
Reconstitution of Human Necrosome Interactions in Saccharomyces cerevisiae


Abstract
1. Introduction
2. Material and Methods
2.1. Yeast Strains and Plasmids
- pGALL-(URA3)-hRIPK3D142N: #1864/#1966 and #1967/#1776
- pGALL-(URA3)-hRIPK3S227A: #1864/#1968 and #1969/#1776
- pGALL-(URA3)-hRIPK3VQ/AA: #1864/#1989 and #1990/#1776
2.2. Antibodies and Reagents
2.3. Yeast Transformation
2.4. Proliferation Assays
2.5. Membrane Integrity Assays
2.6. Clonogenicity
2.7. Western Blot
2.8. Microscopy
3. Results
3.1. Reconstitution of the Necrosome in Yeast
3.2. The RHIM Interacting Domain Is Not Required for MLKL Activation by RIPK3 in Yeast
3.3. RIPK3 Induces Aggregation of MLKL in Yeast
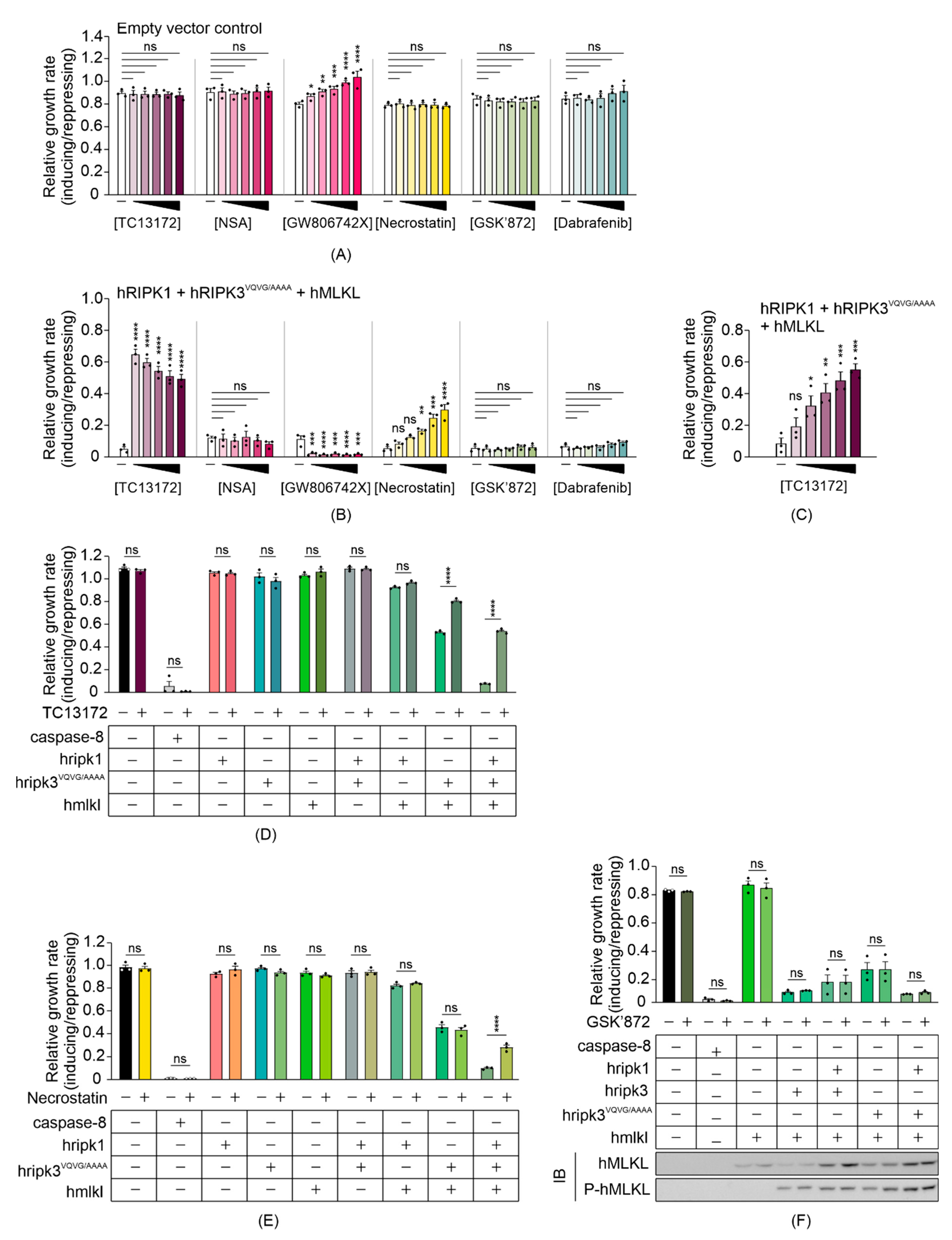
3.4. Some MLKL and RIPK1 Chemical Inhibitors Interfere with Necrosome Function in Yeast
3.5. MLKL and RIPK3 Viral Inhibitors Interfere with Necrosome Function in Yeast
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, M.E.; Price, D.R.; Ryter, S.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, 128834. [Google Scholar] [CrossRef] [PubMed]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflamm. 2018, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kearney, C.J.; Martin, S.J. An Inflammatory Perspective on Necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Hassannia, B.; Vandenabeele, P. An outline of necrosome triggers. Cell. Mol. Life Sci. 2016, 73, 2137–2152. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.-M. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-N.; Yang, Z.-H.; Wang, X.-K.; Zhang, Y.; Wan, H.; Song, Y.; Chen, X.; Shao, J.; Han, J. Distinct roles of RIP1–RIP3 hetero- and RIP3–RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014, 21, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 Complexes with RIP3 to Mediate Virus-Induced Programmed Necrosis that Is Targeted by Murine Cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.-G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.-G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The Pseudokinase MLKL Mediates Necroptosis via a Molecular Switch Mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.-M.; Dobson, R.C.J.; Webb, A.I.; et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef]
- Petrie, E.J.; Sandow, J.J.; Jacobsen, A.V.; Smith, B.J.; Griffin, M.D.W.; Lucet, I.S.; Dai, W.; Young, S.N.; Tanzer, M.C.; Wardak, A.; et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Quarato, G.; Guy, C.S.; Grace, C.R.; Llambi, F.; Nourse, A.; Rodriguez, D.A.; Wakefield, R.; Frase, S.; Moldoveanu, T.; Green, D.R. Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol. Cell 2016, 61, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Tanzer, M.C.; Matti, I.; Hildebrand, J.M.; Young, S.N.; Wardak, A.; Tripaydonis, A.; Petrie, E.J.; Mildenhall, A.L.; Vaux, D.L.; Vince, J.; et al. Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ. 2016, 23, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.-L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Kim, J.W.; Choi, E.