
Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes

 ,
,
Abstract
1. Introduction
2. Old and New Pathophysiological Mechanisms in AD
2.1. The Amyloid Cascade Hypothesis
2.2. Neurofibrillary Tangles
2.3. Unfolded Protein Response and Defective Proteostasis
2.4. Complement Cascade and Neuroinflammation
2.5. The Neuroenergetic Hypothesis
3. Astrocytes as Targets for AD Therapeutics
3.1. Targeting Astrocyte Senescence
3.2. Targeting Astrocyte Glutamate Transporters
3.3. Targeting the Astrocytic Metabolic System
3.4. Upregulation of Astrocytic Neurotrophins and Growth Factors
3.5. Targeting Astrocytes-Driven Amyloid Aggregation and Clearance
3.6. Targeting Astrocytic Reactivity, Complement Cascade, Neuroinflammation, and Oxidative Stress
3.7. Modulation of Astrocytes According to Their Morphofunctional State: The Case of Palmitoylethanolamide
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- FDA. Peripheral and Central Nervous System Drugs Advisory Committee (PCNS) Meeting. 2020. Available online: https://www.fda.gov/advisorycommittees/advisory-committee-calendar/november-6-2020-meeting-peripheral-and-central-nervous-system-drugs-advisory-committee-meeting (accessed on 6 November 2020).
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Ayton, S.; Bush, A.I. beta-amyloid: The known unknowns. Ageing Res. Rev. 2021, 65, 101212. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- Reitz, C.; Rogaeva, E.; Beecham, G.W. Late-onset vs nonmendelian early-onset Alzheimer disease: A distinction without a difference? Neurol. Genet. 2020, 6, e512. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Sasaki, K.; Tanizaki, Y.; Hata, J.; Fujimi, K.; Matsui, Y.; Sekita, A.; Suzuki, S.O.; Kanba, S.; Kiyohara, Y.; et al. Insulin resistance is associated with the pathology of Alzheimer disease: The Hisayama study. Neurology 2010, 75, 764–770. [Google Scholar] [CrossRef]
- Ott, B.R.; Lafleche, G.; Whelihan, W.M.; Buongiorno, G.W.; Albert, M.S.; Fogel, B.S. Impaired awareness of deficits in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1996, 10, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Mangialasche, F.; Ngandu, T. Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 653–666. [Google Scholar] [CrossRef]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef]
- Crane, P.K.; Walker, R.; Hubbard, R.A.; Li, G.; Nathan, D.M.; Zheng, H.; Haneuse, S.; Craft, S.; Montine, T.J.; Kahn, S.E.; et al. Glucose levels and risk of dementia. N. Engl. J. Med. 2013, 369, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Tini, G.; Scagliola, R.; Monacelli, F.; La Malfa, G.; Porto, I.; Brunelli, C.; Rosa, G.M. Alzheimer’s Disease and Cardiovascular Disease: A Particular Association. Cardiol. Res. Pract. 2020, 2020, 2617970. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef]
- Meng, Q.; Lin, M.S.; Tzeng, I.S. Relationship Between Exercise and Alzheimer’s Disease: A Narrative Literature Review. Front. Neurosci. 2020, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.H.; Chang, C.H.; Gean, P.W. Impact of social relationships on Alzheimer’s memory impairment: Mechanistic studies. J. Biomed. Sci. 2018, 25, 3. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, T.C.; Mattsson, N.; Weiner, M.W.; Alzheimer’s Disease Neuroimaging, I. Smoking and increased Alzheimer’s disease risk: A review of potential mechanisms. Alzheimers Dement. 2014, 10, S122–S145. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Olabarria, M.; Chvatal, A.; Verkhratsky, A. Astroglia in dementia and Alzheimer’s disease. Cell Death Differ. 2009, 16, 378–385. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta 2016, 1862, 483–491. [Google Scholar] [CrossRef]
- Aisen, P.S.; Cummings, J.; Jack, C.R., Jr.; Morris, J.C.; Sperling, R.; Frolich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J.; et al. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimers Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef]
- Beason-Held, L.L.; Goh, J.O.; An, Y.; Kraut, M.A.; O’Brien, R.J.; Ferrucci, L.; Resnick, S.M. Changes in brain function occur years before the onset of cognitive impairment. J. Neurosci. 2013, 33, 18008–18014. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- NIA. Alzheimer‘s Disease Diagnostic Guidelines; NIH National Institute on Aging: Bethesda, MD, USA, 2011. [Google Scholar]
- Edgar, C.J.; Vradenburg, G.; Hassenstab, J. The 2018 Revised FDA Guidance for Early Alzheimer’s Disease: Establishing the Meaningfulness of Treatment Effects. J. Prev. Alzheimers Dis. 2019, 6, 223–227. [Google Scholar] [CrossRef]
- FDA. Alzheimer’s Disease: Developing Drugs for Treatment Guidance for Industy. 2018. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/alzheimers-disease-developing-drugs-treatment-guidance-industy (accessed on 10 April 2021).
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Irwin, K.; Sexton, C.; Daniel, T.; Lawlor, B.; Naci, L. Healthy Aging and Dementia: Two Roads Diverging in Midlife? Front. Aging Neurosci. 2018, 10, 275. [Google Scholar] [CrossRef]
- Tan, C.C.; Yu, J.T.; Tan, L. Biomarkers for preclinical Alzheimer’s disease. J. Alzheimers Dis. 2014, 42, 1051–1069. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Iadecola, C. Metabolic and Non-Cognitive Manifestations of Alzheimer’s Disease: The Hypothalamus as Both Culprit and Target of Pathology. Cell Metab. 2015, 22, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Lanctot, K.L.; Amatniek, J.; Ancoli-Israel, S.; Arnold, S.E.; Ballard, C.; Cohen-Mansfield, J.; Ismail, Z.; Lyketsos, C.; Miller, D.S.; Musiek, E.; et al. Neuropsychiatric signs and symptoms of Alzheimer’s disease: New treatment paradigms. Alzheimers Dement. 2017, 3, 440–449. [Google Scholar] [CrossRef]
- Birks, J. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev. 2006, CD005593. [Google Scholar] [CrossRef]
- McShane, R.; Areosa Sastre, A.; Minakaran, N. Memantine for dementia. Cochrane Database Syst. Rev. 2006, CD003154. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Crous-Bou, M.; Minguillon, C.; Gramunt, N.; Molinuevo, J.L. Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimers Res. Ther. 2017, 9, 71. [Google Scholar] [CrossRef]
- Facchinetti, R.; Valenza, M.; Bronzuoli, M.R.; Menegoni, G.; Ratano, P.; Steardo, L.; Campolongo, P.; Scuderi, C. Looking for a Treatment for the Early Stage of Alzheimer’s Disease: Preclinical Evidence with Co-Ultramicronized Palmitoylethanolamide and Luteolin. Int. J. Mol. Sci. 2020, 21, 3802. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Alzheimer, A. Über einen eigenartigen schweren Erkrankungsprozeβ der Hirnrincle. Neurol. Central. 1906, 25, 1134. [Google Scholar]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369. [Google Scholar] [PubMed]
- Van Vickle, G.D.; Esh, C.L.; Kokjohn, T.A.; Patton, R.L.; Kalback, W.M.; Luehrs, D.C.; Beach, T.G.; Newel, A.J.; Lopera, F.; Ghetti, B.; et al. Presenilin-1 280Glu-->Ala mutation alters C-terminal APP processing yielding longer abeta peptides: Implications for Alzheimer’s disease. Mol. Med. 2008, 14, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Burnouf, S.; Gorsky, M.K.; Dols, J.; Gronke, S.; Partridge, L. Abeta43 is neurotoxic and primes aggregation of Abeta40 in vivo. Acta Neuropathol. 2015, 130, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Arnold, S.E.; Hyman, B.T.; Flory, J.; Damasio, A.R.; Van Hoesen, G.W. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex. 1991, 1, 103–116. [Google Scholar] [CrossRef]
- Puig, K.L.; Combs, C.K. Expression and function of APP and its metabolites outside the central nervous system. Exp. Gerontol. 2013, 48, 608–611. [Google Scholar] [CrossRef]
- Coronel, R.; Bernabeu-Zornoza, A.; Palmer, C.; Muniz-Moreno, M.; Zambrano, A.; Cano, E.; Liste, I. Role of Amyloid Precursor Protein (APP) and Its Derivatives in the Biology and Cell Fate Specification of Neural Stem Cells. Mol. Neurobiol. 2018, 55, 7107–7117. [Google Scholar] [CrossRef]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the beta-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Caille, I.; Allinquant, B.; Dupont, E.; Bouillot, C.; Langer, A.; Muller, U.; Prochiantz, A. Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 2004, 131, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, Z.; Li, H.; Wiese, M.; Zheng, H. APP physiological and pathophysiological functions: Insights from animal models. Cell Res. 2012, 22, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, X.; Huang, T.; Bu, G.; Xu, H. Dysregulation of protein trafficking in neurodegeneration. Mol. Neurodegener. 2014, 9, 31. [Google Scholar] [CrossRef]
