2021, 23, 85. [Google Scholar] [CrossRef]


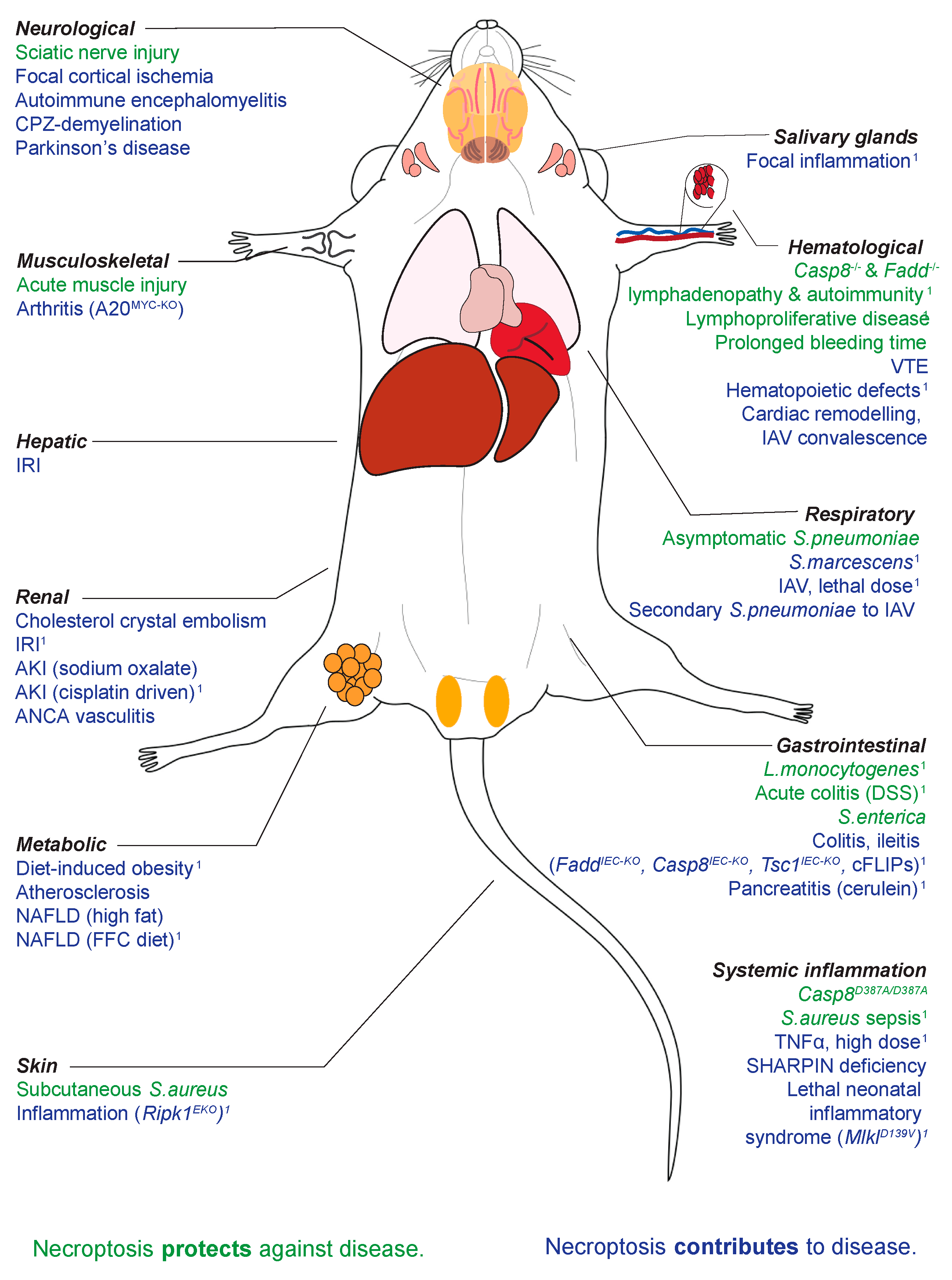{kind=link}
| Challenge | Method | Outcome in Mlkl−/− (Mutant or Knock-Down) Mouse Relative to Wild-Type Control | Mlkl−/− Mouse and Wild-Type Control Details | Reference |
|---|---|---|---|---|
| Ischemia and reperfusion injury (IRI) | ||||
| Hepatic IRI | 45 min ischemia in chow or Western diet fed mice. | Reduced TUNEL positive hepatocytes and serum ALT levels in Mlkl−/− mice, irrespective of diet. Hepatic neutrophil infiltration and TNF, IL-1, MIP-2, and IL-6 mRNA reduced post I/R in Mlkl−/− mice. | Sex-matched Mlkl−/− (CRISPR-Cas9) and Wt mice utilised. | [17] |
| Renal infarction, no reperfusion | Cholesterol crystal embolism. | Mlkl−/− mice protected from infarction (measured by infarct size, kidney injury and neutrophil infiltration). No difference in the extent of acute kidney injury (measured by eGFR) and kidney failure. | Mlkl−/− [5] and Wt mice utilised. | [18] |
| Renal IRI | 40 min bilateral renal pedicle clamp. | Mlkl−/− mice protected from acute kidney injury (measured using serum creatinine and urea). Ripk3−/−Mlkl−/− double knockout mice were only somewhat protected. | Sex- and weight-matched Wt controls for Mlkl−/− [5] mice. | [19] |
| 30 min renal pedicle clamp. | Mlkl−/− prolonged survival by ~4 days over Wt mice. | Male Wt and Mlkl−/− [5] littermates. | [20] | |
| Focal cortical ischemia | Photothrombosis of the cortical microvessels. | Reduced infarction (fewer TUNEL/NeuN-positive cells) in Mlkl−/− mice and improved locomotion from day 7. Fewer F4/F80+ cells and reduced expression of iNOS, TNFα, IL-12 and IL-18. | Mlkl−/− sourced from Xiamen University * | [21] |
| Sterile Inflammatory | ||||
| Acute kidney injury (AKI) | Intraperitoneal injection of sodium oxalate. | Mlkl−/− mice show reduced serum creatinine, neutrophil recruitment, and tubular necrosis (TUNEL staining and tubular injury score). | Sex matched male Wt controls for Mlkl−/− [5] mice. | [22] |
| Intraperitoneal injection of cisplatin. | Mlkl−/− mice resistant to tubular necrosis (histology, blood urea nitrogen and serum creatinine). Reduced TNFα, IL-1β, IFN-γ, IL-6 in proximal tubules despite similar levels to Wt at baseline. | Male Wt and Mlkl−/− (TALEN) mice are littermates. | [23] | |
| Hepatitis | ConA-induced. | Mlkl−/− mice show reduced hepatocyte necrosis (TUNEL staining and ALT/AST). | Wt and Mlkl−/− [5] a combination of C57BL/6J or littermate controls used. | [24] |
| Hepatocyte necrosis in MlklLPC-KO mice, with specifically ablated MLKL in liver parenchymal cells, are indistinguishable from Wt (ALT and H&E). | MlklLPC-KO and Mlklfl/fl are littermates. | [25] | ||
| LPS/GalN-driven (model of apoptotic hepatitis). | Mlkl−/− mice are indistinguishable from Wt (assessed using ALT/AST and histological staining for cleaved Casp3). | Wt and Mlkl−/− [5] a combination of C57BL/6J or littermate controls used. | [24] | |
| Acetaminophen (model of drug-induced liver injury). | MLKL deficiency does not prevent liver injury (assessed using AST/ALT levels and TUNEL staining). | Wt and Mlkl−/− [5] a combination of C57BL/6J or littermate controls used. | [24] | |
| Male Wt used for Mlkl−/− [5] mice. Bred as separate cohorts. | [26] | |||
