
Sublingual AKBA Exerts Antidepressant Effects in the Aβ-Treated Mouse Model


 , and
, and 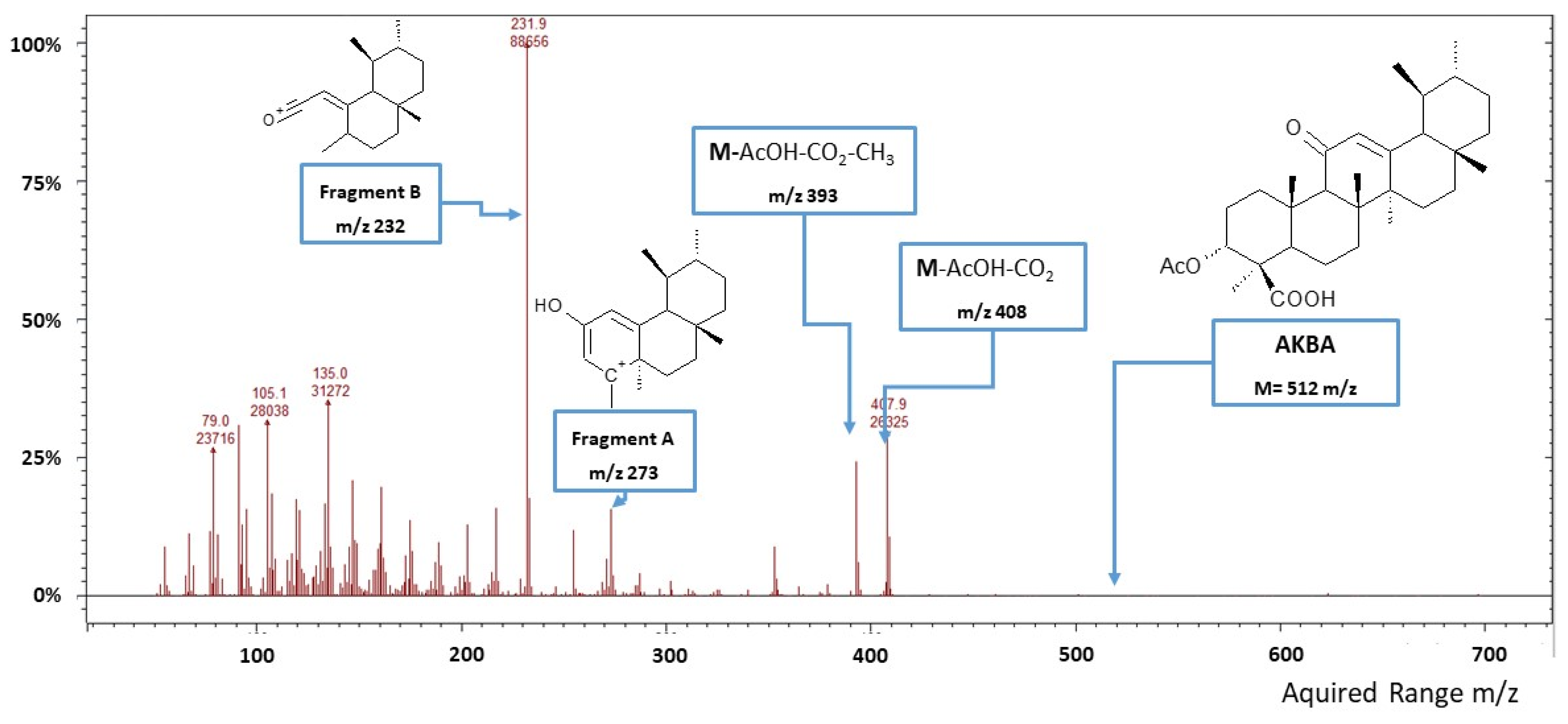{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Surgery and Amyloid Beta Administration
2.3. AKBA Administration and Central Bioavailability Study
AKBA Quantification by GC-MS/IT
2.4. Behavioural Tests
2.4.1. Open Field Test
2.4.2. Splash Test
2.4.3. Tail Suspension Test
2.5. Post-Mortem Tissue Analysis
2.5.1. Neurochemical Quantifications
2.5.2. Western Blotting Quantification
2.6. Statistical Analyses
3. Results
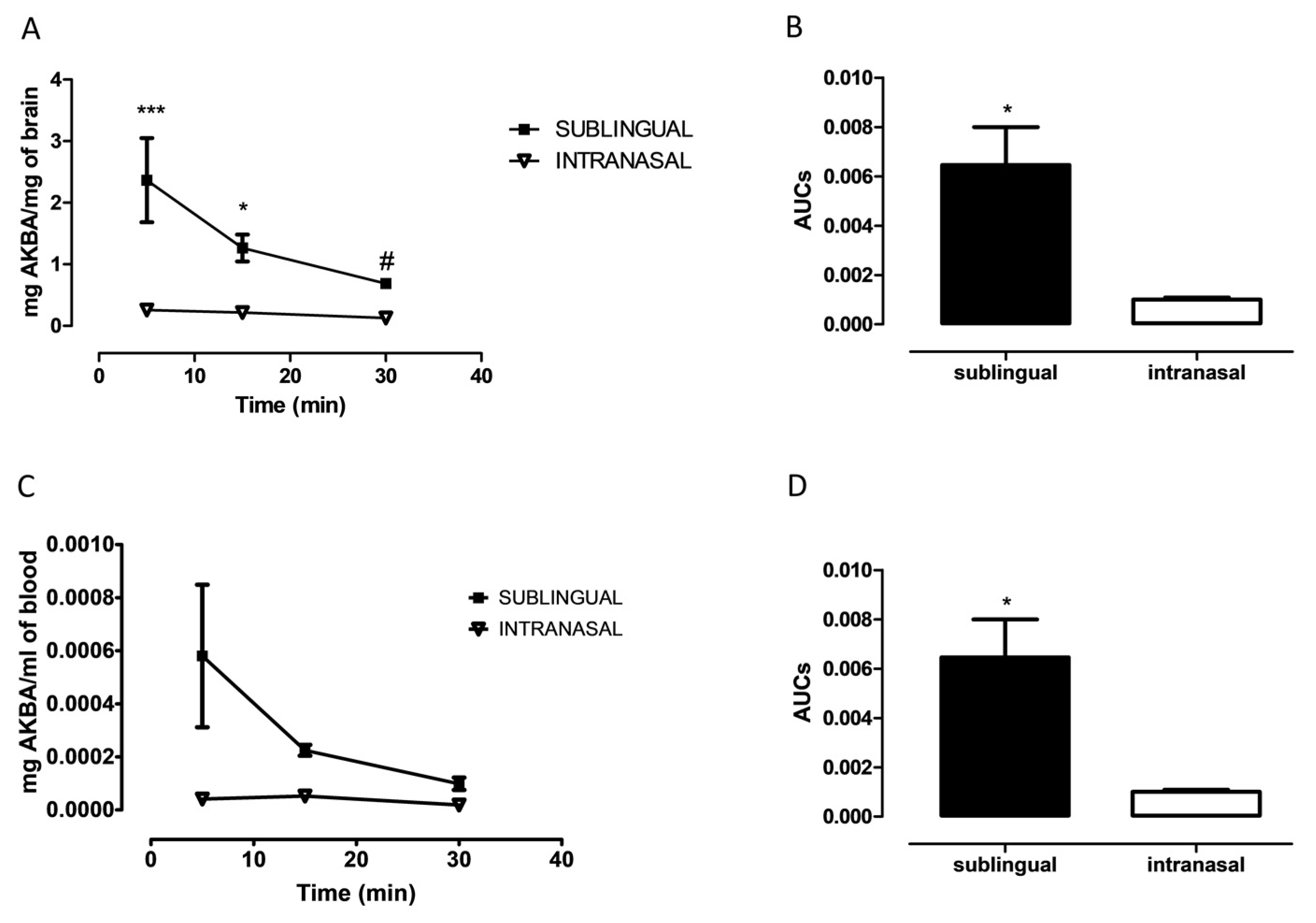
3.1. Evaluation of AKBA in Brain after Sublingual or Intranasal Administration
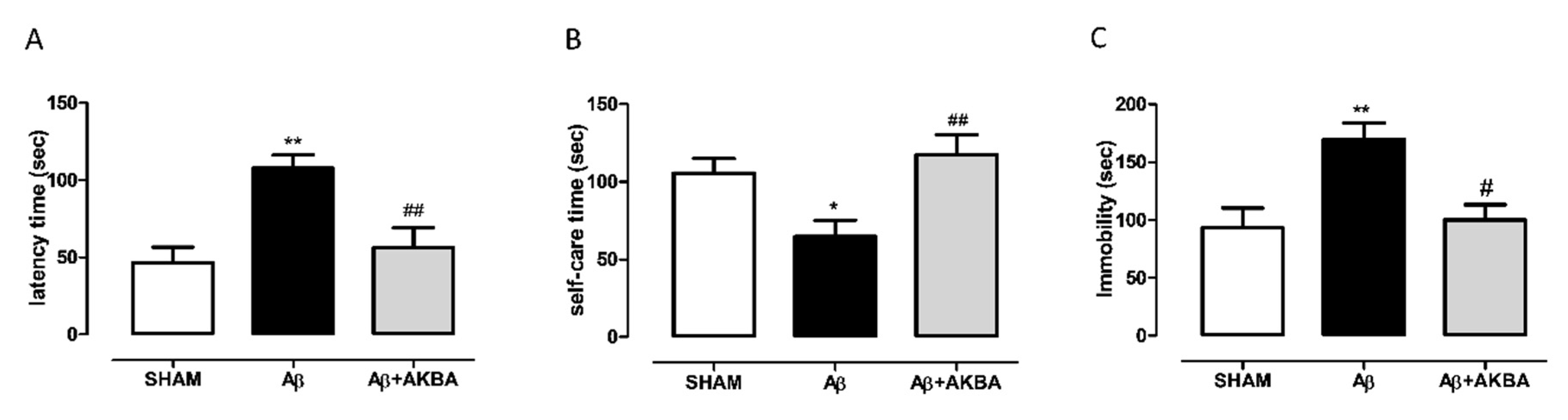
3.2. Antidepressant-Like Effect of Sublingual AKBA in Aβ-Treated Mice
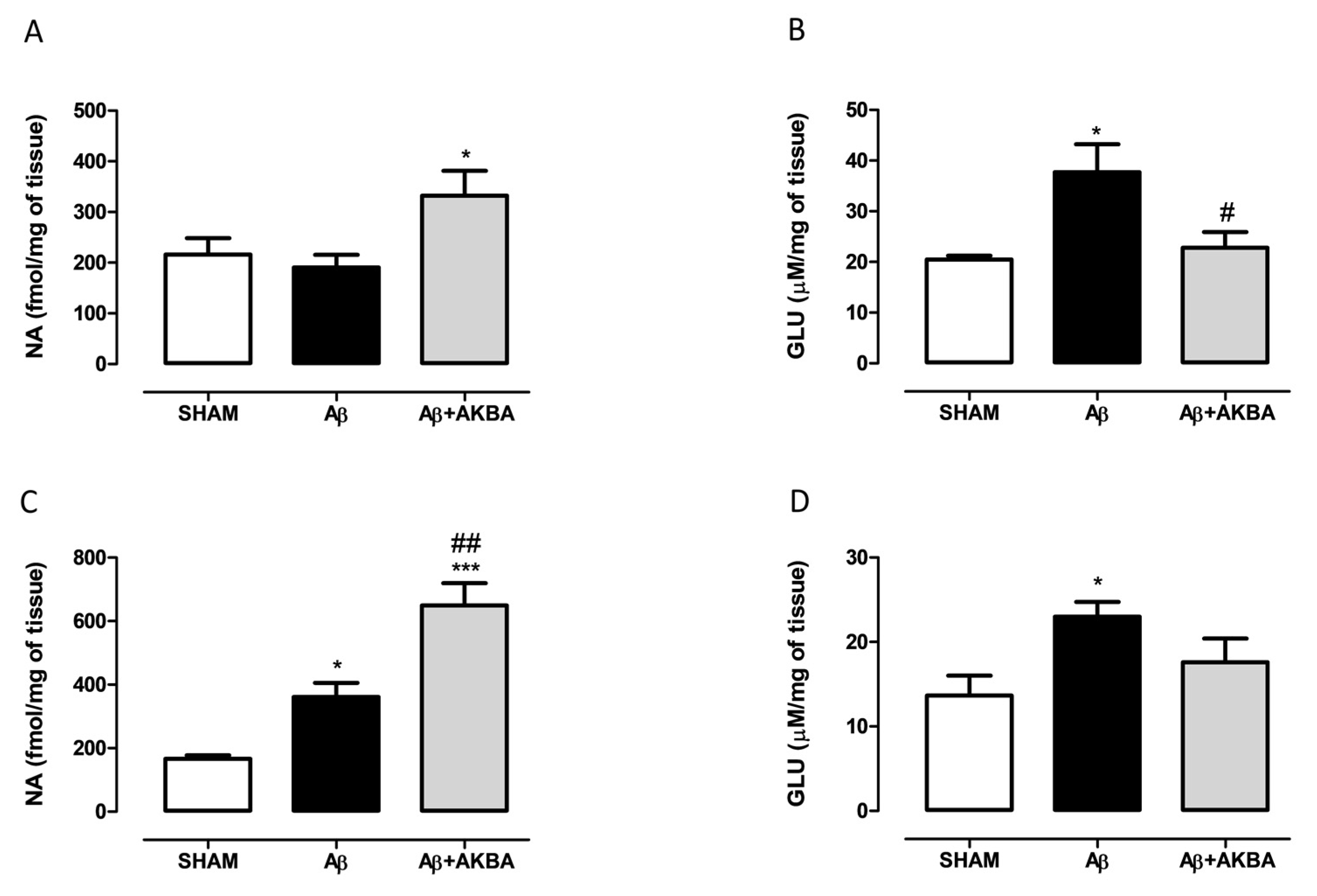
3.3. Effect of Sublingual AKBA on Noradrenaline and Glutamate Content
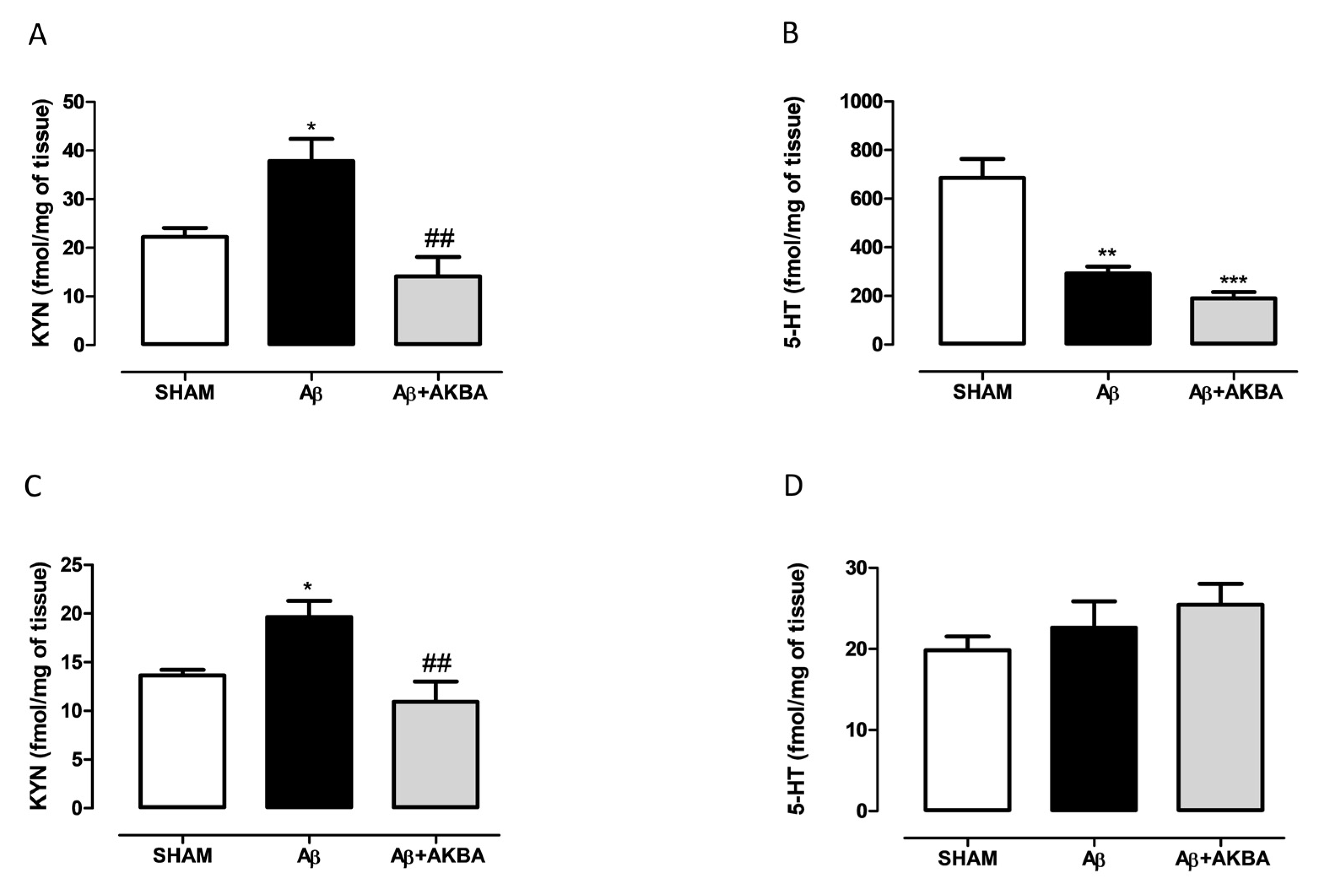
3.4. Effect of Sublingual AKBA on Serotonin and Kynurenine Content
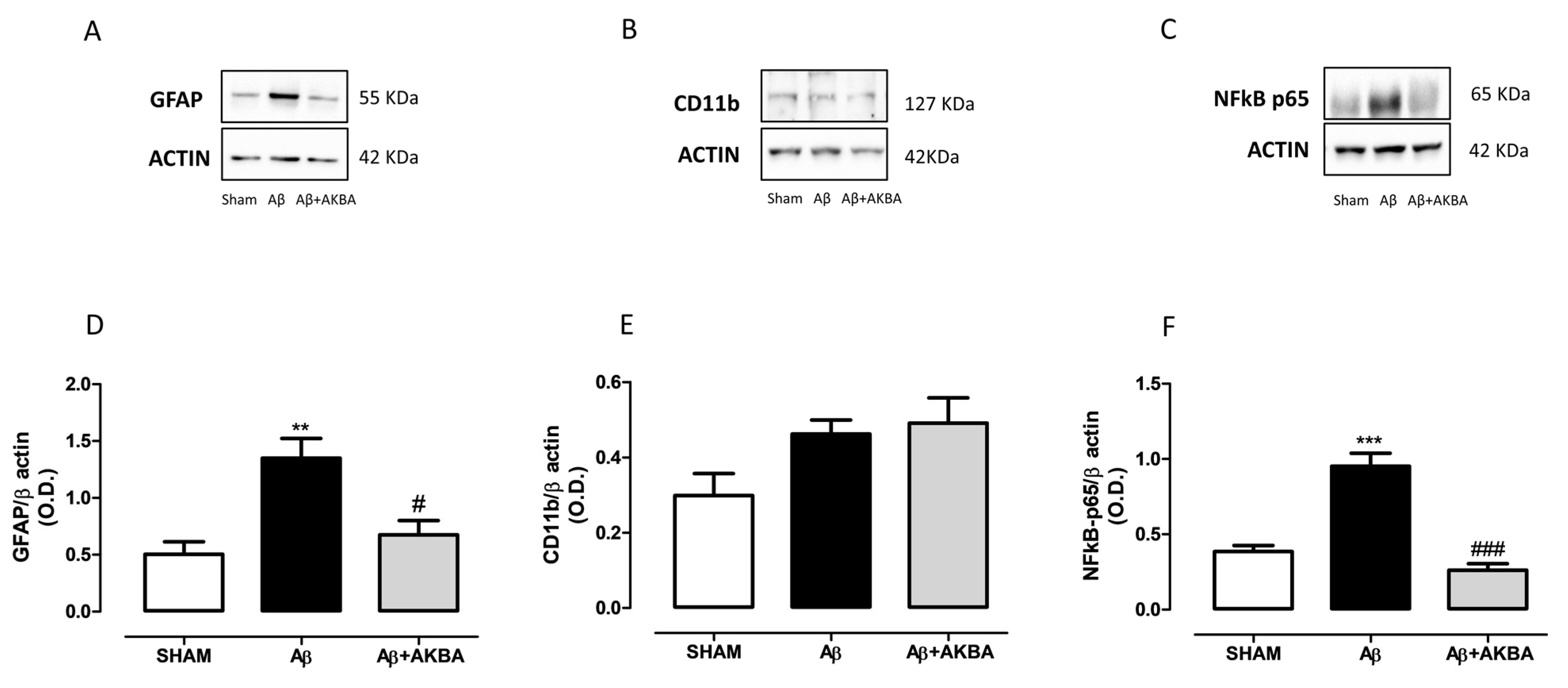
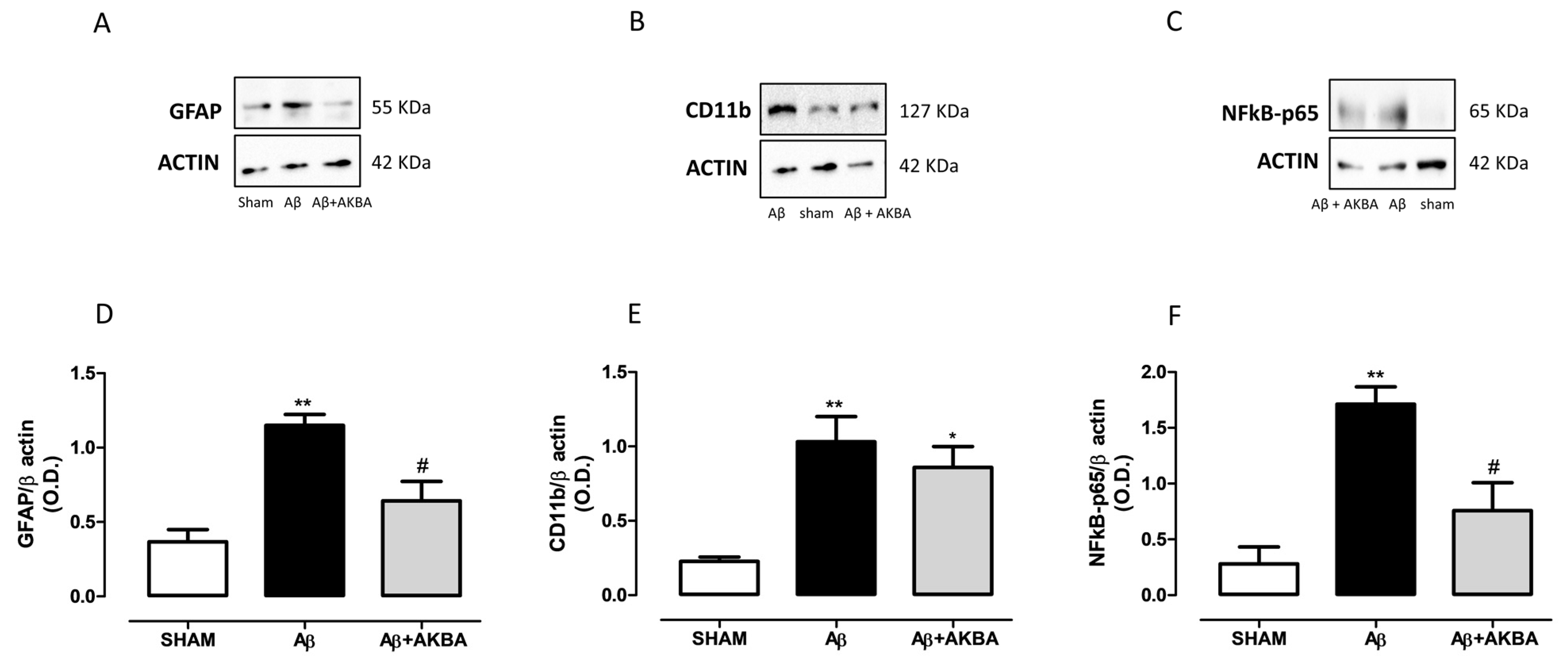
3.5. Effect of Sublingual AKBA on GFAP, CD11b and NFkB Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ammon, H.P. Boswellic Acids and Their Role in Chronic Inflammatory Diseases. Adv. Exp. Med. Biol. 2016, 928, 291–327. [Google Scholar]
- Rajabian, A.; Sadeghnia, H.; Fanoudi, S.; Hosseini, A. Genus Boswellia as a new candidate for neurodegenerative disorders. Iran. J. Basic Med Sci. 2020, 23, 277–286. [Google Scholar]
- Wei, C.; Fan, J.; Sun, X.; Yao, J.; Guo, Y.; Zhou, B.; Shang, Y. Acetyl-11-keto-beta-boswellic acid ameliorates cognitive deficits and reduces amyloid-beta levels in APPswe/PS1dE9 mice through antioxidant and anti-inflammatory pathways. Free Radic. Biol. Med. 2020, 150, 96–108. [Google Scholar] [CrossRef]
- Haghaei, H.; Aref Hosseini, S.R.; Soltani, S.; Fathi, F.; Mokhtari, F.; Karima, S.; Rashidi, M.R. Kinetic and thermodynamic study of beta-Boswellic acid interaction with Tau protein investigated by surface plasmon resonance and molecular modeling methods. Bioimpacts Bi 2020, 10, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, M.I.; den Heijer, T.