
Retinal Dysfunction in Alzheimer’s Disease and Implications for Biomarkers


Abstract
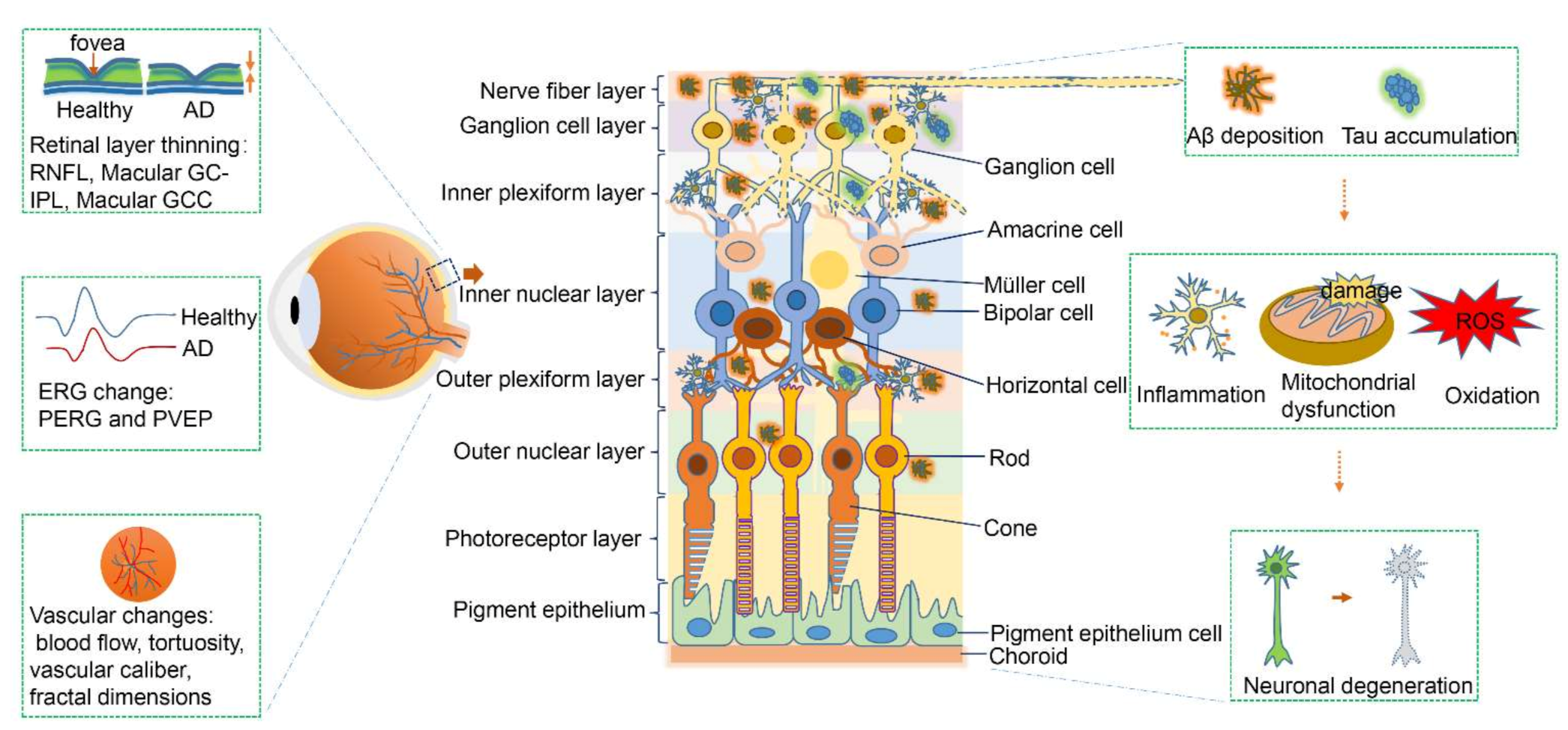
1. Introduction
2. Retinal Changes in Alzheimer’s Disease and Implications for Biomarkers
2.1. Structural Degeneration of the Retina in Early-Stage Alzheimer’s Disease
2.2. Electrophysiological Changes of the Retina in Early-Stage Alzheimer’s Disease
2.3. Amyloid-Beta Deposition in the Retina in Alzheimer’s Disease
2.4. Hyperphosphorylated Tau Aggregation
2.5. Blood Vessels and Retinal Microcirculation
2.6. Apoptosis in the Retina in Alzheimer’s Disease
3. Alzheimer’s Disease and Other Retinal Diseases
3.1. Retinal Dysfunction in Age-Related Macular Degeneration
3.2. Retinal Dysfunction in Glaucoma
3.3. Strategies to Distinguish Alzheimer’s Disease from Age-Related Macular Degeneration and Glaucoma
4. Future Directions and Challenges
Funding
Acknowledgments
Conflicts of Interest
References
- Goedert, M.; Spillantini, M.G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Albert, M.S.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carrillo, M.C.; Thies, B.; Phelps, C.H. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef]
- Gupta, V.B.; Chitranshi, N.; den Haan, J.; Mirzaei, M.; You, Y.; Lim, J.K.; Basavarajappa, D.; Godinez, A.; Di Angelantonio, S.; Sachdev, P.; et al. Retinal changes in Alzheimer’s disease- integrated prospects of imaging, functional and molecular advances. Prog. Retin. Eye Res. 2020, 82, 100899. [Google Scholar] [CrossRef] [PubMed]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.; Mikulskis, A.; et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef]
- Bacioglu, M.; Maia, L.F.; Preische, O.; Schelle, J.; Apel, A.; Kaeser, S.A.; Schweighauser, M.; Eninger, T.; Lambert, M.; Pilotto, A.; et al. Neurofilament Light Chain in Blood and CSF as Marker of Disease Progression in Mouse Models and in Neurodegenerative Diseases. Neuron 2016, 91, 56–66. [Google Scholar] [CrossRef]
- Mattsson, N.; Schöll, M.; Strandberg, O.; Smith, R.; Palmqvist, S.; Insel, P.S.; Hägerström, D.; Ohlsson, T.; Zetterberg, H.; Jögi, J.; et al. 18 F-AV-1451 and CSF T-tau and P-tau as biomarkers in Alzheimer’s disease. EMBO Mol. Med. 2017, 9, 1212–1223. [Google Scholar] [CrossRef]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-tau181 in Alzheimer’s disease: Relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer’s dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef]
- Jindal, V. Interconnection Between Brain and Retinal Neurodegenerations. Mol. Neurobiol. 2014, 51, 885–892. [Google Scholar] [CrossRef]
- Barnstable, C.J. A molecular view of vertebrate retinal development. Mol. Neurobiol. 1987, 1, 9–46. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia–neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef]
- Kolb, H. How the retina works—Much of the construction of an image takes place in the retina itself through the use of specialized neural circuits. Am. Sci. 2003, 91, 28–35. [Google Scholar] [CrossRef]
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain—from eye research to CNS disorders. Nat. Rev. Neurol. 2012, 9, 44–53. [Google Scholar] [CrossRef]
- Alber, J.; Goldfarb, D.; Thompson, L.I.; Arthur, E.; Hernandez, K.; Cheng, D.; DeBuc, D.C.; Cordeiro, F.; Provetti-Cunha, L.; Haan, J.D.; et al. Developing retinal biomarkers for the earliest stages of Alzheimer’s disease: What we know, what we don’t, and how to move forward. Alzheimer’s Dement. 2020, 16, 229–243. [Google Scholar] [CrossRef]
- Snyder, P.J.; Johnson, L.N.; Lim, Y.Y.; Santos, C.Y.; Alber, J.; Maruff, P.; Fernández, B. Nonvascular retinal imaging markers of preclinical Alzheimer’s disease. Alzheimer’s Dement. 2016, 4, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Czakó, C.; Kovács, T.; Ungvari, Z.; Csiszar, A.; Yabluchanskiy, A.; Conley, S.; Csipo, T.; Lipecz, A.; Horváth, H.; Sándor, G.L.; et al. Retinal biomarkers for Alzheimer’s disease and vascular cognitive impairment and dementia (VCID): Implication for early diagnosis and prognosis. GeroScience 2020, 42, 1499–1525. [Google Scholar] [CrossRef]
- Townes-Anderson, E.; Vogt, B.A. Distribution of muscarinic acetylcholine receptors on processes of isolated retinal cells. J. Comp. Neurol. 1989, 290, 369–383. [Google Scholar] [CrossRef]
- Krasodomska, K.; Lubiński, W.; Potemkowski, A.; Honczarenko, K. Pattern electroretinogram (PERG) and pattern visual evoked potential (PVEP) in the early stages of Alzheimer’s disease. Doc. Ophthalmol. 2010, 121, 111–121. [Google Scholar] [CrossRef]
- Ngoo, Q.Z.; Hitam, W.H.W.; Ab Razak, A. Evaluation of Retinal Nerve Fiber Layer Thickness, Electroretinogram and Visual Evoked Potential in Patients with Alzheimer’s Disease. J. Ophthalmol. 2019, 2019, 16248185. [Google Scholar] [CrossRef] [PubMed]
- Hinton, D.R.; Sadun, A.A.; Blanks, J.C.; Miller, C.A. Optic-Nerve Degeneration in Alzheimer’s Disease. N. Engl. J. Med. 1986, 315, 485–487. [Google Scholar] [CrossRef]
- Blanks, J.C.; Hinton, D.R.; Sadun, A.A.; Miller, C.A. Retinal ganglion cell degeneration in Alzheimer’s disease. Brain Res. 1989, 501, 364–372. [Google Scholar] [CrossRef]
- Coppola, G.; di Renzo, A.; Ziccardi, L.; Martelli, F.; Fadda, A.; Manni, G.; Barboni, P.; Pierelli, F.; Sadun, A.A.; Parisi, V. Optical Coherence Tomography in Alzheimer’s Disease: A Meta-Analysis. PLoS ONE 2015, 10, e0134750. [Google Scholar] [CrossRef]
- Danesh-Meyer, H.V.; Birch, H.; Ku, J.Y.-F.; Carroll, S.; Gamble, G. Reduction of optic nerve fibers in patients with Alzheimer disease identified by laser imaging. Neurology 2006, 67, 1852–1854. [Google Scholar] [CrossRef]
- Berisha, F.; Feke, G.T.; Trempe, C.L.; McMeel, J.W.; Schepens, C.L. Retinal Abnormalities in Early Alzheimer’s Disease. Investig. Opthalmol. Vis. Sci. 2007, 48, 2285–2289. [Google Scholar] [CrossRef]
- Gao, L.; Liu, Y.; Li, X.; Bai, Q.; Liu, P. Abnormal retinal nerve fiber layer thickness and macula lutea in patients with mild cognitive impairment and Alzheimer’s disease. Arch. Gerontol. Geriatr. 2015, 60, 162–167. [Google Scholar] [CrossRef]
