Impact of Conventional and Atypical MAPKs on the Development of Metabolic Diseases


Abstract
1. Introduction
2. The Conventional MAPKs
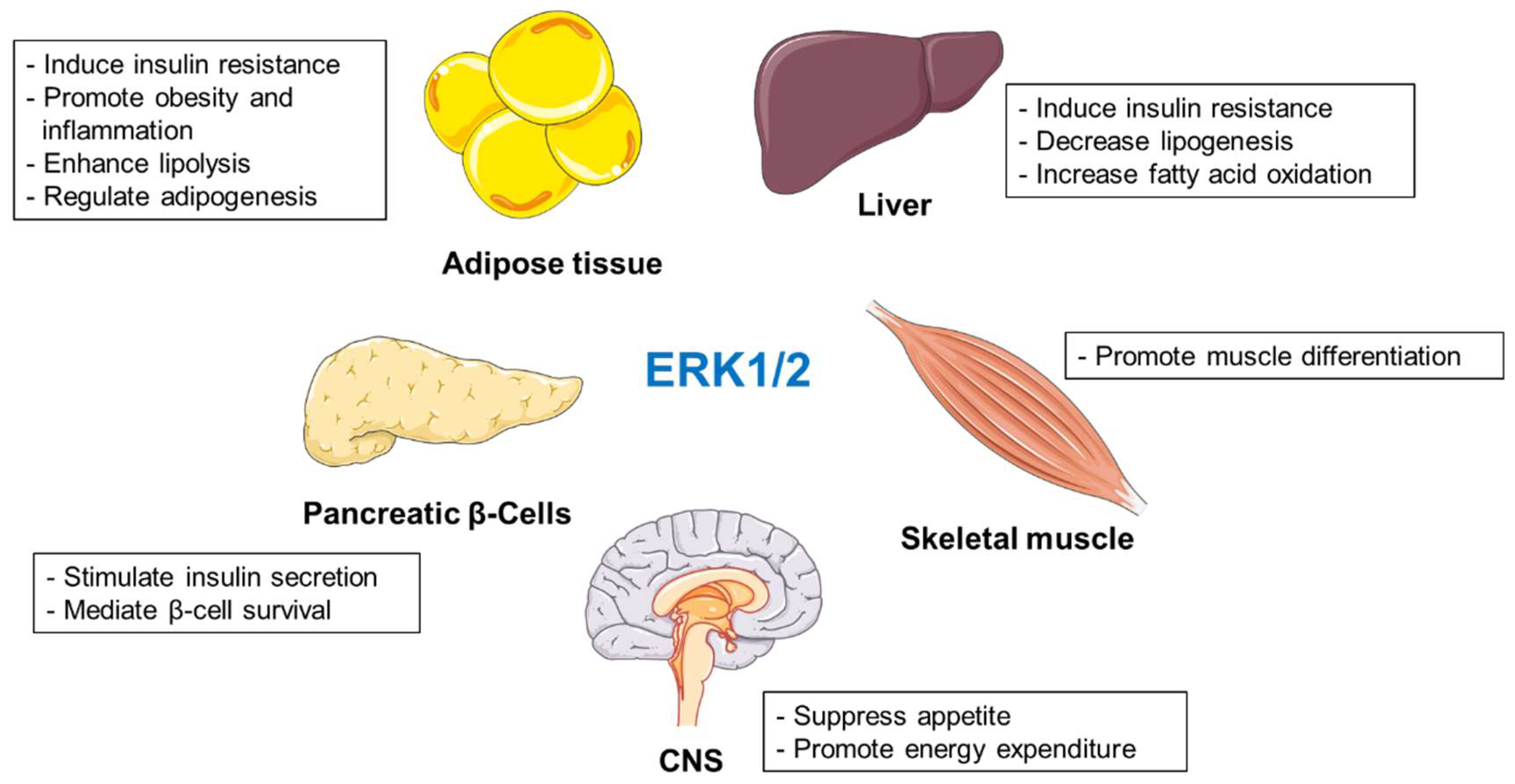
2.1. ERK1/2 Kinases
2.1.1. The Role of ERK1/2 in the Liver
2.1.2. ERK1/2 Promote Adipocyte Acquisition and Preserves Their Function
2.1.3. ERK1/2 Promote Inflammation during Obesity
2.1.4. ERK1/2 Drive Insulin Production in Pancreatic β-Cells
2.1.5. ERK1/2 Promote Skeletal Muscle Acquisition and Metabolism
2.1.6. The Central Role of ERK1/2 in Regulation of Appetite and Energy Dissipation
2.1.7. Summary
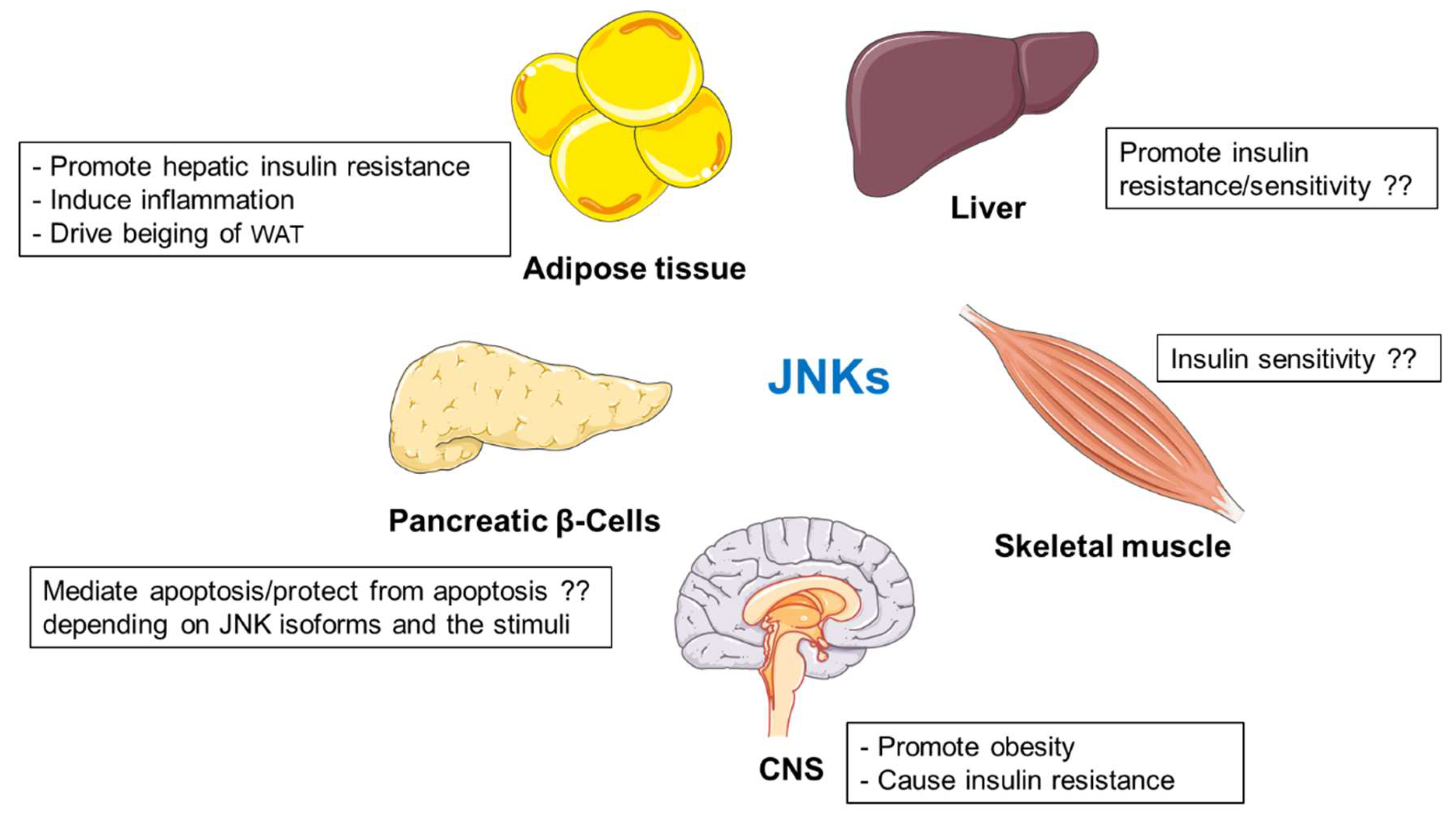
2.2. JNK Kinases
2.2.1. Functions of JNKs in the Liver
2.2.2. JNKs Promote Inflammatory Mediators in Adipose Tissue
2.2.3. The role of JNKs in Immune Cells
2.2.4. The Impact of JNKs on Pancreatic β-Cells during Type 1 and Type 2 Diabetes
2.2.5. JNKs in Skeletal Muscle Metabolism
2.2.6. Central Regulation of Metabolism by JNKs
2.2.7. Summary
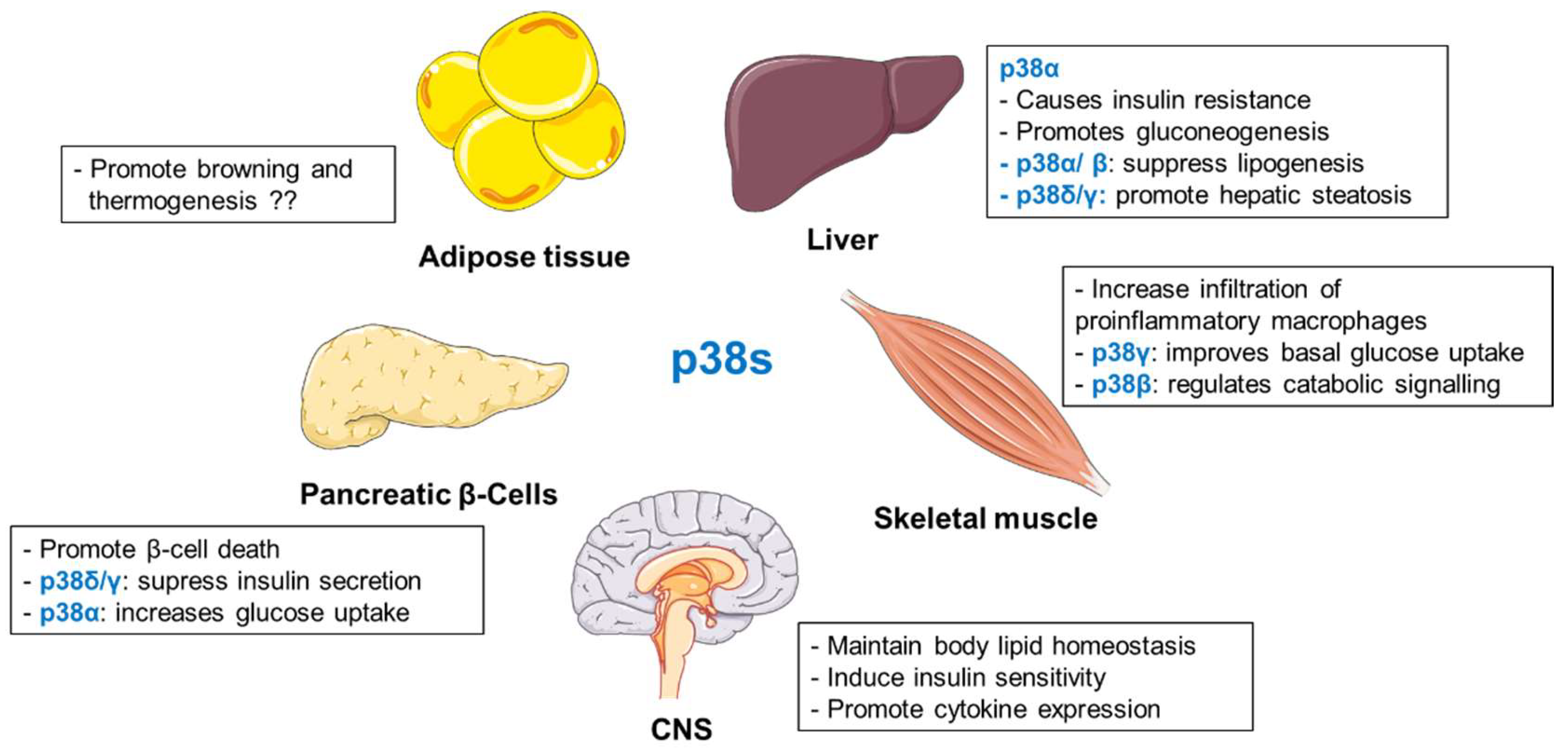
2.3. p38 Kinases
2.3.1. The Role of p38s in the Liver
2.3.2. Functions of p38s in Adipose Tissue
2.3.3. The Role of p38s in Pancreatic β-Cells
2.3.4. p38s Define Inflammatory Response to Control Metabolism
2.3.5. p38s in Regulation of Skeletal Muscle Function
2.3.6. p38s in Central Regulation of Metabolic Homeostasis
2.3.7. Summary
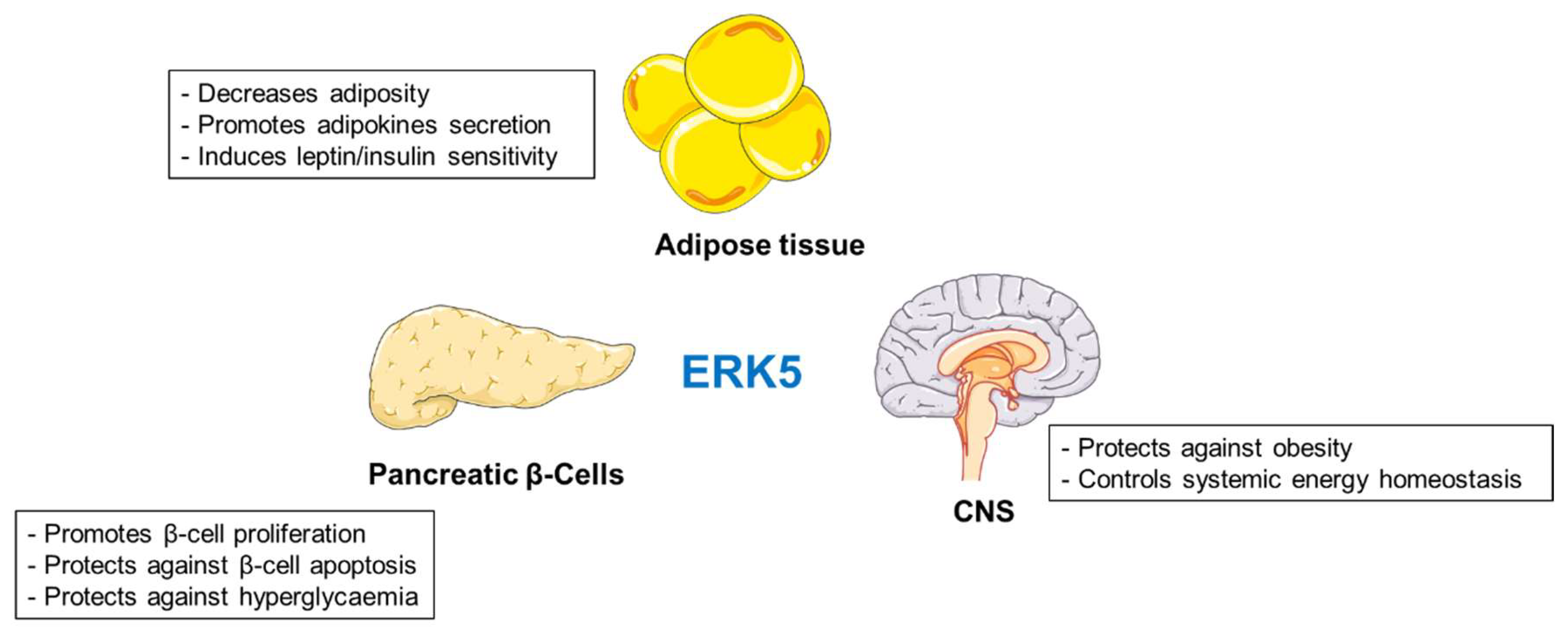
2.4. ERK5 Kinase
Summary
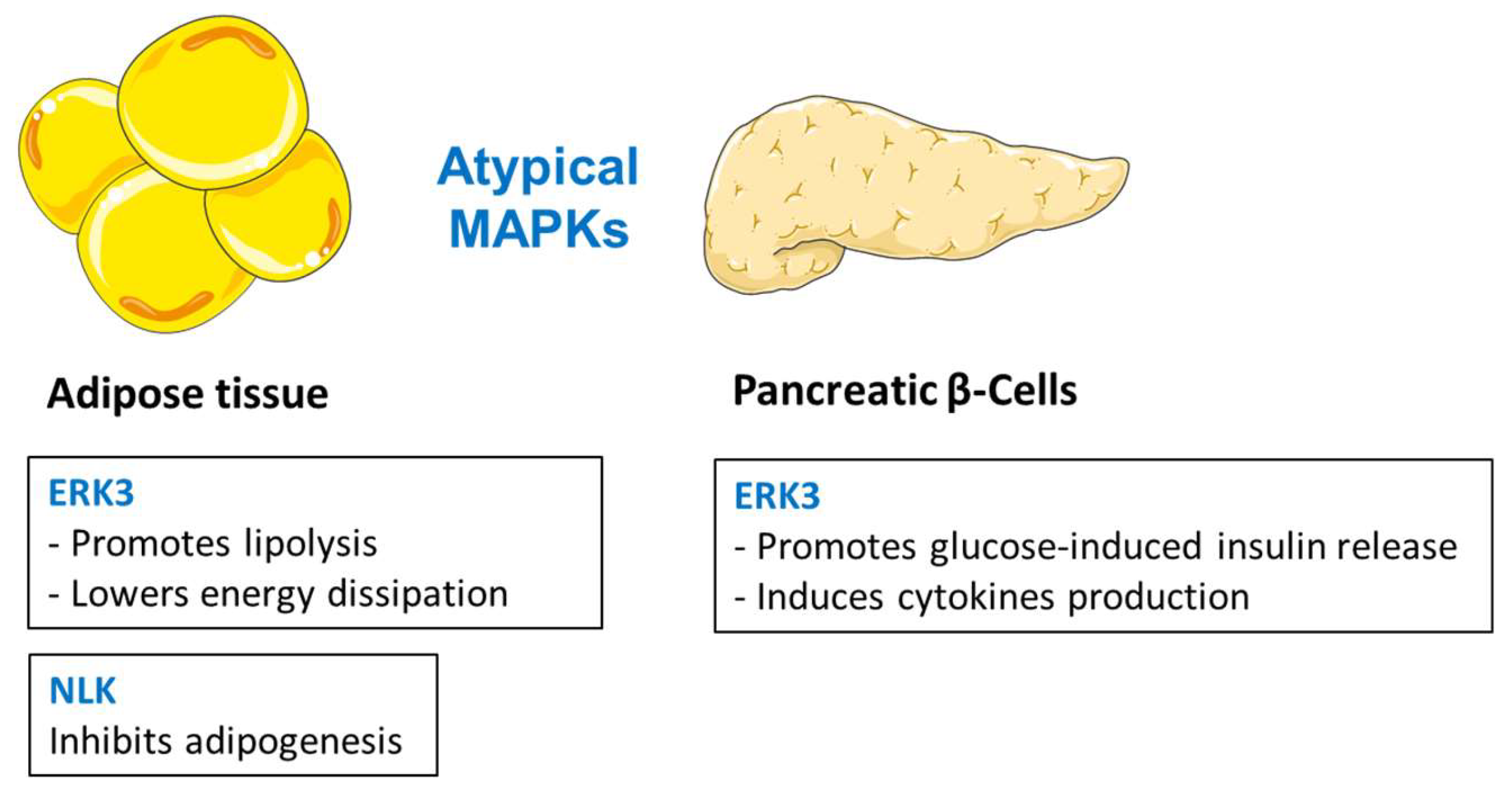
3. The Atypical MAPKs
Summary
4. Conclusions, Future Perspective and Therapeutic Implications
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Coulombe, P.; Meloche, S. Atypical mitogen-activated protein kinases: Structure, regulation and functions. Biochim. Biophys. Acta 2007, 1773, 1376–1387. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Gehart, H.; Kumpf, S.; Ittner, A.; Ricci, R. MAPK signaling in cellular metabolism: Stress or wellness? EMBO Rep. 2010, 11, 834–840. [Google Scholar] [CrossRef]