| Acute pancreatitis | Cerulein-induced. | Mlkl−/− mice experience less severe acinar cell necrosis than Wt mice (as measured by histological and quantitative analysis). | Male Mlkl−/− (TALEN) and Wt mice are littermates. | [7] |
| ANCA-driven necrotising crescent glomerulonephritis | αMPO IgG transfer. | Mlkl−/− mice were protected against NCGN (as measured by reduced leukocyturia, erythrocyturia and less crescents and necrotic change on histology). | Mlkl−/− [5]. * | [27] |
| Arthritis | K/B X N serum transfer. | No significant difference in disease progression between Mlkl−/− and Wt mice (clinical severity scale and myeloperoxidase average radiance). | Sex matched Wt for Mlkl−/− mice. | [28] |
| Myeloid-cell-specific A20 deficiency (A20MYC-KO). | A20MYC-KOMlkl−/− mice were protected against inflammatory arthritis (thickness of rear ankles, histological scores of inflammation, cartilage and bone destruction), splenomegaly and showed reduced expression of IL-1β and TNFα. | Mice with loxP-flanked A20 crossed with Mlkl−/− [29] aged to 23-to-27 weeks old. | [30] | |
| Colitis and ileitis | IEC-specific FADD and Caspase-8 deficiency. | Mlkl−/−FaddIEC-KO and Mlkl−/−Casp8IEC-KO resistant to colitis. Mlkl−/−Casp8IEC-KO are also resistant to ileitis whilst Mlkl−/−FaddIEC-KO are only partially protected. Assessed using histological analysis of colonic and ileal tissues and microarray. | Littermate control mice with homozygous or heterozygous loxP-flanked Fadd or Casp8. | [31] |
| IEC-specific highly active mTORC1 pathway (Tsc1IEC-KO). | Tsc1IEC-KOMlkl−/− profoundly alleviates epithelial cell death and intestinal barrier dysfunction that is seen in Tsc1IEC-KO. | Tsc1IEC-KO and Tsc1IEC-KOMlkl−/− were age- and sex-matched littermate controls. | [32] | |
| Lethal ileitis | X-linked transgene -short form of cellular FLICE-inhibitory protein (cFLIPs). | MLKL deficiency partially rescues male mice from lethal in-utero ileitis (improved survival, small intestine villous architecture, fewer CC3-positive cells). | Male Wt and Mlkl−/− [33]. | [34] |
| Acute colitis | 1.5% DSS in drinking water, 5 days, followed by 3 days of normal drinking water. | Mlkl−/− mice display increased weight loss, but only in some cohorts. Day 5 and 8, indistinguishable murine endoscopic index of colitis (MEIC) scores. Increased inflammation in the proximal colon and decreased submucosal inflammation in the distal colon (H&E). | Wt and Mlkl−/− [5] are littermates. | [35] |
| Inflammatory skin disease | RIPK1 deficient keratinocytes Ripk1EKO. | Specific MLKL deficiency in keratinocytes profoundly ablated the development of the inflammatory skin lesion that invariably develops in Ripk1EKO mice. | Ripk1FL/FL (Takahashi et al., 2014) MlklFL/FL (Murphy et al., 2013) Wt littermates utilised. | [36] |
| Systemic inflammatory response syndrome (SIRS) | 300 μg/kg TNF intravenous/kg TND. | Mlkl−/− mice suffer a hypothermia reaction like that of Wt controls. | Wt and Mlkl−/− [5] are littermates. | [20] |
| 500 μg/kg TNF intravenous. | Mlkl−/− mouse core body temperature higher at 6, 8 and 10 h post dose than Wt controls, indicating moderate protection. | Wt and Mlkl−/− [5] are littermates. | [20] | |
| 30 mg/kg LPS-intraperitoneal. | Mlkl−/− mice show similar serum TNFα and IL-1β levels as Wt (measured at 0, 2, 4 and 8 h by ELISA). | Male Mlkl−/− (TALEN) and Wt mice are littermates. | [7] | |
