
Targeting RIP Kinases in Chronic Inflammatory Disease

 ,
,
Abstract
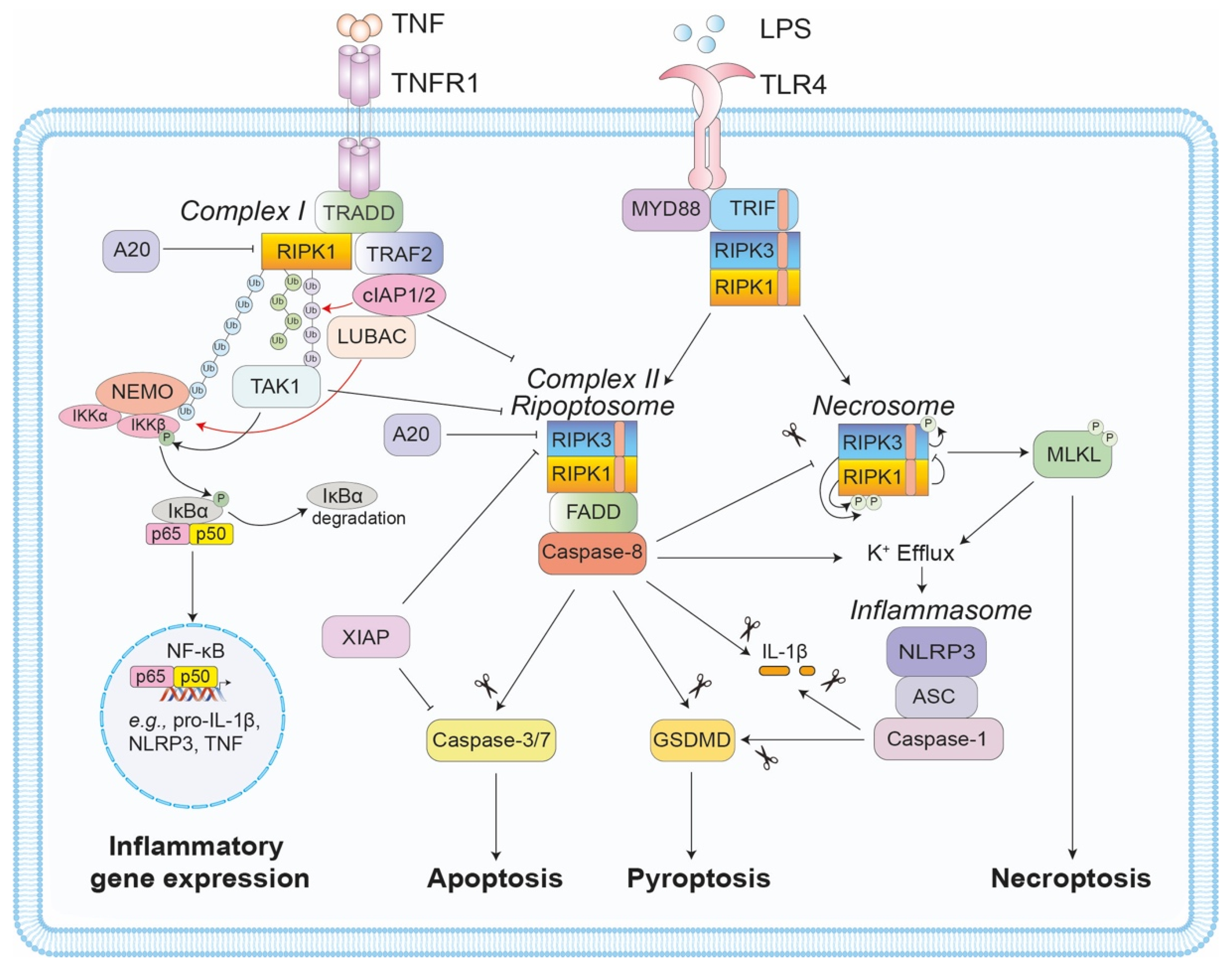
1. Introduction
2. Multi-Organ—Autoinflammatory Syndromes
3. Joints—Rheumatoid Arthritis
4. Skin—Psoriasis and Dermatitis
5. Gut—Inflammatory Bowel Disease
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASC | apoptosis-associated speck-like containing a CARD |
| CAIA | collagen antibody-induced arthritis |
| CAPS | cryopyrin-associated periodic syndromes |
| CARD | caspase activation and recruitment domain |
| caspase | cysteine-dependent aspartic acid-specific protease |
| CD | Crohn’s Disease |
| cFLIP | cellular FLICE-like inhibitory protein |
| cIAP | cellular inhibitor of apoptosis protein |
| CRIA | cleavage-resistant RIPK1-induced autoinflammatory syndrome |
| CYLD | cylindromatosis |
| DAI | DNA-dependent activator of IFN regulatory transcription factors |
| DAMP | damage-associated molecular pattern |
| DNA | deoxyribonucleic acid |
| EDA-ID | ecto-dermal dysplasia with immunodeficiency |
| ER | endoplasmic reticulum |
| GI | gastrointestinal |
| GSDMD | gasdermin D |
| HA20 | haploinsufficiency in A20 |
| HLH | hemophagocytic lymphohistiocytosis |
| HMGB1 | high-mobility group box 1 |
| HOIL-1 | haeme-oxidized IRP2 ubiquitin ligase 1 |
| HOIP | HOIL1-interacting protein |
| HSP | heat shock protein |
| IBD | inflammatory bowel disease |
| IAV | Influenza A virus |
| IEC | intestinal epithelial cell |
| IFN | interferon |
| IL | interleukin |
| IMQ | imiquimod |
| LOF | loss of function |
| LRR | leucine-rich repeat |
| LUBAC | linear ubiquitin chain assembly complex |
| MAPK | mitogen-activated protein kinase |
| MDP | muramyl dipeptide |
| MLKL | mixed lineage kinase domain-like |
| NEMO | NF-κB essential modulator (IKK) |
| NF-κB | nuclear factor-kappa B |
| NLR | NOD-like receptor |
| NLRP3 | NOD-like protein receptor 3 |
| NOD | nucleotide-binding oligomerization domain |
| NSA | necrosulfonamide |
| PAMP | pathogen-associated molecular pattern |
| PRR | pattern recognition receptor |
| RA | rheumatoid arthritis |
| RIPK | receptor-interacting protein kinase |
| RNA | ribonucleic acid |
| ROS | reactive oxygen species |
| RSV | respiratory syncytial virus |
| SHARPIN | SHANK-associated RH domain-interacting protein |
| PTPN6 | protein tyrosine phosphatase-6 |
| SMAC | second mitochondrial activator of caspases |
| SM | Smac mimetic |
| TAK1 | transforming growth factor—activated kinase |
| TLR | Toll-like receptor |
| TNF | tumour necrosis factor |
| TNFR | tumour necrosis factor receptor |
| TRADD | TNFR1-associated death domain protein |
| TRAF2 | TNF receptor-associated factor 2 |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TRAPS | TNFR-associated periodic syndrome |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
| UC | ulcerative colitis |
| VEO | very-early onset |
| XIAP | X-linked inhibitor of apoptosis protein |
| XPL2 | X-linked lymphoproliferative disease 2 |
| ZBP1 | Z-DNA binding protein 1 |
References
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Humphries, F.; Yang, S.; Wang, B.; Moynagh, P.N. RIP kinases: Key decision makers in cell death and innate immunity. Cell Death Differ. 2015, 22, 225–236. [Google Scholar] [CrossRef]
- He, S.; Wang, X. RIP kinases as modulators of inflammation and immunity. Nat. Immunol. 2018, 19, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Eng, V.V.; Wemyss, M.A.; Pearson, J.S. The diverse roles of RIP kinases in host-pathogen interactions. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Meylan, E.; Burns, K.; Hofmann, K.; Blancheteau, V.; Martinon, F.; Kelliher, M.; Tschopp, J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 2004, 5, 503–507. [Google Scholar] [CrossRef]
- Kelliher, M.A.; Grimm, S.; Ishida, Y.; Kuo, F.; Stanger, B.Z.; Leder, P. The Death Domain Kinase RIP Mediates the TNF-Induced NF-kB Signal. Immunity 1998, 8, 297–303. [Google Scholar] [CrossRef]
- Ting, A.T.; Pimentel-Muinos, F.X.; Seed, B. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996, 15, 6189–6196. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Hsu, H.; Huang, J.; Shu, H.B.; Baichwal, V.; Goeddel, D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996, 4, 387–396. [Google Scholar] [CrossRef]
- Park, Y.H.; Jeong, M.S.; Park, H.H.; Jang, S.B. Formation of the death domain complex between FADD and RIP1 proteins In Vitro. Biochim. Biophys. Acta 2013, 1834, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Stanger, B.Z.; Leder, P.; Lee, T.H.; Kim, E.; Seed, B. RIP: A novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 1995, 81, 513–523. [Google Scholar] [CrossRef]
- Sun, X.; Yin, J.; Starovasnik, M.A.; Fairbrother, W.J.; Dixit, V.M. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 2002, 277, 9505–9511. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Offermann, M.K. Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 2005, 174, 4942–4952. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, W.J.; Upton, J.W.; Mocarski, E.S. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 2008, 181, 6427–6434. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Shu, H.B.; Takeuchi, M.; Goeddel, D.V. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc. Natl. Acad. Sci. USA 1996, 93. [Google Scholar] [CrossRef]
- Vince, J.E.; Pantaki, D.; Feltham, R.; Mace, P.D.; Cordier, S.M.; Schmukle, A.C.; Davidson, A.J.; Callus, B.A.; Wong, W.W.; Gentle, I.E.; et al. TRAF2 Must Bind to Cellular Inhibitors of Apoptosis for Tumor Necrosis Factor (TNF) to Efficiently Activate NF-kB and to Prevent TNF-induced Apoptosis. J. Biol. Chem. 2009, 284, 35906–35915. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, B.; Cordier, S.M.; Schmukle, A.C.; Emmerich, C.H.; Rieser, E.; Haas, T.L.; Webb, A.I.; Rickard, J.A.; Anderton, H.; Wong, W.W.; et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011, 471, 591–596. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Tokunaga, F.; Nakagawa, T.; Nakahara, M.; Saeki, Y.; Taniguchi, M.; Sakata, S.; Tanaka, K.; Nakano, H.; Iwai, K. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature 2011, 471, 633–636. [Google Scholar] [CrossRef]
