
Improving the Substrate Affinity and Catalytic Efficiency of β-Glucosidase Bgl3A from Talaromyces leycettanus JCM12802 by Rational Design



Abstract
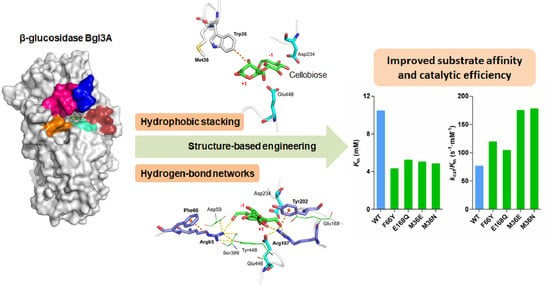
:
1. Introduction
2. Materials and Methods
2.1. Strains, Plasmids, and Materials
2.2. Sequence Analysis, Homology Modeling, and Docking
2.3. Identification of Mutagenesis Sites and Mutant Construction
2.4. Enzyme Expression and Purification
2.5. Enzymatic Assays and Kinetic Parameters
2.6. MD Simulation and Calculation of Binding Energy
3. Results and Discussion
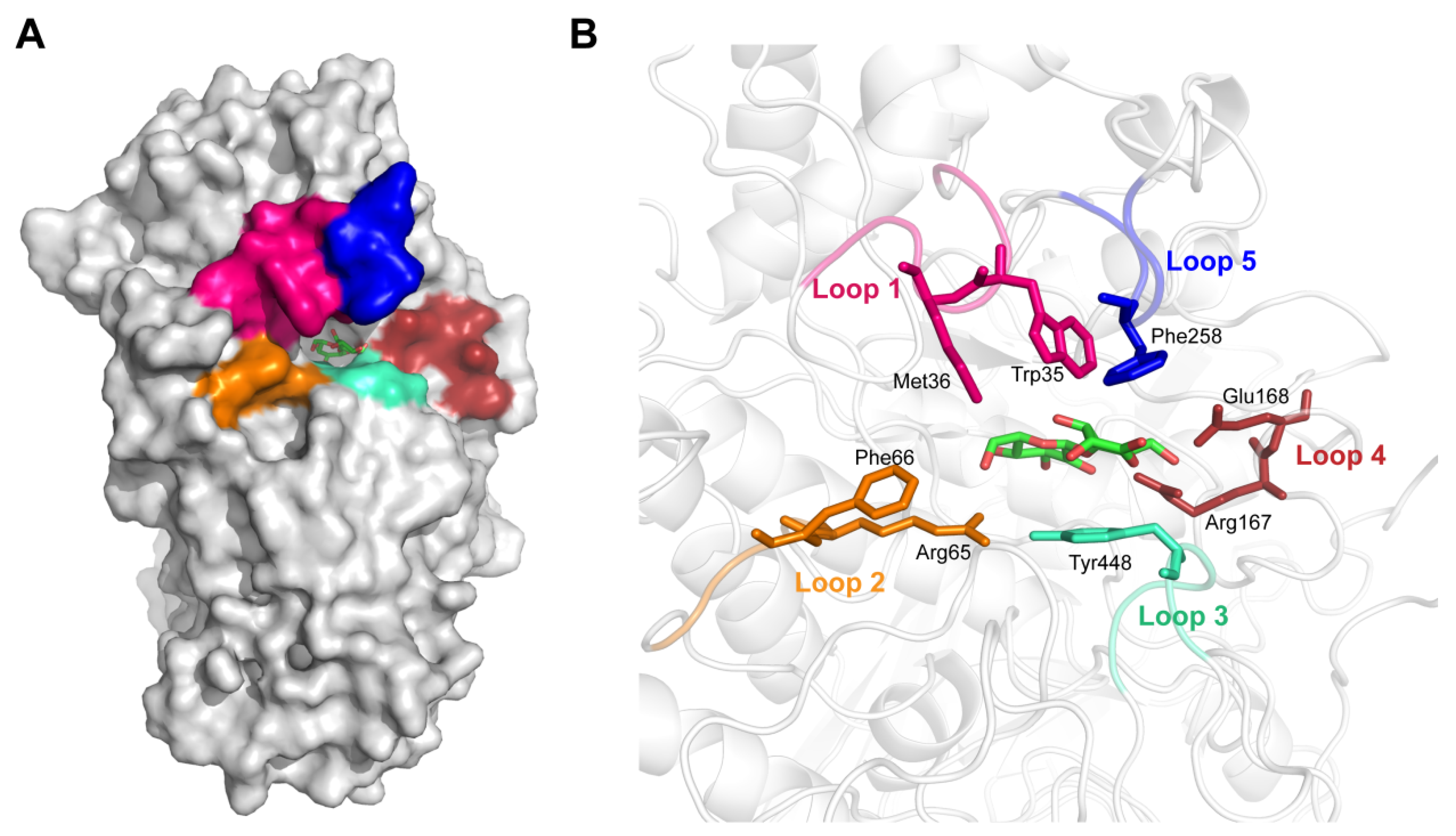
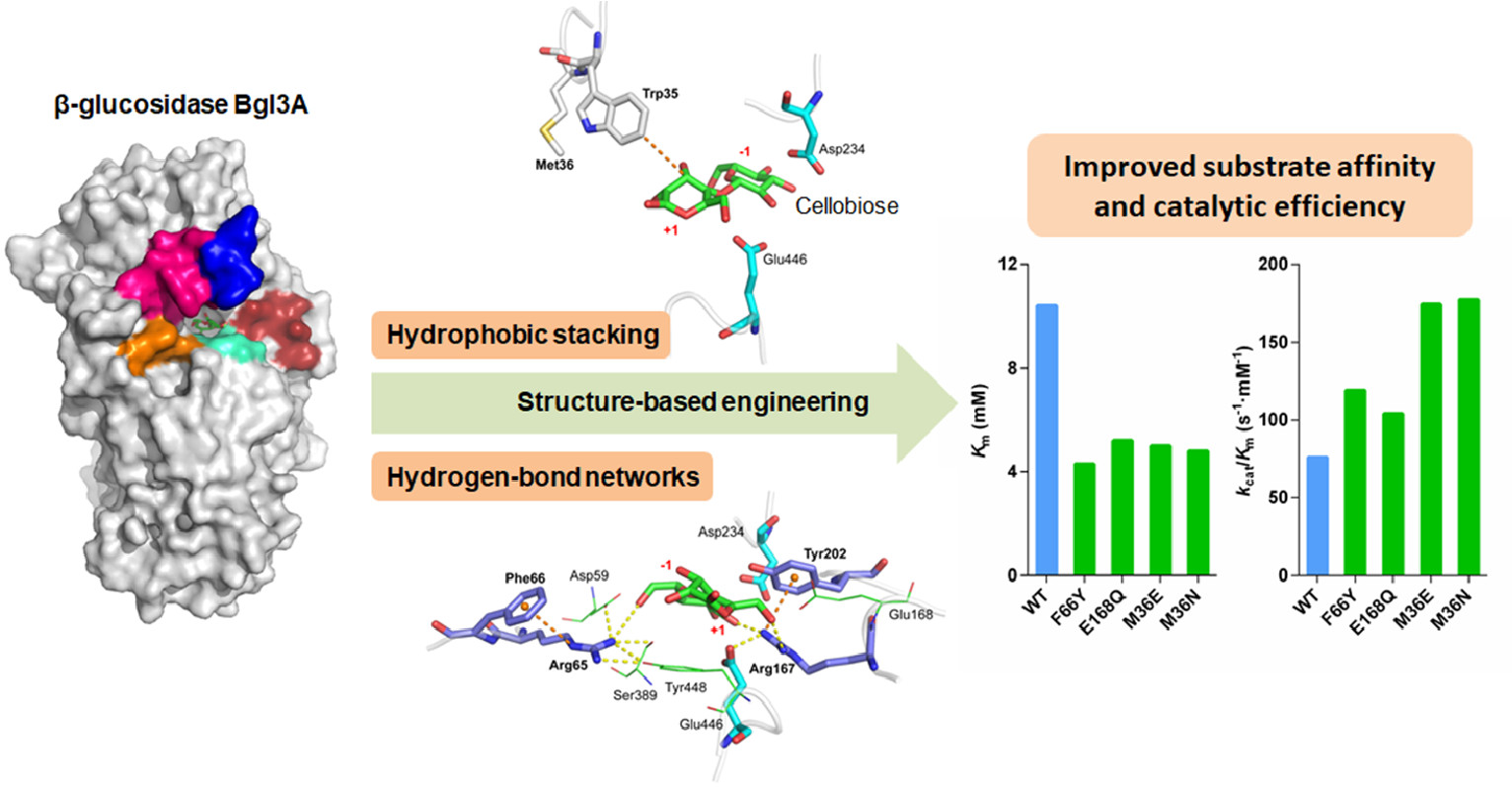
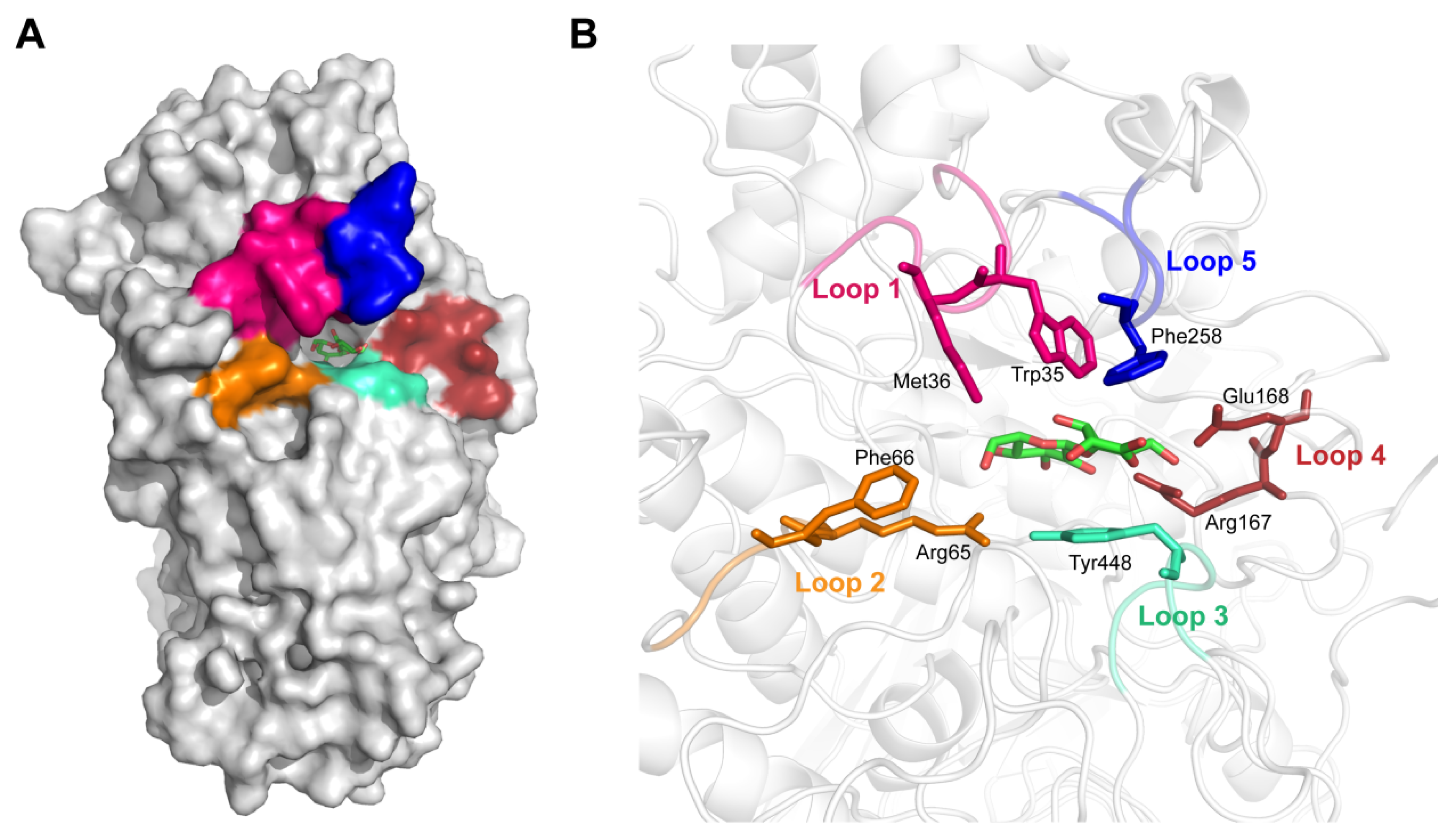
3.1. Substrate Binding Analysis
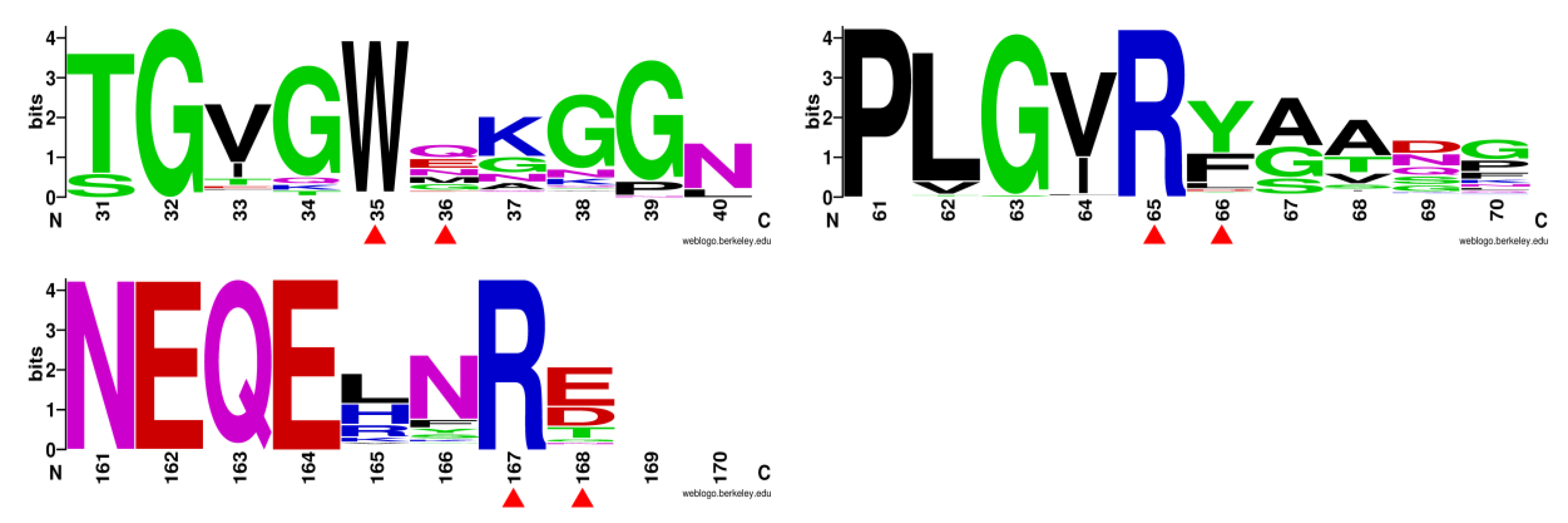
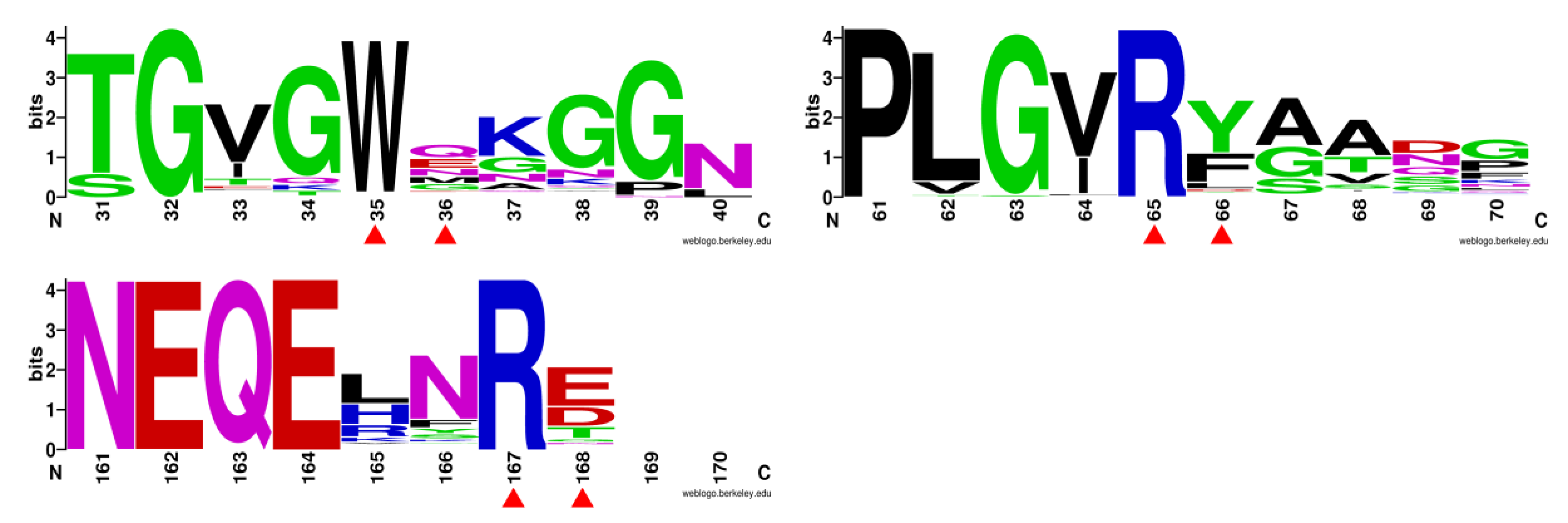
3.2. Mutant Design
3.3. Catalytic Performances of Wild-Type Bgl3A and Its Mutants
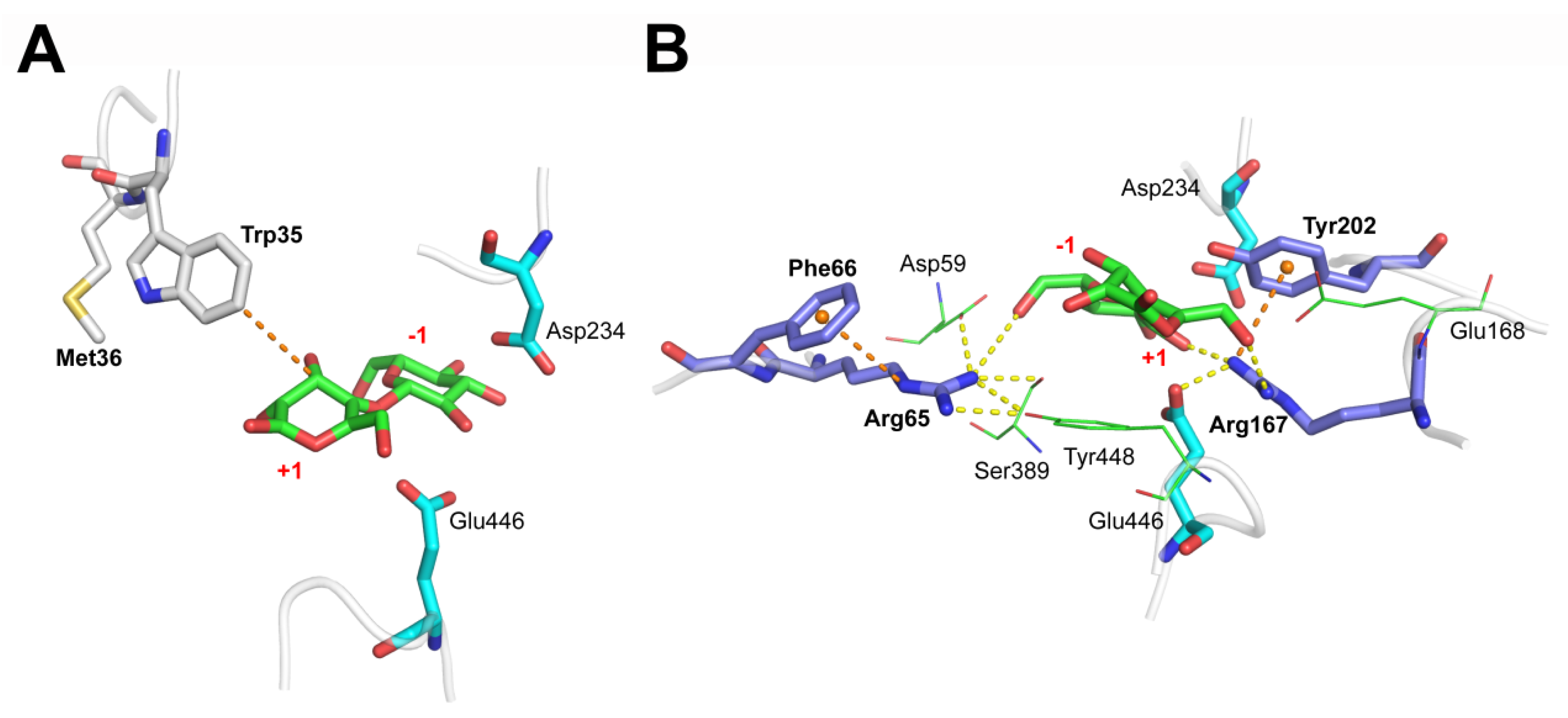
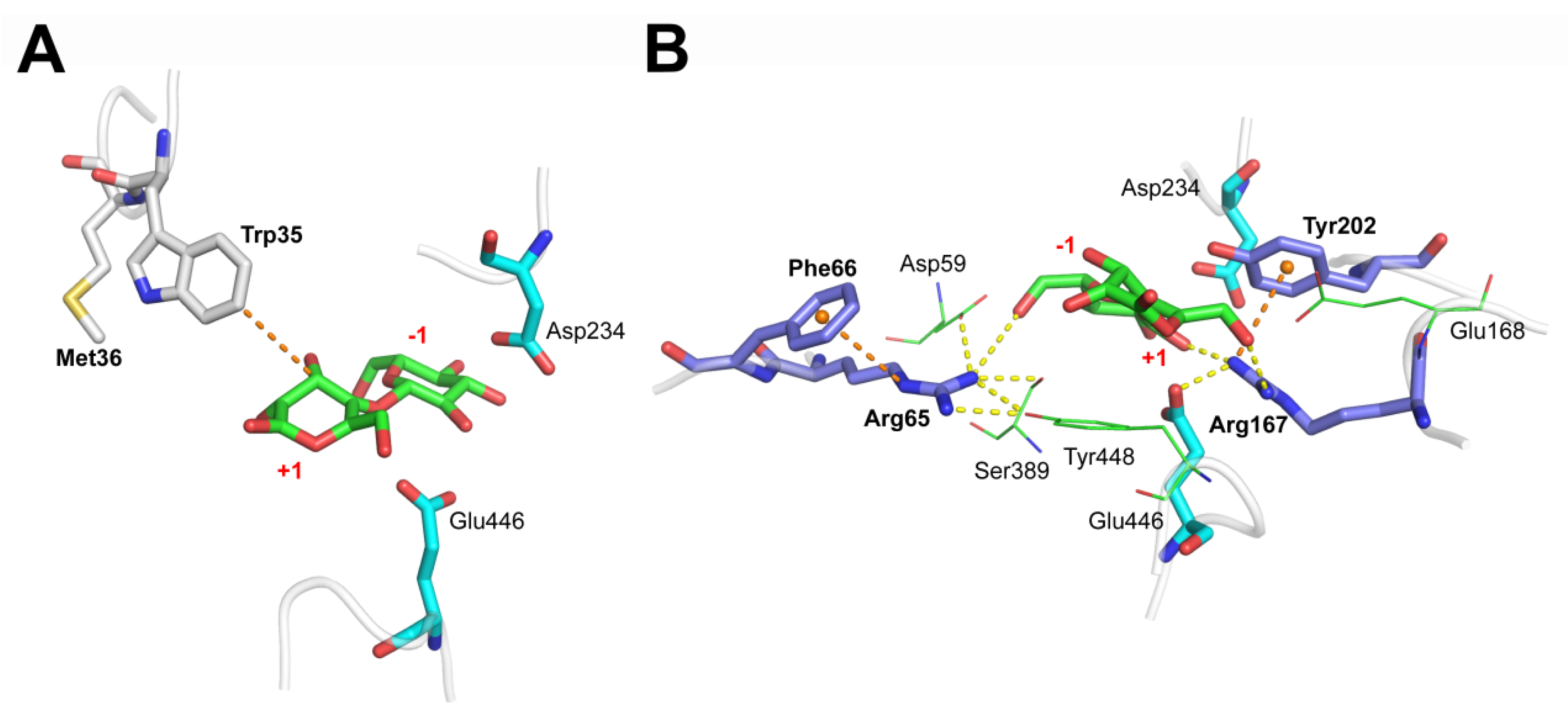
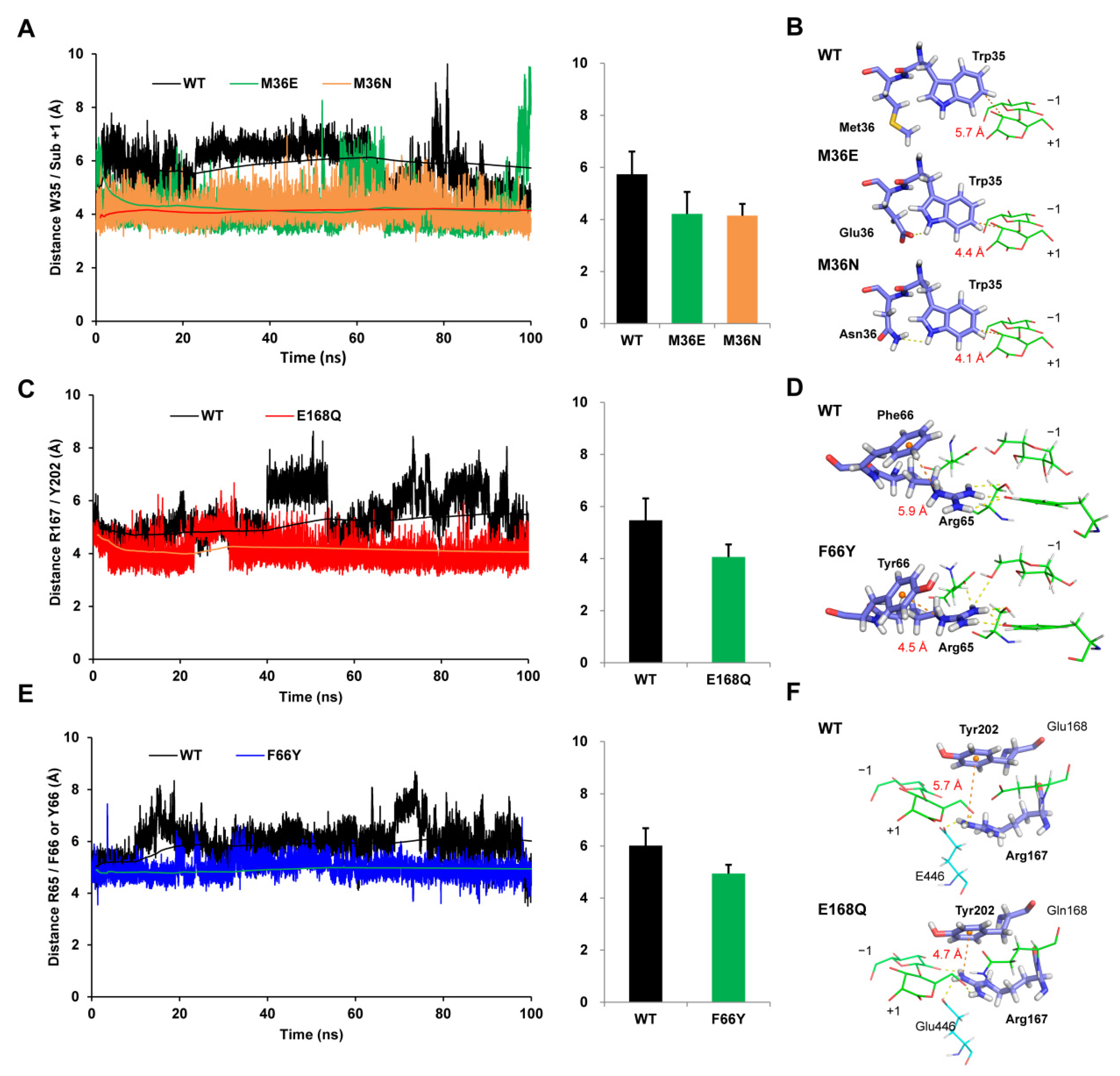
3.4. M36E and M36N Mutations Stabilized Trp35 Conformation by Introducing Hydrogen Bond Interactions
3.5. F66Y and E168Q Mutations Enhanced Substrate Binding by Strengthening the Cation-π Interactions
3.6. Binding Free Energy Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Debnath, B.; Haldar, D.; Purkait, M.K. A critical review on the techniques used for the synthesis and applications of crystalline cellulose derived from agricultural wastes and forest residues. Carbohydr. Polym. 2021, 273, 118537. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Li, Y.; Tan, X.; Wu, H.; Li, H.; Yang, S. Advances in Pretreatment of Straw Biomass for Sugar Production. Front. Chem. 2021, 9, 1–28. [Google Scholar] [CrossRef]
