
Role of Serum Amyloid A in Abdominal Aortic Aneurysm and Related Cardiovascular Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
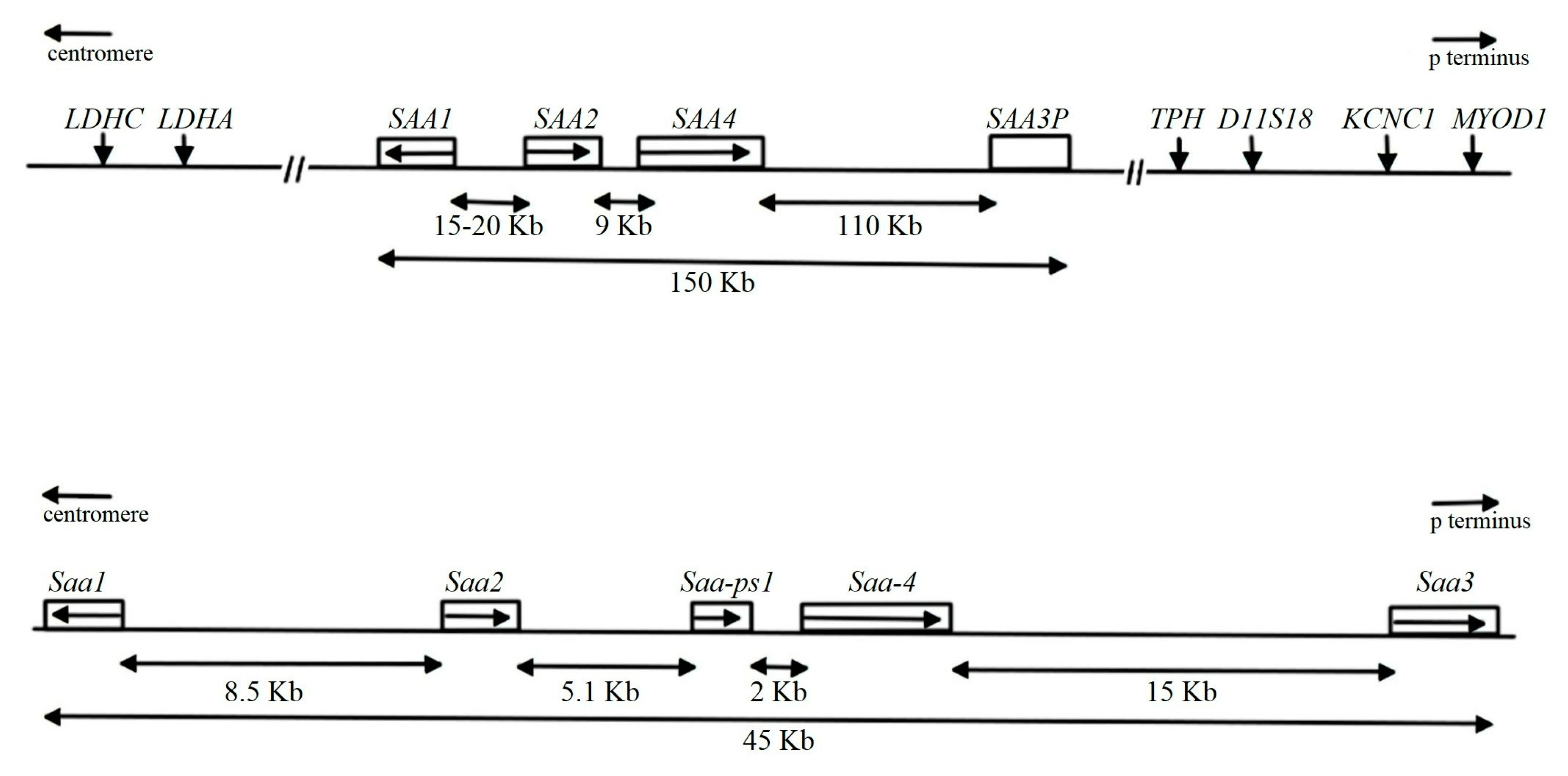
The SAA Family
2. SAA Biology
2.1. Regulation of SAA Production
2.2. The Current Challenges and Controversies in the Study of SAA
3. HDL Association
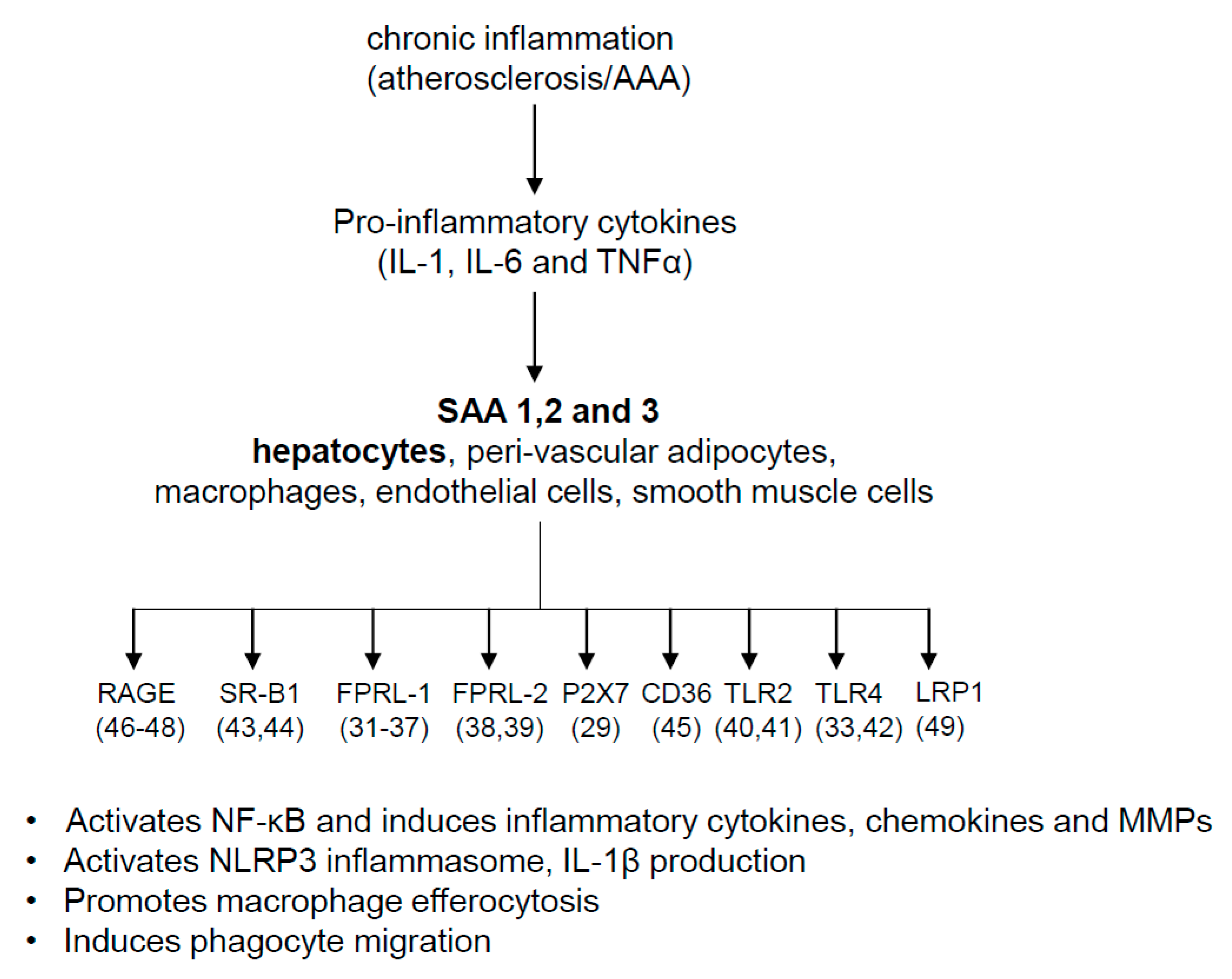
4. Pathophysiologic Roles of SAA
5. SAA and Cardiovascular Diseases
SAA and Abdominal Aortic Aneurysms
6. SAA and Atherosclerosis
7. Possible Mechanisms for SAA’s Role in AAA and Atherosclerosis
8. Systemic vs. Local Production of SAA
9. Conclusions
10. Key Points
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sack, G.H., Jr. Serum amyloid A—A review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef] [PubMed]
- Benditt, E.P.; Meek, R.L. Expression of the third member of the serum amyloid A gene family in mouse adipocytes. J. Exp. Med. 1989, 169, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.E.; Whitehead, A.S. Regulation of serum amyloid A protein expression during the acute-phase response. Biochem. J. 1998, 334 Pt 3, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Sellar, G.C.; Jordan, S.A.; Bickmore, W.A.; Fantes, J.A.; van Heyningen, V.; Whitehead, A.S. The human serum amyloid A protein (SAA) superfamily gene cluster: Mapping to chromosome 11p15.1 by physical and genetic linkage analysis. Genomics 1994, 19, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Whitehead, A.S. Mapping of the mouse serum amyloid A gene cluster by long-range polymerase chain reaction. Immunogenetics 1996, 44, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Kluve-Beckerman, B.; Drumm, M.L.; Benson, M.D. Nonexpression of the human serum amyloid A three (SAA3) gene. DNA Cell Biol. 1991, 10, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, C.S.; Lunden, G.O.; Felin, J.; Backhed, F. Regulation of serum amyloid A3 (SAA3) in mouse colonic epithelium and adipose tissue by the intestinal microbiota. PLoS ONE 2009, 4, e5842. [Google Scholar] [CrossRef] [PubMed]
- Den Hartigh, L.J.; Wang, S.; Goodspeed, L.; Ding, Y.; Averill, M.; Subramanian, S.; Wietecha, T.; O’Brien, K.D.; Chait, A. Deletion of serum amyloid A3 improves high fat high sucrose diet-induced adipose tissue inflammation and hyperlipidemia in female mice. PLoS ONE 2014, 9, e108564. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C.; Wilson, P.G.; Shridas, P.; Ji, A.; de Beer, M.; de Beer, F.C.; Webb, N.R.; Tannock, L.R. Serum amyloid A3 is pro-atherogenic. Atherosclerosis 2018, 268, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.S.; Ericsson, L.H.; Eriksen, N.; Walsh, K.A.; Benditt, E.P. Murine tissue amyloid protein AA. NH2-terminal sequence identity with only one of two serum amyloid protein (ApoSAA) gene products. J. Exp. Med. 1984, 159, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Liepnieks, J.J.; Kluve-Beckerman, B.; Benson, M.D. Characterization of amyloid A protein in human secondary amyloidosis: The predominant deposition of serum amyloid A1. Biochim. Biophys. Acta 1995, 1270, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Uhlar, C.M.; Whitehead, A.S. Serum amyloid A, the major vertebrate acute-phase reactant. Eur. J. Biochem. 1999, 265, 501–523. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Kushner, I. The phenomenon of the acute phase response. Ann. N. Y. Acad. Sci. 1982, 389, 39–48. [Google Scholar] [CrossRef] [PubMed]
- De Buck, M.; Gouwy, M.; Wang, J.M.; Van Snick, J.; Opdenakker, G.; Struyf, S.; Van Damme, J. Structure and Expression of Different Serum Amyloid A (SAA) Variants and their Concentration-Dependent Functions During Host Insults. Curr. Med. Chem. 2016, 23, 1725–1755. [Google Scholar] [CrossRef] [PubMed]