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. History of depression, depressive symptoms, and medial temporal lobe atrophy and the risk of Alzheimer disease. Neurology 2008, 70, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, M.I.; Schoevers, R.A.; Beekman, A.T.; Jonker, C.; Deeg, D.J.; Schmand, B.; Ader, H.J.; Bouter, L.M.; Van Tilburg, W. Depression and risk of cognitive decline and Alzheimer’s disease. Results of two prospective community-based studies in The Netherlands. Br. J. Psychiatry 2000, 176, 568–575. [Google Scholar] [CrossRef]
- Ledo, J.H.; Azevedo, E.P.; Beckman, D.; Ribeiro, F.C.; Santos, L.E.; Razolli, D.S.; Kincheski, G.C.; Melo, H.M.; Bellio, M.; Teixeira, A.L.; et al. Cross Talk Between Brain Innate Immunity and Serotonin Signaling Underlies Depressive-Like Behavior Induced by Alzheimer’s Amyloid-beta Oligomers in Mice. J. Neurosci. 2016, 36, 12106–12116. [Google Scholar] [CrossRef] [PubMed]
- Colaianna, M.; Tucci, P.; Zotti, M.; Morgese, M.G.; Schiavone, S.; Govoni, S.; Cuomo, V.; Trabace, L. Soluble beta amyloid(1–42): A critical player in producing behavioural and biochemical changes evoking depressive-related state? Br. J. Pharmacol. 2010, 159, 1704–1715. [Google Scholar] [CrossRef]
- Bove, M.; Mhillaj, E.; Tucci, P.; Giardino, I.; Schiavone, S.; Morgese, M.G.; Trabace, L. Effects of n-3 PUFA enriched and n-3 PUFA deficient diets in naïve and Aβ-treated female rats. Biochem. Pharmacol. 2018, 155, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Morgese, M.G.; Schiavone, S.; Bove, M.; Mhillaj, E.; Tucci, P.; Trabace, L. Sub-chronic celecoxib prevents soluble beta amyloid-induced depressive-like behaviour in rats. J. Affect. Disord. 2018, 238, 118–121. [Google Scholar] [CrossRef]
- Bauer, M.E.; Teixeira, A.L. Inflammation in psychiatric disorders: What comes first? Ann. N. Y. Acad. Sci. 2019, 1437, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.; Kiss, A.; Lanctot, K.; Herrmann, N. Depression with inflammation: Longitudinal analysis of a proposed depressive subtype in community dwelling older adults. Int. J. Geriatr. Psychiatry 2017, 32, e18–e24. [Google Scholar] [CrossRef] [PubMed]
- Shahidpour, F.; Mehrjerdi, F.Z.; Mozayan, M.R.; Marefati, N.; Hosseini, M. The effects of frankincense extract on depression and anxiety-like behaviors induced by lipopolysaccharide in rats. Learn. Motiv. 2021, 73, 101708. [Google Scholar] [CrossRef]