- Dadwal, A.; Sharma, S.; Satyanarayana, T. Thermostable cellulose saccharifying microbial enzymes: Characteristics, recent advances and biotechnological applications. Int. J. Biol. Macromol. 2021, 188, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Rajnish, K.N.; Samuel, M.S.; John, A.; Datta, S.; Chandrasekar, N.; Balaji, R.; Jose, S.; Selvarajan, E. Immobilization of cellulase enzymes on nano and micro-materials for breakdown of cellulose for biofuel production—A narrative review. Int. J. Biol. Macromol. 2021, 182, 1793–1802. [Google Scholar] [CrossRef]
- Paul, M.; Mohapatra, S.; Das Mohapatra, P.K.; Thatoi, H. Microbial cellulases—An update towards its surface chemistry, genetic engineering and recovery for its biotechnological potential. Bioresour. Technol. 2021, 340, 125710. [Google Scholar] [CrossRef]
- Bischof, R.H.; Ramoni, J.; Seiboth, B. Cellulases and beyond: The first 70 years of the enzyme producer Trichoderma reesei. Microb. Cell Factories 2016, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Wang, K.; Li, H.; Yi, S.; Zhao, X. Enhancing cellulosic ethanol production through coevolution of multiple enzymatic characteristics of β-glucosidase from Penicillium oxalicum 16. Appl. Microbiol. Biotechnol. 2020, 104, 1–10. [Google Scholar] [CrossRef]
- Srivastava, N.; Rathour, R.; Jha, S.; Pandey, K.; Srivastava, M.; Thakur, V.K.; Sengar, R.S.; Gupta, V.K.; Mazumder, P.B.; Khan, A.F.; et al. Microbial Beta Glucosidase Enzymes: Recent Advances in Biomass Conversation for Biofuels Application. Biomolecules 2019, 9, 220. [Google Scholar] [CrossRef] [Green Version]
- Teugjas, H.; Väljamäe, P. Selecting β-glucosidases to support cellulases in cellulose saccharification. Biotechnol. Biofuels 2013, 6, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haven, M.O.; Jørgensen, H. Adsorption of β-glucosidases in two commercial preparations onto pretreated biomass and lignin. Biotechnol. Biofuels 2013, 6, 165. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Bai, Y.; Cui, Y.; Xu, X.; Qian, L.; Shi, P.; Zhang, W.; Luo, H.; Zhan, X.; Yao, B. Functional diversity of family 3 β-glucosidases from thermophilic cellulolytic fungus Humicola insolens Y1. Sci. Rep. 2016, 6, 27062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrissat, B.; Davies, G. Structural and sequence-based classification of glycoside hydrolases. Curr. Opin. Struct. Biol. 1997, 7, 637–644. [Google Scholar] [CrossRef]