- Urieli-Shoval, S.; Cohen, P.; Eisenberg, S.; Matzner, Y. Widespread expression of serum amyloid A in histologically normal human tissues. Predominant localization to the epithelium. J. Histochem. Cytochem. 1998, 46, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Sjöholm, K.; Palming, J.; Olofsson, L.E.; Gummesson, A.; Svensson, P.-A.; Lystig, T.; Jennische, E.; Brandberg, J.; Torgerson, J.S.; Carlsson, B.; et al. A microarray search for genes predominantly expressed in human omental adipocytes: Adipose tissue as a major production site of serum amyloid A. J. Clin. Endocrinol. Metab. 2005, 90, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.-Z.; Lee, M.-J.; Hu, H.; Pollin, T.I.; Ryan, A.S.; Nicklas, B.J.; Snitker, S.; Horenstein, R.B.; Hull, K.; Goldberg, N.H.; et al. Acute-phase serum amyloid A: An inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. 2006, 3, e287. [Google Scholar] [CrossRef] [PubMed]
- Meek, R.L.; Eriksen, N.; Benditt, E.P. Murine serum amyloid A3 is a high density apolipoprotein and is secreted by macrophages. Proc. Natl. Acad. Sci. USA 1992, 89, 7949–7952. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Han, C.Y.; Vaisar, T.; Shimokado, K.; Kargi, A.; Chen, M.-H.; Wang, S.; McDonald, T.O.; O’Brien, K.; Heinecke, J.W.; et al. Serum amyloid A3 does not contribute to circulating SAA levels. J. Lipid Res. 2009, 50, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Tannock, L.R.; De Beer, M.C.; Ji, A.; Shridas, P.; Noffsinger, V.P.; Hartigh, L.D.; Chait, A.; De Beer, F.C.; Webb, N.R. Serum amyloid A3 is a high density lipoprotein-associated acute-phase protein. J. Lipid Res. 2018, 59, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Shridas, P. Tannock LR. Role of serum amyloid A in atherosclerosis. Curr. Opin. Lipidol. 2019, 30, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Meek, R.L.; Urieli-Shoval, S.; Benditt, E.P. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: Implications for serum amyloid A function. Proc. Natl. Acad. Sci. USA 1994, 91, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.R. High-Density Lipoproteins and Serum Amyloid A (SAA). Curr. Atheroscler. Rep. 2021, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhou, H.; Zhu, Z.; Ziyan, Z.; Wang, L.; Liang, Q.; Ye, R.D. Ex vivo and in vitro effect of serum amyloid a in the induction of macrophage M2 markers and efferocytosis of apoptotic neutrophils. J. Immunol. 2015, 194, 4891–4900. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Whitehead, A.S. Differential glucocorticoid enhancement of the cytokine-driven transcriptional activation of the human acute phase serum amyloid A genes, SAA1 and SAA2. J. Immunol. 2002, 169, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Lu, Z.Y.; Whitehead, A.S. Tissue-specific regulation of the human acute-phase serum amyloid A genes, SAA1 and SAA2, by glucocorticoids in hepatic and epithelial cells. Eur. J. Immunol. 2003, 33, 2630–2639. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, L.; Raynes, J.G.; Shah, C.; Karlsson, A.; Dahlgren, C.; Bylund, J. The proinflammatory activity of recombinant serum amyloid A is not shared by the endogenous protein in the circulation. Arthritis Rheum. 2010, 62, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Christenson, K.; Bjorkman, L.; Tangemo, C.; Bylund, J. Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7-sensitive pathway independent of formyl peptide receptor-like 1. J. Leukoc. Biol. 2008, 83, 139–148. [Google Scholar] [CrossRef] [PubMed]