- Pal, M.; Febbraio, M.A.; Lancaster, G.I. The roles of c-Jun NH2-terminal kinases (JNKs) in obesity and insulin resistance. J. Physiol. 2016, 594, 267–279. [Google Scholar] [CrossRef]
- Manieri, E.; Sabio, G. Stress kinases in the modulation of metabolism and energy balance. J. Mol. Endocrinol. 2015, 55, R11–R22. [Google Scholar] [CrossRef]
- Boulton, T.G.; Yancopoulos, G.D.; Gregory, J.S.; Slaughter, C.; Moomaw, C.; Hsu, J.; Cobb, M.H. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 1990, 249, 64–67. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef]
- Carriere, A.; Ray, H.; Blenis, J.; Roux, P.P. The RSK factors of activating the Ras/MAPK signaling cascade. Front. Biosci. 2008, 13, 4258–4275. [Google Scholar] [CrossRef]
- Reyskens, K.M.; Arthur, J.S. Emerging Roles of the Mitogen and Stress Activated Kinases MSK1 and MSK2. Front. Cell Dev. Biol 2016, 4, 56. [Google Scholar] [CrossRef]
- Joshi, S.; Platanias, L.C. Mnk kinase pathway: Cellular functions and biological outcomes. World J. Biol. Chem. 2014, 5, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Babu, G.J.; Lalli, M.J.; Sussman, M.A.; Sadoshima, J.; Periasamy, M. Phosphorylation of elk-1 by MEK/ERK pathway is necessary for c-fos gene activation during cardiac myocyte hypertrophy. J. Mol. Cell Cardiol. 2000, 32, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Whitmarsh, A.J. Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim. Biophys. Acta 2007, 1773, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Viala, E.; Pouysségur, J. Regulation of tumor cell motility by ERK mitogen-activated protein kinases. Ann. N. Y. Acad. Sci. 2004, 1030, 208–218. [Google Scholar] [CrossRef]
- Kalwat, M.A.; Thurmond, D.C. Signaling mechanisms of glucose-induced F-actin remodeling in pancreatic islet β cells. Exp. Mol. Med. 2013, 45, e37. [Google Scholar] [CrossRef]
- Smorodinsky-Atias, K.; Soudah, N.; Engelberg, D. Mutations That Confer Drug-Resistance, Oncogenicity and Intrinsic Activity on the ERK MAP Kinases-Current State of the Art. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2019, 142, 151–168. [Google Scholar] [CrossRef]