- Berkiks, I.; Boulbaroud, S.; Garcia-Segura, L.M.; Mesfioui, A.; Ouichou, A.; Mouden, S.; Benmhammed, H.; El hasnaoui, A.; Nakache, R.; Bahbiti, Y.; et al. Thymelaea lythroides extract attenuates microglial activation and depressive-like behavior in LPS-induced inflammation in adult male rats. Biomed. Pharmacother. 2018, 99, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, M.; Wang, M.; Wang, M.; Zhang, T.; Park, J.; Zhu, Y.; Guo, C.; Jia, Y.; Li, Y.; et al. Neuroprotection by Acetyl-11-Keto-β-Boswellic Acid, in Ischemic Brain Injury Involves the Nrf2/HO-1 defense Pathway. Sci. Rep. 2014, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hiruma, H.; Katakura, T.; Takahashi, S.; Ichikawa, T.; Kawakami, T. Glutamate and Amyloid β-Protein Rapidly Inhibit Fast Axonal Transport in Cultured Rat Hippocampal Neurons by Different Mechanisms. J. Neurosci. 2003, 23, 8967–8977. [Google Scholar] [CrossRef] [PubMed]
- Kopeikina, K.; Hyman, B.; Spires-Jones, T. Soluble forms of tau are toxic in Alzheimer’s disease. Transl. Neurosci. 2012, 3, 223–233. [Google Scholar] [CrossRef]
- Sanacora, G.; Treccani, G.; Popoli, M. Towards a glutamate hypothesis of depression. Neuropharmacology 2012, 62, 63–77. [Google Scholar] [CrossRef]
- Oxenkrug, G. Serotonin-kynurenine hypothesis of depression: Historical overview and recent developments. Curr. Drug Targets 2013, 14, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R. Kynurenines and Glutamate: Multiple Links and Therapeutic Implications. Adv. Pharmacol. 2016, 76, 13–37. [Google Scholar]
- Takada, Y.; Ichikawa, H.; Badmaev, V.; Aggarwal, B.B. Acetyl-11-keto-beta-boswellic acid potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis by suppressing NF-kappa B and NF-kappa B-regulated gene expression. J. Immunol. 2006, 176, 3127–3140. [Google Scholar] [CrossRef]
- Orban, Z.; Mitsiades, N.; Burke, T.R., Jr.; Tsokos, M.; Chrousos, G.P. Caffeic acid phenethyl ester induces leukocyte apoptosis, modulates nuclear factor-kappa B and suppresses acute inflammation. Neuroimmunomodulation 2000, 7, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates; Elsevier Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Abdel-Tawab, M.; Werz, O.; Schubert-Zsilavecz, M. Boswellia serrata: An overall assessment of in vitro, preclinical, pharmacokinetic and clinical data. Clin. Pharmacokinet. 