- De Beer, M.C.; Wroblewski, J.M.; Noffsinger, V.P.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Ji, A.; Shridas, P.; Thompson, J.C.; van der Westhuyzen, D.R.; et al. Deficiency of endogenous acute phase serum amyloid A does not affect atherosclerotic lesions in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Lee, S.Y.; Lee, H.; Cho, K.-H.; Yun, J.; Bae, Y.-S. Serum amyloid A induces CCL2 production via formyl peptide receptor-like 1-mediated signaling in human monocytes. J. Immunol. 2008, 181, 4332–4339. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, M.-K.; Park, K.S.; Bae, Y.H.; Yun, J.; Park, J.-I.; Kwak, J.-Y.; Bae, Y.-S. Serum amyloid A stimulates matrix-metalloproteinase-9 upregulation via formyl peptide receptor like-1-mediated signaling in human monocytic cells. Biochem. Biophys. Res. Commun. 2005, 330, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ooi, S.Q.; Heng, C.K. The role of NF-kB in SAA-induced peroxisome proliferator-activated receptor gamma activation. Atherosclerosis 2013, 227, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Decker, Y.; McBean, G.; Godson, C. Lipoxin A4 inhibits IL-1beta-induced IL-8 and ICAM-1 expression in 1321N1 human astrocytoma cells. Am. J. Physiol. Cell Physiol. 2009, 296, C1420-7. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; An, F.; Wu, T.; Zhang, C.; Zhang, M.; Zhang, Y.; An, G.; An, F. PTX3, a key component of innate immunity, is induced by SAA via FPRL1-mediated signaling in HAECs. J. Cell Biochem. 2011, 112, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Sang, H.; Ye, R.D. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood 2003, 101, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Su, S.B.; Gong, W.; Gao, J.-L.; Shen, W.; Murphy, P.M.; Oppenheim, J.J.; Wang, J.M. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J. Exp. Med. 1999, 189, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Dufton, N.; Hannon, R.; Brancaleone, V.; Dalli, J.; Patel, H.B.; Gray, M.; D’Acquisto, F.; Buckingham, J.C.; Perretti, M.; Flower, R.J. Anti-inflammatory role of the murine formyl-peptide receptor 2: Ligand-specific effects on leukocyte responses and experimental inflammation. J. Immunol. 2010, 184, 2611–2619. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Kim, H.J.; Yun, J.; Baek, S.-H.; Kim, K.; Bae, Y.-S. A pertussis toxin sensitive G-protein-independent pathway is involved in serum amyloid A-induced formyl peptide receptor 2-mediated CCL2 production. Exp. Mol. Med. 2010, 42, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; He, R.; Tian, J.; Ye, P.P.; Ye, R.D. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol. 2008, 181, 22–26. [Google Scholar] [CrossRef] [PubMed]
- He, R.L.; Zhou, J.; Hanson, C.Z.; Chen, J.; Cheng, N.; Ye, R.D. Serum amyloid A induces G-CSF expression and neutrophilia via Toll-like receptor 2. Blood 2009, 113, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Sandri, S.; Rodriguez, D.; Gomes, E.; Monteiro, H.P.; Russo, M.; Campa, A. Is serum amyloid A an endogenous TLR4 agonist? J. Leukoc. Biol. 2008, 83, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Baranova, I.N.; Vishnyakova, T.G.; Bocharov, A.V.; Kurlander, R.; Chen, Z.; Kimelman, M.L.; Remaley, A.T.; Csako, G.; Thomas, F.; Eggerman, T.L.; et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J. Biol. Chem. 2005, 280, 8031–8040. [Google Scholar] [CrossRef] [PubMed]
- Mullan, R.H.; McCormick, J.; Connolly, M.; Bresnihan, B.; Veale, D.J.; Fearon, U. A role for the high-density lipoprotein receptor SR-B1 in synovial inflammation via serum amyloid-A. Am. J. Pathol. 2010, 176, 1999–2008. [Google Scholar] [CrossRef] [PubMed]
- Baranova, I.N.; Bocharov, A.V.; Vishnyakova, T.G.; Kurlander, R.; Chen, Z.; Fu, D.; Arias, I.M.; Csako, G.; Patterson, A.P.; Eggerman, T.L. CD36 is a novel serum amyloid A (SAA) receptor mediating SAA binding and SAA-induced signaling in human and rodent cells. J. Biol. Chem. 2010, 285, 8492–8506. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, S.; Li, J.; D’Amore, J.; D’Angelo, J.; Yang, H.; Wang, P.; Tracey, K.J.; Wang, H. Serum Amyloid A Stimulates PKR Expression and HMGB1 Release Possibly through TLR4/RAGE Receptors. Mol. Med. 2015, 21, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Du Yan, S.; Zhu, H.; Zhu, A.; Golabek, A.; Du, H.; Roher, A.; Yu, J.; Soto, C.; Schmidt, A.M.; Stern, D.; et al. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat. Med. 2000, 6, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Röcken, C.; Kientsch-Engel, R.; Mansfeld, S.; Stix, B.; Stubenrauch, K.; Weigle, B.; Bühling, F.; Schwan, M.; Saeger, W. Advanced glycation end products and receptor for advanced glycation end products in AA amyloidosis. Am. J. Pathol. 2003, 162, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Hu, Z.; Li, Y.; Gattu, S.; Ruhn, K.A.; Raj, P.; Herz, J.; Hooper, L.V. Serum amyloid A delivers retinol to intestinal myeloid cells to promote adaptive immunity. Science 2021, 373, eabf9232. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; de Beer, M.C.; Wroblewski, J.M.; Webb, N.R.; de Beer, F.C. SAA does not induce cytokine production in physiological conditions. Cytokine 2013, 61, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Simons, J.P.; Al-Shawi, R.; Ellmerich, S.; Speck, I.; Aslam, S.; Hutchinson, W.L.; Mangione, P.P.; Disterer, P.; Gilbertson, J.A.; Hunt, T.; et al. Pathogenetic mechanisms of amyloid A amyloidosis. Proc. Natl. Acad. Sci. USA 2013, 110, 16115–16120. [Google Scholar] [CrossRef] [PubMed]
- Benditt, E.P.; Eriksen, N. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc. Natl. Acad. Sci. USA 1977, 74, 4025–4028. [Google Scholar] [CrossRef] [PubMed]
- Kisilevsky, R.; Manley, P.N. Acute-phase serum amyloid A: Perspectives on its physiological and pathological roles. Amyloid 2012, 19, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.G.; Thompson, J.C.; Shridas, P.; McNamara, P.J.; de Beer, M.C.; de Beer, F.C.; Webb, N.R.; Tannock, L.R. Serum Amyloid A Is an Exchangeable Apolipoprotein. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; de Beer, M.C.; Noffsinger, V.; Tannock, L.R.; Ramaiah, C.; Webb, N.R.; van der Westhuyzen, D.R.; de Beer, F.C. HDL remodeling during the acute phase response. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Jahangiri, A.; Wilson, P.G.; Hou, T.; Brown, A.; King, V.L.; Tannock, L.R. SAA is found on apoB-containing lipoproteins in obese diabetic humans. Obesity 2013, 21, 993–996. [Google Scholar] [CrossRef]
- Cunnane, G. Amyloid precursors and amyloidosis in inflammatory arthritis. Curr. Opin. Rheumatol. 2001, 13, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Van Lenten, B.J.; Hama, S.Y.; De Beer, F.C.; Stafforini, D.M.; McIntyre, T.M.; Prescott, S.M.; La Du, B.N.; Fogelman, A.M.; Navab, M. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J. Clin. Investig. 1995, 96, 2758–2767. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Y.; Tang, C.; Guevara, M.E.; Wei, H.; Wietecha, T.; Shao, B.; Subramanian, S.; Omer, M.; Wang, S.; O’Brien, K.D.; et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J. Clin. Investig. 2016, 126, 266–281. [Google Scholar] [CrossRef] [PubMed]
- McGillicuddy, F.C.; Moya, M.D.L.L.; Hinkle, C.C.; Joshi, M.R.; Chiquoine, E.H.; Billheimer, J.T.; Rothblat, G.H.; Reilly, M.P. Inflammation impairs reverse cholesterol transport in vivo. Circulation 2009, 119, 1135–1145. [Google Scholar] [CrossRef]
- De Beer, M.C.; Wroblewski, J.M.; Noffsinger, V.P.; Ji, A.; Meyer, J.M.; Van Der Westhuyzen, D.R.; De Beer, F.C.; Webb, N.R. The Impairment of Macrophage-to-Feces Reverse Cholesterol Transport during Inflammation Does Not Depend on Serum Amyloid A. J. Lipids 2013, 2013, 283486. [Google Scholar] [CrossRef] [PubMed]
- Pamir, N.; Hutchins, P.; Ronsein, G.E.; Vaisar, T.; Reardon, C.A.; Getz, G.S.; Lusis, A.J.; Heinecke, J.W. Proteomic analysis of HDL from inbred mouse strains implicates APOE associated with HDL in reduced cholesterol efflux capacity via the ABCA1 pathway. J. Lipid Res. 2016, 57, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Banka, C.L.; Yuan, T.; de Beer, M.C.; Kindy, M.; Curtiss, L.K.; de Beer, F.C. Serum amyloid A (SAA): Influence on HDL-mediated cellular cholesterol efflux. J. Lipid Res. 1995, 36, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.P.; Flexman, A.; Hulme, J.; Kisilevsky, R. Promoting export of macrophage cholesterol: The physiological role of a major acute-phase protein, serum amyloid A 2.1. J. Lipid Res. 2002, 43, 1410–1420. [Google Scholar] [CrossRef] [PubMed]