- Buoso, E.; Lanni, C.; Schettini, G.; Govoni, S.; Racchi, M. beta-Amyloid precursor protein metabolism: Focus on the functions and degradation of its intracellular domain. Pharmacol. Res. 2010, 62, 308–317. [Google Scholar] [CrossRef]
- Matsui, T.; Ingelsson, M.; Fukumoto, H.; Ramasamy, K.; Kowa, H.; Frosch, M.P.; Irizarry, M.C.; Hyman, B.T. Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 2007, 1161, 116–123. [Google Scholar] [CrossRef]
- Hilbich, C.; Kisters-Woike, B.; Reed, J.; Masters, C.L.; Beyreuther, K. Aggregation and secondary structure of synthetic amyloid beta A4 peptides of Alzheimer’s disease. J. Mol. Biol. 1991, 218, 149–163. [Google Scholar] [CrossRef]
- McGowan, E.; Pickford, F.; Kim, J.; Onstead, L.; Eriksen, J.; Yu, C.; Skipper, L.; Murphy, M.P.; Beard, J.; Das, P.; et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 2005, 47, 191–199. [Google Scholar] [CrossRef]
- Konietzko, U. AICD nuclear signaling and its possible contribution to Alzheimer’s disease. Curr. Alzheimer. Res. 2012, 9, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.O.; Mett, J.; Stahlmann, C.P.; Haupenthal, V.J.; Zimmer, V.C.; Hartmann, T. Neprilysin and Abeta Clearance: Impact of the APP Intracellular Domain in NEP Regulation and Implications in Alzheimer’s Disease. Front. Aging Neurosci. 2013, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Roberts, B.R.; Lind, M.; Wagen, A.Z.; Rembach, A.; Frugier, T.; Li, Q.X.; Ryan, T.M.; McLean, C.A.; Doecke, J.D.; Rowe, C.C.; et al. Biochemically-defined pools of amyloid-beta in sporadic Alzheimer’s disease: Correlation with amyloid PET. Brain 2017, 140, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Porayette, P.; Gallego, M.J.; Kaltcheva, M.M.; Bowen, R.L.; Vadakkadath Meethal, S.; Atwood, C.S. Differential processing of amyloid-beta precursor protein directs human embryonic stem cell proliferation and differentiation into neuronal precursor cells. J. Biol. Chem. 2009, 284, 23806–23817. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.S.; Jung, K.; Kim, I.S.; Park, K.I. Amyloid-beta oligomers regulate the properties of human neural stem cells through GSK-3beta signaling. Exp. Mol. Med. 2013, 45, 60. [Google Scholar] [CrossRef]
- Chen, Y.; Dong, C. Abeta40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ. 2009, 16, 386–394. [Google Scholar] [CrossRef]
- Fonseca, M.B.; Sola, S.; Xavier, J.M.; Dionisio, P.A.; Rodrigues, C.M. Amyloid beta peptides promote autophagy-dependent differentiation of mouse neural stem cells: Abeta-mediated neural differentiation. Mol. Neurobiol. 2013, 48, 829–840. [Google Scholar] [CrossRef]
- Lopez-Toledano, M.A.; Shelanski, M.L. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J. Neurosci. 2004, 24, 5439–5444. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug. Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef]
- Maloney, J.A.; Bainbridge, T.; Gustafson, A.; Zhang, S.; Kyauk, R.; Steiner, P.; van der Brug, M.; Liu, Y.; Ernst, J.A.; Watts, R.J.; et al. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J. Biol. Chem. 2014, 289, 30990–31000. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.A.; Klutkowski, J.A.; Freret, T.; Wolfe, M.S. Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid beta-peptides (Abeta) by gamma-secretase to increase 42-to-40-residue Abeta. J. Biol. Chem. 2014, 289, 31043–31052. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed]
- Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.; George-Hyslop, P.H.; Pericak-Vance, M.A.; Joo, S.H.; Rosi, B.L.; Gusella, J.F.; Crapper-MacLachlan, D.R.; Alberts, M.J.; et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 1993, 43, 1467–1472. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Metaxas, A.; Kempf, S.J. Neurofibrillary tangles in Alzheimer’s disease: Elucidation of the molecular mechanism by immunohistochemistry and tau protein phospho-proteomics. Neural. Regen. Res. 2016, 11, 1579–1581. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Hirokawa, N.; Funakoshi, T.; Sato-Harada, R.; Kanai, Y. Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J. Cell Biol. 1996, 132, 667–679. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef]
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 2001, 114, 1179–1187. [Google Scholar]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef]
- Cash, A.D.; Aliev, G.; Siedlak, S.L.; Nunomura, A.; Fujioka, H.; Zhu, X.; Raina, A.K.; Vinters, H.V.; Tabaton, M.; Johnson, A.B.; et al. Microtubule reduction in Alzheimer’s disease and aging is independent of tau filament formation. Am. J. Pathol. 2003, 162, 1623–1627. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Weitz, T.M.; Town, T. Microglia in Alzheimer’s Disease: It’s All About Context. Int. J. Alzheimers Dis. 2012, 2012, 314185. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; LaFerla, F.M. Linking calcium to Abeta and Alzheimer’s disease. Neuron 2008, 59, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Honda, K.; Liu, Q.; Santos, M.S.; Oliveira, C.R.; Aliev, G.; Nunomura, A.; Zhu, X.; Smith, M.A.; Perry, G. Oxidative stress: The old enemy in Alzheimer’s disease pathophysiology. Curr. Alzheimer Res. 2005, 2, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Llanos-Gonzalez, E.; Henares-Chavarino, A.A.; Pedrero-Prieto, C.M.; Garcia-Carpintero, S.; Frontinan-Rubio, J.; Sancho-Bielsa, F.J.; Alcain, F.J.; Peinado, J.R.; Rabanal-Ruiz, Y.; Duran-Prado, M. Interplay Between Mitochondrial Oxidative Disorders and Proteostasis in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 1444. [Google Scholar] [CrossRef]
- Cornejo, V.H.; Hetz, C. The unfolded protein response in Alzheimer’s disease. Semin. Immunopathol. 2013, 35, 277–292. [Google Scholar] [CrossRef]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef]
- Lilienbaum, A. Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 2013, 4, 1–26. [Google Scholar]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.S.; Stanley, L.C.; Ling, C.; White, L.; MacLeod, V.; Perrot, L.J.; White, C.L., 3rd; Araoz, C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 7611–7615. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Abeta clearance through NLRP3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Strohmeyer, R.; Liang, Z.; Lue, L.F.; Rogers, J. CCAAT/enhancer binding protein delta (C/EBPdelta) expression and elevation in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 991–999. [Google Scholar] [CrossRef]
- Ndoja, A.; Reja, R.; Lee, S.H.; Webster, J.D.; Ngu, H.; Rose, C.M.; Kirkpatrick, D.S.; Modrusan, Z.; Chen, Y.J.; Dugger, D.L.; et al. Ubiquitin Ligase COP1 Suppresses Neuroinflammation by Degrading c/EBPbeta in Microglia. Cell 2020, 182, 1156–1169.e12. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Uherek, M.; Volk, B.; Baeuerle, P.A.; Kaltschmidt, C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1997, 94, 2642–2647. [Google Scholar] [CrossRef]
- Bradt, B.M.; Kolb, W.P.; Cooper, N.R. Complement-dependent proinflammatory properties of the Alzheimer’s disease beta-peptide. J. Exp. Med. 1998, 188, 431–438. [Google Scholar] [CrossRef]
- Shen, Y.; Lue, L.; Yang, L.; Roher, A.; Kuo, Y.; Strohmeyer, R.; Goux, W.J.; Lee, V.; Johnson, G.V.; Webster, S.D.; et al. Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 2001, 305, 165–168. [Google Scholar] [CrossRef]