- Paquet, C.; Boissonnot, M.; Roger, F.; Dighiero, P.; Gil, R.; Hugon, J. Abnormal retinal thickness in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci. Lett. 2007, 420, 97–99. [Google Scholar] [CrossRef]
- Kesler, A.; Vakhapova, V.; Korczyn, A.D.; Naftaliev, E.; Neudorfer, M. Retinal thickness in patients with mild cognitive impairment and Alzheimer’s disease. Clin. Neurol. Neurosurg. 2011, 113, 523–526. [Google Scholar] [CrossRef]
- Blanks, J.C.; Torigoe, Y.; Hinton, D.R.; Blanks, R.H. Retinal pathology in Alzheimer’s disease. I. Ganglion cell loss in foveal/parafoveal retina. Neurobiol. Aging 1996, 17, 377–384. [Google Scholar] [CrossRef]
- Blanks, J.C.; Schmidt, S.Y.; Torigoe, Y.; Porrello, K.V.; Hinton, D.R.; Blanks, R.H. Retinal pathology in Alzheimer’s disease. II. Regional neuron loss and glial changes in GCL. Neurobiol. Aging 1996, 17, 385–395. [Google Scholar] [CrossRef]
- Cheung, C.Y.-L.; Ong, Y.T.; Hilal, S.; Ikram, M.K.; Low, S.; Venketasubramanian, N.; Yap, P.; Seow, D.; Chen, C.L.H.; Wong, T.Y. Retinal Ganglion Cell Analysis Using High-Definition Optical Coherence Tomography in Patients with Mild Cognitive Impairment and Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 45–56. [Google Scholar] [CrossRef]
- López-De-Eguileta, A.; Lage, C.; López-García, S.; Pozueta, A.; García-Martínez, M.; Kazimierczak, M.; Bravo, M.; De Arcocha-Torres, M.; Banzo, I.; Jimenez-Bonilla, J.; et al. Ganglion cell layer thinning in prodromal Alzheimer’s disease defined by amyloid PET. Alzheimer’s Dement. 2019, 5, 570–578. [Google Scholar] [CrossRef]
- Chan, V.T.; Sun, Z.; Tang, S.; Chen, L.J.; Wong, A.; Tham, C.Y.C.; Wong, T.Y.; Chen, C.; Ikram, M.K.; Whitson, H.E.; et al. Spectral-Domain OCT Measurements in Alzheimer’s Disease. Ophthalmology 2018, 126, 497–510. [Google Scholar] [CrossRef]
- Bayhan, H.A.; Bayhan, S.A.; Celikbilek, A.; Tanık, N.; Gürdal, C. Evaluation of the chorioretinal thickness changes in Alzheimer’s disease using spectral-domain optical coherence tomography. Clin. Exp. Ophthalmol. 2014, 43, 145–151. [Google Scholar] [CrossRef]
- Van De Kreeke, J.A.; Nguyen, H.; Haan, J.D.; Konijnenberg, E.; Tomassen, J.; Braber, A.D.; Kate, M.T.; Collij, L.; Yaqub, M.; Van Berckel, B.; et al. Retinal layer thickness in preclinical Alzheimer’s disease. Acta Ophthalmol. 2019, 97, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Pillai, J.A.; Bermel, R.; Bonner-Jackson, A.; Rae-Grant, A.; Fernandez, H.; Bena, J.; Jones, S.E.; Ehlers, J.P.; Leverenz, J.B. Retinal Nerve Fiber Layer Thinning in Alzheimer’s Disease: A Case-Control Study in Comparison to Normal Aging, Parkinson’s Disease, and Non-Alzheimer’s Dementia. Am. J. Alzheimer’s Dis. Other Dement. 2016, 31, 430–436. [Google Scholar] [CrossRef]
- Strack, R. Improving optical coherence tomography. Nat. Methods 2019, 16, 957. [Google Scholar] [CrossRef]
- Martínez-Lapiscina, E.H.; Arnow, S.; Wilson, J.A.; Saidha, S.; Preiningerova, J.L.; Oberwahrenbrock, T.; Brandt, A.U.; Pablo, L.E.; Guerrieri, S.; González-Suárez, I.; et al. Retinal thickness measured with optical coherence tomography and risk of disability worsening in multiple sclerosis: A cohort study. Lancet Neurol. 2016, 15, 574–584. [Google Scholar] [CrossRef]
- Lamirel, C.; Newman, N.; Biousse, V. The use of optical coherence tomography in neurology. Rev. Neurol. Dis. 2009, 6, E105–E120. [Google Scholar] [PubMed]
- Doustar, J.; Torbati, T.; Black, K.L.; Koronyo, Y.; Koronyo-Hamaoui, M. Optical Coherence Tomography in Alzheimer’s Disease and Other Neurodegenerative Diseases. Front. Neurol. 2017, 8, 701. [Google Scholar] [CrossRef]
- Garcia-Martin, E.S.; Rojas, B.; Ramírez, A.I.; De Hoz, R.; Salazar, J.J.; Yubero, R.; Gil, P.; Triviño, A.; Ramirez, J.M. Macular Thickness as a Potential Biomarker of Mild Alzheimer’s Disease. Ophthalmology 2014, 121, 1149–1151.e3. [Google Scholar] [CrossRef]
- Curcio, C.A.; Allen, K.A. Topography of ganglion cells in human retina. J. Comp. Neurol. 1990, 300, 5–25. [Google Scholar] [CrossRef]
- Williams, P.A.; Thirgood, R.A.; Oliphant, H.; Frizzati, A.; Littlewood, E.; Votruba, M.; Good, M.A.; Williams, J.; Morgan, J.E. Retinal ganglion cell dendritic degeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1799–1806. [Google Scholar] [CrossRef]
- Sadun, A.A.; Borchert, M.; DeVita, E.; Hinton, D.R.; Bassi, C.J. Assessment of Visual Impairment in Patients With Alzheimer’s Disease. Am. J. Ophthalmol. 1987, 104, 113–120. [Google Scholar] [CrossRef]
- Kirby, E.; Bandelow, S.; Hogervorst, E. Visual Impairment in Alzheimer’s Disease: A Critical Review. J. Alzheimer’s Dis. 2010, 21, 15–34. [Google Scholar] [CrossRef]
- Asanad, S.; Karanjia, R. Pattern Electroretinogram; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Parisi, V.; Restuccia, R.; Fattapposta, F.; Mina, C.; Bucci, M.G.; Pierelli, F. Morphological and functional retinal impairment in Alzheimer’s disease patients. Clin. Neurophysiol. 2001, 112, 1860–1867. [Google Scholar] [CrossRef]
- Fiorentini, A.; Maffei, L.; Pirchio, M.; Spinelli, D.; Porciatti, V. The ERG in response to alternating gratings in patients with diseases of the peripheral visual pathway. Investig. Ophthalmol. Vis. Sci. 1981, 21, 490–493. [Google Scholar]
- Parisi, V. Correlation between morphological and functional retinal impairment in patients affected by ocular hypertension, glaucoma, demyelinating optic neuritis and Alzheimer’s disease. Semin. Ophthalmol. 2003, 18, 50–57. [Google Scholar] [CrossRef]
- Celesia, G.G.; Kaufman, D.; Cone, S.B. Simultaneous recording of pattern electroretinography and visual evoked potentials in multiple sclerosis. A method to separate demyelination from axonal damage to the optic nerve. Arch. Neurol. 1986, 43, 1247–1252. [Google Scholar] [CrossRef]
- You, Y.; Thie, J.; Klistorner, A.; Gupta, V.K.; Graham, S.L. Normalization of Visual Evoked Potentials Using Underlying Electroencephalogram Levels Improves Amplitude Reproducibility in Rats. Investig. Opthalmol. Vis. Sci. 2012, 53, 1473–1478. [Google Scholar] [CrossRef]