-J.; Joe, c. Activation of death-inducing signaling complex (DISC) by pro-apoptotic C-terminal fragment of RIP. Oncogene 2000, 19, 4491–4499. [Google Scholar] [CrossRef]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.-G. Cleavage of the death domain kinase RIP by Caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.-S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 Necrosome Forms a Functional Amyloid Signaling Complex Required for Programmed Necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Rickard, J.A.; O’Donnell, J.A.; Evans, J.M.; Lalaoui, N.; Poh, A.R.; Rogers, T.; Vince, J.E.; Lawlor, K.E.; Ninnis, R.L.; Anderton, H.; et al. RIPK1 Regulates RIPK3-MLKL-Driven Systemic Inflammation and Emergency Hematopoiesis. Cell 2014, 157, 1175–1188. [Google Scholar] [CrossRef]
- Huang, D.; Zheng, X.; Wang, Z.-A.; Chen, X.; He, W.-T.; Zhang, Y.; Xu, J.-G.; Zhao, H.; Shi, W.; Wang, X.; et al. The MLKL Channel in Necroptosis Is an Octamer Formed by Tetramers in a Dyadic Process. Mol. Cell. Biol. 2017, 37, 00497-16. [Google Scholar] [CrossRef]
- Petrie, E.J.; Birkinshaw, R.W.; Koide, A.; Denbaum, E.; Hildebrand, J.M.; Garnish, S.E.; Davies, K.A.; Sandow, J.J.; Samson, A.L.; Gavin, X.; et al. Identification of MLKL membrane translocation as a checkpoint in necroptotic cell death using Monobodies. Proc. Natl. Acad. Sci. USA 2020, 117, 8468–8475. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.-T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, H.; Johnston, A.; Hanna-Addams, S.; Reynoso, E.; Xiang, Y.; Wang, Z. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, E7450–E7459. [Google Scholar] [CrossRef] [PubMed]
- Kondylis, V.; Kumari, S.; Vlantis, K.; Pasparakis, M. The interplay of IKK, NF-κB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev. 2017, 277, 113–127. [Google Scholar] [CrossRef]
- Zhang, X.; Dowling, J.P.; Zhang, J. RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell Death Dis. 2019, 10, 245. [Google Scholar] [CrossRef]
- Orozco, S.; Oberst, A. RIPK3 in cell death and inflammation: The good, the bad, and the ugly. Immunol. Rev. 2017, 277, 102–112. [Google Scholar] [CrossRef]
- Cao, W.-X.; Li, T.; Tang, Z.-H.; Zhang, L.-L.; Wang, Z.-Y.; Guo, X.; Su, M.-X.; Chen, X.; Lu, J.-J. MLKL mediates apoptosis via a mutual regulation with PERK/eIF2α pathway in response to reactive oxygen species generation. Apoptosis 2018, 23, 521–531. [Google Scholar] [CrossRef]
- Vince, J.E.; Wong, W.W.-L.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef]
- Sharma, D.; Malik, A.; Balakrishnan, A.; Malireddi, R.K.S.; Kanneganti, T.-D. RIPK3 Promotes Mefv Expression and Pyrin Inflammasome Activation via Modulation of mTOR Signaling. J. Immunol. 2020, 205, 2778–2785. [Google Scholar] [CrossRef]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Spall, S.K.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [PubMed]