- Eikelenboom, P.; Stam, F.C. Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol. 1982, 57, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fonseca, M.I.; Pisalyaput, K.; Tenner, A.J. Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. J. Neurochem. 2008, 106, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Afagh, A.; Cummings, B.J.; Cribbs, D.H.; Cotman, C.W.; Tenner, A.J. Localization and cell association of C1q in Alzheimer’s disease brain. Exp. Neurol. 1996, 138, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.; Lue, L.F.; Brachova, L.; Tenner, A.J.; McGeer, P.L.; Terai, K.; Walker, D.G.; Bradt, B.; Cooper, N.R.; Rogers, J. Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer’s disease. Neurobiol. Aging 1997, 18, 415–421. [Google Scholar] [CrossRef]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflamm. 2012, 9, 179. [Google Scholar] [CrossRef] [PubMed]
- Tenner, A.J. Complement-Mediated Events in Alzheimer’s Disease: Mechanisms and Potential Therapeutic Targets. J. Immunol. 2020, 204, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Krance, S.H.; Wu, C.Y.; Zou, Y.; Mao, H.; Toufighi, S.; He, X.; Pakosh, M.; Swardfager, W. The complement cascade in Alzheimer’s disease: A systematic review and meta-analysis. Mol. Psychiatry 2019. [CrossRef]
- Schartz, N.D.; Tenner, A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflamm. 2020, 17, 354. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Lui, H.; Zhang, J.; Makinson, S.R.; Cahill, M.K.; Kelley, K.W.; Huang, H.Y.; Shang, Y.; Oldham, M.C.; Martens, L.H.; Gao, F.; et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 2016, 165, 921–935. [Google Scholar] [CrossRef]
- Vasek, M.J.; Garber, C.; Dorsey, D.; Durrant, D.M.; Bollman, B.; Soung, A.; Yu, J.; Perez-Torres, C.; Frouin, A.; Wilton, D.K.; et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 2016, 534, 538–543. [Google Scholar] [CrossRef]
- Lee, J.D.; Coulthard, L.G.; Woodruff, T.M. Complement dysregulation in the central nervous system during development and disease. Semin. Immunol. 2019, 45, 101340. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Ager, R.R.; Chu, S.H.; Yazan, O.; Sanderson, S.D.; LaFerla, F.M.; Taylor, S.M.; Woodruff, T.M.; Tenner, A.J. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J. Immunol. 2009, 183, 1375–1383. [Google Scholar] [CrossRef]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019, 10, 362. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Davis, K.L.; Berg, J.D.; Schafer, K.; Campbell, K.; Thomas, R.G.; Weiner, M.F.; Farlow, M.R.; Sano, M.; Grundman, M.; et al. A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative Study. Neurology 2000, 54, 588–593. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schafer, K.A.; Grundman, M.; Pfeiffer, E.; Sano, M.; Davis, K.L.; Farlow, M.R.; Jin, S.; Thomas, R.G.; Thal, L.J.; et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: A randomized controlled trial. JAMA 2003, 289, 2819–2826. [Google Scholar] [CrossRef]
- Thal, L.J.; Ferris, S.H.; Kirby, L.; Block, G.A.; Lines, C.R.; Yuen, E.; Assaid, C.; Nessly, M.L.; Norman, B.A.; Baranak, C.C.; et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 2005, 30, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Scharf, S.; Mander, A.; Ugoni, A.; Vajda, F.; Christophidis, N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer’s disease. Neurology 1999, 53, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Group, A.D.C.; Bentham, P.; Gray, R.; Sellwood, E.; Hills, R.; Crome, P.; Raftery, J. Aspirin in Alzheimer’s disease (AD2000): A randomised open-label trial. Lancet Neurol. 2008, 7, 41–49. [Google Scholar] [CrossRef]
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a020420. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting neuroinflammation in Alzheimer’s disease. J. Inflamm. Res. 2016, 9, 199–208. [Google Scholar] [CrossRef]
- Kalaria, R.N.; Harik, S.I. Abnormalities of the glucose transporter at the blood-brain barrier and in brain in Alzheimer’s disease. Prog. Clin. Biol. Res. 1989, 317, 415–421. [Google Scholar]
- Hooijmans, C.R.; Graven, C.; Dederen, P.J.; Tanila, H.; van Groen, T.; Kiliaan, A.J. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 2007, 1181, 93–103. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef]
- Jagust, W.J.; Landau, S.M.; Alzheimer’s Disease Neuroimaging Initiative. Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J. Neurosci. 2012, 32, 18227–18233. [Google Scholar] [CrossRef]
- Griffith, C.M.; Eid, T.; Rose, G.M.; Patrylo, P.R. Evidence for altered insulin receptor signaling in Alzheimer’s disease. Neuropharmacology 2018, 136, 202–215. [Google Scholar] [CrossRef]
- Ragozzino, M.E.; Unick, K.E.; Gold, P.E. Hippocampal acetylcholine release during memory testing in rats: Augmentation by glucose. Proc. Natl. Acad. Sci. USA 1996, 93, 4693–4698. [Google Scholar] [CrossRef]
- Andersen, J.V.; Christensen, S.K.; Westi, E.W.; Diaz-delCastillo, M.; Tanila, H.; Schousboe, A.; Aldana, B.I.; Waagepetersen, H.S. Deficient astrocyte metabolism impairs glutamine synthesis and neurotransmitter homeostasis in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 148, 105198. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed]
- Rouach, N.; Koulakoff, A.; Abudara, V.; Willecke, K.; Giaume, C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 2008, 322, 1551–1555. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, N.C.; Sabbagh, M.N.; Beach, T.G.; Kim, R.C.; Cribbs, D.H.; Cotman, C.W. Brain gene expression patterns differentiate mild cognitive impairment from normal aged and Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1961–1972. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1500–1513. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Kim, Y.S.; Jung, H.M.; Yoon, B.E. Exploring glia to better understand Alzheimer’s disease. Anim. Cells Syst. 2018, 22, 213–218. [Google Scholar] [CrossRef]
- Lau, S.F.; Cao, H.; Fu, A.K.Y.; Ip, N.Y. Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 25800–25809. [Google Scholar] [CrossRef]
- Jakel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef]
- Di Benedetto, B.; Rupprecht, R. Targeting glia cells: Novel perspectives for the treatment of neuropsychiatric diseases. Curr. Neuropharmacol. 2013, 11, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xie, L.; Chung, C.Y. Signaling Pathways Controlling Microglia Chemotaxis. Mol. Cells 2017, 40, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Bergles, D.E.; Richardson, W.D. Oligodendrocyte Development and Plasticity. Cold Spring Harb. Perspect. Biol. 2015, 8, a020453. [Google Scholar] [CrossRef]
- Fields, R.D. A new mechanism of nervous system plasticity: Activity-dependent myelination. Nat. Rev. Neurosci. 2015, 16, 756–767. [Google Scholar] [CrossRef]
- Lasiene, J.; Matsui, A.; Sawa, Y.; Wong, F.; Horner, P.J. Age-related myelin dynamics revealed by increased oligodendrogenesis and short internodes. Aging Cell 2009, 8, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Ingrassia, D.; Sarvadio, M.; Schiavi, S.; Steardo, L.; Verkhratsky, A.; Trezza, V.; Scuderi, C. Neuroglia in the autistic brain: Evidence from a preclinical model. Mol. Autism. 2018, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Papuc, E.; Rejdak, K. The role of myelin damage in Alzheimer’s disease pathology. Arch. Med. Sci. 2020, 16, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Cartocci, V.; Tonini, C.; Di Pippo, T.; Vuono, F.; Schiavi, S.; Marino, M.; Trezza, V.; Pallottini, V. Prenatal exposure to valproate induces sex-, age-, and tissue-dependent alterations of cholesterol metabolism: Potential implications on autism. J. Cell Physiol. 2019, 234, 4362–4374. [Google Scholar] [CrossRef]
- Cainelli, E.; Arrigoni, F.; Vedovelli, L. White matter injury and neurodevelopmental disabilities: A cross-disease (dis)connection. Prog. Neurobiol. 2020, 193, 101845. [Google Scholar] [CrossRef] [PubMed]
- Cartocci, V.; Catallo, M.; Tempestilli, M.; Segatto, M.; Pfrieger, F.W.; Bronzuoli, M.R.; Scuderi, C.; Servadio, M.; Trezza, V.; Pallottini, V. Altered Brain Cholesterol/Isoprenoid Metabolism in a Rat Model of Autism Spectrum Disorders. Neuroscience 2018, 372, 27–37. [Google Scholar] [CrossRef]
- Arthur, V.A.a.B. Astroglia. In Glial Physiology and Pathophysiology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; pp. 105–244. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why are astrocytes important? Neurochem. Res. 2015, 40, 389–401. [Google Scholar] [CrossRef]