- Liu, Z.; Chan, F.K. Regulatory mechanisms of RIPK1 in cell death and inflammation. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Simpson, D.S.; Gabrielyan, A.; Feltham, R. RIPK1 ubiquitination: Evidence, correlations and the undefined. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Jaco, I.; Annibaldi, A.; Lalaoui, N.; Wilson, R.; Tenev, T.; Laurien, L.; Kim, C.; Jamal, K.; Wicky John, S.; Liccardi, G.; et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol. Cell 2017, 66, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Dynek, J.N.; Goncharov, T.; Dueber, E.C.; Fedorova, A.V.; Izrael-Tomasevic, A.; Phu, L.; Helgason, E.; Fairbrother, W.J.; Deshayes, K.; Kirkpatrick, D.S.; et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 2010, 29, 4198–4209. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Aguileta, M.A.; Goossens, V.; Dubuisson, C.; Grootjans, S.; Dejardin, E.; Vandenabeele, P.; Bertrand, M.J. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013, 20, 1381–1392. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Jouan-Lanhouet, S.; Divert, T.; Theatre, E.; Bertin, J.; Gough, P.J.; Giansanti, P.; Heck, A.J.; Dejardin, E.; Vandenabeele, P.; et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol. Cell 2015, 60, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Dannappel, M.; Vlantis, K.; Kumari, S.; Polykratis, A.; Kim, C.; Wachsmuth, L.; Eftychi, C.; Lin, J.; Corona, T.; Hermance, N.; et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 2014, 513, 90–94. [Google Scholar] [CrossRef]
- Dillon, C.P.; Weinlich, R.; Rodriguez, D.A.; Cripps, J.G.; Quarato, G.; Gurung, P.; Verbist, K.C.; Brewer, T.L.; Llambi, F.; Gong, Y.N.; et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 2014, 157, 1189–1202. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Daley-Bauer, L.P.; Thapa, R.J.; Mandal, P.; Berger, S.B.; Huang, C.; Sundararajan, A.; Guo, H.; Roback, L.; Speck, S.H.; et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. USA 2014, 111, 7753–7758. [Google Scholar] [CrossRef] [PubMed]
- Kearney, C.J.; Cullen, S.P.; Clancy, D.; Martin, S.J. RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis. FEBS J. 2014, 281, 4921–4934. [Google Scholar] [CrossRef] [PubMed]
- Orozco, S.; Yatim, N.; Werner, M.R.; Tran, H.; Gunja, S.Y.; Tait, S.W.; Albert, M.L.; Green, D.R.; Oberst, A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014, 21, 1511–1521. [Google Scholar] [CrossRef]
- Rickard, J.A.; O’Donnell, J.A.; Evans, J.M.; Lalaoui, N.; Poh, A.R.; Rogers, T.; Vince, J.E.; Lawlor, K.E.; Ninnis, R.L.; Anderton, H.; et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 2014, 157, 1175–1188. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Vereecke, L.; Bertrand, M.J.; Duprez, L.; Berger, S.B.; Divert, T.; Goncalves, A.; Sze, M.; Gilbert, B.; Kourula, S.; et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 2014, 513, 95–99. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Hacker, G.; Leverkus, M. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar] [CrossRef]
- Mihaly, S.R.; Ninomiya-Tsuji, J.; Morioka, S. TAK1 control of cell death. Cell Death Differ. 2014, 21, 1667–1676. [Google Scholar] [CrossRef]
- Feltham, R.; Vince, J.E.; Lawlor, K.E. Caspase-8: Not so silently deadly. Clin. Transl. Immunol. 2017, 6, e124. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Khan, N.; Mildenhall, A.; Gerlic, M.; Croker, B.A.; D’Cruz, A.A.; Hall, C.; Kaur Spall, S.; Anderton, H.; Masters, S.L.; et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 2015, 6, 6282. [Google Scholar] [CrossRef] [PubMed]
- Yabal, M.; Muller, N.; Adler, H.; Knies, N.; Gross, C.J.; Damgaard, R.B.; Kanegane, H.; Ringelhan, M.; Kaufmann, T.; Heikenwalder, M.; et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014, 7, 1796–1808. [Google Scholar] [CrossRef]
- Wicki, S.; Gurzeler, U.; Wei-Lynn Wong, W.; Jost, P.J.; Bachmann, D.; Kaufmann, T. Loss of XIAP facilitates switch to TNFalpha-induced necroptosis in mouse neutrophils. Cell Death Dis. 2016, 7, e2422. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; Feltham, R.; Yabal, M.; Conos, S.A.; Chen, K.W.; Ziehe, S.; Grass, C.; Zhan, Y.; Nguyen, T.A.; Hall, C.; et al. XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1beta Activation and Cell Death as a Consequence of TLR-MyD88-Induced cIAP1-TRAF2 Degradation. Cell Rep. 2017, 20, 668–682. [Google Scholar] [CrossRef]
- Chen, K.W.; Lawlor, K.E.; von Pein, J.B.; Boucher, D.; Gerlic, M.; Croker, B.A.; Bezbradica, J.S.; Vince, J.E.; Schroder, K. Cutting Edge: Blockade of Inhibitor of Apoptosis Proteins Sensitizes Neutrophils to TNF- but Not Lipopolysaccharide-Mediated Cell Death and IL-1beta Secretion. J. Immunol. 2018, 200, 3341–3346. [Google Scholar] [CrossRef]
- He, S.; Liang, Y.; Shao, F.; Wang, X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 20054–20059. [Google Scholar] [CrossRef]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef]
- Mompean, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244–1253. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.M.; Dobson, R.C.; Webb, A.I.; et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef] [PubMed]
- Shlomovitz, I.; Erlich, Z.; Speir, M.; Zargarian, S.; Baram, N.; Engler, M.; Edry-Botzer, L.; Munitz, A.; Croker, B.A.; Gerlic, M. Necroptosis directly induces the release of full-length biologically active IL-33 In Vitro and in an inflammatory disease model. FEBS J. 2019, 286, 507–522. [Google Scholar] [CrossRef]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Strasser, A.; Pham, V.C.; Lill, J.R.; Roose-Girma, M.; Warming, S.; Solon, M.; et al. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 2016, 540, 129–133. [Google Scholar] [CrossRef]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef]
- Lin, J.; Kumari, S.; Kim, C.; Van, T.M.; Wachsmuth, L.; Polykratis, A.; Pasparakis, M. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 2016, 540, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Lawlor, K.E.; Murphy, J.M.; Vince, J.E. More to life than death: Molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling. Curr. Opin. Immunol. 2014, 26, 76–89. [Google Scholar] [CrossRef]
- Speir, M.; Lawlor, K.E. RIP-roaring inflammation: RIPK1 and RIPK3 driven NLRP3 inflammasome activation and autoinflammatory disease. Semin. Cell Dev. Biol. 2020. [Google Scholar] [CrossRef]
- Gurung, P.; Anand, P.K.; Malireddi, R.K.; Vande Walle, L.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Allam, R.; Lawlor, K.E.; Yu, E.C.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J.; et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef]