- Meylan, E.; Burns, K.; Hofmann, K.; Blancheteau, V.; Martinon, F.; Kelliher, M.; Tschopp, J. RIP1 is an essential mediator of Toll-like receptor 3–induced NF-κB activation. Nat. Immunol. 2004, 5, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Ea, C.-K.; Deng, L.; Xia, Z.-P.; Pineda, G.; Chen, Z. Activation of IKK by TNFα Requires Site-Specific Ubiquitination of RIP1 and Polyubiquitin Binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, C.J.; Wang, S.L.; Hay, B.A. A cloning method to identify caspases and their regulators in yeast: Identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc. Natl. Acad. Sci. USA 1999, 96, 2885–2890. [Google Scholar] [CrossRef]
- Hawkins, C.J.; Silke, J.; Verhagen, A.M.; Foster, R.; Ekert, P.G.; Ashley, D.M. Analysis of candidate antagonists of IAP-mediated caspase inhibition using yeast reconstituted with the mammalian Apaf-1-activated apoptosis mechanism. Apoptosis 2001, 6, 331–338. [Google Scholar] [CrossRef]
- Puryer, M.A.; Hawkins, C.J. Human, insect and nematode caspases kill Saccharomyces cerevisiae independently of YCA1 and Aif1p. Apoptosis 2006, 11, 509–517. [Google Scholar] [CrossRef]
- Hussain, H.; Chong, N.F.-M. Combined Overlap Extension PCR Method for Improved Site Directed Mutagenesis. BioMed Res. Int. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Bloomer, D.T.; Kitevska, T.; Brand, I.L.; Jabbour, A.M.; Nguyen, H.; Hawkins, C.J. Modeling Metazoan Apoptotic Pathways in Yeast. Adv. Struct. Saf. Stud. 2016, 1419, 161–183. [Google Scholar] [CrossRef]
- Petrie, E.J.; Sandow, J.J.; Lehmann, W.I.; Liang, L.-Y.; Coursier, D.; Young, S.N.; Kersten, W.J.; FitzGibbon, C.; Samson, A.L.; Jacobsen, A.V.; et al. Viral MLKL Homologs Subvert Necroptotic Cell Death by Sequestering Cellular RIPK3. Cell Rep. 2019, 28, 3309–3319.e5. [Google Scholar] [CrossRef]
- Kim, J.-H.; Seo, Y.; Jo, M.; Jeon, H.; Lee, W.-H.; Yachie, N.; Zhong, Q.; Vidal, M.; Roth, F.P.; Suk, K. Yeast-Based Genetic Interaction Analysis of Human Kinome. Cells 2020, 9, 1156. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.-F.; Wang, F.-S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Orozco, S.; Yatim, N.; Werner, M.; Tran, H.; Gunja, S.Y.; Tait, S.W.G.; Albert, M.L.; Green, D.R.; Oberst, A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014, 21, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-α. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yin, J.; Starovasnik, M.A.; Fairbrother, W.J.; Dixit, V.M. Identification of a Novel Homotypic Interaction Motif Required for the Phosphorylation of Receptor-interacting Protein (RIP) by RIP3. J. Biol. Chem. 2002, 277, 9505–9511. [Google Scholar] [CrossRef]
- Yan, B.; Liu, L.; Huang, S.; Ren, Y.; Wang, H.; Yao, Z.; Li, L.; Chen, S.; Wang, X.; Zhang, Z. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem. Commun. 2017, 53, 3637–3640. [Google Scholar] [CrossRef]