- Parpura, V.; Verkhratsky, A. The astrocyte excitability brief: From receptors to gliotransmission. Neurochem. Int. 2012, 61, 610–621. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V.; Vardjan, N.; Zorec, R. Physiology of Astroglia. Adv. Exp. Med. Biol. 2019, 1175, 45–91. [Google Scholar] [CrossRef]
- Stallcup, W.B. The NG2 proteoglycan: Past insights and future prospects. J. Neurocytol. 2002, 31, 423–435. [Google Scholar] [CrossRef]
- Butt, A.M.; Hamilton, N.; Hubbard, P.; Pugh, M.; Ibrahim, M. Synantocytes: The fifth element. J. Anat. 2005, 207, 695–706. [Google Scholar] [CrossRef]
- Bergles, D.E.; Roberts, J.D.; Somogyi, P.; Jahr, C.E. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 2000, 405, 187–191. [Google Scholar] [CrossRef]
- Scuderi, C.; Verkhratsky, A. The role of neuroglia in autism spectrum disorders. Prog. Mol. Biol. Transl. Sci. 2020, 173, 301–330. [Google Scholar] [CrossRef] [PubMed]
- Bernaus, A.; Blanco, S.; Sevilla, A. Glia Crosstalk in Neuroinflammatory Diseases. Front. Cell Neurosci. 2020, 14, 209. [Google Scholar] [CrossRef]
- Jha, M.K.; Kim, J.H.; Song, G.J.; Lee, W.H.; Lee, I.K.; Lee, H.W.; An, S.S.A.; Kim, S.; Suk, K. Functional dissection of astrocyte-secreted proteins: Implications in brain health and diseases. Prog. Neurobiol. 2018, 162, 37–69. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Parpura, V.; Rodriguez-Arellano, J.J.; Zorec, R. Astroglia in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 273–324. [Google Scholar] [CrossRef]
- Cassano, T.; Magini, A.; Giovagnoli, S.; Polchi, A.; Calcagnini, S.; Pace, L.; Lavecchia, M.A.; Scuderi, C.; Bronzuoli, M.R.; Ruggeri, L.; et al. Early intrathecal infusion of everolimus restores cognitive function and mood in a murine model of Alzheimer’s disease. Exp. Neurol. 2019, 311, 88–105. [Google Scholar] [CrossRef]
- Maftei, D.; Ratano, P.; Fusco, I.; Marconi, V.; Squillace, S.; Negri, L.; Severini, C.; Balboni, G.; Steardo, L.; Bronzuoli, M.R.; et al. The prokineticin receptor antagonist PC1 rescues memory impairment induced by beta amyloid administration through the modulation of prokineticin system. Neuropharmacology 2019, 158, 107739. [Google Scholar] [CrossRef] [PubMed]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging 2014, 35, 2746–2760. [Google Scholar] [CrossRef]
- Guillemaud, O.; Ceyzeriat, K.; Saint-Georges, T.; Cambon, K.; Petit, F.; Ben Haim, L.; Carrillo-de Sauvage, M.A.; Guillermier, M.; Bernier, S.; Herard, A.S.; et al. Complex roles for reactive astrocytes in the triple transgenic mouse model of Alzheimer disease. Neurobiol. Aging 2020, 90, 135–146. [Google Scholar] [CrossRef]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Cota-Coronado, A.; Diaz-Martinez, N.F.; Padilla-Camberos, E.; Diaz-Martinez, N.E. Editing the Central Nervous System Through CRISPR/Cas9 Systems. Front. Mol. Neurosci. 2019, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Yabluchanskiy, A.; Tarantini, S.; Balasubramanian, P.; Kiss, T.; Csipo, T.; Fulop, G.A.; Lipecz, A.; Ahire, C.; DelFavero, J.; Nyul-Toth, A.; et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation-induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience 2020, 42, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Turnquist, C.; Beck, J.A.; Horikawa, I.; Obiorah, I.E.; Von Muhlinen, N.; Vojtesek, B.; Lane, D.P.; Grunseich, C.; Chahine, J.J.; Ames, H.M.; et al. Radiation-induced astrocyte senescence is rescued by Delta133p53. Neuro. Oncol. 2019, 21, 474–485. [Google Scholar] [CrossRef]
- Hou, J.; Cui, C.; Kim, S.; Sung, C.; Choi, C. Ginsenoside F1 suppresses astrocytic senescence-associated secretory phenotype. Chem. Biol. Interact. 2018, 283, 75–83. [Google Scholar] [CrossRef]
- Fan, S.; Xian, X.; Li, L.; Yao, X.; Hu, Y.; Zhang, M.; Li, W. Ceftriaxone Improves Cognitive Function and Upregulates GLT-1-Related Glutamate-Glutamine Cycle in APP/PS1 Mice. J. Alzheimers Dis. 2018, 66, 1731–1743. [Google Scholar] [CrossRef]
- Okamoto, M.; Gray, J.D.; Larson, C.S.; Kazim, S.F.; Soya, H.; McEwen, B.S.; Pereira, A.C. Riluzole reduces amyloid beta pathology, improves memory, and restores gene expression changes in a transgenic mouse model of early-onset Alzheimer’s disease. Transl. Psychiatry 2018, 8, 153. [Google Scholar] [CrossRef]
- Konttinen, H.; Gureviciene, I.; Oksanen, M.; Grubman, A.; Loppi, S.; Huuskonen, M.T.; Korhonen, P.; Lampinen, R.; Keuters, M.; Belaya, I.; et al. PPARbeta/delta-agonist GW0742 ameliorates dysfunction in fatty acid oxidation in PSEN1DeltaE9 astrocytes. Glia 2019, 67, 146–159. [Google Scholar] [CrossRef]
- van Gijsel-Bonnello, M.; Baranger, K.; Benech, P.; Rivera, S.; Khrestchatisky, M.; de Reggi, M.; Gharib, B. Metabolic changes and inflammation in cultured astrocytes from the 5xFAD mouse model of Alzheimer’s disease: Alleviation by pantethine. PLoS ONE 2017, 12, e0175369. [Google Scholar] [CrossRef]
- Crespo, M.C.; Tome-Carneiro, J.; Pintado, C.; Davalos, A.; Visioli, F.; Burgos-Ramos, E. Hydroxytyrosol restores proper insulin signaling in an astrocytic model of Alzheimer’s disease. Biofactors 2017, 43, 540–548. [Google Scholar] [CrossRef]
- Zheng, J.; Xie, Y.; Ren, L.; Qi, L.; Wu, L.; Pan, X.; Zhou, J.; Chen, Z.; Liu, L. GLP-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer’s disease. Mol. Metab. 2021, 47, 101180. [Google Scholar] [CrossRef]
- Wang, G.; Cui, W.; Chen, S.; Shao, Z.; Li, Y.; Wang, W.; Mao, L.; Li, J.; Mei, X. Metformin alleviates high glucose-induced ER stress and inflammation by inhibiting the interaction between caveolin1 and AMPKalpha in rat astrocytes. Biochem. Biophys. Res. Commun. 2021, 534, 908–913. [Google Scholar] [CrossRef]
- Sawamoto, A.; Okuyama, S.; Nakajima, M.; Furukawa, Y. Citrus flavonoid 3,5,6,7,8,3′,4′-heptamethoxyflavone induces BDNF via cAMP/ERK/CREB signaling and reduces phosphodiesterase activity in C6 cells. Pharmacol. Rep. 2019, 71, 653–658. [Google Scholar] [CrossRef]
- Luo, G.; Huang, Y.; Jia, B.; Zhang, X.; Mo, D.; Ma, N.; Gao, F.; Song, L.; Wang, B.; Miao, Z. Quetiapine prevents Abeta25-35-induced cell death in cultured neuron by enhancing brain-derived neurotrophic factor release from astrocyte. Neuroreport 2018, 29, 92–98. [Google Scholar] [CrossRef]
- de Pins, B.; Cifuentes-Diaz, C.; Farah, A.T.; Lopez-Molina, L.; Montalban, E.; Sancho-Balsells, A.; Lopez, A.; Gines, S.; Delgado-Garcia, J.M.; Alberch, J.; et al. Conditional BDNF Delivery from Astrocytes Rescues Memory Deficits, Spine Density, and Synaptic Properties in the 5xFAD Mouse Model of Alzheimer Disease. J. Neurosci. 2019, 39, 2441–2458. [Google Scholar] [CrossRef]
- Bali, P.; Banik, A.; Nehru, B.; Anand, A. Neurotrophic Factors Mediated Activation of Astrocytes Ameliorate Memory Loss by Amyloid Clearance after Transplantation of Lineage Negative Stem Cells. Mol. Neurobiol. 2019, 56, 8420–8434. [Google Scholar] [CrossRef]
- Chen, X.; Li, Z.; Cheng, Y.; Kardami, E.; Loh, Y.P. Low and High Molecular Weight FGF-2 Have Differential Effects on Astrocyte Proliferation, but Are Both Protective Against Abeta-Induced Cytotoxicity. Front. Mol. Neurosci. 2019, 12, 328. [Google Scholar] [CrossRef]
- Chernick, D.; Ortiz-Valle, S.; Jeong, A.; Swaminathan, S.K.; Kandimalla, K.K.; Rebeck, G.W.; Li, L. High-density lipoprotein mimetic peptide 4F mitigates amyloid-beta-induced inhibition of apolipoprotein E secretion and lipidation in primary astrocytes and microglia. J. Neurochem. 2018, 147, 647–662. [Google Scholar] [CrossRef] [PubMed]
- Wojtas, A.M.; Sens, J.P.; Kang, S.S.; Baker, K.E.; Berry, T.J.; Kurti, A.; Daughrity, L.; Jansen-West, K.R.; Dickson, D.W.; Petrucelli, L.; et al. Astrocyte-derived clusterin suppresses amyloid formation in vivo. Mol. Neurodegener. 2020, 15, 71. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Swartzlander, D.B.; Ghosh, A.; Fryer, J.D.; Wang, B.; Zheng, H. Clusterin secreted from astrocyte promotes excitatory synaptic transmission and ameliorates Alzheimer’s disease neuropathology. Mol. Neurodegener. 2021, 16, 5. [Google Scholar] [CrossRef]