| 1 mg/kg TNF in 200 μL PBS via the tail vein. | Mlkl−/− mice showed increased survival. Ripk3−/−Mlkl−/− double knockout mice were not similarly protected. | Female Wt and Mlkl−/− [5] mice bred as separate cohorts. | [19] | |
| 50 μg/kg LPS intraperitoneal. | Mlkl−/− mice showed comparable levels of TNFα and IL-6 (ELISA analysis) 1 h post-treatment. Pre-treatment with necrostatin-1 (IV), 15 min prior to LPS, significantly reduced Mlkl−/− mice ability to produce TNFα and IL-6. | * | [37] | |
| Generalised inflammation | A20 gene deficiency. | Mlkl−/−A20−/− mice display similar inflammation (hepatic neutrophil infiltration, RANTES secretion and dermatitis) and lifespan. | Wt are A20−/− littermates. | [20] |
| TNF-induced multi-organ inflammation | SHARPIN deficiency shpnm/m. | 12-week-old Shpnm/mMlkl−/− mice show reduced liver inflammation, splenomegaly and leucocytosis in comparison to Shpm/m controls (histology, ADVIA automated hematological analysis). | C57BL/Ka Shpncpdm/cpdm backcrossed to C57BL/6J one or two times or crossed with C56BL/6J Mlkl−/− [5] mice. | [38] |
| Casp8D387A/D387A (non-cleavable caspase 8)-induced systemic inflammation | Casp8D387A/D387A mutation. | Mlkl−/− Casp8DA/DA do not develop LPR disease. Mlkl−/− Casp8DA/DA show an exacerbated inflammatory phenotype with significant splenomegaly, liver damage (serum ALT, AST) and premature death. | Casp8D387A/D387A (CRISPR-Cas9) and Mlkl−/− [5] mice are sex-matched. | [39] |
| MlklD139V-induced lethal neonatal inflammatory syndrome | Whole body homozygosity for constitutively active MlklD139V mutation. | MlklD139V homozygotes are born normal but develop acute multifocal inflammation of the head, neck, and mediastinum by day P2/P3. Maximum lifespan observed 6 days. | Age and sex matched littermate controls. | [40] |
| Infection | ||||
| Bacterial | ||||
| Staphylococcus aureus | Subcutaneous 2 × 106 CFU of S. aureus strain MRSA USA300. | 5-day p.i. Mlkl−/− mice suffer greater bacterial burdens, larger skin lesions, significantly increased neutrophil, macrophage, γδ T cell infiltrate and enhanced pro-inflammatory cytokine expression. | Sex-matched Wt and Mlkl−/− [5]. | [41] |
| Retro-orbital 1 × 108 CFU S. aureus strain MRSA USA300. | Mlkl−/− mice show increased mortality rates compared to WT (a mean survival of 4 days). | Sex-matched Wt and Mlkl−/− [5]. | [41] | |
| Intravenous 1 × 107 CFU of S. aureus strain MRSA. | Accelerated weight loss and morbidity in Mlkl−/− mice. Circulating neutrophil numbers elevated 24 h p.i. Increased CFU in the blood and MRSA burden in the kidney of Mlkl−/− mice. | Mlkl−/− [5] mice littermates utilised. | [42] | |
| Retro-orbital 1 × 106–7 CFU of S. aureus strain MRSA. | Mlkl−/− mice show increased MRSA burden in blood and kidneys and greater number of peripheral neutrophils (flow cytometry). | Mlkl−/− [5] mice littermates utilised. | [42] | |
| Intraperitoneal 1 × 107 CFU of S. aureus strain MRSA. | Mlkl−/− mice display more severe bacteraemia 24 h p.i. | Mlkl−/− [5] mice littermates utilised. | [42] | |
| Mycobacterium tuberculosis | ~100–200 CFU of Mtb via aerosol strain H37Rv. | Mlkl−/− mice are indistinguishable from Wt controls in terms of splenic and respiratory bacterial burden, gross lung histopathology of inflammatory lesions (number and size), organisation of granulomatous inflammation, immune cell counts, TNFα and IL-1β. | Sex-matched Wt and Mlkl−/− [5] mice. | [43] |