- Weng, D.; Marty-Roix, R.; Ganesan, S.; Proulx, M.K.; Vladimer, G.I.; Kaiser, W.J.; Mocarski, E.S.; Pouliot, K.; Chan, F.K.; Kelliher, M.A.; et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fan, C.; Zhang, H.; Zhao, Q.; Liu, Y.; Xu, C.; Xie, Q.; Wu, X.; Yu, X.; Zhang, J.; et al. MLKL and FADD Are Critical for Suppressing Progressive Lymphoproliferative Disease and Activating the NLRP3 Inflammasome. Cell Rep. 2016, 16, 3247–3259. [Google Scholar] [CrossRef]
- Philip, N.H.; DeLaney, A.; Peterson, L.W.; Santos-Marrero, M.; Grier, J.T.; Sun, Y.; Wynosky-Dolfi, M.A.; Zwack, E.E.; Hu, B.; Olsen, T.M.; et al. Activity of Uncleaved Caspase-8 Controls Anti-bacterial Immune Defense and TLR-Induced Cytokine Production Independent of Cell Death. PLoS Pathog. 2016, 12, e1005910. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6, 7515. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Maltzman, A.; Dugger, D.L.; Reja, R.; Zhang, Y.; Roose-Girma, M.; Modrusan, Z.; Sagolla, M.S.; Webster, J.D.; et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019, 575, 679–682. [Google Scholar] [CrossRef]
- Antonopoulos, C.; Russo, H.M.; El Sanadi, C.; Martin, B.N.; Li, X.; Kaiser, W.J.; Mocarski, E.S.; Dubyak, G.R. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J. Biol. Chem. 2015, 290, 20167–20184. [Google Scholar] [CrossRef]
- Sagulenko, V.; Thygesen, S.J.; Sester, D.P.; Idris, A.; Cridland, J.A.; Vajjhala, P.R.; Roberts, T.L.; Schroder, K.; Vince, J.E.; Hill, J.M.; et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013, 20, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.S.; Gross, C.J.; Dreier, R.F.; Saller, B.S.; Mishra, R.; Gorka, O.; Heilig, R.; Meunier, E.; Dick, M.S.; Cikovic, T.; et al. The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep. 2017, 21, 3846–3859. [Google Scholar] [CrossRef]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef]
- Moriwaki, K.; Bertin, J.; Gough, P.J.; Chan, F.K. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J. Immunol. 2015, 194, 1938–1944. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Delanghe, T.; Priem, D.; Wynosky-Dolfi, M.A.; Sorobetea, D.; Rojas-Rivera, D.; Giansanti, P.; Roelandt, R.; Gropengiesser, J.; Ruckdeschel, K.; et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 2019, 10, 1729. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Gropengiesser, J.; Fischer, J.; Novikova, L.; Deuretzbacher, A.; Lafera, J.; Schimmeck, H.; Czymmeck, N.; Ronkina, N.; Kotlyarov, A.; et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 2017, 19, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Malireddi, R.K.S.; Gurung, P.; Mavuluri, J.; Dasari, T.K.; Klco, J.M.; Chi, H.; Kanneganti, T.D. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 2018, 215, 1023–1034. [Google Scholar] [CrossRef]
- Duong, B.H.; Onizawa, M.; Oses-Prieto, J.A.; Advincula, R.; Burlingame, A.; Malynn, B.A.; Ma, A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015, 42, 55–67. [Google Scholar] [CrossRef]
- Vande Walle, L.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Malireddi, R.K.S.; Gurung, P.; Kesavardhana, S.; Samir, P.; Burton, A.; Mummareddy, H.; Vogel, P.; Pelletier, S.; Burgula, S.; Kanneganti, T.D. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef]
- Conos, S.A.; Chen, K.W.; De Nardo, D.; Hara, H.; Whitehead, L.; Núñez, G.; Masters, S.L.; Murphy, J.M.; Schroder, K.; Vaux, D.L.; et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. USA 2017, 114, E961–E969. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.B.; Yang, S.H.; Toth, B.; Kovalenko, A.; Wallach, D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 2013, 38, 27–40. [Google Scholar] [CrossRef]
- Knop, J.; Spilgies, L.M.; Rufli, S.; Reinhart, R.; Vasilikos, L.; Yabal, M.; Owsley, E.; Jost, P.J.; Marsh, R.A.; Wajant, H.; et al. TNFR2 induced priming of the inflammasome leads to a RIPK1-dependent cell death in the absence of XIAP. Cell Death Dis. 2019, 10, 700. [Google Scholar] [CrossRef]
- Gottlieb, A.B.; Blauvelt, A.; Thaci, D.; Leonardi, C.L.; Poulin, Y.; Drew, J.; Peterson, L.; Arendt, C.; Burge, D.; Reich, K. Certolizumab pegol for the treatment of chronic plaque psoriasis: Results through 48 weeks from 2 phase 3, multicenter, randomized, double-blinded, placebo-controlled studies (CIMPASI-1 and CIMPASI-2). J. Am. Acad. Dermatol. 2018, 79, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, M.; Blauvelt, A.; Paul, C.; Sofen, H.; Weglowska, J.; Piguet, V.; Burge, D.; Rolleri, R.; Drew, J.; Peterson, L.; et al. Certolizumab pegol for the treatment of chronic plaque psoriasis: Results through 48 weeks of a phase 3, multicenter, randomized, double-blind, etanercept- and placebo-controlled study (CIMPACT). J. Am. Acad. Dermatol. 2018, 79, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, P.S.; Bissonnette, R.; Teixeira, H.D.; Valdecantos, W.C. Systematic review of efficacy of anti-tumor necrosis factor (TNF) therapy in patients with psoriasis previously treated with a different anti-TNF agent. J. Am. Acad. Dermatol. 2016, 75, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.; Seidelin, J.B.; LaCasse, E.C.; Nielson, O.H. SMAC mimetics and RIPK inhibitors as therapeutics for chronic inflammatory diseases. Sci. Signal. 2020, 13, eaax8295. [Google Scholar] [CrossRef] [PubMed]
- Martens, S.; Hofmans, S.; Declercq, W.; Augustyns, K.; Vandenabeele, P. Inhibitors Targeting RIPK1/RIPK3: Old and New Drugs. Trends Pharmacol. Sci. 2020, 41, 209–224. [Google Scholar] [CrossRef]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef]
- Newton, K.; Dugger, D.L.; Wickliffe, K.E.; Kapoor, N. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014, 343, 1357–1360. [Google Scholar] [CrossRef]
- Polykratis, A.; Hermance, N.; Zelic, M.; Roderick, J.; Kim, C.; Van, T.M.; Lee, T.H.; Chan, F.K.M.; Pasparakis, M.; Kelliher, M.A. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 2014, 193, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Cuchet-Lourenco, D.; Eletto, D.; Wu, C.; Plagnol, V.; Papapietro, O.; Curtis, J.; Ceron-Gutierrez, L.; Bacon, C.M.; Hackett, S.; Alsaleem, B.; et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 2018, 361, 810–813. [Google Scholar] [CrossRef]
- Li, Y.; Fuhrer, M.; Bahrami, E.; Socha, P.; Klaudel-Dreszler, M.; Bouzidi, A.; Liu, Y.; Lehle, A.S.; Magg, T.; Hollizeck, S.; et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 970–975. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef] [PubMed]