- Lima, R.A.T.; Oliveira, G.M.; Souza, A.A.; Lopes, F.A.C.; Santana, R.H.; Istvan, P.; Quirino, B.F.; Barbosa, J.A.; Freitas, S.M.; Garay, A.V.; et al. Functional and structural characterization of a novel GH3 β-glucosidase from the gut metagenome of the Brazilian Cerrado termite Syntermes wheeleri. Int. J. Biol. Macromol. 2020, 165, 822–834. [Google Scholar] [CrossRef]
- Xia, W.; Xu, X.; Qian, L.; Shi, P.; Bai, Y.; Luo, H.; Ma, R.; Yao, B. Engineering a highly active thermophilic β-glucosidase to enhance its pH stability and saccharification performance. Biotechnol. Biofuels 2016, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Sumitani, J.-I.; Nam, Y.-W.; Nishimaki, T.; Tani, S.; Wakagi, T.; Kawaguchi, T.; Fushinobu, S. Crystal structures of glycoside hydrolase family 3 β-glucosidase 1 from Aspergillus aculeatus. Biochem. J. 2013, 452, 211–221. [Google Scholar] [CrossRef]
- Karkehabadi, S.; Helmich, K.E.; Kaper, T.; Hansson, H.; Mikkelsen, N.-E.; Gudmundsson, M.; Piens, K.; Fujdala, M.; Banerjee, G.; Scott-Craig, J.S.; et al. Biochemical Characterization and Crystal Structures of a Fungal Family 3 β-Glucosidase, Cel3A from Hypocrea jecorina. J. Biol. Chem. 2014, 289, 31624–31637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, X.-Q.; Yi, Z.-L.; Tang, C.-G.; Wu, Z.-L. Three amino acid changes contribute markedly to the thermostability of β-glucosidase BglC from Thermobifida fusca. Bioresour. Technol. 2011, 102, 3337–3342. [Google Scholar] [CrossRef] [PubMed]
- Marana, S.R.; Mendonça, L.M.F.; Andrade, E.H.P.; Terra, W.R.; Ferreira, C. The role of residues R97 and Y331 in modulating the pH optimum of an insect β-glycosidase of family 1. JBIC J. Biol. Inorg. Chem. 2003, 270, 4866–4875. [Google Scholar] [CrossRef]