| Polymicrobial septic shock | Cecal ligation and puncture (CLP). | Mlkl−/− and Wt mice showed same survival profile (mortality was monitored from 24 h to 144 h). | Age- and sex-matched littermates of Wt and Mlkl−/−. | [7] |
| Mlkl−/− have better survival (~50%) than Wt controls (~25%) at 180 h and are moderately protected against hypothermia. Mlkl−/− experience less severe lung, small intestine (H&E) injury with reduced levels of ALT, serum BUN, HMGB1, TF, IL-1β, TNFα. | Age- and sex-matched Wt and Mlkl−/−. | [44] | ||
| Acute kidney injury (AKI) following polymicrobial septic shock | Cecal ligation and puncture (CLP). | Mlkl−/− mice suffer same extent of AKI. Ratio of lipocalin-2/urine creatinine levels were lower in Mlkl−/− mice than Wt, however not as reduced as Ripk3−/− mice. | Wt and Mlkl−/− (Jiahuai Han laboratory) littermates. | [45] |
| Asymptomatic chronic nasopharyngeal colonisation with S. pneumoniae | Nasal instillation of ~1 × 105 CFU of serotype 4 strain TIGR4 m. | Mlkl−/− mice demonstrate reduced nasopharyngeal epithelial cell sloughing and increased LDH, Il-33, IL-1α, CXCL2 levels and decreased Il-6, Il-17 and polymorphonuclear cells than Wt. Mlkl−/− mice cleared S. pneumoniae colonisation at a slower rate than Wt controls. Less anti-PspA IgG than Wt controls despite comparable total serum IgG concentration. | Wt and Mlkl−/− [5] mice utilised. | [46] |
| S. marcescens hemorrhagic pneumonia | Intratracheal infection with strain MB383. | Mlkl−/− mice have increased alveolar macrophages and suffer less lung damage (histological analysis). | Wt and Mlkl−/− littermates (Douglas Green). | [47] |
| Listeria monocytogenes | Oral 1 × 108 CFU L. monocytogenes 1/2b strain 2011L-2858. | 3 days p.i. Mlkl−/− mice have a moderate increase in liver bacterial colonisation compared to Wt littermate controls. | Female Wt and Mlkl−/− [5] littermates. | [48] |
| Salmonella enterica | Oral 5 × 107 CFU −1 × 108 CFU Subsp. Enterica serovar Typhimurium strain SL1344. | Mlkl−/− mice show greater submucosal oedema, loss of goblet cells, PMN infiltration, loss of epithelial barrier integrity and salmonella colonisation despite comparable bacterial faecal loads. Also have increased body and cecal weight loss. | Wt and Mlkl−/− (Dr. Jia-Huai Han, Xiamen University, China) were sex-matched. | [49] |
| Viral | ||||
| Influenza A strain PR8 | Instranasal 4000 EID50. | Mlkl−/− mice indistinguishable from Wt in terms of survival (75% of both Mlkl−/− and Wt mice survived and recovered) and lung progeny virion output. | Sex-matched Wt controls *. | [50] |
| Instranasal 2500 EID50. Intranasal 6000 EID50. | At moderate dose, Mlkl−/− mice are indistinguishable from Wt in terms of survival. At lethal dose, Mlkl−/− mice show increased survival; fewer disrupted epithelial cells despite same viral titre 6 days p.i. Influx of neutrophils into the lungs in Mlkl−/− mice both delayed and diminished. | Sex-matched littermates used for Mlkl−/− mice [5]. | [13] | |
| Instranasal 2500 EID50. Instranasal 1500 EID50. | Mlkl−/− are indistinguishable from Wt controls in terms of survival, viral titres, morphometry of viral spread and percentage of infected lung. IAV-mediated alveolar inflammation; septal thickening, inflamed alveoli, and hyaline membranes in Mlkl−/− mice comparable to that of Wt mice. | Sex matched Wt littermate or Wt C57BL/6 mice were used as controls for Mlkl−/− mice [5]. | [51] | |