- Pagès, G.; Guérin, S.; Grall, D.; Bonino, F.; Smith, A.; Anjuere, F.; Auberger, P.; Pouysségur, J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science 1999, 286, 1374–1377. [Google Scholar] [CrossRef]
- Frémin, C.; Saba-El-Leil, M.K.; Lévesque, K.; Ang, S.L.; Meloche, S. Functional Redundancy of ERK1 and ERK2 MAP Kinases during Development. Cell Rep. 2015, 12, 913–921. [Google Scholar] [CrossRef]
- Han, H.S.; Kang, G.; Kim, J.S.; Choi, B.H.; Koo, S.H. Regulation of glucose metabolism from a liver-centric perspective. Exp. Mol. Med. 2016, 48, e218. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Feng, B.; Li, Y.; He, Q.; Xu, H. Hepatic ERK activity plays a role in energy metabolism. Mol. Cell Endocrinol. 2013, 375, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, W.; Pendleton, E.; Leng, S.; Wu, J.; Chen, R.; Sun, X.J. Improved insulin sensitivity by calorie restriction is associated with reduction of ERK and p70S6K activities in the liver of obese Zucker rats. J. Endocrinol. 2009, 203, 337–347. [Google Scholar] [CrossRef]
- Khan, A.S.; Subramaniam, S.; Dramane, G.; Khelifi, D.; Khan, N.A. ERK1 and ERK2 activation modulates diet-induced obesity in mice. Biochimie 2017, 137, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Jiao, P.; Yang, Z.; Xu, H. MEK/ERK pathway mediates insulin-promoted degradation of MKP-3 protein in liver cells. Mol. Cell Endocrinol. 2012, 361, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Manowsky, J.; Camargo, R.G.; Kipp, A.P.; Henkel, J.; Püschel, G.P. Insulin-induced cytokine production in macrophages causes insulin resistance in hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E938–E946. [Google Scholar] [CrossRef] [PubMed]
- Fisher, F.M.; Estall, J.L.; Adams, A.C.; Antonellis, P.J.; Bina, H.A.; Flier, J.S.; Kharitonenkov, A.; Spiegelman, B.M.; Maratos-Flier, E. Integrated regulation of hepatic metabolism by fibroblast growth factor 21 (FGF21) in vivo. Endocrinology 2011, 152, 2996–3004. [Google Scholar] [CrossRef]