- Kisilevsky, R.; Tam, S.P. Macrophage cholesterol efflux and the active domains of serum amyloid A 2.1. J. Lipid Res. 2003, 44, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- van der Westhuyzen, D.R.; Cai, L.; de Beer, M.C.; de Beer, F.C. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J. Biol. Chem. 2005, 280, 35890–35895. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; de Beer, M.C.; de Beer, F.C.; van der Westhuyzen, D.R. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J. Biol. Chem. 2005, 280, 2954–2961. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.S.; Benditt, E.P. Changes in high density lipoprotein content following endotoxin administration in the mouse. Formation of serum amyloid protein-rich subfractions. J. Biol. Chem. 1982, 257, 10510–10517. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-H.; de Beer, M.C.; Wroblewski, J.M.; Charnigo, R.J.; Ji, A.; Webb, N.R.; de Beer, F.C.; van der Westhuyzen, D.R. Impact of individual acute phase serum amyloid A isoforms on HDL metabolism in mice. J. Lipid Res. 2016, 57, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Webb, N.R.; De Beer, M.C.; Wroblewski, J.M.; Ji, A.; Bailey, W.; Shridas, P.; Charnigo, R.J.; Noffsinger, V.P.; Witta, J.; Howatt, D.A.; et al. Deficiency of Endogenous Acute-Phase Serum Amyloid A Protects apoE−/− Mice from Angiotensin II-Induced Abdominal Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Bang, Y.J.; Ruhn, K.A.; Hooper, L.V. Molecular basis for retinol binding by serum amyloid A during infection. Proc. Natl. Acad. Sci. USA 2019, 116, 19077–19082. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Liang, Y.; Du, X.; Ye, R.D. Serum amyloid A promotes LPS clearance and suppresses LPS-induced inflammation and tissue injury. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sheng, J.; Fan, Y.; Zhu, X.; Tao, Q.; He, Y.; Wang, S. Association between serum amyloid A levels and cancers: A systematic review and meta-analysis. Postgrad. Med. J. 2018, 94, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Lederle, F.A.; Johnson, G.R.; Wilson, S.E.; Ballard, D.J.; Jordan, J.W.D.; Blebea, J.; Littooy, F.N.; Freischlag, J.A.; Bandyk, D.; Rapp, J.H.; et al. Rupture rate of large abdominal aortic aneurysms in patients refusing or unfit for elective repair. JAMA 2002, 287, 2968–2972. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.C.; Gardner, J.B.; Meissner, M.H.; Johansen, H.K. Rupture in small abdominal aortic aneurysms. J. Vasc. Surg. 1998, 28, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Cosford, P.A.; Leng, G.C. Screening for abdominal aortic aneurysm. Cochrane Database Syst. Rev. 2007, CD002945. [Google Scholar] [CrossRef] [PubMed]
- Hackam, D.G.; Thiruchelvam, D.; Redelmeier, D.A. Angiotensin-converting enzyme inhibitors and aortic rupture: A population-based case-control study. Lancet 2006, 368, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Lederle, F.A.; Noorbaloochi, S.; Nugent, S.; Taylor, B.; Grill, J.P.; Kohler, T.R.; Cole, L. Multicentre study of abdominal aortic aneurysm measurement and enlargement. Br. J. Surg. 2015, 102, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Investig. 2000, 105, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, C.; Paradis, P.; Schiffrin, E.L. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol. Sci. 2008, 29, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.D.; Kip, K.E.; Marroquin, O.C.; Ridker, P.M.; Kelsey, S.F.; Shaw, L.J.; Pepine, C.J.; Sharaf, B.; Merz, C.N.B.; Sopko, G.; et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: The National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE). Circulation 2004, 109, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, M.; Ebina, T.; Ishikawa, T.; Hibi, K.; Tsukahara, K.; Okuda, J.; Iwahashi, N.; Ozaki, H.; Yano, H.; Kusama, I.; et al. Serum amyloid A is a better predictor of clinical outcomes than C-reactive protein in non-ST-segment elevation acute coronary syndromes. Circ. J. 2007, 71, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, K.; Mashiba, S.; Wada, Y.; Sahara, M.; Uchida, K.; Aizawa, T.; Kodama, T. A serum amyloid A and LDL complex as a new prognostic marker in stable coronary artery disease. Atherosclerosis 2004, 174, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Liuzzo, G.; Biasucci, L.M.; Gallimore, J.R.; Grillo, R.L.; Rebuzzi, A.G.; Pepys, M.B.; Maseri, A. The prognostic value of C-reactive protein and serum amyloid a protein in severe unstable angina. N. Engl. J. Med. 1994, 331, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Shen, Y.; Yamen, E.; Hsu, K.; Yan, W.; Witting, P.K.; Geczy, C.L.; Ben Freedman, S. Serum amyloid A may potentiate prothrombotic and proinflammatory events in acute coronary syndromes. Atherosclerosis 2009, 202, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Maier, W.; Altwegg, L.A.; Corti, R.; Gay, S.; Hersberger, M.; Maly, F.E.; Sütsch, G.; Roffi, M.; Neidhart, M.; Eberli, F.R.; et al. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: Locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation 2005, 111, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.D.; McDonald, T.O.; Kunjathoor, V.; Eng, K.; Knopp, E.A.; Lewis, K.; Lopez, R.; Kirk, E.A.; Chait, A.; Wight, T.N.; et al. Serum amyloid A and lipoprotein retention in murine models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Boren, J.; Williams, K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: A triumph of simplicity. Curr. Opin. Lipidol. 2016, 27, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Wu, T.; Qin, W.; An, C.; Wang, Z.; Zhang, M.; Zhang, Y.; Zhang, C.; An, F. Serum Amyloid A Directly Accelerates the Progression of Atherosclerosis in Apolipoprotein E-Deficient Mice. Mol. Med. 2011, 17, 1357–1364. [Google Scholar] [CrossRef]
- Thompson, J.C.; Jayne, C.; Thompson, J.; Wilson, P.G.; Yoder, M.H.; Webb, N.; Tannock, L.R. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J. Lipid Res. 2015, 56, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Krishack, P.A.; Bhanvadia, C.V.; Lukens, J.; Sontag, T.J.; De Beer, M.C.; Getz, G.S.; Reardon, C.A. Serum Amyloid A Facilitates Early Lesion Development in Ldlr−/− Mice. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Niemi, K.; Teirilä, L.; Lappalainen, J.; Rajamäki, K.; Baumann, M.; Öörni, K.; Wolff, H.; Kovanen, P.T.; Matikainen, S.; Eklund, K.K. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 2011, 186, 6119–6128. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Izumi, Y.; Jiuchi, Y.; Kozuru, H.; Kawahara, C.; Nakamura, M.; Nakamura, T.; Agematsu, K.; Masumoto, J.; Yasunami, M.; et al. Serum amyloid A induces NLRP-3-mediated IL-1beta secretion in neutrophils. PLoS ONE 2014, 9, e96703. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Liu, S.; Yi, X.; Zhang, S.; Ding, Y. Serum amyloid A induces interleukin-1beta secretion from keratinocytes via the NACHT, LRR and PYD domains-containing protein 3 inflammasome. Clin. Exp. Immunol. 2015, 179, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Ather, J.; Ckless, K.; Martin, R.; Foley, K.L.; Suratt, B.T.; Boyson, J.E.; Fitzgerald, K.; Flavell, R.A.; Eisenbarth, S.C.; Poynter, M.E. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J. Immunol. 