- Weisel, K.; Scott, N.E.; Tompson, D.J.; Votta, B.J.; Madhavan, S.; Povey, K.; Wolstenholme, A.; Simeoni, M.; Rudo, T.; Richards-Peterson, L.; et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Aksentijevich, I.; Galon, J.; Soares, M.; Mansfield, E.; Hull, K.; Oh, H.; Goldbach-Mansky, R.; Dean, B.; Athreya, A.; Reginato, J.; et al. The tumor-necrosis-factor receptor-associated periodic syndrome: New mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am. J. Hum. Genet. 2001, 69, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Shabani, M.; Razaghian, A.; Alimadadi, H.; Shiari, R.; Shahrooei, M.; Parvaneh, N. Different phenotypes of the same XIAP mutation in a family: A case of XIAP deficiency with juvenile idiopathic arthritis. Pediatr. Blood Cancer 2019, 66, e27593. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yu, X.; Demirkaya, E.; Deuitch, N.; Stone, D.; Tsai, W.L.; Kuehn, H.S.; Wang, H.; Yang, D.; Park, Y.H.; et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA 2016, 113, 10127–10132. [Google Scholar] [CrossRef] [PubMed]
- Oda, H.; Beck, D.B.; Kuehn, H.S.; Sampaio Moura, N.; Hoffmann, P.; Ibarra, M.; Stoddard, J.; Tsai, W.L.; Gutierrez-Cruz, G.; Gadina, M.; et al. Second Case of HOIP Deficiency Expands Clinical Features and Defines Inflammatory Transcriptome Regulated by LUBAC. Front. Immunol. 2019, 10, 479. [Google Scholar] [CrossRef]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.R.; McHale, D.; Maher, E.R.; McKenzie, A.N.J.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215–1230. [Google Scholar] [CrossRef]
- Tsuchida, N.; Kirino, Y.; Soejima, Y.; Onodera, M.; Arai, K.; Tamura, E.; Ishikawa, T.; Kawai, T.; Uchiyama, T.; Nomura, S.; et al. Haploinsufficiency of A20 caused by a novel nonsense variant or entire deletion of TNFAIP3 is clinically distinct from Behcet’s disease. Arthritis Res. Ther. 2019, 21, 137. [Google Scholar] [CrossRef]
- Moulin, M.; Anderton, H.; Voss, A.K.; Thomas, T.; Wong, W.W.; Bankovacki, A.; Feltham, R.; Chau, D.; Cook, W.D.; Silke, J.; et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development. EMBO J. 2012, 31, 1679–1691. [Google Scholar] [CrossRef]
- Onizawa, M.; Oshima, S.; Schulze-Topphoff, U.; Oses-Prieto, J.A.; Lu, T.; Tavares, R.; Prodhomme, T.; Duong, B.; Whang, M.I.; Advincula, R.; et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 2015, 16, 618–627. [Google Scholar] [CrossRef]
- Turer, E.E.; Tavares, R.M.; Mortier, E.; Hitotsumatsu, O.; Advincula, R.; Lee, B.; Shifrin, N.; Malynn, B.A.; Ma, A. Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J. Exp. Med. 2008, 205, 451–464. [Google Scholar] [CrossRef]
- Peltzer, N.; Rieser, E.; Taraborrelli, L.; Draber, P.; Darding, M.; Pernaute, B.; Shimizu, Y.; Sarr, A.; Draberova, H.; Montinaro, A.; et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 2014, 9, 153–165. [Google Scholar] [CrossRef]
- Rickard, J.A.; Anderton, H.; Etemadi, N.; Nachbur, U.; Darding, M.; Peltzer, N.; Lalaoui, N.; Lawlor, K.E.; Vanyai, H.; Hall, C.; et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Ostrov, B.E. Immunotherapeutic Biologic Agents in Autoimmune and Autoinflammatory Diseases. Immunol. Investig. 2015, 44, 777–802. [Google Scholar] [CrossRef]
- Matsumoto, S.; Müller-Ladner, U.; Gay, R.E.; Nishioka, K.; Gay, S. Ultrastructural demonstration of apoptosis, Fas and Bcl-2 expression of rheumatoid synovial fibroblasts. J. Rheumatol. 1996, 23, 1345–1352. [Google Scholar] [PubMed]
- Liu, H.; Huang, Q.; Shi, B.; Eksarko, P.; Temkin, V.; Pope, R.M. Regulation of Mcl-1 expression in rheumatoid arthritis synovial macrophages. Arthritis Rheum. 2006, 54, 3174–3181. [Google Scholar] [CrossRef] [PubMed]
- Dharmapatni, A.A.; Smith, M.D.; Findlay, D.M.; Holding, C.A.; Evdokiou, A.; Ahern, M.J.; Weedon, H.; Chen, P.; Screaton, G.; Xu, X.N.; et al. Elevated expression of caspase-3 inhibitors, survivin and xIAP correlates with low levels of apoptosis in active rheumatoid synovium. Arthritis Res. Ther. 2009, 11, R13. [Google Scholar] [CrossRef]
- Bai, S.; Liu, H.; Chen, K.H.; Eksarko, P.; Perlman, H.; Moore, T.L.; Pope, R.M. NF-kappaB-regulated expression of cellular FLIP protects rheumatoid arthritis synovial fibroblasts from tumor necrosis factor alpha-mediated apoptosis. Arthritis Rheum. 2004, 50, 3844–3855. [Google Scholar] [CrossRef]
- Liu, H.; Eksarko, P.; Temkin, V.; Haines, G.K., 3rd; Perlman, H.; Koch, A.E.; Thimmapaya, B.; Pope, R.M. Mcl-1 is essential for the survival of synovial fibroblasts in rheumatoid arthritis. J. Immunol. 2005, 175, 8337–8345. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Rosloniec, E.; Price, J.; Boothby, M.; Chen, J. Constitutive expression of BCL-X(L) in the T lineage attenuates collagen-induced arthritis in Bcl-X(L) transgenic mice. Arthritis Rheum. 2002, 46, 514–521. [Google Scholar] [CrossRef]
- Zheng, B.; Marinova, E.; Switzer, K.; Wansley, D.; He, H.; Bheekha-Escura, R.; Behrens, T.W.; Han, S. Overexpression of Bcl(XL) in B cells promotes Th1 response and exacerbates collagen-induced arthritis. J. Immunol. 2007, 179, 7087–7092. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.E.; van Nieuwenhuijze, A.; Parker, K.L.; Drake, S.F.; Campbell, I.K.; Smith, S.D.; Vince, J.E.; Strasser, A.; Wicks, I.P. Bcl-2 overexpression ameliorates immune complex-mediated arthritis by altering FcgammaRIIb expression and monocyte homeostasis. J. Leukoc. Biol. 2013, 93, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.Q.; Birkett, R.; Koessler, R.E.; Cuda, C.M.; Haines, G.K., 3rd; Jin, J.P.; Perlman, H.; Pope, R.M. Fas signaling in macrophages promotes chronicity in K/BxN serum-induced arthritis. Arthritis Rheumatol. 2014, 66, 68–77. [Google Scholar] [CrossRef]
- Bardwell, P.D.; Gu, J.; McCarthy, D.; Wallace, C.; Bryant, S.; Goess, C.; Mathieu, S.; Grinnell, C.; Erickson, J.; Rosenberg, S.H.; et al. The Bcl-2 family antagonist ABT-737 significantly inhibits multiple animal models of autoimmunity. J. Immunol. 2009, 182, 7482–7489. [Google Scholar] [CrossRef] [PubMed]
- Dharmapatni, A.A.; Cantley, M.D.; Marino, V.; Perilli, E.; Crotti, T.N.; Smith, M.D.; Haynes, D.R. The X-Linked Inhibitor of Apoptosis Protein Inhibitor Embelin Suppresses Inflammation and Bone Erosion in Collagen Antibody Induced Arthritis Mice. Mediat. Inflamm. 2015, 2015, 564042. [Google Scholar] [CrossRef]