- Wu, H.T.; Lu, F.H.; Ou, H.Y.; Su, Y.C.; Hung, H.C.; Wu, J.S.; Yang, Y.C.; Wu, C.L.; Chang, C.J. The role of hepassocin in the development of non-alcoholic fatty liver disease. J. Hepatol. 2013, 59, 1065–1072. [Google Scholar] [CrossRef]
- Wu, H.T.; Ou, H.Y.; Hung, H.C.; Su, Y.C.; Lu, F.H.; Wu, J.S.; Yang, Y.C.; Wu, C.L.; Chang, C.J. A novel hepatokine, HFREP1, plays a crucial role in the development of insulin resistance and type 2 diabetes. Diabetologia 2016, 59, 1732–1742. [Google Scholar] [CrossRef]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef]
- Cao, R.; Cronk, Z.X.; Zha, W.; Sun, L.; Wang, X.; Fang, Y.; Studer, E.; Zhou, H.; Pandak, W.M.; Dent, P.; et al. Bile acids regulate hepatic gluconeogenic genes and farnesoid X receptor via G(alpha)i-protein-coupled receptors and the AKT pathway. J. Lipid Res. 2010, 51, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Zhu, W.; Wang, S.; Xie, C.; Li, X.; Wu, J.; Li, Y.; Chen, Y.; Wang, X.; Meng, Y.; et al. P53 modulates hepatic insulin sensitivity through NF-κB and p38/ERK MAPK pathways. Biochem. Biophys. Res. Commun. 2018, 495, 2139–2144. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yu, J.; Xia, T.; Xiao, Y.; Zhang, Q.; Liu, B.; Guo, Y.; Deng, J.; Deng, Y.; Chen, S.; et al. Hepatic serum- and glucocorticoid-regulated protein kinase 1 (SGK1) regulates insulin sensitivity in mice via extracellular-signal-regulated kinase 1/2 (ERK1/2). Biochem. J. 2014, 464, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Spiegelman, B.M. Cell biology of fat storage. Mol. Biol Cell 2016, 27, 2523–2527. [Google Scholar] [CrossRef]
- Hong, S.; Song, W.; Zushin, P.H.; Liu, B.; Jedrychowski, M.P.; Mina, A.I.; Deng, Z.; Cabarkapa, D.; Hall, J.A.; Palmer, C.J.; et al. Phosphorylation of Beta-3 adrenergic receptor at serine 247 by ERK MAP kinase drives lipolysis in obese adipocytes. Mol. Metab. 2018, 12, 25–38. [Google Scholar] [CrossRef]