- Yamamoto, N.; Shibata, M.; Ishikuro, R.; Tanida, M.; Taniguchi, Y.; Ikeda-Matsuo, Y.; Sobue, K. Epigallocatechin gallate induces extracellular degradation of amyloid beta-protein by increasing neprilysin secretion from astrocytes through activation of ERK and PI3K pathways. Neuroscience 2017, 362, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Luo, C.; Feng, Y.; Yao, X.; Shi, Z.; Liang, F.; Kang, J.X.; Wan, J.B.; Pei, Z.; Su, H. Omega-3 polyunsaturated fatty acids promote amyloid-beta clearance from the brain through mediating the function of the glymphatic system. FASEB J. 2017, 31, 282–293. [Google Scholar] [CrossRef]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- McManus, R.M.; Finucane, O.M.; Wilk, M.M.; Mills, K.H.G.; Lynch, M.A. FTY720 Attenuates Infection-Induced Enhancement of Abeta Accumulation in APP/PS1 Mice by Modulating Astrocytic Activation. J. Neuroimmune Pharmacol. 2017, 12, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Stancu, I.C.; Schakman, O.; Pierrot, N.; Huaux, F.; Kienlen-Campard, P.; Dewachter, I.; Octave, J.N. Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J. Neuroinflammation 2016, 13, 20. [Google Scholar] [CrossRef]
- Dekens, D.W.; De Deyn, P.P.; Sap, F.; Eisel, U.L.M.; Naude, P.J.W. Iron chelators inhibit amyloid-beta-induced production of lipocalin 2 in cultured astrocytes. Neurochem. Int. 2020, 132, 104607. [Google Scholar] [CrossRef]
- Kim, M.; Park, M.H.; Nam, G.; Lee, M.; Kang, J.; Song, I.S.; Choi, M.K.; Jin, H.K.; Bae, J.S.; Lim, M.H. A Glycosylated Prodrug to Attenuate Neuroinflammation and Improve Cognitive Deficits in Alzheimer’s Disease Transgenic Mice. Mol. Pharm. 2021, 18, 101–112. [Google Scholar] [CrossRef]
- Ceyzeriat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef]
- Beggiato, S.; Borelli, A.C.; Ferraro, L.; Tanganelli, S.; Antonelli, T.; Tomasini, M.C. Palmitoylethanolamide Blunts Amyloid-beta42-Induced Astrocyte Activation and Improves Neuronal Survival in Primary Mouse Cortical Astrocyte-Neuron Co-Cultures. J. Alzheimers Dis. 2018, 61, 389–399. [Google Scholar] [CrossRef]
- Beggiato, S.; Cassano, T.; Ferraro, L.; Tomasini, M.C. Astrocytic palmitoylethanolamide pre-exposure exerts neuroprotective effects in astrocyte-neuron co-cultures from a triple transgenic mouse model of Alzheimer’s disease. Life Sci. 2020, 257, 118037. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L., Jr.; Romano, A.; Stecca, C.; Passarella, S.; Steardo, L.; Cassano, T.; Scuderi, C. Palmitoylethanolamide Dampens Reactive Astrogliosis and Improves Neuronal Trophic Support in a Triple Transgenic Model of Alzheimer’s Disease: In Vitro and In Vivo Evidence. Oxid. Med. Cell Longev. 2018, 2018, 4720532. [Google Scholar] [CrossRef]
- Casili, G.; Lanza, M.; Campolo, M.; Siracusa, R.; Paterniti, I.; Ardizzone, A.; Scuderi, S.A.; Cuzzocrea, S.; Esposito, E. Synergic Therapeutic Potential of PEA-Um Treatment and NAAA Enzyme Silencing In the Management of Neuroinflammation. Int. J. Mol. Sci. 2020, 21, 7486. [Google Scholar] [CrossRef]
- Tsoy, A.; Saliev, T.; Abzhanova, E.; Turgambayeva, A.; Kaiyrlykyzy, A.; Akishev, M.; Saparbayev, S.; Umbayev, B.; Askarova, S. The Effects of Mobile Phone Radiofrequency Electromagnetic Fields on beta-Amyloid-Induced Oxidative Stress in Human and Rat Primary Astrocytes. Neuroscience 2019, 408, 46–57. [Google Scholar] [CrossRef]
- Sohanaki, H.; Baluchnejadmojarad, T.; Nikbakht, F.; Roghani, M. Pelargonidin improves memory deficit in amyloid beta25-35 rat model of Alzheimer’s disease by inhibition of glial activation, cholinesterase, and oxidative stress. Biomed. Pharmacother. 2016, 83, 85–91. [Google Scholar] [CrossRef]
- Shi, Y.C.; Pan, T.M.; Liao, V.H. Monascin from Monascus-Fermented Products Reduces Oxidative Stress and Amyloid-beta Toxicity via DAF-16/FOXO in Caenorhabditis elegans. J. Agric. Food Chem. 2016, 64, 7114–7120. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Wang, Y.; Xu, W.; Liu, Y.; Chen, H.; Yu, Z. KCa3.1 deficiency attenuates neuroinflammation by regulating an astrocyte phenotype switch involving the PI3K/AKT/GSK3beta pathway. Neurobiol. Dis. 2019, 132, 104588. [Google Scholar] [CrossRef]
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2Y1 receptor blockade normalizes network dysfunction and cognition in an Alzheimer’s disease model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef]
- Lee, C.C.; Chang, C.P.; Lin, C.J.; Lai, H.L.; Kao, Y.H.; Cheng, S.J.; Chen, H.M.; Liao, Y.P.; Faivre, E.; Buee, L.; et al. Adenosine Augmentation Evoked by an ENT1 Inhibitor Improves Memory Impairment and Neuronal Plasticity in the APP/PS1 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8936–8952. [Google Scholar] [CrossRef]
- Orr, A.G.; Lo, I.; Schumacher, H.; Ho, K.; Gill, M.; Guo, W.; Kim, D.H.; Knox, A.; Saito, T.; Saido, T.C.; et al. Istradefylline reduces memory deficits in aging mice with amyloid pathology. Neurobiol. Dis. 2018, 110, 29–36. [Google Scholar] [CrossRef]
- Scuderi, C.; Bronzuoli, M.R.; Facchinetti, R.; Pace, L.; Ferraro, L.; Broad, K.D.; Serviddio, G.; Bellanti, F.; Palombelli, G.; Carpinelli, G.; et al. Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer’s disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry 2018, 8, 32. [Google Scholar] [CrossRef]
- Kritsilis, M.; Rizou, S.V.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Valenza, M.; Cassano, T.; Steardo, L.; Scuderi, C. Astrocyte Function Is Affected by Aging and Not Alzheimer’s Disease: A Preliminary Investigation in Hippocampi of 3xTg-AD Mice. Front. Pharmacol. 2019, 10, 644. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell 2016, 15, 416–427. [Google Scholar] [CrossRef]
- Crowe, E.P.; Tuzer, F.; Gregory, B.D.; Donahue, G.; Gosai, S.J.; Cohen, J.; Leung, Y.Y.; Yetkin, E.; Nativio, R.; Wang, L.S.; et al. Changes in the Transcriptome of Human Astrocytes Accompanying Oxidative Stress-Induced Senescence. Front. Aging Neurosci. 2016, 8, 208. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Rane, A.; Demaria, M.; Campisi, J.; Andersen, J.K. Cellular senescence and the aging brain. Exp Gerontol 2015, 68, 3–7. [Google Scholar] [CrossRef]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Capoccia, E.; Cirillo, C.; Marchetto, A.; Tiberi, S.; Sawikr, Y.; Pesce, M.; D’Alessandro, A.; Scuderi, C.; Sarnelli, G.; Cuomo, R.; et al. S100B-p53 disengagement by pentamidine promotes apoptosis and inhibits cellular migration via aquaporin-4 and metalloproteinase-2 inhibition in C6 glioma cells. Oncol. Lett. 2015, 9, 2864–2870. [Google Scholar] [CrossRef]
- Cirillo, C.; Capoccia, E.; Iuvone, T.; Cuomo, R.; Sarnelli, G.; Steardo, L.; Esposito, G. S100B Inhibitor Pentamidine Attenuates Reactive Gliosis and Reduces Neuronal Loss in a Mouse Model of Alzheimer’s Disease. Biomed. Res. Int. 2015, 2015, 508342. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef]
- Mookherjee, P.; Green, P.S.; Watson, G.S.; Marques, M.A.; Tanaka, K.; Meeker, K.D.; Meabon, J.S.; Li, N.; Zhu, P.; Olson, V.G.; et al. GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. J. Alzheimers Dis. 2011, 26, 447–455. [Google Scholar] [CrossRef]
- Garaschuk, O.; Verkhratsky, A. GABAergic astrocytes in Alzheimer’s disease. Aging 2019, 11, 1602–1604. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Brawek, B.; Chesters, R.; Klement, D.; Muller, J.; Lerdkrai, C.; Hermes, M.; Garaschuk, O. A bell-shaped dependence between amyloidosis and GABA accumulation in astrocytes in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 61, 187–197. [Google Scholar] [CrossRef]
- Kwakowsky, A.; Calvo-Flores Guzman, B.; Pandya, M.; Turner, C.; Waldvogel, H.J.; Faull, R.L. GABAA receptor subunit expression changes in the human Alzheimer’s disease hippocampus, subiculum, entorhinal cortex and superior temporal gyrus. J. Neurochem. 2018, 145, 374–392. [Google Scholar] [CrossRef]
- Fontana, A.C. Current approaches to enhance glutamate transporter function and expression. J. Neurochem. 2015, 134, 982–1007. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Su, Z.Z.; Emdad, L.; Gupta, P.; Sarkar, D.; Borjabad, A.; Volsky, D.J.; Fisher, P.B. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J. Biol. Chem. 2008, 283, 13116–13123. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Dykes Hoberg, M.; Vidensky, S.; Chung, D.S.; et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef]