- Yadav, S.; Pandey, A.K.; Dubey, S.K. Molecular modeling, docking and simulation dynamics of β-glucosidase reveals high-efficiency, thermo-stable, glucose tolerant enzyme in Paenibacillus lautus BHU3 strain. Int. J. Biol. Macromol. 2020, 168, 371–382. [Google Scholar] [CrossRef] [PubMed]
- LaRue, K.; Melgar, M.; Martin, V.J.J. Directed evolution of a fungal β-glucosidase in Saccharomyces cerevisiae. Biotechnol. Biofuels 2016, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Baba, Y.; Sumitani, J.-I.; Tanaka, K.; Tani, S.; Kawaguchi, T. Site-saturation mutagenesis for β-glucosidase 1 from Aspergillus aculeatus to accelerate the saccharification of alkaline-pretreated bagasse. Appl. Microbiol. Biotechnol. 2016, 100, 10495–10507. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.-C.; Hong, S.-H.; Seo, M.-J.; Oh, D.-K. An amino acid at position 512 in β-glucosidase from Clavibacter michiganensis determines the regioselectivity for hydrolyzing gypenoside XVII. Appl. Microbiol. Biotechnol. 2015, 99, 7987–7996. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xue, Y.; Lin, Y. Enhanced Catalytic Efficiency in Quercetin-4′-glucoside Hydrolysis of Thermotoga maritima β-Glucosidase A by Site-Directed Mutagenesis. J. Agric. Food Chem. 2014, 62, 6763–6770. [Google Scholar] [CrossRef] [PubMed]
- Tina, K.G.; Bhadra, R.; Srinivasan, N. PIC: Protein Interactions Calculator. Nucleic Acids Res. 2007, 35, W473–W476. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 3, 198–210. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Geronimo, I.; Payne, C.M.; Sandgren, M. The role of catalytic residue pKa on the hydrolysis/transglycosylation partition in family 3 β-glucosidases. Org. Biomol. Chem. 2017, 16, 316–324. [Google Scholar] [CrossRef] [Green Version]