- Rimmer, S.; Iragui, V.; Klauber, M.R.; Katz, B. Retinocortical time exhibits spatial selectivity. Investig. Ophthalmol. Vis. Sci. 1989, 30, 2045–2049. [Google Scholar] [CrossRef]
- Odom, J.V.; Bach, M.; Barber, C.; Brigell, M.; Marmor, M.F.; Tormene, A.P.; Holder, G.E. Visual evoked potentials standard (2004). Doc. Ophthalmol. 2004, 108, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Celesia, G.G.; Kaufman, D. Pattern ERGs and visual evoked potentials in maculopathies and optic nerve diseases. Investig. Ophthalmol. Vis. Sci. 1985, 26, 726–735. [Google Scholar]
- Katz, B.; Rimmer, S.; Iragui, V.; Katzman, R. Abnormal pattern electroretinogram in Alzheimer’s disease: Evidence for retinal ganglion cell degeneration? Ann. Neurol. 1989, 26, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Partanen, J.; Hartikainen, P.; Könönen, M.; Jousmäki, V.; Soininen, H.; Riekkinen, P. Prolonged Latencies of Pattern Reversal Visual Evoked Early Potentials in Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 1994, 8, 250–258. [Google Scholar] [CrossRef]
- Lorenz, R.; Dodt, E.; Heider, W. Pattern electroretinogram peak times as a clinical means of discriminating retinal from optic nerve disease. Doc. Ophthalmol. 1989, 71, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Pollock, V.E.; Schneider, L.S.; Chui, H.C.; Henderson, V.; Zemansky, M.; Sloane, R.B. Visual evoked potentials in dementia: A meta-analysis and empirical study of Alzheimer’s disease patients. Biol. Psychiatry 1989, 25, 1003–1013. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef]
- Wang, H.; Huang, J.-F.; Li, L.; Luo, J.; Chen, D.; Tong, J.-B.; Zeng, L.-P.; Cao, Y.-Q.; Xiang, J.; Luo, X.-G.; et al. BACE1 in the retina: A sensitive biomarker for monitoring early pathological changes in Alzheimer′s disease. Neural Regen. Res. 2016, 11, 447–453. [Google Scholar] [CrossRef]
- Koronyo-Hamaoui, M.; Koronyo, Y.; Ljubimov, A.V.; Miller, C.A.; Ko, M.K.; Black, K.L.; Schwartz, M.; Farkas, D.L. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. NeuroImage 2011, 54, S204–S217. [Google Scholar] [CrossRef]
- Grimaldi, A.; Brighi, C.; Peruzzi, G.; Ragozzino, D.; Bonanni, V.; Limatola, C.; Ruocco, G.; Di Angelantonio, S. Inflammation, neurodegeneration and protein aggregation in the retina as ocular biomarkers for Alzheimer’s disease in the 3xTg-AD mouse model. Cell Death Dis. 2018, 9, 685. [Google Scholar] [CrossRef]
- Alexandrov, P.N.; Pogue, A.; Bhattacharjee, S.; Lukiw, W.J. Retinal amyloid peptides and complement factor H in transgenic models of Alzheimer’s disease. NeuroReport 2011, 22, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, M.; Veys, L.; Lemmens, S.; Hadoux, X.; Gelders, G.; Masin, L.; Serneels, L.; Theunis, J.; Saito, T.; Saido, T.C.; et al. The AppNL-G-F mouse retina is a site for preclinical Alzheimer’s disease diagnosis and research. Acta Neuropathol. Commun. 2021, 9, 6. [Google Scholar] [CrossRef]
- Hart, N.; Koronyo, Y.; Black, K.L.; Koronyo-Hamaoui, M. Ocular indicators of Alzheimer’s: Exploring disease in the retina. Acta Neuropathol. 2016, 132, 767–787. [Google Scholar] [CrossRef]
- Koronyo, Y.; Biggs, D.; Barron, E.; Boyer, D.S.; Pearlman, J.A.; Au, W.J.; Kile, S.J.; Blanco, A.; Fuchs, D.-T.; Ashfaq, A.; et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight 2017, 2, e93621. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Yang, L.-B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.-L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Lindholm, K.; Yang, L.-B.; Yue, X.; Citron, M.; Yan, R.; Beach, T.; Sue, L.; Sabbagh, M.; Cai, H.; et al. Amyloid peptide load is correlated with increased -secretase activity in sporadic Alzheimer’s disease patients. Proc. Natl. Acad. Sci. USA 2004, 101, 3632–3637. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; He, P.; Lee, T.; Yao, H.; Li, R.; Shen, Y. High Activities of BACE1 in Brains with Mild Cognitive Impairment. Am. J. Pathol. 2014, 184, 141–147. [Google Scholar] [CrossRef]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and Aβ. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.T.; Bruno, M.A.; Ducatenzeiler, A.; Klein, W.L.; Cuello, C. Intracellular Aβ-oligomers and early inflammation in a model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Amyloid-β–induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of Amyloid beta-Protein: Synaptic and Network Dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Pushpitha, K.; Joseph, C.; Gupta, V.; Rajput, R.; Chitranshi, N.; Dheer, Y.; Amirkhani, A.; Kamath, K.; Pascovici, D.; et al. Amyloid β Induces Early Changes in the Ribosomal Machinery, Cytoskeletal Organization and Oxidative Phosphorylation in Retinal Photoreceptor Cells. Front. Mol. Neurosci. 2019, 12, 24. [Google Scholar] [CrossRef]
- Ning, A.; Cui, J.; To, E.; Ashe, K.H.; Matsubara, J. Amyloid-β Deposits Lead to Retinal Degeneration in a Mouse Model of Alzheimer Disease. Investig. Opthalmol. Vis. Sci. 2008, 49, 5136–5143. [Google Scholar] [CrossRef]
- Liu, B.; Rasool, S.; Yang, Z.; Glabe, C.G.; Schreiber, S.S.; Ge, J.; Tan, Z. Amyloid-Peptide Vaccinations Reduce β-Amyloid Plaques but Exacerbate Vascular Deposition and Inflammation in the Retina of Alzheimer’s Transgenic Mice. Am. J. Pathol. 2009, 175, 2099–2110. [Google Scholar] [CrossRef]
- Perez, S.E.; Lumayag, S.; Kovacs, B.; Mufson, E.J.; Xu, S. β-Amyloid Deposition and Functional Impairment in the Retina of the APPswe/PS1ΔE9 Transgenic Mouse Model of Alzheimer’s Disease. Investig. Opthalmol. Vis. Sci. 2009, 50, 793–800. [Google Scholar] [CrossRef]
- Mirzaei, M.; Pushpitha, K.; Deng, L.; Chitranshi, N.; Gupta, V.; Rajput, R.; Mangani, A.B.; Dheer, Y.; Godinez, A.; McKay, M.J.; et al. Upregulation of Proteolytic Pathways and Altered Protein Biosynthesis Underlie Retinal Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6017–6034. [Google Scholar] [CrossRef]
- Lim, J.; Li, Q.-X.; He, Z.; Vingrys, A.J.; Chinnery, H.R.; Mullen, J.; Bui, B.; Nguyen, C.T.O. Retinal Functional and Structural Changes in the 5xFAD Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 862. [Google Scholar] [CrossRef]