- Li, J.-X.; Feng, J.-M.; Wang, Y.; Li, X.-H.; Chen, X.-X.; Su, Y.; Shen, Y.-Y.; Chen, Y.; Xiong, B.; Yang, C.-H.; et al. The B-RafV600E inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like Receptor 3-mediated Necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’En, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Sammond, D.M.; Nailor, K.E.; Veal, J.M.; Nolte, R.T.; Wang, L.; Knick, V.B.; Rudolph, S.K.; Truesdale, A.T.; Nartey, E.N.; Stafford, J.A.; et al. Discovery of a novel and potent series of dianilinopyrimidineurea and urea isostere inhibitors of VEGFR2 tyrosine kinase. Bioorganic Med. Chem. Lett. 2005, 15, 3519–3523. [Google Scholar] [CrossRef]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.-E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Daley-Bauer, L.P.; Roback, L.; Crosby, L.N.; McCormick, A.L.; Feng, Y.; Kaiser, W.J.; Mocarski, E.S. Mouse cytomegalovirus M36 and M45 death suppressors cooperate to prevent inflammation resulting from antiviral programmed cell death pathways. Proc. Natl. Acad. Sci. USA 2017, 114, E2786–E2795. [Google Scholar] [CrossRef] [PubMed]
- Amor, I.Y.H.; Smaoui, K.; Chaabane, I.; Mabrouk, I.; Djemal, L.; Elleuch, H.; Allouche, M.; Mokdad-Gargouri, R.; Gargouri, A.; Chaabène, I.; et al. Human p53 induces cell death and downregulates thioredoxin expression in Saccharomyces cerevisiae. FEMS Yeast Res. 2008, 8, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Perkins, E.; Sun, D.; Nguyen, A.; Tulac, S.; Francesco, M.; Tavana, H.; Nguyen, H.; Tugendreich, S.; Barthmaier, P.; Couto, J.; et al. Novel inhibitors of poly(ADP-ribose) polymerase/PARP1 and PARP2 identified using a cell-based screen in yeast. Cancer Res. 2001, 61, 4175–4183. [Google Scholar]
- Rencus-Lazar, S.; DeRowe, Y.; Adsi, H.; Gazit, E.; Bar-Yosef, D.L. Yeast Models for the Study of Amyloid-Associated Disorders and Development of Future Therapy. Front. Mol. Biosci. 2019, 6, 15. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. Virus Inhibition of RIP3-Dependent Necrosis. Cell Host Microbe 2010, 7, 302–313. [Google Scholar] [CrossRef]
- Moujalled, D.M.; Cook, W.D.; Murphy, J.M.; Vaux, D.L. Necroptosis induced by RIPK3 requires MLKL but not Drp1. Cell Death Dis. 2014, 5, e1086. [Google Scholar] [CrossRef]
- Arnež, K.H.; Kindlova, M.; Bokil, N.J.; Murphy, J.M.; Sweet, M.J.; Gunčar, G. Analysis of the N-terminal region of human MLKL, as well as two distinct MLKL isoforms, reveals new insights into necroptotic cell death. Biosci. Rep. 2016, 36, e00291. [Google Scholar] [CrossRef]
- Lin, J.; Kumari, S.; Kim, C.; Van, T.-M.; Wachsmuth, L.; Polykratis, A.; Pasparakis, M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nat. Cell Biol. 2016, 540, 124–128. [Google Scholar] [CrossRef]
- Weber, K.; Roelandt, R.; Bruggeman, I.; Estornes, Y.; Vandenabeele, P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun. Biol. 2018, 1, 1–13. [Google Scholar] [CrossRef]
- Degterev, A.; Maki, J.L.; Yuan, J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 2012, 20, 366. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 Induces Apoptosis Independent of Pronecrotic Kinase Activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, R.K.; Vaidhyanathan, S.; Dudek, A.Z.; Elmquist, W.F. Mechanisms limiting distribution of the threonine-protein kinase B-RaF(V600E) inhibitor dabrafenib to the brain: Implications for the treatment of melanoma brain metastases. J. Pharmacol. Exp. Ther. 2013, 344, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Balzi, E.; Wang, M.; Leterme, S.; Van Dyck, L.; Goffeau, A. PDR5, a novel yeast multidrug resistance conferring transporter controlled by the transcription regulator PDR1. J. Biol. Chem. 1994, 269, 2206–2214. [Google Scholar] [CrossRef]