- Geronimo, I.; Payne, C.M.; Sandgren, M. Hydrolysis and Transglycosylation Transition States of Glycoside Hydrolase Family 3 β-Glucosidases Differ in Charge and Puckering Conformation. J. Phys. Chem. B 2018, 122, 9452–9459. [Google Scholar] [CrossRef] [PubMed]
- Agirre, J.; Ariza, A.; Offen, W.A.; Turkenburg, J.; Roberts, S.M.; McNicholas, S.; Harris, P.V.; McBrayer, B.; Dohnalek, J.; Cowtan, K.D.; et al. Three-dimensional structures of two heavily N-glycosylatedAspergillussp. family GH3 β-d-glucosidases. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 254–265. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsson, M.; Hansson, H.; Karkehabadi, S.; Larsson, A.; Stals, I.; Kim, S.; Sunux, S.; Fujdala, M.; Larenas, E.; Kaper, T.; et al. Structural and functional studies of the glycoside hydrolase family 3 β-glucosidase Cel3A from the moderately thermophilic fungusRasamsonia emersonii. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkehabadi, S.; Hansson, H.; Mikkelsen, N.E.; Kim, S.; Kaper, T.; Sandgren, M.; Gudmundsson, M. Structural studies of a glycoside hydrolase family 3 β-glucosidase from the model fungusNeurospora crassa. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, D.A. Cation-π interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, J.P.; Dougherty, D.A. Cation-πinteractions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 17, 9459–9464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, D.A. The Cation−π Interaction. Acc. Chem. Res. 2012, 46, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Kronberg, B. The hydrophobic effect. Curr. Opin. Colloid Interface Sci. 2016, 22, 14–22. [Google Scholar] [CrossRef]
- Privalov, P.L.; Gill, S.J. Stability of Protein Structure and Hydrophobic Interaction. Adv. Protein Chem. 1988, 39, 191–234. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Optima | pNPG | Cellobiose | Fold Change in kcat/Km on Cellobiose | Activity Ratio Cellobiose/pNPG | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T (°C) | pH | Specific Actvitity (U·mg–1) | Km (mM) | kcat (s–1) | kcat/Km (s–1·mM–1) | Specific Actvitity (U·mg–1) | Km (mM) | kcat (s–1) | kcat/Km (s–1·mM–1) | |||