| Influenza strain A/California/7/2009 | 250 PFU of IAV. | At baseline, Mlkl−/− mouse myocardium showed increased mitochondrial and antioxidant activity (proteome analysis), increased survival and reduced weight loss. | Wt (B6NTac) mice and Mlkl−/− mice [5] were bred independently. | [52] |
| Secondary S. pneumoniae following influenza A viral infection | Intranasal 250 PFU pdmH1N1. Intratracheal 1 × 103 CFU of S. pneumoniae. | Mlkl−/− mice showed reduced pulmonary cell death (TUNEL-staining), bacterial burden, lung consolidation and IFN-α, -β expression. No changes in the amount of oxidative-stress-induced DNA damage (immunofluorescence of 8-OHdG). | Wt and Mlkl−/− [5]. | [53] |
| West Nile Virus (WNV) encephalitis | Subcutaneous injection with 100 pfu of WNV-TX 2002-HC strain. | Mlkl−/− mice are indistinguishable from Wt in terms of survival and viral titres. | Sex matched Wt C57BL/6J used for Mlkl−/− mice [5]. | [54] |
| Metabolic | ||||
| Non-alcoholic fatty liver disease | Choline-deficient, methionine-supplemented (CDE), fed once. | Mlkl−/− mice were indistinguishable from Wt controls in terms of hepatic necrosis (serum AST and PI-positive cells). Reduced systemic levels of Il-6 and IL-1β (RT-PCR), comparable levels of TNFα. | Male C57BL/6J (CLEA Japan) including Mlkl−/− (M.Pasparakis) were utilised. | [55] |
| 12 weeks of high fat diet. | Mlkl−/− mice gain body weight comparable to Wt. At baseline, Mlkl−/− mice had increased serum AST/ALT and decreased serum fasting blood glucose. Mlkl−/− mice demonstrate reduced NAFLD activity score, steatosis score, hepatocyte ballooning, lobular inflammation, serum AST/ALT, triglyceride levels and de novo fat synthesis. | C57BL/6N Wt and Mlkl−/− (Jiahuai Han) mice utilised. | [56] | |
| 8 weeks of Western diet. | Mlkl−/− are indistinguishable from Wt in terms of level of steatosis and liver triglyceride accumulation. | Sex-matched Mlkl−/− (CRISPR-Cas9) on C57BL/6N and Wt utilised. | [17] | |
| 12 weeks of fat, fructose and cholesterol (FFC) diet. | Mlkl−/− mice are protected from liver injury (measured using AST/ALT, hepatic triglyceride accumulation, macrovesicular and microvesicular steatosis (H&E). Mlkl−/− mice were protected from FFC-induced apoptosis (measured using M30 levels, cleaved caspase-3, and TUNEL-positive cells) and inflammation (measured using mRNA levels of TNFα, Il-1β, MCP-1 and F4/80). | Littermates utilised. | [57] | |
| Alcoholic fatty liver disease (ALFD) | Chronic ethanol-induced. | Mlkl−/− mice are indistinguishable from Wt in terms of ALT/AST, hepatic triglycerides, macrovesicular and microvesicular steatosis (H&E). | Sex-matched littermates utilised. | [58] |
| Gao-binge. | Mlkl−/− mice are indistinguishable from Wt in terms of body weight, food intake, ALT/AST, hepatic triglycerides, macrovesicular and microvesicular steatosis (H&E). Mlkl−/− mice have similar levels of CYP2E1, ER stress and hepatocyte apoptosis but mildly reduced levels of some hepatic inflammatory markers. | Sex-matched littermates utilised. | [58] | |