- Schön, C.; Hoffmann, N.A.; Ochs, S.M.; Burgold, S.; Filser, S.; Steinbach, S.; Seeliger, W.M.; Arzberger, T.; Goedert, M.; Kretzschmar, H.A.; et al. Long-Term In Vivo Imaging of Fibrillar Tau in the Retina of P301S Transgenic Mice. PLoS ONE 2012, 7, e53547. [Google Scholar] [CrossRef]
- Ho, C.-Y.; Troncoso, J.C.; Knox, D.; Stark, W.; Eberhart, C.G. Beta-Amyloid, Phospho-Tau and Alpha-Synuclein Deposits Similar to Those in the Brain Are Not Identified in the Eyes of Alzheimer’s and Parkinson’s Disease Patients. Brain Pathol. 2013, 24, 25–32. [Google Scholar] [CrossRef]
- Haan, J.D.; Morrema, T.H.J.; Verbraak, F.D.; de Boer, J.; Scheltens, P.; Rozemuller, A.J.; Bergen, A.A.B.; Bouwman, F.H.; Hoozemans, J.J. Amyloid-beta and phosphorylated tau in post-mortem Alzheimer’s disease retinas. Acta Neuropathol. Commun. 2018, 6, 147. [Google Scholar] [CrossRef]
- Hadoux, X.; Hui, F.; Lim, J.K.H.; Masters, C.; Pébay, A.; Chevalier, S.; Ha, J.; Loi, S.; Fowler, C.J.; Rowe, C.; et al. Non-invasive in vivo hyperspectral imaging of the retina for potential biomarker use in Alzheimer’s disease. Nat. Commun. 2019, 10, 4227. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin Inhibits Formation of Amyloid β Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in Vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathologyin vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II Trial of Curcumin in Patients with Advanced Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef]
- More, S.S.; Vince, R. Hyperspectral Imaging Signatures Detect Amyloidopathy in Alzheimer’s Mouse Retina Well before Onset of Cognitive Decline. ACS Chem. Neurosci. 2014, 6, 306–315. [Google Scholar] [CrossRef]
- Lu, G.; Fei, B. Medical hyperspectral imaging: A review. J. Biomed. Opt. 2014, 19, 010901. [Google Scholar] [CrossRef] [PubMed]
- More, S.S.; Beach, J.M.; McClelland, C.; Mokhtarzadeh, A.; Vince, R. In Vivo Assessment of Retinal Biomarkers by Hyperspectral Imaging: Early Detection of Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 4492–4501. [Google Scholar] [CrossRef]
- Gupta, V.B.; Anitha, S.; Hegde, M.; Zecca, L.; Garruto, R.M.; Ravid, R.; Shankar, S.K.; Stein, R.; Shanmugavelu, P.; Rao, K.S.J. Aluminium in Alzheimer?s disease: Are we still at a crossroad? Cell. Mol. Life Sci. 2005, 62, 143–158. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Buerger, K.; Ewers, M.; Pirttilä, T.; Zinkowski, R.; Alafuzoff, I.; Teipel, S.J.; Debernardis, J.; Kerkman, D.; McCulloch, C.; Soininen, H.; et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain 2006, 129, 3035–3041. [Google Scholar] [CrossRef]
- Bejanin, A.; Schonhaut, D.; La Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M.; et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef]
- Cowan, C.M.; Bossing, T.; Page, A.; Shepherd, D.; Mudher, A. Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 2010, 120, 593–604. [Google Scholar] [CrossRef]
- Andreasen, N.; Minthon, L.; Davidsson, P.; Vanmechelen, E.; Vanderstichele, H.; Winblad, B.; Blennow, K. Evaluation of CSF-tau and CSF-Aβ42 as Diagnostic Markers for Alzheimer Disease in Clinical Practice. Arch. Neurol. 2001, 58, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Crowther, R.A.; Martin, K.R.; Berg, N.; Coleman, M.; Goedert, M.; Spillantini, M.G. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: Effects on axonal viability. Neurobiol. Aging 2011, 32, 419–433. [Google Scholar] [CrossRef]
- Lim, J.; Li, Q.-X.; He, Z.; Vingrys, A.; Wong, V.H.Y.; Currier, N.; Mullen, J.; Bui, B.; Nguyen, C.T.O. The Eye As a Biomarker for Alzheimer’s Disease. Front. Neurosci. 2016, 10, 536. [Google Scholar] [CrossRef]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Velde, C.V.; Panayi, F.; Louis, C.; Di Polo, A. Tau accumulation in the retina promotes early neuronal dysfunction and precedes brain pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Chang, R.; Che, H.; Wang, J.; Yang, L.; Fang, W.; Xia, Y.; Li, N.; Ma, Q.; Wang, X. Hyperphosphorylation of tau protein by calpain regulation in retina of Alzheimer’s disease transgenic mouse. Neurosci. Lett. 2013, 551, 12–16. [Google Scholar] [CrossRef]
- Nilson, A.N.; English, K.C.; Gerson, J.E.; Whittle, T.B.; Crain, C.N.; Xue, J.; Sengupta, U.; Castillo-Carranza, D.L.; Zhang, W.; Gupta, P.; et al. Tau Oligomers Associate with Inflammation in the Brain and Retina of Tauopathy Mice and in Neurodegenerative Diseases. J. Alzheimer’s Dis. 2016, 55, 1083–1099. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Fong, J.; Ang, L.C.; Yücel, Y.H. Retinal tau pathology in human glaucomas. Can. J. Ophthalmol. 2008, 43, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Knopman, D.S. Dementia and Cerebrovascular Disease. Mayo Clin. Proc. 2006, 81, 223–230. [Google Scholar] [CrossRef]
- Toledo, J.; Arnold, S.E.; Raible, K.; Brettschneider, J.; Xie, S.X.; Grossman, M.; Monsell, S.E.; Kukull, W.A.; Trojanowski, J.Q. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013, 136, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.N. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol. 2016, 131, 659–685. [Google Scholar] [CrossRef]
- Gorelick, P.B.; Scuteri, A.; Black, S.; DeCarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef]
- Cheung, C.Y.-L.; Ong, Y.T.; Ikram, M.K.; Ong, S.Y.; Li, X.; Hilal, S.; Catindig, J.-A.S.; Venketasubramanian, N.; Yap, P.; Seow, D.; et al. Microvascular network alterations in the retina of patients with Alzheimer’s disease. Alzheimer’s Dement. 2014, 10, 135–142. [Google Scholar] [CrossRef]
- Patton, N.; Aslam, T.; MacGillivray, T.; Pattie, A.; Deary, I.J.; Dhillon, B. Retinal vascular image analysis as a potential screening tool for cerebrovascular disease: A rationale based on homology between cerebral and retinal microvasculatures. J. Anat. 2005, 206, 319–348. [Google Scholar] [CrossRef]