| WT | 75 | 4.5 | 905.0 ± 11.3 | 0.18 ± 0.03 | 1664.3 ± 26.9 | 9096.0 | 265.5 ± 3.9 | 10.4 ± 0.4 | 786.0 ± 9.6 | 75.8 | 1.0 | 0.29 |
| F66Y | 75 | 4.5 | 892.9 ± 19.6 | 0.24 ± 0.02 | 1773.3 ± 11.9 | 7451.0 | 341.0 ± 4.7 | 4.3 ± 0.3 | 511.7 ± 11.2 | 119.0 | 1.6 | 0.38 |
| E168Q | 75 | 4.5 | 858.9 ± 12.4 | 0.21 ± 0.03 | 1744.2 ± 27.8 | 8385.6 | 236.1 ± 6.8 | 5.2 ± 0.2 | 540.6 ± 8.6 | 104.0 | 1.4 | 0.27 |
| M36E | 75 | 4.5 | 766.0 ± 21.2 | 0.25 ± 0.03 | 1798.0 ± 31.6 | 7088.0 | 256.6 ± 4.6 | 5.0 ± 0.7 | 875.4 ± 6.9 | 174.2 | 2.3 | 0.33 |
| M36N | 75 | 4.5 | 850.8 ± 17.1 | 0.59 ± 0.07 | 1067.3 ± 33.7 | 1794.1 | 247.9 ± 3.7 | 4.8 ± 0.4 | 843.5 ± 7.1 | 177.2 | 2.3 | 0.29 |
| M36G | 75 | 4.5 | 907.8 ± 12.1 | 1.10 ± 0.06 | 2566.4 ± 15.9 | 2320.0 | 75.2 ± 2.8 | 5.4 ± 1.3 | 115.0 ± 3.1 | 21.5 | 0.3 | 0.08 |
| M36A | 75 | 4.5 | 876.9 ± 26.8 | 1.40 ± 0.13 | 1561.1 ± 16.8 | 1080.9 | 29.4 ± 1.2 | 5.3 ± 0.7 | 70.8 ± 2.7 | 13.3 | 0.2 | 0.03 |
| M36V | 70 | 4.5 | 478.4 ± 11.7 | 0.80 ± 0.04 | 817.8 ± 21.6 | 1038.6 | 25.8 ± 1.6 | 5.2 ± 1.2 | 54.4 ± 1.6 | 10.5 | 0.1 | 0.05 |
| M36L | 75 | 4.5 | 483.6 ± 15.4 | 0.66 ± 0.12 | 1279.1 ± 14.8 | 1932.4 | 54.5 ± 1.7 | 3.9 ± 0.2 | 89.9 ± 3.9 | 23.2 | 0.3 | 0.11 |
| M36I | 75 | 4.5 | 390.2 ± 11.8 | 1.24 ± 0.09 | 1304.5 ± 29.7 | 1053.2 | 34.9 ± 1.2 | 4.8 ± 0.5 | 88.8 ± 3.7 | 18.4 | 0.2 | 0.09 |
| M36P | 75 | 4.5 | 23.9 ± 3.4 | 2.10 ± 0.13 | 195.5 ± 11.2 | 93.0 | 2.9 ± 0.3 | 25.1 ± 1.4 | 22.5 ± 1.4 | 0.9 | 0.0 | 0.12 |
| M36S | 70 | 4.5 | 32.8 ± 2.7 | 2.26 ± 0.27 | 105.8 ± 7.9 | 46.8 | 2.4 ± 0.5 | 5.7 ± 0.4 | 5.6 ± 0.6 | 1.0 | 0.0 | 0.07 |
| M36C | 70 | 4.5 | 614.6 ± 9.7 | 0.97 ± 0.15 | 1888.5 ± 51.3 | 1956.9 | 57.8 ± 2.9 | 4.1 ± 0.7 | 116.9 ± 6.4 | 28.9 | 0.4 | 0.09 |
| M36T | 75 | 4.5 | 593.2 ± 13.8 | 0.75 ± 0.07 | 1688.0 ± 15.9 | 2250.7 | 63.6 ± 3.7 | 5.7 ± 1.1 | 185.5 ± 5.1 | 32.7 | 0.4 | 0.11 |