2011, 187, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Koga, T.; Satomura, K.; Izumi, M.; Torigoshi, T.; Maeda, Y.; Izumi, Y.; Jiuchi, Y.; Miyashita, T.; Yamasaki, S.; et al. Serum amyloid A triggers the mosodium urate -mediated mature interleukin-1beta production from human synovial fibroblasts. Arthritis Res. Ther. 2012, 14, R119. [Google Scholar] [CrossRef] [PubMed]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Yoshimura, K.; Aoki, H.; Tsutsui, H.; Noda, T.; et al. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Johnston, W.F.; Salmon, M.; Su, G.; Lu, G.; Stone, M.L.; Zhao, Y.; Owens, G.K.; Upchurch, G.R., Jr.; Ailawadi, G. Genetic and pharmacologic disruption of interleukin-1beta signaling inhibits experimental aortic aneurysm formation. Arterioscler. Thromb. Vasc Biol. 2013, 33, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Dihlmann, S.; Erhart, P.; Mehrabi, A.; Nickkholgh, A.; Lasitschka, F.; Böckler, D.; Hakimi, M. Increased expression and activation of absent in melanoma 2 inflammasome components in lymphocytic infiltrates of abdominal aortic aneurysms. Mol. Med. 2014, 20, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Newman, K.M.; Jean-Claude, J.; Li, H.; Ramey, W.G.; Tilson, M.D. Cytokines that activate proteolysis are increased in abdominal aortic aneurysms. Circulation 1994, 90 Pt 2, II224-7. [Google Scholar] [PubMed]
- Keen, R.R.; Nolan, K.D.; Cipollone, M.; Scott, E.; Shively, V.P.; Yao, J.S.; Pearce, W.H. Interleukin-1 beta induces differential gene expression in aortic smooth muscle cells. J. Vasc. Surg. 1994, 20, 774–784, discussion 84–66. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Song, C.; Hsu, K.; Yamen, E.; Yan, W.; Fock, J.; Witting, P.K.; Geczy, C.L.; Ben Freedman, S. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis 2009, 207, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Furlaneto, C.J.; Campa, A. A novel function of serum amyloid A: A potent stimulus for the release of tumor necrosis factor-alpha, interleukin-1beta, and interleukin-8 by human blood neutrophil. Biochem. Biophys. Res. Commun. 2000, 268, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Badolato, R.; A Johnston, J.; Wang, J.M.; McVicar, D.; Xu, L.L.; Oppenheim, J.J.; Kelvin, D.J. Serum amyloid A induces calcium mobilization and chemotaxis of human monocytes by activating a pertussis toxin-sensitive signaling pathway. J. Immunol. 1995, 155, 4004–4010. [Google Scholar]
- Wilson, P.G.; Thompson, J.C.; Webb, N.R.; De Beer, F.C.; King, V.L.; Tannock, L.R. SAA, but not CRP, stimulates vascular proteoglycan synthesis in a pro-atherogenic manner. Am. J. Pathol. 2008, 173, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Kawabe, Y.; Tominaga, M.; Origuchi, T.; Aoyagi, T.; Eguchi, K. Serum amyloid A protein induces production of matrix metalloproteinases by human synovial fibroblasts. Lab. Investig. 1998, 78, 535–539. [Google Scholar] [PubMed]
- Mullan, R.H.; Bresnihan, B.; Golden-Mason, L.; Markham, T.; O’Hara, R.; FitzGerald, O.; Veale, D.J.; Fearon, U. Acute-phase serum amyloid A stimulation of angiogenesis, leukocyte recruitment, and matrix degradation in rheumatoid arthritis through an NF-kappaB-dependent signal transduction pathway. Arthritis Rheum. 2006, 54, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.; Mullan, R.H.; McCormick, J.; Matthews, C.; Sullivan, O.; Kennedy, A.; FitzGerald, O.; Poole, A.R.; Bresnihan, B.; Veale, D.; et al. Acute-phase serum amyloid A regulates tumor necrosis factor alpha and matrix turnover and predicts disease progression in patients with inflammatory arthritis before and after biologic therapy. Arthritis Rheum. 2012, 64, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Z.; Ooi, D.S.; Shen, H.M.; Heng, C.K. The atherogenic effects of serum amyloid A are potentially mediated via inflammation and apoptosis. J. Atheroscler. Thromb. 2014, 21, 854–867. [Google Scholar] [CrossRef] [PubMed]