- Williams, M.; McGowan, A.J.; Cardwell, C.; Cheung, C.; Craig, D.W.; Passmore, P.; Silvestri, G.; Maxwell, A.P.; McKay, G.J. Retinal microvascular network attenuation in Alzheimer’s disease. Alzheimer’s Dement. 2015, 1, 229–235. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, X.; Azhati, G.; Li, T.; Xu, G.; Liu, F. Retinal microvascular attenuation in mental cognitive impairment and Alzheimer’s disease by optical coherence tomography angiography. Acta Ophthalmol. 2020, 98, e781–e787. [Google Scholar] [CrossRef] [PubMed]
- Sharafi, S.M.; Sylvestre, J.P.; Chevrefils, C.; Soucy, J.P.; Beaulieu, S.; Pascoal, T.A.; Arbour, J.D.; Rhéaume, M.; Robillard, A.; Chayer, C.; et al. Vascular retinal biomarkers improves the detection of the likely cerebral amyloid status from hyperspectral retinal images. Alzheimer’s Dement. 2019, 5, 610–617. [Google Scholar] [CrossRef]
- Frost, S.; the AIBL Research Group; Kanagasingam, Y.; Sohrabi, H.R.; Vignarajan, J.; Bourgeat, P.; Salvado, O.; Villemagne, V.; Rowe, C.C.; Macaulay, S.L.; et al. Retinal vascular biomarkers for early detection and monitoring of Alzheimer’s disease. Transl. Psychiatry 2013, 3, e233. [Google Scholar] [CrossRef]
- Ellis, R.J.; Olichney, J.M.; Thal, L.J.; Mirra, S.S.; Morris, J.C.; Beekly, D.; Heyman, A. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: The CERAD experience, Part XV. Neurology 1996, 46, 1592–1596. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Leurgans, S.E.; Wang, Z.; Wilson, R.S.; Bennett, D.A.; Schneider, J.A. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 2010, 69, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; Van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—one peptide, two pathways. Nat. Rev. Neurol. 2019, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Trost, A.; Lange, S.; Schroedl, F.; Bruckner, D.; Motloch, K.A.; Bogner, B.; Kaser-Eichberger, A.; Strohmaier, C.; Runge, C.; Aigner, L.; et al. Brain and Retinal Pericytes: Origin, Function and Role. Front. Cell. Neurosci. 2016, 10, 20. [Google Scholar] [CrossRef]
- Shi, H.; Koronyo, Y.; Rentsendorj, A.; Regis, G.C.; Sheyn, J.; Fuchs, D.-T.; Kramerov, A.A.; Ljubimov, A.V.; Dumitrascu, O.M.; Rodriguez, A.R.; et al. Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol. 2020, 139, 813–836. [Google Scholar] [CrossRef]
- Tisi, A.; Feligioni, M.; Passacantando, M.; Ciancaglini, M.; Maccarone, R. The Impact of Oxidative Stress on Blood-Retinal Barrier Physiology in Age-Related Macular Degeneration. Cells 2021, 10, 64. [Google Scholar] [CrossRef]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Giebel, S.J.; Menicucci, G.; McGuire, P.G.; Das, A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood–retinal barrier. Lab. Investig. 2005, 85, 597–607. [Google Scholar] [CrossRef]
- Xu, H.; Forrester, J.V.; Liversidge, J.; Crane, I.J. Leukocyte trafficking in experimental autoimmune uveitis: Breakdown of blood-retinal barrier and upregulation of cellular adhesion molecules. Investig. Opthalmol. Vis. Sci. 2003, 44, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, J.; Levin, B.E.; Baumel, B.S.; Camargo, C.J.; Signorile, J.F.; Rundek, T. Retinal Microvascular Alterations as the Biomarkers for Alzheimer Disease: Are We There Yet? J. Neuro-Ophthalmol. 2020, 41, 251–260. [Google Scholar] [CrossRef]
- Querques, G.; Borrelli, E.; Sacconi, R.; De Vitis, L.; Leocani, L.; Santangelo, R.; Magnani, G.; Comi, G.; Bandello, F. Functional and morphological changes of the retinal vessels in Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 2019, 9, 63. [Google Scholar] [CrossRef]
- Tian, J.; Smith, G.; Guo, H.; Liu, B.; Pan, Z.; Wang, Z.; Xiong, S.; Fang, R. Modular machine learning for Alzheimer’s disease classification from retinal vasculature. Sci. Rep. 2021, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Ong, Y.-T.; Hilal, S.; Cheung, C.; Xu, X.; Chen, C.; Venketasubramanian, N.; Wong, T.Y.; Ikram, M.K. Retinal Vascular Fractals and Cognitive Impairment. Dement. Geriatr. Cogn. Disord. Extra 2014, 4, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Dorr, A.; Thomason, L.A.M.; Koletar, M.M.; Sled, J.G.; Stefanovic, B.; McLaurin, J. Venular degeneration leads to vascular dysfunction in a transgenic model of Alzheimer’s disease. Brain 2015, 138, 1046–1058. [Google Scholar] [CrossRef]
- Cheung, C.; Ikram, M.K.; Chen, C.; Wong, T.Y. Imaging retina to study dementia and stroke. Prog. Retin. Eye Res. 2017, 57, 89–107. [Google Scholar] [CrossRef]
- Liao, H.; Zhu, Z.; Peng, Y. Potential Utility of Retinal Imaging for Alzheimer’s Disease: A Review. Front. Aging Neurosci. 2018, 10, 188. [Google Scholar] [CrossRef]
- Ikram, M.K.; Cheung, C.; Wong, T.Y.; Chen, C.P.L.H. Retinal pathology as biomarker for cognitive impairment and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2012, 83, 917–922. [Google Scholar] [CrossRef]
- Jiang, H.; Wei, Y.; Shi, Y.; Wright, C.B.; Sun, X.; Gregori, G.; Zheng, F.; Vanner, E.A.; Lam, B.L.; Rundek, T.; et al. Altered Macular Microvasculature in Mild Cognitive Impairment and Alzheimer Disease. J. Neuroophthalmol. 2018, 38, 292–298. [Google Scholar] [CrossRef]
- Leitgeb, R.A.; Werkmeister, R.M.; Blatter, C.; Schmetterer, L. Doppler Optical Coherence Tomography. Prog. Retin. Eye Res. 2014, 41, 26–43. [Google Scholar] [CrossRef] [PubMed]
- Feke, G.T.; Hyman, B.T.; Stern, R.A.; Pasquale, L.R. Retinal blood flow in mild cognitive impairment and Alzheimer’s disease. Alzheimer’s Dement. 2015, 1, 144–151. [Google Scholar] [CrossRef]
- Szegedi, S.; Dal-Bianco, P.; Stögmann, E.; Traub-Weidinger, T.; Rainer, M.; Masching, A.; Schmidl, D.; Werkmeister, R.M.; Chua, J.; Schmetterer, L.; et al. Anatomical and functional changes in the retina in patients with Alzheimer’s disease and mild cognitive impairment. Acta Ophthalmol. 2020, 98, e914–e921. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Chen, L.; Chen, X.; Liu, X. Soluble amyloid β oligomers may contribute to apoptosis of retinal ganglion cells in glaucoma. Med. Hypotheses 2008, 71, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Smale, G.; Nichols, N.R.; Brady, D.R.; Finch, C.E.; Horton, W.E. Evidence for Apoptotic Cell Death in Alzheimer’s Disease. Exp. Neurol. 1995, 133, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Anderson, A.J.; Cummings, B.J.; Cotman, C.W. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. NeuroReport 1994, 5, 2529–2533. [Google Scholar] [CrossRef]