| M36Q | 75 | 4.5 | 408.6 ± 10.6 | 0.83 ± 0.13 | 860.4 ± 21.8 | 1039.8 | 27.4 ± 0.8 | 3.0 ± 0.2 | 41.0 ± 2.8 | 13.9 | 0.2 | 0.07 |
| M36W | 75 | 4.5 | 212.6 ± 8.7 | 1.23 ± 0.22 | 1701.8 ± 15.7 | 1386.2 | 63.8 ± 0.9 | 7.3 ± 0.2 | 83.4 ± 3.4 | 11.4 | 0.2 | 0.30 |
| M36Y | 75 | 4.5 | 348.6 ± 16.9 | 1.84 ± 0.18 | 2684.3 ± 19.4 | 1458.9 | 35.8 ± 1.3 | 6.1 ± 0.1 | 48.5 ± 1.8 | 7.9 | 0.1 | 0.10 |
| M36F | 75 | 4.5 | 170.9 ± 3.7 | 1.67 ± 0.17 | 918.9 ± 21.9 | 551.3 | 24.3 ± 0.6 | 6.7 ± 0.3 | 48.5 ± 3.1 | 7.3 | 0.1 | 0.14 |
| M36R | 70 | 4.5 | 589.1 ± 16.9 | 0.99 ± 0.11 | 1110.0 ± 11.7 | 1114.7 | 21.8 ± 1.1 | 2.5 ± 0.2 | 37.6 ± 1.1 | 14.9 | 0.2 | 0.04 |
| M36H | 75 | 4.5 | 194.5 ± 21.3 | 1.24 ± 0.18 | 1382.1 ± 10.8 | 1114.6 | 27.8 ± 1.1 | 29.0 ± 3.7 | 737.8 ± 21.3 | 25.1 | 0.3 | 0.14 |
| M36K | 70 | 4.5 | 208.9 ± 19.7 | 1.18 ± 0.12 | 1854.5 ± 26.9 | 1571.6 | 25.9 ± 0.9 | 27.1 ± 2.9 | 730.7 ± 33.7 | 26.7 | 0.4 | 0.12 |
| M36D | 70 | 4.5 | 93.5 ± 8.6 | 1.94 ± 0.07 | 633.2 ± 9.1 | 325.8 | 16.5 ± 1.7 | 6.8 ± 0.7 | 31.1 ± 2.7 | 4.6 | 0.1 | 0.18 |
| Acceptor | Donor H | Donor | Hydrogen Bond Occupancy (%) a | ||||
|---|---|---|---|---|---|---|---|
| WT | M36E | M36N | F66Y | E168Q | |||
| Trp35 | |||||||
| Asn36@ND2 | Trp35@HE1 | Trp35@NE1 | - b | - | 22.46 ± 2.13% | - | - |
| Glu36@OE1 | Trp35@HE1 | Trp35@NE1 | - | 38.30 ± 1.34% | - | - | - |
| Arg65 | |||||||
| Asp59@OD2 | Arg65@HH11 | Arg65@NH1 | 35.93 ± 2.37% | 39.34 ± 1.47% | 32.46 ± 1.68% | 71.48 ± 5.16% | 27.67 ± 0.94% |
| Subsite −1@O6 | Arg65@HH11 | Arg65@NH1 | 7.09 ± 2.11% | 13.93 ± 2.13% | 10.14 ± 1.59% | 44.64 ± 3.45% | 8.11 ± 1.12% |
| Ser389@OG | Arg65@HH12 | Arg65@NH1 | 45.20 ± 4.69% | 47.08 ± 3.18% | 39.74 ± 2.26% | 86.03 ± 5.34% | 43.61 ± 3.17% |
| Tyr448@OH | Arg65@HH12 | Arg65@NH1 | 79.88 ± 3.97% | 74.71 ± 2.26% | 79.16 ± 4.13% | 97.85 ± 3.89% | 86.82 ± 5.33% |
| Tyr448@OH | Arg65@HH22 | Arg65@NH2 | 12.86 ± 1.26% | 19.25 ± 3.14% | 18.95 ± 2.33% | 53.11 ± 4.11% | 15.14 ± 2.37% |