- Scatizzi, J.C.; Hutcheson, J.; Pope, R.M.; Firestein, G.S.; Koch, A.E.; Mavers, M.; Smason, A.; Agrawal, H.; Haines, G.K., 3rd; Chandel, N.S.; et al. Bim-Bcl-2 homology 3 mimetic therapy is effective at suppressing inflammatory arthritis through the activation of myeloid cell apoptosis. Arthritis Rheum. 2010, 62, 441–451. [Google Scholar] [CrossRef]
- Park, J.S.; Oh, Y.; Park, O.; Foss, C.A.; Lim, S.M.; Jo, D.G.; Na, D.H.; Pomper, M.G.; Lee, K.C.; Lee, S. PEGylated TRAIL ameliorates experimental inflammatory arthritis by regulation of Th17 cells and regulatory T cells. J. Control. Release 2017, 267, 163–171. [Google Scholar] [CrossRef]
- Matmati, M.; Jacques, P.; Maelfait, J.; Verheugen, E.; Kool, M.; Sze, M.; Geboes, L.; Louagie, E.; Mc Guire, C.; Vereecke, L.; et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat. Genet. 2011, 43, 908–912. [Google Scholar] [CrossRef]
- Huang, Q.Q.; Perlman, H.; Birkett, R.; Doyle, R.; Fang, D.; Haines, G.K.; Robinson, W.; Datta, S.; Huang, Z.; Li, Q.Z.; et al. CD11c-mediated deletion of Flip promotes autoreactivity and inflammatory arthritis. Nat. Commun. 2015, 6, 7086. [Google Scholar] [CrossRef]
- Polykratis, A.; Martens, A.; Eren, R.O.; Shirasaki, Y.; Yamagishi, M.; Yamaguchi, Y.; Uemura, S.; Miura, M.; Holzmann, B.; Kollias, G.; et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 2019, 21, 731–742. [Google Scholar] [CrossRef]
- Dominguez, S.; Montgomery, A.B.; Haines, G.K., 3rd; Bloomfield, C.L.; Cuda, C.M. The caspase-8/RIPK3 signaling axis in antigen presenting cells controls the inflammatory arthritic response. Arthritis Res. Ther. 2017, 19, 224. [Google Scholar] [CrossRef]
- Cuda, C.M.; Misharin, A.V.; Khare, S.; Saber, R.; Tsai, F.; Archer, A.M.; Homan, P.J.; Haines, G.K., 3rd; Hutcheson, J.; Dorfleutner, A.; et al. Conditional deletion of caspase-8 in macrophages alters macrophage activation in a RIPK-dependent manner. Arthritis Res. Ther. 2015, 17, 291. [Google Scholar] [CrossRef]
- Vince, J.E.; De Nardo, D.; Gao, W.; Vince, A.J.; Hall, C.; McArthur, K.; Simpson, D.; Vijayaraj, S.; Lindqvist, L.M.; Bouillet, P.; et al. The Mitochondrial Apoptotic Effectors BAX/BAK Activate Caspase-3 and -7 to Trigger NLRP3 Inflammasome and Caspase-8 Driven IL-1beta Activation. Cell Rep. 2018, 25, 2339–2353. [Google Scholar] [CrossRef] [PubMed]
- Jhun, J.; Lee, S.H.; Kim, S.Y.; Ryu, J.; Kwon, J.Y.; Na, H.S.; Jung, K.; Moon, S.J.; Cho, M.L.; Min, J.K. RIPK1 inhibition attenuates experimental autoimmune arthritis via suppression of osteoclastogenesis. J. Transl. Med. 2019, 17, 84. [Google Scholar] [CrossRef]
- Patel, S.; Webster, J.D.; Varfolomeev, E.; Kwon, Y.C.; Cheng, J.H.; Zhang, J.; Dugger, D.L.; Wickliffe, K.E.; Maltzman, A.; Sujatha-Bhaskar, S.; et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 2019, 27, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Weisel, K.; Berger, S.; Thorn, K.; Taylor, P.C.; Peterfy, C.; Siddall, H.; Tompson, D.; Wang, S.; Quattrocchi, E.; Burriss, S.W.; et al. A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res. Ther. 2021, 23, 85. [Google Scholar] [CrossRef] [PubMed]
- Devos, M.; Tanghe, G.; Gilbert, B.; Dierick, E.; Verheirstraeten, M.; Nemegeer, J.; de Reuver, R.; Lefebvre, S.; De Munck, J.; Rehwinkel, J.; et al. Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Lalaoui, N.; Boyden, S.E.; Oda, H.; Wood, G.M.; Stone, D.L.; Chau, D.; Liu, L.; Stoffels, M.; Kratina, T.; Lawlor, K.E.; et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 2019, 577, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Anderton, H.; Rickard, J.A.; Varigos, G.A.; Lalaoui, N.; Silke, J. Inhibitor of Apoptosis Proteins (IAPs) Limit RIPK1-Mediated Skin Inflammation. J. Investig. Dermatol. 2017, 137, 2371–2379. [Google Scholar] [CrossRef]
- Berger, S.B.; Kasparcova, V.; Hoffman, S.; Swift, B.; Dare, L.; Schaeffer, M.; Capriotti, C.; Cook, M.; Finger, J.; Hughes-Earle, A.; et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 2014, 192, 5476–5480. [Google Scholar] [CrossRef]
- Rendon, A.; Schakel, K. Psoriasis Pathogenesis and Treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Dobbs, K.; Cobat, A.; Tarantino, N.; Hazen, M.; Lidov, H.G.; Hopkins, G.; Du, L.; Belkadi, A.; et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 2015, 212, 939–951. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israel, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef]
- Peltzer, N.; Darding, M.; Montinaro, A.; Draber, P.; Draberova, H.; Kupka, S.; Rieser, E.; Fisher, A.; Hutchinson, C.; Taraborrelli, L.; et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 2018, 557, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Taraborrelli, L.; Peltzer, N.; Montinaro, A.; Kupka, S.; Rieser, E.; Hartwig, T.; Sarr, A.; Darding, M.; Draber, P.; Haas, T.L.; et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat. Commun. 2018, 9, 3910. [Google Scholar] [CrossRef]
- Seymour, R.E.; Hasham, M.G.; Cox, G.A.; Shultz, L.D.; Hogenesch, H.; Roopenian, D.C.; Sundberg, J.P. Spontaneous mutations in the mouse Sharpin gene result in multiorgan inflammation, immune system dysregulation and dermatitis. Genes Immun. 2007, 8, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, F.; Deribe, Y.L.; Skanland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; van Wijk, S.J.; Goswami, P.; Nagy, V.; Terzic, J.; et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Douglas, T.; Champagne, C.; Morizot, A.; Lapointe, J.M.; Saleh, M. The Inflammatory Caspases-1 and -11 Mediate the Pathogenesis of Dermatitis in Sharpin-Deficient Mice. J. Immunol. 2015, 195, 2365–2373. [Google Scholar] [CrossRef]
- Gurung, P.; Lamkanfi, M.; Kanneganti, T.D. Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J. Immunol. 2015, 194, 2064–2067. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Sharma, B.R.; Kanneganti, T.D. Distinct role of IL-1beta in instigating disease in Sharpin(cpdm) mice. Sci. Rep. 2016, 6, 36634. [Google Scholar] [CrossRef]
- Kumari, S.; Redouane, Y.; Lopez-Mosqueda, J.; Shiraishi, R.; Romanowska, M.; Lutzmayer, S.; Kuiper, J.; Martinez, C.; Dikic, I.; Pasparakis, M.; et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.D.; Kwon, Y.C.; Park, S.; Zhang, H.; Corr, N.; Ljumanovic, N.; Adedeji, A.O.; Varfolomeev, E.; Goncharov, T.; Preston, J.; et al. RIP1 kinase activity is critical for skin inflammation but not for viral propagation. J. Leukoc. Biol. 2020, 107, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Morrish, E.; Brumatti, G.; Silke, J. Future Therapeutic Directions for Smac-Mimetics. Cells 2020, 9, 406. [Google Scholar] [CrossRef]