- Bost, F.; Aouadi, M.; Caron, L.; Even, P.; Belmonte, N.; Prot, M.; Dani, C.; Hofman, P.; Pagès, G.; Pouysségur, J.; et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes 2005, 54, 402–411. [Google Scholar] [CrossRef]
- Jager, J.; Corcelle, V.; Grémeaux, T.; Laurent, K.; Waget, A.; Pagès, G.; Binétruy, B.; Le Marchand-Brustel, Y.; Burcelin, R.; Bost, F.; et al. Deficiency in the extracellular signal-regulated kinase 1 (ERK1) protects leptin-deficient mice from insulin resistance without affecting obesity. Diabetologia 2011, 54, 180–189. [Google Scholar] [CrossRef]
- Robidoux, J.; Kumar, N.; Daniel, K.W.; Moukdar, F.; Cyr, M.; Medvedev, A.V.; Collins, S. Maximal beta3-adrenergic regulation of lipolysis involves Src and epidermal growth factor receptor-dependent ERK1/2 activation. J. Biol. Chem. 2006, 281, 37794–37802. [Google Scholar] [CrossRef]
- Rydén, M.; Arvidsson, E.; Blomqvist, L.; Perbeck, L.; Dicker, A.; Arner, P. Targets for TNF-alpha-induced lipolysis in human adipocytes. Biochem. Biophys. Res. Commun. 2004, 318, 168–175. [Google Scholar] [CrossRef]
- Martin, S.; Parton, R.G. Lipid droplets: A unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 2006, 7, 373–378. [Google Scholar] [CrossRef]
- Greenberg, A.S.; Shen, W.J.; Muliro, K.; Patel, S.; Souza, S.C.; Roth, R.A.; Kraemer, F.B. Stimulation of lipolysis and hormone-sensitive lipase via the extracellular signal-regulated kinase pathway. J. Biol. Chem. 2001, 276, 45456–45461. [Google Scholar] [CrossRef] [PubMed]
- Rapold, R.A.; Wueest, S.; Knoepfel, A.; Schoenle, E.J.; Konrad, D. Fas activates lipolysis in a Ca2+-CaMKII-dependent manner in 3T3-L1 adipocytes. J. Lipid Res. 2013, 54, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Bost, F.; Caron, L.; Marchetti, I.; Dani, C.; Le Marchand-Brustel, Y.; Binétruy, B. Retinoic acid activation of the ERK pathway is required for embryonic stem cell commitment into the adipocyte lineage. Biochem. J. 2002, 361, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Cho, Y.S.; Park, J.M.; Yoon, S.O.; Kim, K.W.; Chung, A.S. Pro-MMP-2 activation by the PPARgamma agonist, ciglitazone, induces cell invasion through the generation of ROS and the activation of ERK. FEBS Lett. 2007, 581, 3303–3310. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Kim, J.B.; Sarraf, P.; Spiegelman, B.M. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science 1996, 274, 2100–2103. [Google Scholar] [CrossRef]
- Wang, N.; Zhao, T.T.; Li, S.M.; Sun, X.; Li, Z.C.; Li, Y.H.; Li, D.S.; Wang, W.F. Fibroblast Growth Factor 21 Exerts its Anti-inflammatory Effects on Multiple Cell Types of Adipose Tissue in Obesity. Obesity (Silver Spring) 2019, 27, 399–408. [Google Scholar] [CrossRef]
- Chang, C.C.; Chen, C.Y.; Wen, H.C.; Huang, C.Y.; Hung, M.S.; Lu, H.C.; Chen, W.L.; Chang, C.H. Caveolin-1 Secreted from Adipose Tissues and Adipocytes Functions as an Adipogenesis Enhancer. Obesity (Silver Spring) 2017, 25, 1932–1940. [Google Scholar] [CrossRef]
- Wu, H.T.; Chen, S.C.; Fan, K.C.; Kuo, C.H.; Lin, S.Y.; Wang, S.H.; Chang, C.J.; Li, H.Y. Targeting fibrinogen-like protein 1 is a novel therapeutic strategy to combat obesity. FASEB J. 2020, 34, 2958–2967. [Google Scholar] [CrossRef]
- Lindquist, J.M.; Rehnmark, S. Ambient temperature regulation of apoptosis in brown adipose tissue. Erk1/2 promotes norepinephrine-dependent cell survival. J. Biol. Chem. 1998, 273, 30147–30156. [Google Scholar] [CrossRef]
- Than, A.; Xu, S.; Li, R.; Leow, M.K.; Sun, L.; Chen, P. Erratum: Author Correction: Angiotensin type 2 receptor activation promotes browning of white adipose tissue and brown adipogenesis. Signal. Transduct Target Ther. 2018, 3, 1. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Fujishiro, M.; Gotoh, Y.; Katagiri, H.; Sakoda, H.; Ogihara, T.; Anai, M.; Onishi, Y.; Ono, H.; Abe, M.; Shojima, N.; et al. Three mitogen-activated protein kinases inhibit insulin signaling by different mechanisms in 3T3-L1 adipocytes. Mol. Endocrinol. 2003, 17, 487–497. [Google Scholar] [CrossRef]