- Holmes, A.R.; Cardno, T.S.; Strouse, J.J.; Ivnitski-Steele, I.; Keniya, M.V.; Lackovic, K.; Monk, B.C.; Sklar, L.A.; Cannon, R.D. Targeting efflux pumps to overcome antifungal drug resistance. Futur. Med. Chem. 2016, 8, 1485–1501. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Plasmid | Primer Sequence 5′–3′ | Template | Number |
|---|---|---|---|
| pGALL-(LEU2)-hMLKL | AGAAAAAACCCCGGATCCATGGAAAATTTGAAGCATATTATCACC | Human mlkl | #1 |
| AAGCAGAGATTATCTAGACTACTTAGAAAAGGTGGAGAGTTTCT | #2 | ||
| TCTAGATAATCTCTGCTTTTGTGCG | pGALL-(LEU2) | #3 | |
| GGATCCGGGGTTTTTTCTCCTTG | #4 | ||
| pGALL-(TRP1)-hRIPK1 | TTTGAATTCATGCAACCAGACATGTCCT | Human ripk1 | #2014 |
| TTTCTCGAGTTAGTTCTGGCTGACGTAA | #2015 | ||
| pGALL-(URA3)-hRIPK3D142N | CCACTTTAACTAATACTTTCAACATTTTCGG | pGALL-(URA3)-hRIPK3 | #1864 |
| ATGGCTTCAAATTTCTATGCAACAA | #1966 | ||
| TTGTTGCATAGAAATTTGAAGCCAT | #1967 | ||
| CTTTATTATTTTTATTTTATTGAGAGGGTGG | #1776 | ||
| pGALL-(URA3)-hRIPK3S227A | CCACTTTAACTAATACTTTCAACATTTTCGG | pGALL-(URA3)-hRIPK3 | #1864 |
| CGTAAACTAAAGCTGGTTCAGTTGG | #1968 | ||
| CCAACTGAACCAGCTTTAGTTTACG | #1969 | ||
| CTTTATTATTTTTATTTTATTGAGAGGGTGG | #1776 | ||
| pGALL-(URA3)-hRIPK3VQ/AA | CCACTTTAACTAATACTTTCAACATTTTCGG | pGALL-(URA3)-hRIPK3 | #1864 |
| CACCAACAGCAGCACCAGA | #1989 | ||
| TCTGGTGCTGCTGTTGGTG | #1990 | ||
| CTTTATTATTTTTATTTTATTGAGAGGGTGG | #1776 | ||
| pGALL-(URA3)-hRIPK3VQVG/AAAA | CCACTTTAACTAATACTTTCAACATTTTCGG | pGALL-(URA3)-hRIPK3VQ/AA | #1864 |
| GTTGTCAGCAGCAGCAGC | #1991 | ||
| GCTGCTGCTGCTGACAAC | #1992 | ||
| CTTTATTATTTTTATTTTATTGAGAGGGTGG | #1776 | ||
| pGALL-(LEU2)-GFPS65T | GCTGGATCCGCCTCTAGAATGGGTAAAGGAGAAGAAC | pGALL-(HIS3)-GFPS65T | #2024 |
| TTCTTGCTAGCTTATTTGTATAGTTCATC | #2025 | ||
| pGALL-(LEU2)-hMLKLGFP | GCTGGATCCATGGAAAATTTGAAGCATATT | pGALL-(LEU2)-hMLKL | #2026 |
| GCCTCTAGACTTAGAAAAGGTGGAGAGTTT | #2027 | ||
| pGALL-(HIS3)-BAV_Rmil | GCGGATCCACCATGACTGATCCCCTGTTGCACAA | pFTRE3G PGK Puro-BAV_Rmil | #2017 |
| TTTTCTAGATTACTCAATCTTCTTGTTGAACTTGTAGATT | #2018 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Ward, L.A.; Hawkins, C.J. Reconstitution of Human Necrosome Interactions in Saccharomyces cerevisiae. Biomolecules 2021, 11, 153. https://doi.org/10.3390/biom11020153
Ji Y, Ward LA, Hawkins CJ. Reconstitution of Human Necrosome Interactions in Saccharomyces cerevisiae. Biomolecules. 2021; 11(2):153. https://doi.org/10.3390/biom11020153
Chicago/Turabian StyleJi, Y., L. A. Ward, and C. J. Hawkins. 2021. "Reconstitution of Human Necrosome Interactions in Saccharomyces cerevisiae" Biomolecules 11, no. 2: 153. https://doi.org/10.3390/biom11020153
APA StyleJi, Y., Ward, L. A., & Hawkins, C. J. (2021). Reconstitution of Human Necrosome Interactions in Saccharomyces cerevisiae. Biomolecules, 11(2), 153. https://doi.org/10.3390/biom11020153