- Pereira, A.C.; Lambert, H.K.; Grossman, Y.S.; Dumitriu, D.; Waldman, R.; Jannetty, S.K.; Calakos, K.; Janssen, W.G.; McEwen, B.S.; Morrison, J.H. Glutamatergic regulation prevents hippocampal-dependent age-related cognitive decline through dendritic spine clustering. Proc. Natl. Acad. Sci. USA 2014, 111, 18733–18738. [Google Scholar] [CrossRef]
- Fumagalli, E.; Funicello, M.; Rauen, T.; Gobbi, M.; Mennini, T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur. J. Pharmacol. 2008, 578, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.C.; Gray, J.D.; Kogan, J.F.; Davidson, R.L.; Rubin, T.G.; Okamoto, M.; Morrison, J.H.; McEwen, B.S. Age and Alzheimer’s disease gene expression profiles reversed by the glutamate modulator riluzole. Mol. Psychiatry 2017, 22, 296–305. [Google Scholar] [CrossRef]
- Ciavardelli, D.; Piras, F.; Consalvo, A.; Rossi, C.; Zucchelli, M.; Di Ilio, C.; Frazzini, V.; Caltagirone, C.; Spalletta, G.; Sensi, S.L. Medium-chain plasma acylcarnitines, ketone levels, cognition, and gray matter volumes in healthy elderly, mildly cognitively impaired, or Alzheimer’s disease subjects. Neurobiol. Aging 2016, 43, 1–12. [Google Scholar] [CrossRef]
- Cai, X.S.; Tan, Z.G.; Li, J.J.; Gao, W.H.; Li, S.J.; Li, J.L.; Tang, Y.M.; Li, H.W.; Hui, H.X. Glucagon-Like Peptide-1 (GLP-1) Treatment Ameliorates Cognitive Impairment by Attenuating Arc Expression in Type 2 Diabetic Rats. Med. Sci. Monit. 2017, 23, 4334–4342. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schindowski, K.; Belarbi, K.; Buee, L. Neurotrophic factors in Alzheimer’s disease: Role of axonal transport. Genes Brain Behav. 2008, 7, 43–56. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Banik, A.; Prabhakar, S.; Kalra, J.; Anand, A. Effect of human umbilical cord blood derived lineage negative stem cells transplanted in amyloid-beta induced cognitive impaired mice. Behav. Brain Res. 2015, 291, 46–59. [Google Scholar] [CrossRef]
- Taliyan, R.; Chandran, S.K.; Kakoty, V. Therapeutic Approaches to Alzheimer’s Type of Dementia: A Focus on FGF21 Mediated Neuroprotection. Curr. Pharm. Des. 2019, 25, 2555–2568. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, C.; Lilli, C.; Baroni, T.; Parnetti, L.; Sorbi, S.; Emiliani, C.; Lumare, E.; Calabresi, P.; Balloni, S.; Bodo, M. Differences in extracellular matrix production and basic fibroblast growth factor response in skin fibroblasts from sporadic and familial Alzheimer’s disease. Mol. Med. 2007, 13, 542–550. [Google Scholar] [CrossRef]
- Kato, T.; Sasaki, H.; Katagiri, T.; Sasaki, H.; Koiwai, K.; Youki, H.; Totsuka, S.; Ishii, T. The binding of basic fibroblast growth factor to Alzheimer’s neurofibrillary tangles and senile plaques. Neurosci. Lett. 1991, 122, 33–36. [Google Scholar] [CrossRef]
- Sollvander, S.; Nikitidou, E.; Brolin, R.; Soderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-beta by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 38. [Google Scholar] [CrossRef]
- Pan, J.; Ma, N.; Yu, B.; Zhang, W.; Wan, J. Transcriptomic profiling of microglia and astrocytes throughout aging. J. Neuroinflamm. 2020, 17, 97. [Google Scholar] [CrossRef]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Lalakova, J.; Kuhnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e19. [Google Scholar] [CrossRef] [PubMed]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef]
- Fernandez, C.G.; Hamby, M.E.; McReynolds, M.L.; Ray, W.J. The Role of APOE4 in Disrupting the Homeostatic Functions of Astrocytes and Microglia in Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef]
- DeMattos, R.B.; O’Dell, M.A.; Parsadanian, M.; Taylor, J.W.; Harmony, J.A.K.; Bales, K.R.; Paul, S.M.; Aronow, B.J.; Holtzman, D.M. Clusterin promotes amyloid plaque formation and is critical for neuritic toxicity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10843–10848. [Google Scholar] [CrossRef]
- Narayan, P.; Orte, A.; Clarke, R.W.; Bolognesi, B.; Hook, S.; Ganzinger, K.A.; Meehan, S.; Wilson, M.R.; Dobson, C.M.; Klenerman, D. The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-beta(1-40) peptide. Nat. Struct. Mol. Biol. 2011, 19, 79–83. [Google Scholar] [CrossRef]
- Valenza, M.; Facchinetti, R.; Steardo, L.; Scuderi, C. Altered Waste Disposal System in Aging and Alzheimer’s Disease: Focus on Astrocytic Aquaporin-4. Front. Pharmacol. 2020, 10, 1656. [Google Scholar] [CrossRef]
- Steardo, L., Jr.; Bronzuoli, M.R.; Iacomino, A.; Esposito, G.; Steardo, L.; Scuderi, C. Does neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytes. Front. Neurosci. 2015, 9, 259. [Google Scholar] [CrossRef]
- Scuderi, C.; Stecca, C.; Iacomino, A.; Steardo, L. Role of astrocytes in major neurological disorders: The evidence and implications. IUBMB Life 2013, 65, 957–961. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Facchinetti, R.; Steardo, L.; Valenza, M. Neuroinflammation in Alzheimer’s Disease: Friend or Foe? FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Scuderi, C.; Noda, M.; Verkhratsky, A. Editorial: Neuroglia Molecular Mechanisms in Psychiatric Disorders. Front. Mol. Neurosci. 2018, 11, 407. [Google Scholar] [CrossRef] [PubMed]
- Schwab, C.; Klegeris, A.; McGeer, P.L. Inflammation in transgenic mouse models of neurodegenerative disorders. Biochim. Biophys. Acta 2010, 1802, 889–902. [Google Scholar] [CrossRef]
- Scuderi, C.; Filippis, D.D.; Iuvone, T.; Blasio, A.; Steardo, A.; Esposito, G. Cannabidiol in medicine: A review of its therapeutic potential in CNS disorders. Phytother. Res. 2009, 23, 597–602. [Google Scholar] [CrossRef]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5YAPP+ cells through PPARgamma involvement. Phytother. Res. 2014, 28, 1007–1013. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L., Jr.; De Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V.; Steardo, L. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratu, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Abeta-induced neuroinflammation and promotes hippocampal neurogenesis through PPARgamma involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef]
- Wu, T.; Dejanovic, B.; Gandham, V.D.; Gogineni, A.; Edmonds, R.; Schauer, S.; Srinivasan, K.; Huntley, M.A.; Wang, Y.; Wang, T.M.; et al. Complement C3 Is Activated in Human AD Brain and Is Required for Neurodegeneration in Mouse Models of Amyloidosis and Tauopathy. Cell Rep. 2019, 28, 2111–2123.e6. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Stecca, C.; Bronzuoli, M.R.; Rotili, D.; Valente, S.; Mai, A.; Steardo, L. Sirtuin modulators control reactive gliosis in an in vitro model of Alzheimer’s disease. Front. Pharmacol. 2014, 5, 89. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo, L., Jr.; Carnuccio, R.; De Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by beta-amyloid peptide. J. Cell Mol. Med. 2011, 15, 2664–2674. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Hermann, P.; Villar-Pique, A.; Diaz-Lucena, D.; Nagga, K.; Hansson, O.; Santana, I.; Schmitz, M.; Schmidt, C.; Varges, D.; et al. Cerebrospinal fluid lipocalin 2 as a novel biomarker for the differential diagnosis of vascular dementia. Nat. Commun. 2020, 11, 619. [Google Scholar] [CrossRef]
- Mesquita, S.D.; Ferreira, A.C.; Falcao, A.M.; Sousa, J.C.; Oliveira, T.G.; Correia-Neves, M.; Sousa, N.; Marques, F.; Palha, J.A. Lipocalin 2 modulates the cellular response to amyloid beta. Cell Death Differ. 2014, 21, 1588–1599. [Google Scholar] [CrossRef] [PubMed]