| Arg167 | |||||||
| Glu 446@OE2 | Arg167@HH22 | Arg167@NH2 | 77.88 ± 1.98% | 79.84 ± 6.13% | 84.84 ± 3.11% | 73.48 ± 2.37% | 96.13 ± 3.51% |
| Subsite +1@O6 | Arg167@HH12 | Arg167@NH1 | 29.31 ± 1.31% | 34.44 ± 3.17% | 30.67 ± 3.21% | 23.25 ± 2.15% | 49.32 ± 4.11% |
| Subsite −1@O2 | Arg167@HH22 | Arg167@NH2 | 11.17 ± 2.18% | 13.62 ± 2.31% | 8.26 ± 0.91% | 15.03 ± 1.13% | 37.53 ± 2.34% |
| Energy (kcal/mol) | WT | M36E | M36N | F66Y | E168Q |
|---|---|---|---|---|---|
| ΔEvdW | −34.84 ± 0.39 | −36.48 ± 0.70 | −38.17 ± 0.49 | −34.72 ± 0.99 | −34.29 ± 0.78 |
| ΔEel | −85.13 ± 0.53 | −83.79 ± 1.34 | −83.67 ± 0.67 | −89.11 ± 0.88 | −90.22 ± 1.16 |
| ΔEpol | 103.07 ± 1.75 | 100.21 ± 1.21 | 100.77 ± 1.12 | 97.69 ± 0.92 | 99.40 ± 0.83 |
| ΔEnp | −5.60 ± 0.11 | −7.46 ± 0.21 | −9.06 ± 0.40 | −7.10 ± 0.30 | −5.94 ± 0.51 |
| Gbinding | −22.50 ± 0.72 | −27.52 ± 0.88 | −30.13 ± 0.78 | −33.25 ± 1.08 | −31.05 ± 0.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, W.; Bai, Y.; Shi, P. Improving the Substrate Affinity and Catalytic Efficiency of β-Glucosidase Bgl3A from Talaromyces leycettanus JCM12802 by Rational Design. Biomolecules 2021, 11, 1882. https://doi.org/10.3390/biom11121882
Xia W, Bai Y, Shi P. Improving the Substrate Affinity and Catalytic Efficiency of β-Glucosidase Bgl3A from Talaromyces leycettanus JCM12802 by Rational Design. Biomolecules. 2021; 11(12):1882. https://doi.org/10.3390/biom11121882
Chicago/Turabian StyleXia, Wei, Yingguo Bai, and Pengjun Shi. 2021. "Improving the Substrate Affinity and Catalytic Efficiency of β-Glucosidase Bgl3A from Talaromyces leycettanus JCM12802 by Rational Design" Biomolecules 11, no. 12: 1882. https://doi.org/10.3390/biom11121882
APA StyleXia, W., Bai, Y., & Shi, P. (2021). Improving the Substrate Affinity and Catalytic Efficiency of β-Glucosidase Bgl3A from Talaromyces leycettanus JCM12802 by Rational Design. Biomolecules, 11(12), 1882. https://doi.org/10.3390/biom11121882