- Cordeiro, M.F.; Normando, E.M.; Cardoso, M.J.; Miodragovic, S.; Jeylani, S.; Davis, B.M.; Guo, L.; Ourselin, S.; A’Hern, R.; Bloom, P.A. Real-time imaging of single neuronal cell apoptosis in patients with glaucoma. Brain 2017, 140, 1757–1767. [Google Scholar] [CrossRef]
- Mazzoni, F.; Müller, C.; DeAssis, J.; Lew, D.; Leevy, W.M.; Finnemann, S.C. Non-invasive in vivo fluorescence imaging of apoptotic retinal photoreceptors. Sci. Rep. 2019, 9, 1590. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.M.K.; Hoang, C.; Dukes, R.T.; Yee, R.W.; Gray, B.D.; Pak, K.Y.; Caprioli, J. Bis(Zinc-Dipicolylamine), Zn-DPA, a New Marker for Apoptosis. Investig. Opthalmol. Vis. Sci. 2014, 55, 4913–4921. [Google Scholar] [CrossRef][Green Version]
- Ashok, A.; Singh, N.; Chaudhary, S.; Bellamkonda, V.; Kritikos, A.E.; Wise, A.S.; Rana, N.; McDonald, D.; Ayyagari, R. Retinal Degeneration and Alzheimer’s Disease: An Evolving Link. Int. J. Mol. Sci. 2020, 21, 7290. [Google Scholar] [CrossRef]
- Wang, L.; Mao, X. Role of Retinal Amyloid-β in Neurodegenerative Diseases: Overlapping Mechanisms and Emerging Clinical Applications. Int. J. Mol. Sci. 2021, 22, 2360. [Google Scholar] [CrossRef]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Klaver, C.; Ott, A.; Hofman, A.; Assink, J.J.M.; Breteler, M.M.; De Jong, P.T.V.M. Is Age-related Maculopathy Associated with Alzheimer’s Disease?: The Rotterdam Study. Am. J. Epidemiol. 1999, 150, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Larson, E.B.; Gibbons, L.E.; Lee, A.Y.; McCurry, S.M.; Bowen, J.D.; McCormick, W.C.; Crane, P.K. Associations between recent and established ophthalmic conditions and risk of Alzheimer’s disease. Alzheimer’s Dement. 2018, 15, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.-Y.; Wan, L.; Lai, J.-N.; Chen, C.S.; Chen, J.J.-Y.; Wu, M.-Y.; Hu, K.-C.; Chiu, L.-T.; Tien, P.-T.; Lin, H.-J. Increased risk of Alzheimer’s disease among patients with age-related macular degeneration: A nationwide population-based study. PLoS ONE 2021, 16, e0250440. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.A.; Silvestri, V.; Craig, D.; Passmore, A.P.; Silvestri, G. The Prevalence of Age-Related Macular Degeneration in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 42, 909–914. [Google Scholar] [CrossRef]
- Ohno-Matsui, K. Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog. Retin. Eye Res. 2011, 30, 217–238. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.H.; Talaga, K.C.; Rivest, A.J.; Barron, E.; Hageman, G.S.; Johnson, L.V. Characterization of β amyloid assemblies in drusen: The deposits associated with aging and age-related macular degeneration. Exp. Eye Res. 2004, 78, 243–256. [Google Scholar] [CrossRef]
- Johnson, L.V.; Leitner, W.P.; Rivest, A.J.; Staples, M.K.; Radeke, M.J.; Anderson, D.H. The Alzheimer’s A-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 11830–11835. [Google Scholar] [CrossRef]
- Bevan, R.J.; Hughes, T.R.; Williams, P.A.; Good, M.A.; Morgan, B.P.; Morgan, J.E. Retinal ganglion cell degeneration correlates with hippocampal spine loss in experimental Alzheimer’s disease. Acta Neuropathol. Commun. 2020, 8, 216. [Google Scholar] [CrossRef]
- Bailey, S.T.; Thaware, O.; Wang, J.; Hagag, A.; Zhang, X.; Flaxel, C.J.; Lauer, A.K.; Hwang, T.S.; Lin, P.; Huang, D.; et al. Detection of Nonexudative Choroidal Neovascularization and Progression to Exudative Choroidal Neovascularization Using OCT Angiography. Ophthalmol. Retin. 2019, 3, 629–636. [Google Scholar] [CrossRef]
- Sulzbacher, F.; Pollreisz, A.; Kaider, A.; Kickinger, S.; Sacu, S.; Schmidt-Erfurth, U.; the Vienna Eye Study Center. Identification and clinical role of choroidal neovascularization characteristics based on optical coherence tomography angiography. Acta Ophthalmol. 2017, 95, 414–420. [Google Scholar] [CrossRef]
- Yu, J.-T.; Tan, L.; Hardy, J. Apolipoprotein E in Alzheimer’s Disease: An Update. Annu. Rev. Neurosci. 2014, 37, 79–100. [Google Scholar] [CrossRef]
- Baird, P.N.; Richardson, A.J.; Robman, L.D.; Dimitrov, P.N.; Tikellis, G.; McCarty, C.A.; Guymer, R.H. Apolipoprotein (APOE) gene is associated with progression of age-related macular degeneration (AMD). Hum. Mutat. 2006, 27, 337–342. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.-C.; Parfitt, M.; Legrain, S.; Pérez-Tur, J.; Brousseau, T.; Evans, A.; Berr, C.; Vldal, O.; Roques, P.; Gourlet, V.; et al. Apolipoprotein E, ε4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: Analysis of the 19q13.2 chromosomal region. Hum. Mol. Genet. 1994, 3, 569–574. [Google Scholar] [CrossRef]
- Jiang, S.; Kametani, M.; Chen, D.F. Adaptive Immunity: New Aspects of Pathogenesis Underlying Neurodegeneration in Glaucoma and Optic Neuropathy. Front. Immunol. 2020, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Lin, I.-C.; Wang, Y.-H.; Wang, T.-J.; Wang, I.-J.; Shen, Y.-D.; Chi, N.-F.; Chien, L.-N. Glaucoma, Alzheimer’s Disease, and Parkinson’s Disease: An 8-Year Population-Based Follow-Up Study. PLoS ONE 2014, 9, e108938. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.U.; Ferrari, F.; Erb, C. High Occurrence Rate of Glaucoma among Patients with Alzheimer’s Disease. Eur. Neurol. 2002, 47, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Sadun, A.A.; Bassi, C.J. Optic Nerve Damage in Alzheimer’s Disease. Ophthalmology 1990, 97, 9–17. [Google Scholar] [CrossRef]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; López-Cuenca, I.; Rojas, P.; Triviño, A.; Ramírez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef]
- Goldblum, D.; Kipfer-Kauer, A.; Sarra, G.-M.; Wolf, S.; Frueh, B.E. Distribution of Amyloid Precursor Protein and Amyloid-β Immunoreactivity in DBA/2J Glaucomatous Mouse Retinas. Investig. Opthalmol. Vis. Sci. 2007, 48, 5085–5090. [Google Scholar] [CrossRef][Green Version]
- McKinnon, S.J. Glaucoma ocular Alzheimer s disease. Front. Biosci. 2003, 8, s1140–s1156. [Google Scholar] [CrossRef]