- Nesterovitch, A.B.; Gyorfy, Z.; Hoffman, M.D.; Moore, E.C.; Elbuluk, N.; Tryniszewska, B.; Rauch, T.A.; Simon, M.; Kang, S.; Fisher, G.J.; et al. Alteration in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am. J. Pathol. 2011, 178, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Vogel, P.; Johnson, G.R.; Kelliher, M.A.; Iwakura, Y.; Lamkanfi, M.; Kanneganti, T.D. RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3. Nature 2013, 498, 224–227. [Google Scholar] [CrossRef]
- Abram, C.L.; Roberge, G.L.; Pao, L.I.; Neel, B.G.; Lowell, C.A. Distinct roles for neutrophils and dendritic cells in inflammation and autoimmunity in motheaten mice. Immunity 2013, 38, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Speir, M.; Nowell, C.J.; Chen, A.A.; O’Donnell, J.A.; Shamie, I.S.; Lakin, P.R.; D’Cruz, A.A.; Braun, R.O.; Babon, J.J.; Lewis, R.S.; et al. Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation. Nat. Immunol. 2019, 21, 54–64. [Google Scholar] [CrossRef]
- Saito, N.; Honma, M.; Shibuya, T.; Iinuma, S.; Igawa, S.; Kishibe, M.; Ishida-Yamamoto, A. RIPK1 downregulation in keratinocyte enhances TRAIL signaling in psoriasis. J. Dermatol. Sci. 2018, 91, 79–86. [Google Scholar] [CrossRef]
- Duan, X.; Liu, X.; Liu, N.; Huang, Y.; Jin, Z.; Zhang, S.; Ming, Z.; Chen, H. Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis. 2020, 11, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3, eaat2738. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-McNulty, B.; Carano, R.A.; Cao, T.C.; van Bruggen, N.; Bernstein, L.; et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016, 23, 1565–1576. [Google Scholar] [CrossRef]
- Weisel, K.; Berger, S.; Papp, K.; Maari, C.; Krueger, J.G.; Scott, N.; Tompson, D.; Wang, S.; Simeoni, M.; Bertin, J.; et al. Response to Inhibition of Receptor-Interacting Protein Kinase 1 (RIPK1) in Active Plaque Psoriasis: A Randomized Placebo-Controlled Study. Clin. Pharmacol. Ther. 2020. [Google Scholar] [CrossRef]
- Harris, P.A.; Berger, S.B.; Jeong, J.U.; Nagilla, R.; Bandyopadhyay, D.; Campobasso, N.; Capriotti, C.A.; Cox, J.A.; Dare, L.; Dong, X.; et al. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- de Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, R.; Duckworth, C.A.; Moussata, D.; Gloeckner, A.; Lim, L.G.; Goetz, M.; Pritchard, D.M.; Galle, P.R.; Neurath, M.F.; Watson, A.J. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut 2012, 61, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Buhner, S.; Buning, C.; Genschel, J.; Kling, K.; Herrmann, D.; Dignass, A.; Kuechler, I.; Krueger, S.; Schmidt, H.H.; Lochs, H. Genetic basis for increased intestinal permeability in families with Crohn’s disease: Role of CARD15 3020insC mutation? Gut 2006, 55, 342–347. [Google Scholar] [CrossRef]
- Cifaldi, C.; Chiriaco, M.; Di Matteo, G.; Di Cesare, S.; Alessia, S.; De Angelis, P.; Rea, F.; Angelino, G.; Pastore, M.; Ferradini, V.; et al. Novel X-Linked Inhibitor of Apoptosis Mutation in Very Early-Onset Inflammatory Bowel Disease Child Successfully Treated with HLA-Haploidentical Hemapoietic Stem Cells Transplant after Removal of alphabeta(+) T and B Cells. Front. Immunol. 2017, 8, 1893. [Google Scholar] [CrossRef]
- Lekbua, A.; Ouahed, J.; O’Connell, A.E.; Kahn, S.A.; Goldsmith, J.D.; Imamura, T.; Duncan, C.N.; Kelsen, J.R.; Worthey, E.; Snapper, S.B.; et al. Risk-factors Associated With Poor Outcomes in VEO-IBD Secondary to XIAP Deficiency: A Case Report and Literature Review. J. Pediatr. Gastroenterol. Nutr. 2019, 69, e13–e18. [Google Scholar] [CrossRef] [PubMed]
- Serra, E.G.; Schwerd, T.; Moutsianas, L.; Cavounidis, A.; Fachal, L.; Pandey, S.; Kammermeier, J.; Croft, N.M.; Posovszky, C.; Rodrigues, A.; et al. Somatic mosaicism and common genetic variation contribute to the risk of very-early-onset inflammatory bowel disease. Nat. Commun. 2020, 11, 995. [Google Scholar] [CrossRef] [PubMed]
- Girardelli, M.; Arrigo, S.; Barabino, A.; Loganes, C.; Morreale, G.; Crovella, S.; Tommasini, A.; Bianco, A.M. The diagnostic challenge of very early-onset enterocolitis in an infant with XIAP deficiency. BMC Pediatr. 2015, 15, 208. [Google Scholar] [CrossRef]
- Latour, S.; Aguilar, C. XIAP deficiency syndrome in humans. Semin. Cell Dev. Biol. 2015, 39, 115–123. [Google Scholar] [CrossRef]
- Uhlig, H.H. Monogenic diseases associated with intestinal inflammation: Implications for the understanding of inflammatory bowel disease. Gut 2013, 62, 1795–1805. [Google Scholar] [CrossRef]
- Nielsen, O.H.; LaCasse, E.C. How genetic testing can lead to targeted management of XIAP deficiency-related inflammatory bowel disease. Genet. Med. 2017, 19, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Stafford, C.A.; Lawlor, K.E.; Heim, V.J.; Bankovacki, A.; Bernardini, J.P.; Silke, J.; Nachbur, U. IAPs Regulate Distinct Innate Immune Pathways to Co-ordinate the Response to Bacterial Peptidoglycans. Cell Rep. 2018, 22, 1496–1508. [Google Scholar] [CrossRef]
- Negroni, A.; Stronati, L.; Pierdomenico, M.; Tirindelli, D.; Di Nardo, G.; Mancini, V.; Maiella, G.; Cucchiara, S. Activation of NOD2-mediated intestinal pathway in a pediatric population with Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Stronati, L.; Negroni, A.; Merola, P.; Pannone, V.; Borrelli, O.; Cirulli, M.; Annese, V.; Cucchiara, S. Mucosal NOD2 expression and NF-kappaB activation in pediatric Crohn’s disease. Inflamm. Bowel Dis. 2008, 14, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Stronati, L.; Negroni, A.; Pierdomenico, M.; D’Ottavio, C.; Tirindelli, D.; Di Nardo, G.; Oliva, S.; Viola, F.; Cucchiara, S. Altered expression of innate immunity genes in different intestinal sites of children with ulcerative colitis. Dig. Liver Dis. 2010, 42, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Chirieleison, S.M.; Marsh, R.A.; Kumar, P.; Rathkey, J.K.; Dubyak, G.R.; Abbott, D.W. Nucleotide-binding oligomerization domain (NOD) signaling defects and cell death susceptibility cannot be uncoupled in X-linked inhibitor of apoptosis (XIAP)-driven inflammatory disease. J. Biol. Chem. 2017, 292, 9666–9679. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L. Anti-TNF therapy in inflammatory bowel diseases: A huge review. Minerva Gastroenterol. Dietol. 2010, 56, 233–243. [Google Scholar]