- El Kebir, D.; József, L.; Khreiss, T.; Pan, W.; Petasis, N.; Serhan, C.N.; Filep, J.G. Aspirin-triggered lipoxins override the apoptosis-delaying action of serum amyloid A in human neutrophils: A novel mechanism for resolution of inflammation. J. Immunol. 2007, 179, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Badolato, R.; Wang, J.M.; Murphy, W.J.; Lloyd, A.R.; Michiel, D.F.; Bausserman, L.L.; Kelvin, D.J.; Oppenheim, J.J.; Fava, R.A.; Olsen, N.J.; et al. Serum amyloid A is a chemoattractant: Induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J. Exp. Med. 1994, 180, 203–209. [Google Scholar] [CrossRef] [PubMed]
- De Buck, M.; Gouwy, M.; Wang, J.M.; Van Snick, J.; Proost, P.; Struyf, S.; Van Damme, J. The cytokine-serum amyloid A-chemokine network. Cytokine Growth Factor Rev. 2016, 30, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N.; Dahlén, S.-E.; Drazen, J.M.; Hay, D.W.P.; Rovati, G.; Shimizu, T.; Yokomizo, T.; Brink, C. The lipoxin receptor ALX: Potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 2006, 58, 463–487. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Song, C.; Geczy, C.L.; Freedman, S.B.; Witting, P.K. A role for acute-phase serum amyloid A and high-density lipoprotein in oxidative stress, endothelial dysfunction and atherosclerosis. Redox. Rep. 2009, 14, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Page, M.; Thomson, G.J.A.; Nunes, J.M.; Engelbrecht, A.-M.; Nell, T.A.; De Villiers, W.J.S.; De Beer, M.C.; Engelbrecht, L.; Kell, D.B.; Pretorius, E. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci. Rep. 2019, 9, 3102. [Google Scholar] [CrossRef] [PubMed]
- Lehman, S.J.; Massaro, J.M.; Schlett, C.L.; O’Donnell, C.J.; Hoffmann, U.; Fox, C.S. Peri-aortic fat, cardiovascular disease risk factors, and aortic calcification: The Framingham Heart Study. Atherosclerosis 2010, 210, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Poitou, C.; Viguerie, N.; Cancello, R.; De Matteis, R.; Cinti, S.; Stich, V.; Coussieu, C.; Gauthier, E.; Courtine, M.; Zucker, J.D.; et al. Serum amyloid A: Production by human white adipocyte and regulation by obesity and nutrition. Diabetologia 2005, 48, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Clancy, P.; Jamrozik, K.; Norman, P.E. Obesity, adipokines, and abdominal aortic aneurysm: Health in Men study. Circulation 2007, 116, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Djousse, L.; Song, Y.; Akinkuolie, A.O.; Matsumoto, C.; Manson, J.E.; Gaziano, J.M.; Sesso, H.D. Associations of Diabetes and Obesity with Risk of Abdominal Aortic Aneurysm in Men. J. Obes. 2017, 2017, 3521649. [Google Scholar] [CrossRef] [PubMed]
- Kugo, H.; Tanaka, H.; Moriyama, T.; Zaima, N. Pathological Implication of Adipocytes in AAA Development and the Rupture. Ann. Vasc. Dis. 2018, 11, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Police, S.B.; Thatcher, S.E.; Charnigo, R.; Daugherty, A.; Cassis, L.A. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, S.; Siekmeier, R. Influence of smoking and body weight on adipokines in middle aged women. Eur. J. Med. Res. 2009, 14 (Suppl. S4), 21–26. [Google Scholar] [CrossRef] [PubMed][Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shridas, P.; Patrick, A.C.; Tannock, L.R. Role of Serum Amyloid A in Abdominal Aortic Aneurysm and Related Cardiovascular Diseases. Biomolecules 2021, 11, 1883. https://doi.org/10.3390/biom11121883
Shridas P, Patrick AC, Tannock LR. Role of Serum Amyloid A in Abdominal Aortic Aneurysm and Related Cardiovascular Diseases. Biomolecules. 2021; 11(12):1883. https://doi.org/10.3390/biom11121883
Chicago/Turabian StyleShridas, Preetha, Avery C. Patrick, and Lisa R. Tannock. 2021. "Role of Serum Amyloid A in Abdominal Aortic Aneurysm and Related Cardiovascular Diseases" Biomolecules 11, no. 12: 1883. https://doi.org/10.3390/biom11121883
APA StyleShridas, P., Patrick, A. C., & Tannock, L. R. (2021). Role of Serum Amyloid A in Abdominal Aortic Aneurysm and Related Cardiovascular Diseases. Biomolecules, 11(12), 1883. https://doi.org/10.3390/biom11121883