2011, 50, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Lama, A.; Pirozzi, C.; Annunziata, C.; Morgese, M.G.; Senzacqua, M.; Severi, I.; Calignano, A.; Trabace, L.; Giordano, A.; Meli, R.; et al. Palmitoylethanolamide counteracts brain fog improving depressive-like behaviour in obese mice: Possible role of synaptic plasticity and neurogenesis. Br. J. Pharmacol. 2021, 178, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Can, A.; Dao, D.T.; Terrillion, C.E.; Piantadosi, S.C.; Bhat, S.; Gould, T.D. The tail suspension test. J. Vis. Exp. Jove 2012, 59, e3769. [Google Scholar] [CrossRef] [PubMed]
- Morgese, M.G.; Colaianna, M.; Mhillaj, E.; Zotti, M.; Schiavone, S.; D’Antonio, P.; Harkin, A.; Gigliucci, V.; Campolongo, P.; Trezza, V.; et al. Soluble beta amyloid evokes alteration in brain norepinephrine levels: Role of nitric oxide and interleukin-1. Front. Neurosci. 2015, 9, 428. [Google Scholar] [CrossRef]
- Morgese, M.G.; Mhillaj, E.; Francavilla, M.; Bove, M.; Morgano, L.; Tucci, P.; Trabace, L.; Schiavone, S. Chlorella sorokiniana Extract Improves Short-Term Memory in Rats. Molecules 2016, 21, 1311. [Google Scholar] [CrossRef]
- Francavilla, M.; Colaianna, M.; Zotti, M.; Morgese, M.G.; Trotta, P.; Tucci, P.; Schiavone, S.; Cuomo, V.; Trabace, L. Extraction, characterization and in vivo neuromodulatory activity of phytosterols from microalga Dunaliella tertiolecta. Curr. Med. Chem. 2012, 19, 3058–3067. [Google Scholar] [CrossRef]
- Bove, M.; Ike, K.; Eldering, A.; Buwalda, B.; de Boer, S.F.; Morgese, M.G.; Schiavone, S.; Cuomo, V.; Trabace, L.; Kas, M.J.H. The Visible Burrow System: A behavioral paradigm to assess sociability and social withdrawal in BTBR and C57BL/6J mice strains. Behav. Brain Res. 2018, 344, 9–19. [Google Scholar] [CrossRef]
- Schiavone, S.; Mhillaj, E.; Neri, M.; Morgese, M.G.; Tucci, P.; Bove, M.; Valentino, M.; Di Giovanni, G.; Pomara, C.; Turillazzi, E.; et al. Early Loss of Blood-Brain Barrier Integrity Precedes NOX2 Elevation in the Prefrontal Cortex of an Animal Model of Psychosis. Mol. Neurobiol. 2017, 54, 2031–2044. [Google Scholar] [CrossRef]
- Krüger, P.; Kanzer, J.; Hummel, J.; Fricker, G.; Schubert-Zsilavecz, M.; Abdel-Tawab, M. Permeation of Boswellia extract in the Caco-2 model and possible interactions of its constituents KBA and AKBA with OATP1B3 and MRP2. Eur. J. Pharm. Sci. 2009, 36, 275–284. [Google Scholar] [CrossRef]