| Diet-induced obesity | 16 weeks of high fat diet (HFD) consisting of 60% kcal from fat or chow diet (CD). | Mlkl−/− mice on regular CD are indistinguishable from Wt littermates in terms of body weight, glucose disposal, glucose tolerance, or insulin sensitivity. After 16 weeks on HFD, Mlkl−/− mice have lower body weight and visceral adipose, and better glucose and insulin tolerance. No difference in inflammatory markers or TUNEL positive cells in the liver. | Body weight matched, male Wt and Mlkl−/− littermates are utilised. | [59] |
| Atherosclerosis | Western diet for 8 or 16 weeks. | Apoe−/− mice fed a Western diet while receiving MLKL antisense oligonucleotides (ASOs) demonstrated reduced necrotic core size of aortic sinus plaques, reduced plasma cholesterol, fewer TUNEL positive cells but increased lipid content in atherosclerotic plaque (oil red O staining). | Apoe−/− C57Bl/6N mice administered with control or MLKL antisense oligonucleotides (ASOs). | [60] |
| Neoplasia | ||||
| Acute myeloid leukemia | Retroviral expression of the fusion protein (MLL-ENL) in Mlkl−/− hematopoietic stem cells. | AML cells (MLL-ENL-transduced E14 liver hematopoietic stem cells (HSCs)) transplanted into lethally irradiated Wt mice. AML generated from Mlkl−/− HSCs showed similar leukemia progression and overall survival compared to AML generated from Wt HSCs. | Age matched, both Mlkl−/− and Wt on C57B/6J background. | [61] |
| Colon cancer | Sporadic intestinal adenoma (APCmin mouse). | No significant difference in colonic tumour burden. No difference in colonic inflammation (H&E) or expression of Il-6. | Wt and Mlkl−/− [5] are littermates. Data for both sexes presented. | [35] |
| Colitis-associated cancer (azoxymethane, DSS). | No significant difference in weight loss observed. Similar timing of tumour onset and burden between Mlkl−/− and Wt (endoscopic tumour scores and H&E). | Wt and Mlkl−/− [5] are littermates. Data for both sexes presented. | [35] | |
| Colitis-associated tumorigeneses | AOM injection at 10 mg/kg of body weight. Five days post, fed with three cycles of 3% (w/v) DSS, followed by 14 days of normal water. | Mlkl−/− mice have a significant reduction in body weight, increased clinical severity, shorter colons, and worse survival in comparison to Wt. Mlkl−/− mice exhibit increased burden of anal and colonic polyps with significant increase in inflammation, hyperplasia, and dysplasia (H&E). | Wt and Mlkl−/− (CRISPR-Cas9) were sex-matched (males only). | [62] |
| Apcmin/+ Mlkl−/− mice have a median survival time of 127 days compared with Apcmin/+ survival of 185 days, alongside marked increases in tumour number and load. | Wt and Mlkl−/− (CRISPR-Cas9) were littermate controls. | [63] | ||
| Progressive lymphoproliferative disease | Fadd gene knock out. | Mlkl−/− rescues Fadd−/− mice from embryonic lethality but results in more severe progressive lymphoproliferative disease compared to Ripk3−/−Fadd−/− mice. | Mlkl−/− generated by CRISPR-Cas9 (Bioray Labs). | [64] |
| Littermates derived from Fadd+/−, Mlkl+/− crosses [5] mice utilised. | [65] | |||
| Casp8 gene knock out. | Mlkl−/− rescues Casp8−/− mice from embryonic lethality but results in more severe progressive lymphoproliferative disease compared to Ripk3−/−Casp8−/− mice. | Littermates derived from Casp8+/−, Mlkl+/− crosses [5] mice utilised. | [65] | |
| Neuromuscular | ||||
| Amyotrophic lateral sclerosis | SOD1G93A mice (express mutant human superoxide dismutase 1). | MLKL deficiency does not affect disease onset, progression, or survival in SOD1G93A mice. MLKL ablation has no impact on astrocyte or microglial activation. | Four isogenic genotypes for study: SOD1G93A;Mlkl−/−, SOD1G93A, Mlkl−/− and Wt littermates. Mlkl−/− mice [5]. | [66] |