- Liu, Z.; Kong, F.; Vallance, J.E.; Harmel-Laws, E.; Amarachintha, S.; Steinbrecher, K.A.; Rosen, M.J.; Bhattacharyya, S. Activation of TGF-beta activated kinase 1 promotes colon mucosal pathogenesis in inflammatory bowel disease. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef]
- Vlantis, K.; Wullaert, A.; Polykratis, A.; Kondylis, V.; Dannappel, M.; Schwarzer, R.; Welz, P.; Corona, T.; Walczak, H.; Weih, F.; et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-kappaB-Dependent and -Independent Functions. Immunity 2016, 44, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Chae, S.; Eckmann, L.; Miyamoto, Y.; Pothoulakis, C.; Karin, M.; Kagnoff, M.F. Epithelial cell I kappa B-kinase beta has an important protective role in Clostridium difficile toxin A-induced mucosal injury. J. Immunol. 2006, 177, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Eckmann, L.; Nebelsiek, T.; Fingerle, A.A.; Dann, S.M.; Mages, J.; Lang, R.; Robine, S.; Kagnoff, M.F.; Schmid, R.M.; Karin, M.; et al. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 15058–15063. [Google Scholar] [CrossRef] [PubMed]
- Kajino-Sakamoto, R.; Inagaki, M.; Lippert, E.; Akira, S.; Robine, S.; Matsumoto, K.; Jobin, C.; Ninomiya-Tsuji, J. Enterocyte-derived TAK1 signaling prevents epithelium apoptosis and the development of ileitis and colitis. J. Immunol. 2008, 181, 1143–1152. [Google Scholar] [CrossRef]
- Fish, J.D.; Duerst, R.E.; Gelfand, E.W.; Orange, J.S.; Bunin, N. Challenges in the use of allogeneic hematopoietic SCT for ectodermal dysplasia with immune deficiency. Bone Marrow Transplant. 2009, 43, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Nishikomori, R.; Heike, T. Diagnosis and treatment in anhidrotic ectodermal dysplasia with immunodeficiency. Allergol. Int. 2012, 61, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.Y.; Levy, O.; Jabara, H.H.; Glickman, J.N.; Stoler-Barak, L.; Sachs, J.; Nurko, S.; Orange, J.S.; Geha, R.S. Allogeneic transplantation successfully corrects immune defects, but not susceptibility to colitis, in a patient with nuclear factor-kappaB essential modulator deficiency. J. Allergy Clin. Immunol. 2008, 122, 1113–1118. [Google Scholar] [CrossRef]
- Permaul, P.; Narla, A.; Hornick, J.L.; Pai, S.Y. Allogeneic hematopoietic stem cell transplantation for X-linked ectodermal dysplasia and immunodeficiency: Case report and review of outcomes. Immunol. Res. 2009, 44, 89–98. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, L.; Vieira-Silva, S.; Billiet, T.; van Es, J.H.; Mc Guire, C.; Slowicka, K.; Sze, M.; van den Born, M.; De Hertogh, G.; Clevers, H.; et al. A20 controls intestinal homeostasis through cell-specific activities. Nat. Commun. 2014, 5, 5103. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, L.; Sze, M.; Mc Guire, C.; Rogiers, B.; Chu, Y.; Schmidt-Supprian, M.; Pasparakis, M.; Beyaert, R.; van Loo, G. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J. Exp. Med. 2010, 207, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Kolodziej, L.E.; Lodolce, J.P.; Chang, J.E.; Schneider, J.R.; Grimm, W.A.; Bartulis, S.J.; Zhu, X.; Messer, J.S.; Murphy, S.F.; Reddy, N.; et al. TNFAIP3 maintains intestinal barrier function and supports epithelial cell tight junctions. PLoS ONE 2011, 6, e26352. [Google Scholar] [CrossRef]
- Rhee, L.; Murphy, S.F.; Kolodziej, L.E.; Grimm, W.A.; Weber, C.R.; Lodolce, J.P.; Chang, J.E.; Bartulis, S.J.; Messer, J.S.; Schneider, J.R.; et al. Expression of TNFAIP3 in intestinal epithelial cells protects from DSS- but not TNBS-induced colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G220–G227. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Carbonell, R.; Wong, J.; Kim, J.Y.; Close, L.A.; Boland, B.S.; Wong, T.L.; Harris, P.A.; Ho, S.B.; Das, S.; Ernst, P.B.; et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc. Natl. Acad. Sci. USA 2018, 115, E9192–E9200. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.A.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.C.; O’Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, X.; McQuade, T.; Li, J.; Chan, F.K.; Zhang, J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 2011, 471, 373–376. [Google Scholar] [CrossRef]
- Welz, P.S.; Wullaert, A.; Vlantis, K.; Kondylis, V.; Fernandez-Majada, V.; Ermolaeva, M.; Kirsch, P.; Sterner-Kock, A.; van Loo, G.; Pasparakis, M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 2011, 477, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Gunther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 2011, 477, 335–339. [Google Scholar] [CrossRef]
- Stolzer, I.; Kaden-Volynets, V.; Ruder, B.; Letizia, M.; Bittel, M.; Rausch, P.; Basic, M.; Bleich, A.; Baines, J.F.; Neurath, M.F.; et al. Environmental Microbial Factors Determine the Pattern of Inflammatory Lesions in a Murine Model of Crohn’s Disease-Like Inflammation. Inflamm. Bowel Dis. 2020, 26, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, R.; Jiao, H.; Wachsmuth, L.; Tresch, A.; Pasparakis, M. FADD and Caspase-8 Regulate Gut Homeostasis and Inflammation by Controlling MLKL- and GSDMD-Mediated Death of Intestinal Epithelial Cells. Immunity 2020, 52, 978–993. [Google Scholar] [CrossRef] [PubMed]
- Wittkopf, N.; Gunther, C.; Martini, E.; He, G.; Amann, K.; He, Y.W.; Schuchmann, M.; Neurath, M.F.; Becker, C. Cellular FLICE-like inhibitory protein secures intestinal epithelial cell survival and immune homeostasis by regulating caspase-8. Gastroenterology 2013, 145, 1369–1379. [Google Scholar] [CrossRef]
- Lehle, A.S.; Farin, H.F.; Marquardt, B.; Michels, B.E.; Magg, T.; Li, Y.; Liu, Y.; Ghalandary, M.; Lammens, K.; Hollizeck, S.; et al. Intestinal Inflammation and Dysregulated Immunity in Patients With Inherited Caspase-8 Deficiency. Gastroenterology 2019, 156, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, Y.; Kim, C.A.; Pastorino, A.C.; Ceroni, J.; Lima, P.P.; de Barros Dorna, M.; Honjo, R.S.; Bertola, D.; Hamanaka, K.; Fujita, A.; et al. Primary immunodeficiency with chronic enteropathy and developmental delay in a boy arising from a novel homozygous RIPK1 variant. J. Hum. Genet. 2019. [Google Scholar] [CrossRef]
- Dourmashkin, R.; Davies, H.; Wells, C.; Shah, D.; Price, A.; O’Morain, J.; Levi, J. Epithelial patchy necrosis in Crohn’s disease. Hum. Pathol. 1989, 14, 643–648. [Google Scholar] [CrossRef]