- Sterk, V.; Buchele, B.; Simmet, T. Effect of food intake on the bioavailability of boswellic acids from a herbal preparation in healthy volunteers. Planta Med. 2004, 70, 1155–1160. [Google Scholar] [CrossRef]
- Del Bo, C.; Riso, P.; Gardana, C.; Brusamolino, A.; Battezzati, A.; Ciappellano, S. Effect of two different sublingual dosages of vitamin B12 on cobalamin nutritional status in vegans and vegetarians with a marginal deficiency: A randomized controlled trial. Clin. Nutr. 2019, 38, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Karolewicz, B.; Paul, I.A. Group housing of mice increases immobility and antidepressant sensitivity in the forced swim and tail suspension tests. Eur. J. Pharmacol. 2001, 415, 197–201. [Google Scholar] [CrossRef]
- Crowley, J.J.; Blendy, J.A.; Lucki, I. Strain-dependent antidepressant-like effects of citalopram in the mouse tail suspension test. Psychopharmacology 2005, 183, 257–264. [Google Scholar] [CrossRef]
- Morgese, M.G.; Schiavone, S.; Maffione, A.B.; Tucci, P.; Trabace, L. Depressive-like phenotype evoked by lifelong nutritional omega-3 deficiency in female rats: Crosstalk among kynurenine, Toll-like receptors and amyloid beta oligomers. BrainBehav. Immun. 2020, 87, 444–454. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Maragos, W.F.; Albin, R.L.; Penney, J.B.; Young, A.B. Glutamate transmission and toxicity in Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1988, 12, 421–430. [Google Scholar] [CrossRef]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.F.; de Kort, G.J.; Steggerda, S.; Shigemoto, R.; Ribeiro-da-Silva, A.; Cuello, A.C. Structural involvement of the glutamatergic presynaptic boutons in a transgenic mouse model expressing early onset amyloid pathology. Neurosci. Lett. 2003, 353, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.F.; Claudio Cuello, A. Altered synaptic function in Alzheimer’s disease. Eur. J. Pharmacol. 2006, 545, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, S.; Takamiya, A.; Noda, Y.; Horita, N.; Wada, M.; Tsugawa, S.; Plitman, E.; Sano, Y.; Tarumi, R.; ElSalhy, M.; et al. Glutamatergic neurometabolite levels in major depressive disorder: A systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol. Psychiatry 2019, 24, 952–964. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, S.; Tucci, P.; Mhillaj, E.; Bove, M.; Trabace, L.; Morgese, M.G. Antidepressant drugs for beta amyloid-induced depression: A new standpoint? Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 78, 114–122. [Google Scholar] [CrossRef]