- Chiasseu, M.; Vargas, J.L.C.; Destroismaisons, L.; Velde, C.V.; Leclerc, N.; Di Polo, A. Tau Accumulation, Altered Phosphorylation, and Missorting Promote Neurodegeneration in Glaucoma. J. Neurosci. 2016, 36, 5785–5798. [Google Scholar] [CrossRef]
- Zabel, P.; Kaluzny, J.J.; Zabel, K.; Kaluzna, M.; Lamkowski, A.; Jaworski, D.; Makowski, J.; Gebska-Toloczko, M.; Kucharski, R. Quantitative assessment of retinal thickness and vessel density using optical coherence tomography angiography in patients with Alzheimer’s disease and glaucoma. PLoS ONE 2021, 16, e0248284. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A. Intraocular Pressure and Glaucoma. Am. J. Ophthalmol. 1989, 107, 186–188. [Google Scholar] [CrossRef]
- Kurna, S.A.; Akar, G.; Altun, A.; Agirman, Y.; Gozke, E.; Şengör, T. Confocal scanning laser tomography of the optic nerve head on the patients with Alzheimer’s disease compared to glaucoma and control. Int. Ophthalmol. 2014, 34, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Toschi, N.; Babiloni, C.; Baldacci, F.; Black, K.L.; Bokde, A.L.; Bun, R.S.; Cacciola, F.; Cavedo, E.; Chiesa, P.A.; et al. Revolution of Alzheimer Precision Neurology. Passageway of Systems Biology and Neurophysiology1. J. Alzheimer’s Dis. 2018, 64, S47–S105. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Wu, Y.; Wang, M.; Cao, J.; Feng, W.; Cheng, Y.; Li, C.; Shen, Y. Greater Attenuation of Retinal Nerve Fiber Layer Thickness in Alzheimer’s Disease Patients. J. Alzheimer’s Dis. 2014, 40, 277–283. [Google Scholar] [CrossRef]
- Marziani, E.; Pomati, S.; Ramolfo, P.; Cigada, M.; Giani, A.; Mariani, C.; Staurenghi, G. Evaluation of Retinal Nerve Fiber Layer and Ganglion Cell Layer Thickness in Alzheimer’s Disease Using Spectral-Domain Optical Coherence Tomography. Investig. Opthalmol. Vis. Sci. 2013, 54, 5953–5958. [Google Scholar] [CrossRef] [PubMed]
- Pappuru, R.R.; Ouyang, Y.; Nittala, M.G.; Hemmati, H.D.; Keane, P.A.; Walsh, A.C.; Sadda, S.R. Relationship between Outer Retinal Thickness Substructures and Visual Acuity in Eyes with Dry Age-Related Macular Degeneration. Investig. Opthalmol. Vis. Sci. 2011, 52, 6743–6748. [Google Scholar] [CrossRef]
- Gerth, C. The role of the ERG in the diagnosis and treatment of Age-Related Macular Degeneration. Doc. Ophthalmol. 2008, 118, 63–68. [Google Scholar] [CrossRef]
- Lek, J.J.; Nguyen, B.N.; McKendrick, A.M.; Vingrys, A.J. An Electrophysiological Comparison of Contrast Response Functions in Younger and Older Adults, and Those With Glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, M.F.; Guo, L.; Coxon, K.M.; Duggan, J.; Nizari, S.; Normando, E.M.; Sensi, S.L.; Sillito, A.M.; Fitzke, F.W.; Salt, T.E.; et al. Imaging multiple phases of neurodegeneration: A novel approach to assessing cell death in vivo. Cell Death Dis. 2010, 1, e3. [Google Scholar] [CrossRef]
- Dunaief, J.L.; Dentchev, T.; Ying, G.-S.; Milam, A.H. The Role of Apoptosis in Age-Related Macular Degeneration. Arch. Ophthalmol. 2002, 120, 1435–1442. [Google Scholar] [CrossRef]
- Yap, T.E.; Donna, P.; Almonte, M.T.; Cordeiro, M.F. Real-Time Imaging of Retinal Ganglion Cell Apoptosis. Cells 2018, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Dielemans, I.; Vingerling, J.R.; Algra, D.; Hofman, A.; Grobbee, D.E.; De Jong, P.T. Primary Open-angle Glaucoma, Intraocular Pressure, and Systemic Blood Pressure in the General Elderly Population. Ophthalmology 1995, 102, 54–60. [Google Scholar] [CrossRef]
- Wang, J.J.; Foran, S.; Smith, W.; Mitchell, P. Risk of Age-Related Macular Degeneration in Eyes With Macular Drusen or Hyperpigmentation. Arch. Ophthalmol. 2003, 121, 658–663. [Google Scholar] [CrossRef]
- Spaide, R.F.; Armstrong, D.; Browne, R. Choroidal neovascularization in age-related macular degeneration—What is the cause? Retina 2003, 23, 595–614. [Google Scholar] [CrossRef]
- Kergoat, H.; Kergoat, M.-J.; Justino, L.; Chertkow, H.; Robillard, A.; Bergman, H. Visual Retinocortical Function in Dementia of the Alzheimer Type. Gerontology 2002, 48, 197–203. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Molecular Changes | Retina | Brain |
|---|---|---|
| Aβ deposition | In the retina at 2.5 months in APP/PS1 mice [60] In the GCL of the retina at 5–10 post-natal weeks in 3xTg-AD mice [61] Senile plaque appeared in the retina at 2 months in 5xFAD mice and at 6 months in PSAPP mice [62] Soluble Aβ42 appeared at 3 months in APPNL-G-F knock-in mice [63] In the GCL, RNFL, OPL, ONL, INL, and IPL of the retina in patients with AD [64,65] and in the whole-mounted retinas of patients with definite AD or suspected early-stage AD [60] | Appears at 5 months in APP/PS1 mice [60] Distributes in the neocortex, entorhinal region, hippocampus, diencephalic, brain stem, and cerebellar regions of the brain in patients with AD [66] |
| BACE1 expression | In the GCL of the retina at 3 months and then spreads to the IPL and OPL at 6 and 8 months, respectively, in APP/PS1 mice [59] | In the entorhinal cortex, hippocampus, and prefrontal cortex at 6 and 8 months in APP/PS1 mice [59] Increased expression of BACE1 in the temporal cortex and hippocampus in the AD brain [67,68] High activity of BACE1 in the brain in patients with sporadic AD [67] High activity of BACE1 in the cerebrospinal fluid in patients with MCI or AD [69] |
| Symptom | AD | AMD | Glaucoma |
|---|---|---|---|
| Retinal thickness | Thinning in the RNFL, GC-IPL, and GCC of the retina [29,31,171,172] | Thinning in the macular region and photoreceptor layers [173] | Thinning in the GCL and RNFL of the retina [163] |
| Electrophysiology | PERG and PVEP changes [17,18] | Full-field ERG and multifocal ERG changes [174] | PERG and PVEP changes [175] |
| Aβ deposition | In the inner retina [64,65,143] | Along with drusen [150,151] | In the optic nerve and RGC layer of the retina [164] |
| Tau accumulation | In the OPL, IPL, GCL, and RNFL of the retina [3,83] | N | In the INL, IPL, and GCL of the retina [102,166] |