| Sciatic nerve crush injury | Sciatic nerve cut or crushed unilaterally. | pMLKL (serine 441) is upregulated in damaged sciatic nerve cells of both Wt and Ripk3−/− mouse sciatic nerves. Mlkl−/− mice suffer drastically reduced myelin sheath breakdown and thus reduced sciatic nerve regeneration and function. | Male only Mlkl−/− (Li et al., 2017) and Wt (C57BL/6J) utilised. | [67] |
| Experimental autoimmune encephalomyelitis (EAE) | Intraperitoneal injection of 200ng of pertussis toxin. | Mlkl−/− mice display significantly reduced clinical EAE disease scores with delayed reduction of myelination (MBP immunofluorescence). | Sex matched Wt and Mlkl−/− (CRISPR-Cas9) bred as separate cohorts. Same housing conditions since birth. | [68] |
| Cuprizone CPZ-induced demyelination | Fed 0.2% CPZ in chow for 4 weeks. | Mlkl−/− mice have delayed demyelination in the caudal corpus callosum (MBP immunofluorescence). | Sex matched Wt and Mlkl−/− (CRISPR-Cas9) bred as separate cohorts. Same housing conditions since birth. | [68] |
| Parkinson’s disease (PD) | Intraperitoneal injection of MPTP. | Mlkl−/− show reduced neuroinflammatory markers (TNFα, IL-1β and IL-1 mRNA) and are significantly protected from striatal dopamine reduction and TH-positive neuron loss from the substantia nigra pars compacta compared to Wt controls. | Male Mlkl−/− (Jiahuai Han, Xiamen University, China) and Wt utilised. | [69] |
| Acute muscle injury | Intramuscular injection of cardiotoxin (CTX). | Mlkl−/− mice have fewer active muscle stem cells and thus suffer a significantly impaired capacity to regenerate muscle fibres. Regenerating myofibrils from Mlkl−/− mice express less myogenic factors MyoD, MyoG and nascent myofibril marker MYH3 in contrast to Wt. | Male Mlkl−/− (CRISPR-Cas9) and Wt mice utilised. | [70] |
| Hematological | ||||
| Venous thromboembolic (VTE) disease | 100% flow obstruction of the IVC using ETHICON. | Mlkl−/− mice develop significantly smaller thrombi, with reduced areas of TUNEL+ cells, Ly6b+ neutrophils and F4/80+ macrophages. Systemically, Mlkl−/− mice have fewer circulating neutrophils, monocytes, and serum histone–DNA complexes following IVC ligation. No difference in baseline number of neutrophils, monocytes, DAMPs or bleeding time. | Male Wt (C57BL/6N) and Mlkl−/− [5]. | [71] |
| Baseline | Mlkl−/− mice aged to 50 and 100 days. | Mlkl−/− mice are indistinguishable from Wt for the following blood parameters: WBC and lymphocyte count, thymic weight and cell count, splenic weight and cell count, lymph node weight and cell count. | Littermates derived from Casp8+/−, Mlkl+/− crosses [5] mice utilised. | [65] |
| Casp8 or FADD deficiency | Casp8 or FADD deficient background. | Compared with Casp8−/−Ripk3−/− or Fadd−/−Ripk3−/− mice, Casp8−/−Mlkl−/− or Fadd−/−Mlkl−/− demonstrate more severe lymphadenopathy and autoimmune manifestations. | Littermates derived from Casp8+/−, Mlkl+/− crosses [5] mice utilised. | [65] |
| Reconstitution of the hematopoietic system | Competitive transplantation assay using myeloablated recipients. | Following myeloablation, Mlkl−/− bone marrow stem cells were able to compete effectively with Wt counterparts for reconstitution of the hematopoietic system (blood, bone marrow and spleen). | Littermate controls. | [5] |