- Bishnoi, M.; Patil, C.S.; Kumar, A.; Kulkarni, S.K. Co-administration of acetyl-11-keto-beta-boswellic acid, a specific 5-lipoxygenase inhibitor, potentiates the protective effect of COX-2 inhibitors in kainic acid-induced neurotoxicity in mice. Pharmacology 2007, 79, 34–41. [Google Scholar] [CrossRef]
- Santamaria, A.; Galvan-Arzate, S.; Lisy, V.; Ali, S.F.; Duhart, H.M.; Osorio-Rico, L.; Rios, C.; St’astny, F. Quinolinic acid induces oxidative stress in rat brain synaptosomes. Neuroreport 2001, 12, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitron, R.; Blanco-Ayala, T.; Ugalde-Muniz, P.; Carrillo-Mora, P.; Pedraza-Chaverri, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzon, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Rajabian, A.; Boroushaki, M.T.; Hayatdavoudi, P.; Sadeghnia, H.R. Boswellia serrata Protects Against Glutamate-Induced Oxidative Stress and Apoptosis in PC12 and N2a Cells. Dna Cell Biol. 2016, 35, 666–679. [Google Scholar] [CrossRef]
- Mhillaj, E.; Morgese, M.G.; Tucci, P.; Furiano, A.; Luongo, L.; Bove, M.; Maione, S.; Cuomo, V.; Schiavone, S.; Trabace, L. Celecoxib Prevents Cognitive Impairment and Neuroinflammation in Soluble Amyloid beta-treated Rats. Neuroscience 2018, 372, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Morgese, M.G.; Schiavone, S.; Mhillaj, E.; Bove, M.; Tucci, P.; Trabace, L. N-3 PUFA diet enrichment prevents amyloid beta-induced depressive-like phenotype. Pharmacol. Res. 2018, 129, 526–534. [Google Scholar] [CrossRef]
- López-Gil, X.; Jiménez-Sánchez, L.; Campa, L.; Castro, E.; Frago, C.; Adell, A. Role of Serotonin and Noradrenaline in the Rapid Antidepressant Action of Ketamine. ACS Chem. Neurosci. 2019, 10, 3318–3326. [Google Scholar] [CrossRef]
- Braun, D.; Madrigal, J.L.; Feinstein, D.L. Noradrenergic regulation of glial activation: Molecular mechanisms and therapeutic implications. Curr. Neuropharmacol. 2014, 12, 342–352. [Google Scholar] [CrossRef]