| Apoptosis | RGC apoptosis [176] | Apoptosis of the RPE, photoreceptors, and INL cells [177] | RGC apoptosis [178] |
| Ocular hypertension | Ocular hypertension was absent in patients with AD [161] | N | Elevated IOP in high-tension glaucoma and normal IOP in normal-tension glaucoma [168,179] |
| Hyperpigmentation | N | Hyperpigmentation is present in AMD patients [180] | N |
| Geographic atrophy | N | Geographic atrophy surround and spare the central macula [144] | N |
| Choroidal neovascularization | N | Choroidal neovascularization is present in neovascular AMD [181] | N |
| Techniques | Description | Main Findings |
|---|---|---|
| Spectral-domain optical coherence tomography | A noninvasive method that provides high-resolution images of retinal morphological structures and volumetric parameters [130] | Reduction in RNFL thickness [29,31,171,172] Decrease in macular thickness and volume [31] Reduction in macular GC-IPL thickness [29,31] Reduction in macular GCC thickness [31,32] |
| Optical coherence tomography–angiography | A noninvasive method that can accurately visualize the vascular system of different retinal layers at a 3-dimensional level [110] | Decrease in the microvascular density of deep retinal capillary plexuses [110] |
| Doppler optical coherence tomography and laser Doppler blood flowmeter | Noninvasive methods that can visualize and quantify blood flow [133] | Reduction in venous blood flow [134] Decrease in retinal arterial blood flow [135] |
| Retinal photography (ocular fundus) | A noninvasive method that provides retinal images and vascular signs [130] | Narrowing of venular calibers [107] Reduction in arteriolar and venular fractal dimensions [107,109] Increase in tortuosity of arterioles and venules [107] Detection of retinal Aβ plaques after curcumin administration [60] |
| Pattern electroretinogram and pattern visual evoked potential | Noninvasive methods that reflect the bioelectrical functions of the retina and optic nerve related to visual pathway transmission [17] | Increase in implicit time of P50 [17,18] Amplitude reduction in both P50 and N95 [17,18] Increase in P100-wave latency [17,18] Increase in retinocortical time [17] |
| Hyperspectral imaging | A noninvasive method that detects retinal Aβ without extrinsic fluorescence labeling by scanning within a specific wavelength range [88] | Detection of retinal Aβ signature [84,90] |
| Detection of apoptosing retinal cells | A method that allows real-time observation of retinal apoptotic cells in vivo [139] | RGC apoptosis in 3×Tg-AD mice (a mouse model of AD) [176] |
| Pathology | Detection Methods | Expected Outcomes |
|---|---|---|
| AD pathology | ||
| Amyloid deposition | Retinal imaging Hyperspectral imaging | Detects retinal Aβ plaques after curcumin administration [60] Detects retinal Aβ signature [84,90] |
| BACE1 expression | Currently unavailable in patients with AD | No information |
| Tau aggregation | Currently unavailable in patients with AD | No information |
| Non-AD pathology | ||
| Structural changes | ||
| RNFL thickness | Optical coherence tomography | Decrease in RNFL thickness [29,31,171,172] |
| GC-IPL thickness | Decrease in macular GC-IPL thickness [29,31] | |
| Macular GCC thickness | Decrease in macular GCC thickness [31,32] | |
| Electrophysiological changes | ||
| PERG parameters | PERG examination | Increase in implicit time of P50 [17,18] Amplitude reduction in both P50 and N95 [17,18] |
| PVEP parameters | PVEP examination | Increase in P100-wave latency [17,18] |
| Vascular changes | ||
| RVPs | Retinal photography Doppler optical coherence tomography and laser Doppler blood flowmeter | Narrowing of venular calibers [107] Reduction in arteriolar and venular fractal dimensions [107,109] Increase in tortuosity of arterioles and venules [107] |
| Apoptosis | Detection of apoptosing retinal cells in vivo Currently unavailable in patients with AD | Detected RGC apoptosis in 3×Tg-AD mice (a mouse model of AD) [176] |
| Retinal Indicators | Results | Contradictory Findings |
|---|---|---|
| Retinal thickness | Reduction in RNFL, GC-IPL, and macular GCC thickness [29,31,171,172] | No differences in macular retinal layer thickness and pRNFL thickness [33,34] |
| Aβ plaques | Retinal Aβ plaques in patients with AD [64,65,143] | No Aβ plaques in the retina in patients with AD [20,81,82,83], or APP/PS1 or Tg2576 mice [81] |
| BACE1 expression | Detected in the retina of APP/PS1 mice [59] | N |
| Tau aggregation | Hyperphosphorylated tau and total tau found in the retina in patients with AD [3,83] | No fibrillary tau, paired helical filaments, or neurofibrillary tangles in AD [81,83] |
| PERG and PVEP | Changes in PERG and PVEP parameters [17,18] | No changes in PERG and PVEP parameters [182] |
| Retinal vasculature changes | Decrease in the retinal microvascular density of the DRCPs [110] Narrowing of venular calibers [107] Reduction in arteriolar and venular fractal dimensions [107,109] Increase in tortuosity of arterioles and venules [107] Decrease in venous blood flow and retinal arterial blood flow [134] | Decrease in arteriolar tortuosity [109] No significant change in the density of DVP [124,125] |
| RGC apoptosis | RGC apoptosis in 3×Tg-AD mice (a mouse model of AD) [176] | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, C.; Xu, J.; Chen, Y.; Ip, N.Y. Retinal Dysfunction in Alzheimer’s Disease and Implications for Biomarkers. Biomolecules 2021, 11, 1215. https://doi.org/10.3390/biom11081215
Liao C, Xu J, Chen Y, Ip NY. Retinal Dysfunction in Alzheimer’s Disease and Implications for Biomarkers. Biomolecules. 2021; 11(8):1215. https://doi.org/10.3390/biom11081215
Chicago/Turabian StyleLiao, Chunyan, Jinying Xu, Yu Chen, and Nancy Y. Ip. 2021. "Retinal Dysfunction in Alzheimer’s Disease and Implications for Biomarkers" Biomolecules 11, no. 8: 1215. https://doi.org/10.3390/biom11081215
APA StyleLiao, C., Xu, J., Chen, Y., & Ip, N. Y. (2021). Retinal Dysfunction in Alzheimer’s Disease and Implications for Biomarkers. Biomolecules, 11(8), 1215. https://doi.org/10.3390/biom11081215