- Staurenghi, E.; Cerrato, V.; Gamba, P.; Testa, G.; Giannelli, S.; Leoni, V.; Caccia, C.; Buffo, A.; Noble, W.; Perez-Nievas, B.G.; et al. Oxysterols present in Alzheimer’s disease brain induce synaptotoxicity by activating astrocytes: A major role for lipocalin-2. Redox. Biol. 2021, 39, 101837. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Shah, S.A.; Ali, T.; Chung, J.I.; Kim, M.O. Anthocyanins Reversed D-Galactose-Induced Oxidative Stress and Neuroinflammation Mediated Cognitive Impairment in Adult Rats. Mol. Neurobiol. 2017, 54, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Chen, L.; Pan, X.; Chen, J.; Wang, L.; Wang, W.; Cheng, R.; Wu, F.; Feng, X.; Yu, Y.; et al. The effect of resveratrol on beta amyloid-induced memory impairment involves inhibition of phosphodiesterase-4 related signaling. Oncotarget 2016, 7, 17380–17392. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.F.; Yanagisawa, D.; Durani, L.W.; Hamezah, H.S.; Damanhuri, H.A.; Wan Ngah, W.Z.; Tsuji, M.; Kiuchi, Y.; Ono, K.; Tooyama, I. Tocotrienol-Rich Fraction Modulates Amyloid Pathology and Improves Cognitive Function in AbetaPP/PS1 Mice. J. Alzheimers Dis. 2017, 55, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, E.; Limon, D.; Perez-Severiano, F.; Diaz, A.; Ortega, L.; Zenteno, E.; Guevara, J. Antioxidant effects of epicatechin on the hippocampal toxicity caused by amyloid-beta 25-35 in rats. Eur. J. Pharmacol. 2009, 616, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, M.; Hu, J.; Shen, W.; Hu, J.; Yao, Y.; Wang, X.; Afzal, C.M.; Ma, R.; Li, G. Protective effect of 3H-1, 2-dithiole-3-thione on cellular model of Alzheimer’s disease involves Nrf2/ARE signaling pathway. Eur. J. Pharmacol. 2017, 795, 115–123. [Google Scholar] [CrossRef]
- Sharman, M.J.; Gyengesi, E.; Liang, H.; Chatterjee, P.; Karl, T.; Li, Q.X.; Wenk, M.R.; Halliwell, B.; Martins, R.N.; Munch, G. Assessment of diets containing curcumin, epigallocatechin-3-gallate, docosahexaenoic acid and alpha-lipoic acid on amyloid load and inflammation in a male transgenic mouse model of Alzheimer’s disease: Are combinations more effective? Neurobiol. Dis. 2019, 124, 505–519. [Google Scholar] [CrossRef]
- Delekate, A.; Fuchtemeier, M.; Schumacher, T.; Ulbrich, C.; Foddis, M.; Petzold, G.C. Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat. Commun. 2014, 5, 5422. [Google Scholar] [CrossRef]
- Orr, A.G.; Hsiao, E.C.; Wang, M.M.; Ho, K.; Kim, D.H.; Wang, X.; Guo, W.; Kang, J.; Yu, G.Q.; Adame, A.; et al. Astrocytic adenosine receptor A2A and Gs-coupled signaling regulate memory. Nat. Neurosci. 2015, 18, 423–434. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Marutle, A.; Rodriguez-Arellano, J.J.; Nordberg, A. Glial Asthenia and Functional Paralysis: A New Perspective on Neurodegeneration and Alzheimer’s Disease. Neuroscientist 2015, 21, 552–568. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Steardo, L. Neuroglial roots of neurodegenerative diseases: Therapeutic potential of palmitoylethanolamide in models of Alzheimer’s disease. CNS Neurol. Disord. Drug. Targets. 2013, 12, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Tomasini, M.C.; Ferraro, L. Palmitoylethanolamide (PEA) as a Potential Therapeutic Agent in Alzheimer’s Disease. Front. Pharmacol. 2019, 10, 821. [Google Scholar] [CrossRef] [PubMed]
- Cipriano, M.; Esposito, G.; Negro, L.; Capoccia, E.; Sarnelli, G.; Scuderi, C.; De Filippis, D.; Steardo, L.; Iuvone, T. Palmitoylethanolamide Regulates Production of Pro-Angiogenic Mediators in a Model of beta Amyloid-Induced Astrogliosis In Vitro. CNS Neurol. Disord. Drug Targets 2015, 14, 828–837. [Google Scholar] [CrossRef]
- Scuderi, C.; Stecca, C.; Valenza, M.; Ratano, P.; Bronzuoli, M.R.; Bartoli, S.; Steardo, L.; Pompili, E.; Fumagalli, L.; Campolongo, P.; et al. Palmitoylethanolamide controls reactive gliosis and exerts neuroprotective functions in a rat model of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1419. [Google Scholar] [CrossRef]
- Scuderi, C.; Valenza, M.; Stecca, C.; Esposito, G.; Carratu, M.R.; Steardo, L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-alpha. J. Neuroinflammation 2012, 9, 49. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef]
- D’Agostino, G.; Russo, R.; Avagliano, C.; Cristiano, C.; Meli, R.; Calignano, A. Palmitoylethanolamide protects against the amyloid-beta25-35-induced learning and memory impairment in mice, an experimental model of Alzheimer disease. Neuropsychopharmacology 2012, 37, 1784–1792. [Google Scholar] [CrossRef]
- Esposito, E.; Impellizzeri, D.; Mazzon, E.; Paterniti, I.; Cuzzocrea, S. Neuroprotective activities of palmitoylethanolamide in an animal model of Parkinson’s disease. PLoS ONE 2012, 7, e41880. [Google Scholar] [CrossRef]
- Pertwee, R.G. GPR55: A new member of the cannabinoid receptor clan? Br. J. Pharmacol. 2007, 152, 984–986. [Google Scholar] [CrossRef]
- Zygmunt, P.M.; Ermund, A.; Movahed, P.; Andersson, D.A.; Simonsen, C.; Jonsson, B.A.; Blomgren, A.; Birnir, B.; Bevan, S.; Eschalier, A.; et al. Monoacylglycerols activate TRPV1—A link between phospholipase C and TRPV1. PLoS ONE 2013, 8, e81618. [Google Scholar] [CrossRef]
- Mattace Raso, G.; Russo, R.; Calignano, A.; Meli, R. Palmitoylethanolamide in CNS health and disease. Pharmacol. Res. 2014, 86, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharmacol. 2017, 174, 1349–1365. [Google Scholar] [CrossRef] [PubMed]
- Harcha, P.A.; Garces, P.; Arredondo, C.; Fernandez, G.; Saez, J.C.; van Zundert, B. Mast Cell and Astrocyte Hemichannels and Their Role in Alzheimer’s Disease, ALS, and Harmful Stress Conditions. Int. J. Mol. Sci. 2021, 22, 1924. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, J.K.; Kulka, M. Decoding Mast Cell-Microglia Communication in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 1093. [Google Scholar] [CrossRef]
- Petrosino, S.; Schiano Moriello, A. Palmitoylethanolamide: A Nutritional Approach to Keep Neuroinflammation within Physiological Boundaries-A Systematic Review. Int. J. Mol. Sci. 2020, 21, 9526. [Google Scholar] [CrossRef] [PubMed]
- Paterniti, I.; Cordaro, M.; Campolo, M.; Siracusa, R.; Cornelius, C.; Navarra, M.; Cuzzocrea, S.; Esposito, E. Neuroprotection by association of palmitoylethanolamide with luteolin in experimental Alzheimer’s disease models: The control of neuroinflammation. CNS Neurol. Disord. Drug Targets 2014, 13, 1530–1541. [Google Scholar] [CrossRef]
- Beggiato, S.; Tomasini, M.C.; Cassano, T.; Ferraro, L. Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 428. [Google Scholar] [CrossRef]
- Cassano, T.; Bellanti, F.; Bukke, V.N.; Archana, M.; Scuderi, C.; Ferraro, L.; Serviddio, G.; Cuomo, V. Effects of ultramicronized palmitoylethanolamide treatment on the glutamatergic alterations and mitochondrial impairment in 3×tg-ad mice. Pharmadvances 2021, 3, 87–88. [Google Scholar] [CrossRef]
- Scuderi, C. Unraveling the complex glial response in aging and alzheimer’s disease. Pharmadvances 2021, 3. [Google Scholar] [CrossRef]
- Assogna, M.; Casula, E.P.; Borghi, I.; Bonni, S.; Sama, D.; Motta, C.; Di Lorenzo, F.; D’Acunto, A.; Porrazzini, F.; Minei, M.; et al. Effects of Palmitoylethanolamide Combined with Luteoline on Frontal Lobe Functions, High Frequency Oscillations, and GABAergic Transmission in Patients with Frontotemporal Dementia. J. Alzheimers Dis. 2020, 76, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Calabrò, R.S.; Naro, A.; De Luca, R.; Leonardi, S.; Russo, M.; Marra, A.; Bramanti, P. PEALut efficacy in mild cognitive impairment: Evidence from a SPECT case study! Aging Clin. Exp. Res. 2016, 28, 1279–1282. [Google Scholar] [CrossRef]
- Caltagirone, C.; Cisari, C.; Schievano, C.; Di Paola, R.; Cordaro, M.; Bruschetta, G.; Esposito, E.; Cuzzocrea, S.; Stroke Study, G. Co-ultramicronized Palmitoylethanolamide/Luteolin in the Treatment of Cerebral Ischemia: From Rodent to Man. Transl. Stroke Res. 2016, 7, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Paladini, A.; Fusco, M.; Cenacchi, T.; Schievano, C.; Piroli, A.; Varrassi, G. Palmitoylethanolamide, a Special Food for Medical Purposes, in the Treatment of Chronic Pain: A Pooled Data Meta-analysis. Pain Physician 2016, 19, 11–24. [Google Scholar] [PubMed]


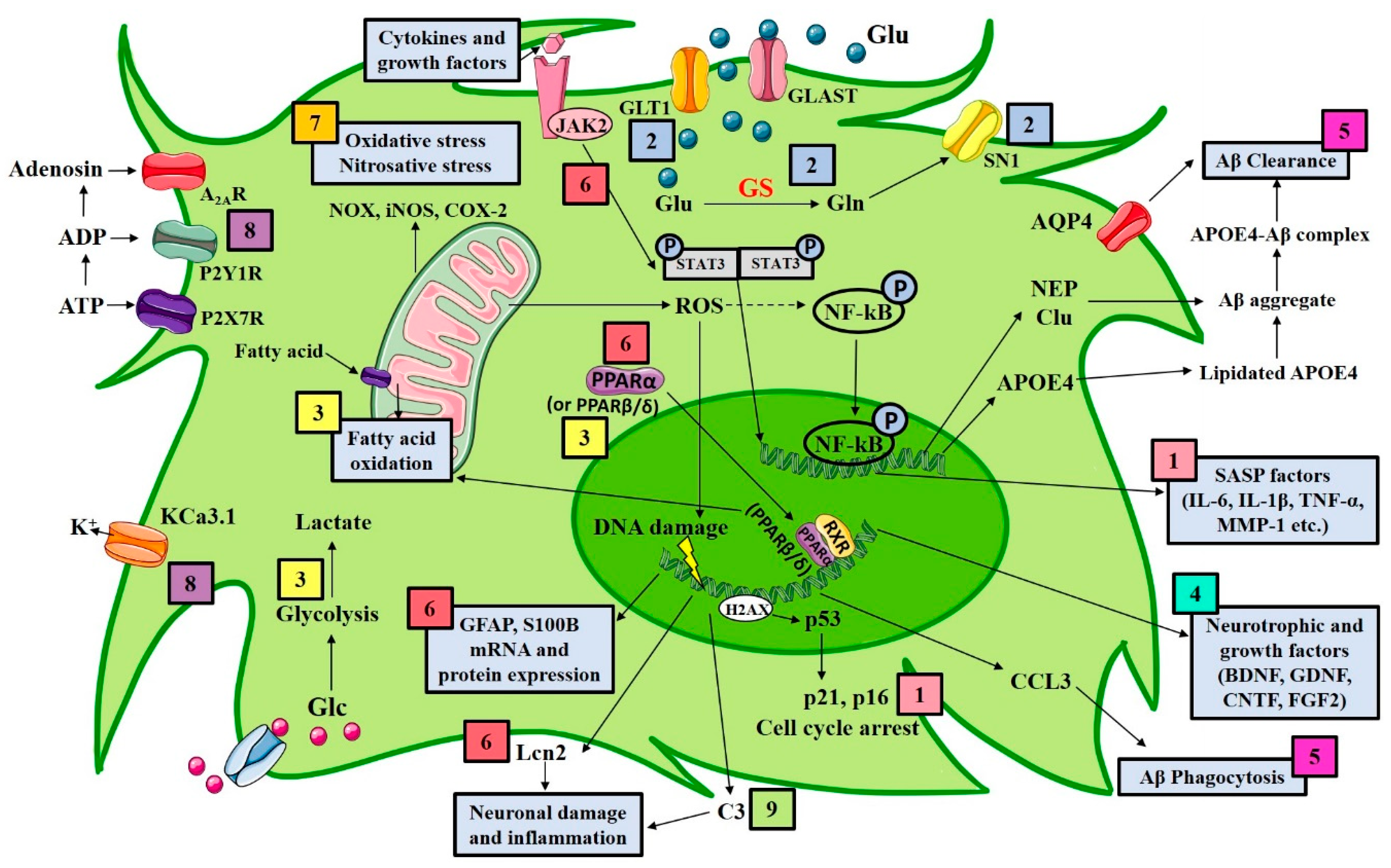{kind=link}
| Astrocytic Target | Experimental Strategy | Results | References |
|---|---|---|---|
| Senescence | Removal of senescent cells in vivo by radiation treatment or by genetic ablation | Reduction in astrogliosis, tau hyperphosphorylation, neuronal degeneration; preservation of cognition | [181,182] |