- Pierdomenico, M.; Negroni, A.; Stronati, L.; Vitali, R.; Prete, E.; Bertin, J.; Gough, P.J.; Aloi, M.; Cucchiara, S. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am. J. Gastroenterol. 2014, 109, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Negroni, A.; Colantoni, E.; Pierdomenico, M.; Palone, F.; Costanzo, M.; Oliva, S.; Tiberti, A.; Cucchiara, S.; Stronati, L. RIP3 AND pMLKL promote necroptosis-induced inflammation and alter membrane permeability in intestinal epithelial cells. Dig. Liver Dis. 2017, 49, 1201–1210. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y.; Xue, L.; Sheng, Y.; Xu, L.; Xue, Y. Expression of receptor interacting protein 3 and mixed lineage kinase domain-like protein-key proteins in necroptosis is upregulated in ulcerative colitis. Ann. Palliat. Med. 2019, 8, 483–489. [Google Scholar] [CrossRef]
- Zhou, M.; He, J.; Shi, Y.; Liu, X.; Luo, S.; Cheng, C.; Ge, W.; Qu, C.; Du, P.; Chen, Y. ABIN3 Negatively Regulates Necroptosis-induced Intestinal Inflammation Through Recruiting A20 and Restricting the Ubiquitination of RIPK3 in Inflammatory Bowel Disease. J. Crohns Colitis. 2021, 15, 99–114. [Google Scholar] [CrossRef]
- Gobbetti, T.; Berger, S.B.; Fountain, K.; Slocombe, T.; Rowles, A.; Pearse, G.; Harada, I.; Bertin, J.; Haynes, A.C.; Beal, A.M. Receptor-interacting protein 1 kinase inhibition therapeutically ameliorates experimental T cell-dependent colitis in mice. Cell Death Dis. 2020, 11, 220. [Google Scholar] [CrossRef]
- Zhang, J.; Qin, D.; Yang, Y.J.; Hu, G.Q.; Qin, X.X.; Du, C.T.; Chen, W. MLKL deficiency inhibits DSS-induced colitis independent of intestinal microbiota. Mol. Immunol. 2019, 107, 132–141. [Google Scholar] [CrossRef]
- Zhao, Q.; Yu, X.; Li, M.; Liu, Y.; Han, Y.; Zhang, X.; Li, X.M.; Wu, X.; Qin, J.; Fang, J.; et al. MLKL attenuates colon inflammation and colitis-tumorigenesis via suppression of inflammatory responses. Cancer Lett. 2019, 459, 100–111. [Google Scholar] [CrossRef]
- Alvarez-Diaz, S.; Preaudet, A.; Samson, A.L.; Nguyen, P.M.; Fung, K.Y.; Garnham, A.L.; Alexander, W.S.; Strasser, A.; Ernst, M.; Putoczki, T.L.; et al. Necroptosis is dispensable for the development of inflammation-associated or sporadic colon cancer in mice. Cell Death Differ. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Li, H.; Fan, C.; Qi, Q.; Yan, Y.; Wu, Y.; Feng, C.; Wu, B.; Gao, Y.; Zuo, J.; et al. RIPK1 inhibitor ameliorates colitis by directly maintaining intestinal barrier homeostasis and regulating following IECs-immuno crosstalk. Biochem. Pharmacol. 2020, 172, 113751. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene Name | Mutation | RIP Kinase Signalling | Joint | Skin | Gut | Ref. |
|---|---|---|---|---|---|---|
| Tnfaip3 Mouse | Myeloid-specific A20 knockout (Tnfaip3LysM.cre) | RIPK1 kinase/RIPK3/MLKL- dependent necroptosis and NLRP3 inflammasome activation | Arthritis | [82,128,130] | ||
| Intestinal epithelium- and myeloid-specific A20 knockout B (Tnfaip3Villin.creTnfaip3LysM.cre) | ⇑ TNF-induced IEC apoptosis C | Severe colitis | [194,195] | |||
| cIAP1, cIAP2 Mouse | Myeloid-specific cIAP1 and global knockout of cIAP2 A,B (cIap1LysM.crecIap2−/−) | ⇑ TNF serum levels | Arthritis | [45] | ||
| Epidermal-specific cIAP1 and global knockout of cIAP2 (cIap1E-KOcIap2−/−) | TNF-mediated RIPK1- dependent apoptosis | Severe dermatitis | [139] | |||
| cIAP1, XIAP Mouse | Epidermal-specific cIAP1 and global knockout of XIAP (cIap1E-KO Xiap−/−) | RIPK1-dependent cell death | Severe dermatitis | [139] | ||
| cIAP1, cIAP2, XIAP Mouse | Myeloid-specific cIAP1 and global knockout of cIAP2 and XIAP B (cIap1LysM.crecIap2−/− Xiap−/−) | ⇑ TNF, IL-1 RIPK3-dependent apoptosis or necroptosis | Arthritis | Inflammation | [45] | |
| Fadd Mouse | Intestinal epithelial cell-specific knockout of FADD (FaddVillin.cre) | TNF-induced RIPK3- dependent necroptosis Caspase-8/GSDMD- dependent pyroptosis | Colitis, ileitis | [201,204] | ||
| Caspase-8 Mouse | IEC-specific knockout of Caspase 8 (Caspase-8Villin.cre) | TNF-induced RIPK3- dependent necroptosis | Ileitis, susceptible to colitis | [202,203] | ||
| Human | Loss of function mutations in Caspase-8 | ⇑ IL-1 (patient-derived monocytes) C | VEO-IBD | [206] | ||
| RIPK1 Mouse | Global RIPK1 knockout A,B (Ripk1−/−) |
Apoptosis and RIPK3- dependent necroptosis | Epidermal hyperplasia | [35,97] | ||
| Epithelial-specific knockout of RIPK1 (Ripk1E-KO) | TNFR1-induced RIPK3- and ZBP1-RIPK3-MLKL- dependent necroptosis | Psoriasis-like disease | [30,61,137] | |||
| RHIM domain mutation A (Ripk1RHIM/RHIM) | Overactive ZBP1-RIPK3-MLKL- dependent necroptosis | Dermatitis | [59] | |||
| IEC-specific knockout of RIPK1 A (Ripk1Villin.cre) |
TNF/FADD-dependent apoptosis or RIPK3- dependent necroptosis (when FADD is absent) | Inflammation | [30,36] | |||
| Caspase-8 cleavage-resistant RIPK1 B (Ripk1D325A/D325A) | TNF-induced apoptosis and necroptosis | Skin hyperplasia | [138] | |||
| Human | Homozygous loss-of-function or missense mutations B | ⇑ IL-1β signalling Aberrant TNFR and TLR signalling | Early- onset poly- arthritis | Skin lesions | IBD | [98,99,138,207] |
| HOIL-1 Mouse | Keratinocyte-specific deletion of HOILA (HoilE-KO) | TNFR1-induced Caspase-8-dependent apoptosis (RIPK1 independent) | Dermatitis | [145] | ||
| Human | Autosomal recessive loss-of-function B | ⇓ RIPK1 polyubiquitination | Dermatitis | IBD | [143] | |
| Hoip Mouse | Keratinocyte-specific deletion of HOIP A (HoipE-KO) | TNFR1-induced Caspase-8-dependent RIPK-independent apoptosis | Lethal dermatitis | [145,112] | ||
| Sharpin Mouse | Global knockout (Sharpincpdm/cpdm) B | TNFR1-induced cell death ⇓ NF-κB signalling | Arthritis | Severe dermatitis | Loss of Peyer’s patches in gut | [19,113,135,140,147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Speir, M.; Djajawi, T.M.; Conos, S.A.; Tye, H.; Lawlor, K.E. Targeting RIP Kinases in Chronic Inflammatory Disease. Biomolecules 2021, 11, 646. https://doi.org/10.3390/biom11050646
Speir M, Djajawi TM, Conos SA, Tye H, Lawlor KE. Targeting RIP Kinases in Chronic Inflammatory Disease. Biomolecules. 2021; 11(5):646. https://doi.org/10.3390/biom11050646
Chicago/Turabian StyleSpeir, Mary, Tirta M. Djajawi, Stephanie A. Conos, Hazel Tye, and Kate E. Lawlor. 2021. "Targeting RIP Kinases in Chronic Inflammatory Disease" Biomolecules 11, no. 5: 646. https://doi.org/10.3390/biom11050646
APA StyleSpeir, M., Djajawi, T. M., Conos, S. A., Tye, H., & Lawlor, K. E. (2021). Targeting RIP Kinases in Chronic Inflammatory Disease. Biomolecules, 11(5), 646. https://doi.org/10.3390/biom11050646