- Counts, S.E.; Mufson, E.J. Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J. Neurochem. 2010, 113, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, K.; Weinshenker, D. Norepinephrine Protects against Amyloid-beta Toxicity via TrkB. J. Alzheimers Dis. 2015, 44, 251–260. [Google Scholar] [CrossRef]
- Noda, M.; Nakanishi, H.; Akaike, N. Glutamate release from microglia via glutamate transporter is enhanced by amyloid-beta peptide. Neuroscience 1999, 92, 1465–1474. [Google Scholar] [CrossRef]
- Martin-Hernandez, D.; Tendilla-Beltran, H.; Madrigal, J.L.M.; Garcia-Bueno, B.; Leza, J.C.; Caso, J.R. Chronic Mild Stress Alters Kynurenine Pathways Changing the Glutamate Neurotransmission in Frontal Cortex of Rats. Mol. Neurobiol. 2019, 56, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Lattke, M.; Reichel, S.N.; Baumann, B. NF-kappaB-mediated astrocyte dysfunction initiates neurodegeneration. Oncotarget 2017, 8, 50329–50330. [Google Scholar] [CrossRef]
- Ghosh, M.; Yang, Y.; Rothstein, J.D.; Robinson, M.B. Nuclear factor-kappaB contributes to neuron-dependent induction of glutamate transporter-1 expression in astrocytes. J. Neurosci. 2011, 31, 9159–9169. [Google Scholar] [CrossRef]
- Du, Y.; Chen, X.; Wei, X.; Bales, K.R.; Berg, D.T.; Paul, S.M.; Farlow, M.R.; Maloney, B.; Ge, Y.W.; Lahiri, D.K. NF-(kappa)B mediates amyloid beta peptide-stimulated activity of the human apolipoprotein E gene promoter in human astroglial cells. Brain Res. Mol. Brain Res. 2005, 136, 177–188. [Google Scholar] [CrossRef]
- Carrero, I.; Gonzalo, M.R.; Martin, B.; Sanz-Anquela, J.M.; Arevalo-Serrano, J.; Gonzalo-Ruiz, A. Oligomers of beta-amyloid protein (Abeta1-42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp. Neurol. 2012, 236, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Sailer, E.R.; Schweizer, S.; Boden, S.E.; Ammon, H.P.; Safayhi, H. Characterization of an acetyl-11-keto-beta-boswellic acid and arachidonate-binding regulatory site of 5-lipoxygenase using photoaffinity labeling. Eur. J. Biochem. FEBS 1998, 256, 364–368. [Google Scholar] [CrossRef]
- Gelosa, P.; Colazzo, F.; Tremoli, E.; Sironi, L.; Castiglioni, L. Cysteinyl Leukotrienes as Potential Pharmacological Targets for Cerebral Diseases. Mediat. Inflamm. 2017, 2017, 3454212. [Google Scholar] [CrossRef]