| MlklD139V-induced hematopoietic defects | Whole body homozygosity for constitutively active MlklD139V mutation. | P3 MlklD139V/D139V mice had significant deficits in lymphocyte and platelet counts in comparison to P3 MlklWT/WT and MlklD139V/WT. | Age and sex matched littermate controls. | [40] |
| Myelosuppressive irradiation. | Recovery was delayed in MlklWT/D139V adult mice. | Age and sex matched littermate controls. | [40] | |
| 5-fluorouracil. | Adult MlklD139V/WT mice had delayed recovery of hematopoietic stem and progenitor cells. | Age and sex matched littermate controls. | [40] | |
| Competitive bone marrow transplants. | Bone marrow-derived HSCs from MlklWT/D139V adults and foetal liver-derived HSCs from MlklWT/D139V and MlklD139V/D139V competed poorly or not at all with co-transplanted Wt bone marrow. | Age and sex matched littermate controls. | [40] | |
| Acute thrombocytopaenia | Single dose of anti-platelet serum. | Mlkl−/− mounted a similar recovery to Wt in terms of magnitude and kinetics. | Wt and Mlkl−/− [5] | [72] |
| Hemostasis | Bleed times into 37 °C saline, after 3-mm tail amputations, measured over 10 min. | Mlkl−/− show prolonged bleeding times compared to Wt and yet equivalent total blood loss. | Wt and Mlkl−/− [5] | [72] |
| Reproductive system | ||||
| Male reproductive system aging | Male mice aged 15 months. | Mlkl−/− mice showed reduced body weight, reduced seminal vesicle weight, increased testosterone, increased fertility and fewer empty seminiferous tubules. | Wt and Mlkl−/− (CRISPR-Cas9) bred as separate cohorts. Same housing conditions since birth. | [73] |
| Male mice aged 18 months. | No significant difference in mortality, body weight, testis weight, seminal vesicle weight or germ cell loss in seminiferous tubules between Mlkl−/− mice and Wt mice. | Wt and Mlkl−/− [5] are litter-mate controls. | [74] | |
| Other | ||||
| Ventilator-induced lung injury (VILI) | Mlkl−/− mice were not protected against VILI at low or high tidal volumes. | Littermate Wt used for Mlkl−/− (Jiahuai Han Laboratory) mice. | [75] | |
| Progressive renal fibrosis | Left urethral obstruction (UUO) by double ligation. | Mlkl−/− mice were not protected from renal damage. | Sex-matched male Wt and Mlkl−/− (Jiahuai Han Laboratory) mice. | [76] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tovey Crutchfield, E.C.; Garnish, S.E.; Hildebrand, J.M. The Role of the Key Effector of Necroptotic Cell Death, MLKL, in Mouse Models of Disease. Biomolecules 2021, 11, 803. https://doi.org/10.3390/biom11060803
Tovey Crutchfield EC, Garnish SE, Hildebrand JM. The Role of the Key Effector of Necroptotic Cell Death, MLKL, in Mouse Models of Disease. Biomolecules. 2021; 11(6):803. https://doi.org/10.3390/biom11060803
Chicago/Turabian StyleTovey Crutchfield, Emma C., Sarah E. Garnish, and Joanne M. Hildebrand. 2021. "The Role of the Key Effector of Necroptotic Cell Death, MLKL, in Mouse Models of Disease" Biomolecules 11, no. 6: 803. https://doi.org/10.3390/biom11060803
APA StyleTovey Crutchfield, E. C., Garnish, S. E., & Hildebrand, J. M. (2021). The Role of the Key Effector of Necroptotic Cell Death, MLKL, in Mouse Models of Disease. Biomolecules, 11(6), 803. https://doi.org/10.3390/biom11060803