| In vivo administration of the senolytic drug ABT263 (navitoclax) | Prevention from the upregulation of senescence-associated genes attenuated tau phosphorylation; cognitive improvements | [181,182] | |
| Overexpression of Δ133p53 in radiation-induced senescent astrocytes | Repression of the irradiation-induced SASP | [183] | |
| Ginsenoside F1 in vitro treatment | SASP suppression by downregulation of p38MAPK-dependent NF-κB pathway | [184] | |
| Glutamate transporters | Ceftriaxone administration in APP/PS1 mice | Raise in GLT1, GS and SN1 protein expression and cognitive performance improvements | [185] |
| Chronic oral administration of riluzole in 5xFAD mice | Prevention of senescent associated gene expression changes; reduction of Aβ oligomers and plaques | [186] | |
| Metabolism | PPARβ/δ agonist treatment of human AD astrocytes (PSEN1ΔE9) | Enhancement of AD-reduced fatty acid oxidation | [187] |
| Pantethine in vitro treatment of astrocytes obtained from 5xFAD mice | Reversal of the altered activity of several metabolic enzymes and of the induced IL-1β expression | [188] | |
| Hydroxytyrosol treatment of glioma cell cultures challenged with Aβ (25–35) | Proper glucose metabolism restoration by Akt activation | [189] | |
| GLP-1 in vitro treatment of Aβ-exposed astrocytes | Reversal of the Aβ-altered glycolysis by activation of the PI3K/Akt pathway | [190] | |
| Metformin in vitro treatment of astrocytes exposed to high glucose concentration | Inhibition of both the ER stress and inflammation induced by high glucose | [191] | |
| Neurotrophins and growth factors | HMF treatment of primary astrocytes and C6 glioma cell line | Raise in BDNF expression induced by both the activation of cAMP/ERK/CREB signaling and the inhibition of PDE4B and PDE4D | [192] |
| Primary neurons exposed to Aβ (25–35) incubated with quetiapine-treated astrocyte conditioned medium | High BDNF release by astrocytes treated with quetiapine promoted viability of primary neurons | [193] | |
| Overexpression of BDNF specifically in GFAP-positive astrocytes by genetic crossing in 5xFAD mice | The raise in BDNF levels that are reduced in 5xFAD mice improved synaptic plasticity and cognition | [194] | |
| In situ stem cell transplant in intrahippocampal Aβ42 infused mice | Reversal of the Aβ42-induced cognitive impairment by BDNF-TrKB pathway activation | [195] | |
| FGF2 treatment of primary astrocytes challenged with Aβ42 | Promotion of astrocyte proliferation through enhanced expression of c-Myc, Cyclin D1, Cyclin E | [196] | |
| Aβ clearance | HDL mimetic peptide in vitro treatment of primary human and murine astrocytes | Raise in apoE4 lipidation lowers its detrimental cellular accumulation | [197] |
| In vivo overexpression or downregulation of Clu specifically in GFAP-positive astrocytes in APP/PS1 mice | Clu overexpression is associated with a reduction in Aβ burden. The opposite phenomenon was found with Clu downregulation | [198] | |
| In vivo overexpression of Clu specifically in GFAP-positive astrocytes in 5xFAD mice | Reduction in plaques number and sizes. Improvement in synaptic function | [199] | |
| EGCG treatment of Aβ40 challenged medium from cultured astrocytes | Elevation of the expression of NEP, an enzyme that degrades Aβ | [200] | |
| PUFAs oral administration in fat-1 transgenic mice and AQP4 knockout mice | PUFAs promoted Aβ clearance in fat-1 transgenic mice, but not in AQP4 knockout mice. PUFAs protected from Aβ-induced AQP4 polarization | [201] | |
| Complement cascade | Genetic ablation of C3 gene in APP/PS1 mice | Reduction of glia at plaques | [202] |
| Phagocytosis | Fingolimod oral administration in APP/PS1 mice infected by B. pertussis | Increase in astrocyte phagocytosis of Aβ; reduction of GFAP immunoreactivity | [203] |
| In vitro and in vivo downregulation of the Aβ-induced inflammasome, respectively in astrocytes and in 5xFAD mice | In vitro Aβ phagocytosis increase due to the release of the chemokine CCL3 and improved memory in vivo | [204] | |
| Cell reactivity | Iron chelators deferoxamine and deferiprone treatment in Aβ-challenged astrocytes | Inhibition of Aβ-induced Lcn2 | [205] |
| Glu-DAPPD chronic administration in APP/PS1 mice | Reduction of Aβ aggregates as well as GFAP and Iba1 immunostaining. Cognitive functions improvement | [206] | |
| Downregulation of the JAK2-STAT3 pathway in hippocampal astrocytes of transgenic APP mice | Reduction of Aβ deposits; mice spatial learning improvement; control of pro-inflammatory genes | [207] | |
| Downregulation of the JAK2-STAT3 pathway in hippocampal astrocytes of transgenic 3xTg-AD mice | Full reversal of early synaptic and LTP alterations; short-term memory and reduced anxiety behavior improvements | [176,207] | |
| In vitro treatment with PEA of Aβ42-challenged primary astrocytes and mixed astrocytes-neurons cultures | Prevention of Aβ-induced neuronal loss and reduction of neuronal viability | [208] | |
| In vitro treatment with PEA of Aβ42-challenged mixed astrocytes-neurons cultures isolated from 3xTg-AD mice | Prevention of Aβ-induced neuronal loss and reduction of neuronal viability | [209] | |
| In vitro treatment with PEA of primary cortical astrocytes and mixed astrocytes-neurons cultures isolated from 3xTg-AD mice | Reduction of astrogliosis and improvement of neuronal viability | [210] | |
| Um-PEA treatment in glioma and neuroblastoma cells challenged by lipopolisaccaride and interferon γ | Improvement of cell viability; reduction of protein expression of both iNOS and COX-2 | [211] | |
| Co-ultra PEALut administration for 14 days starting from the day that rats received a single intrahippocampal Aβ42 infusion | Prevention of Aβ-induced astrocyte hypertrophy, neuroinflammation; and BDNF and GDNF mRNA downregulation | [35] | |
| Oxidative stress | Electromagnetic fields exposure of human and rat primary astrocytes challenged with Aβ or H2O2 | Reduction of both ROS production and NADPH oxidase activity | [212] |
| In vivo pelargonidin administration in rats subjected to an intrahippocampal injection of Aβ(25–35) | Raise in acetylcholinesterase and catalase activities. Improvement in cognitive performance | [213] | |
| In vivo treatment of C. elegans with monascin | Reduction of Aβ-toxicity and activation of the expression of several anti-oxidative genes | [214] | |
| Channels and receptors | In vivo genetic ablation of the Ca2+-activated K+-channel KCa3.1 | Improvements in memory performance and insulin signaling.Reduction of glial hypertrophy and tau hyperphosphorylation | [215] |
| Chronic intracerebroventricular infusion of P2Y1R inhibitors in APP/PS1 mice | Reversal of structural and functional markers of astrocyte activation. Memory performance improvement | [216] | |
| Inhibition of adenosine recycle by J4 hippocampal infusion in APP/PS1 mice | Improvement of memory deficits | [217] | |
| Oral administration of istradefylline, an A2A antagonist, to transgenic APP mice | Memory improvements | [218] | |
| Astrocyte modulation | Chronic um-PEA administration to 6-month-old 3xTg-AD mice | Reduction of cortical astrocyte hypertrophy and reactivity. Reduction in both cortical and hippocampal inflammation | [210,219] |
| Chronic um-PEA administration to 12-month-old 3xTg-AD mice | Support for asthenic/atrophic astrocytes | [219] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenza, M.; Facchinetti, R.; Menegoni, G.; Steardo, L.; Scuderi, C. Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes. Biomolecules 2021, 11, 600. https://doi.org/10.3390/biom11040600
Valenza M, Facchinetti R, Menegoni G, Steardo L, Scuderi C. Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes. Biomolecules. 2021; 11(4):600. https://doi.org/10.3390/biom11040600
Chicago/Turabian StyleValenza, Marta, Roberta Facchinetti, Giorgia Menegoni, Luca Steardo, and Caterina Scuderi. 2021. "Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes" Biomolecules 11, no. 4: 600. https://doi.org/10.3390/biom11040600
APA StyleValenza, M., Facchinetti, R., Menegoni, G., Steardo, L., & Scuderi, C. (2021). Alternative Targets to Fight Alzheimer’s Disease: Focus on Astrocytes. Biomolecules, 11(4), 600. https://doi.org/10.3390/biom11040600