- Ikonomovic, M.D.; Abrahamson, E.E.; Uz, T.; Manev, H.; Dekosky, S.T. Increased 5-lipoxygenase immunoreactivity in the hippocampus of patients with Alzheimer’s disease. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2008, 56, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Firuzi, O.; Zhuo, J.; Chinnici, C.M.; Wisniewski, T.; Pratico, D. 5-Lipoxygenase gene disruption reduces amyloid-beta pathology in a mouse model of Alzheimer’s disease. Faseb J. 2008, 22, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Giannopoulos, P.F.; Ceballos-Diaz, C.; Golde, T.E.; Pratico, D. Adeno-associated virus-mediated brain delivery of 5-lipoxygenase modulates the AD-like phenotype of APP mice. Mol. Neurodegener. 2012, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Hu, M.; Wang, H.; Hu, M.; Long, Y.; Miao, M.X.; Li, J.C.; Wang, X.B.; Kong, L.Y.; Hong, H. Montelukast targeting the cysteinyl leukotriene receptor 1 ameliorates Abeta1-42-induced memory impairment and neuroinflammatory and apoptotic responses in mice. Neuropharmacology 2014, 79, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Manev, R.; Mrazovac, D.; Manev, H. Possible role for interactions between 5-lipoxygenase (5-LOX) and AMPA GluR1 receptors in depression and in antidepressant therapy. Med. Hypotheses 2007, 69, 1076–1079. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgese, M.G.; Bove, M.; Francavilla, M.; Schiavone, S.; Dimonte, S.; Colia, A.L.; Bevilacqua, M.; Trabace, L.; Tucci, P. Sublingual AKBA Exerts Antidepressant Effects in the Aβ-Treated Mouse Model. Biomolecules 2021, 11, 686. https://doi.org/10.3390/biom11050686
Morgese MG, Bove M, Francavilla M, Schiavone S, Dimonte S, Colia AL, Bevilacqua M, Trabace L, Tucci P. Sublingual AKBA Exerts Antidepressant Effects in the Aβ-Treated Mouse Model. Biomolecules. 2021; 11(5):686. https://doi.org/10.3390/biom11050686
Chicago/Turabian StyleMorgese, Maria Grazia, Maria Bove, Matteo Francavilla, Stefania Schiavone, Stefania Dimonte, Anna Laura Colia, Matteo Bevilacqua, Luigia Trabace, and Paolo Tucci. 2021. "Sublingual AKBA Exerts Antidepressant Effects in the Aβ-Treated Mouse Model" Biomolecules 11, no. 5: 686. https://doi.org/10.3390/biom11050686
APA StyleMorgese, M. G., Bove, M., Francavilla, M., Schiavone, S., Dimonte, S., Colia, A. L., Bevilacqua, M., Trabace, L., & Tucci, P. (2021). Sublingual AKBA Exerts Antidepressant Effects in the Aβ-Treated Mouse Model. Biomolecules, 11(5), 686. https://doi.org/10.3390/biom11050686