- Fischer, A.M.; Katayama, C.D.; Pagès, G.; Pouysségur, J.; Hedrick, S.M. The role of erk1 and erk2 in multiple stages of T cell development. Immunity 2005, 23, 431–443. [Google Scholar] [CrossRef]
- Richardson, E.T.; Shukla, S.; Nagy, N.; Boom, W.H.; Beck, R.C.; Zhou, L.; Landreth, G.E.; Harding, C.V. ERK Signaling Is Essential for Macrophage Development. PLoS ONE 2015, 10, e0140064. [Google Scholar] [CrossRef]
- Molgat, A.S.; Gagnon, A.; Sorisky, A. Preadipocyte apoptosis is prevented by macrophage-conditioned medium in a PDGF-dependent manner. Am. J. Physiol. Cell Physiol. 2009, 296, C757–C765. [Google Scholar] [CrossRef]
- Molgat, A.S.; Gagnon, A.; Sorisky, A. Macrophage-induced preadipocyte survival depends on signaling through Akt, ERK1/2, and reactive oxygen species. Exp. Cell Res. 2011, 317, 521–530. [Google Scholar] [CrossRef]
- Constant, V.A.; Gagnon, A.; Yarmo, M.; Sorisky, A. The antiadipogenic effect of macrophage-conditioned medium depends on ERK1/2 activation. Metabolism 2008, 57, 465–472. [Google Scholar] [CrossRef]
- Huang, Z.; Zhong, L.; Lee, J.T.H.; Zhang, J.; Wu, D.; Geng, L.; Wang, Y.; Wong, C.M.; Xu, A. The FGF21-CCL11 Axis Mediates Beiging of White Adipose Tissues by Coupling Sympathetic Nervous System to Type 2 Immunity. Cell Metab. 2017, 26, 493–508.e494. [Google Scholar] [CrossRef]
- Habibian, J.S.; Jefic, M.; Bagchi, R.A.; Lane, R.H.; McKnight, R.A.; McKinsey, T.A.; Morrison, R.F.; Ferguson, B.S. DUSP5 functions as a feedback regulator of TNFα-induced ERK1/2 dephosphorylation and inflammatory gene expression in adipocytes. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Takahashi, N.; Sawaragi, Y.; Naknukool, S.; Yu, R.; Goto, T.; Kawada, T. Inflammation induced by RAW macrophages suppresses UCP1 mRNA induction via ERK activation in 10T1/2 adipocytes. Am. J. Physiol. Cell Physiol. 2013, 304, C729–C738. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Naknukool, S.; Yoshitake, R.; Hanafusa, Y.; Tokiwa, S.; Li, Y.; Sakamoto, T.; Nitta, T.; Kim, M.; Takahashi, N.; et al. Proinflammatory cytokine interleukin-1β suppresses cold-induced thermogenesis in adipocytes. Cytokine 2016, 77, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Šrámek, J.; Němcová-Fürstová, V.; Kovář, J. Kinase Signaling in Apoptosis Induced by Saturated Fatty Acids in Pancreatic β-Cells. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Sidarala, V.; Kowluru, A. The Regulatory Roles of Mitogen-Activated Protein Kinase (MAPK) Pathways in Health and Diabetes: Lessons Learned from the Pancreatic β-Cell. Recent Pat. Endocr. Metab. Immune Drug Discov. 2017, 10, 76–84. [Google Scholar] [CrossRef]
- Benes, C.; Roisin, M.P.; Van Tan, H.; Creuzet, C.; Miyazaki, J.; Fagard, R. Rapid activation and nuclear translocation of mitogen-activated protein kinases in response to physiological concentration of glucose in the MIN6 pancreatic beta cell line. J. Biol. Chem. 1998, 273, 15507–15513. [Google Scholar] [CrossRef]
- Benes, C.; Poitout, V.; Marie, J.C.; Martin-Perez, J.; Roisin, M.P.; Fagard, R. Mode of regulation of the extracellular signal-regulated kinases in the pancreatic beta-cell line MIN6 and their implication in the regulation of insulin gene transcription. Biochem. J. 1999, 340, 219–225. [Google Scholar] [CrossRef]
- Longuet, C.; Broca, C.; Costes, S.; Hani, E.H.; Bataille, D.; Dalle, S. Extracellularly regulated kinases 1/2 (p44/42 mitogen-activated protein kinases) phosphorylate synapsin I and regulate insulin secretion in the MIN6 beta-cell line and islets of Langerhans. Endocrinology 2005, 146, 643–654. [Google Scholar] [CrossRef]
- Leduc, M.; Richard, J.; Costes, S.; Muller, D.; Varrault, A.; Compan, V.; Mathieu, J.; Tanti, J.F.; Pagès, G.; Pouyssegur, J.; et al. ERK1 is dispensable for mouse pancreatic beta cell function but is necessary for glucose-induced full activation of MSK1 and CREB. Diabetologia 2017, 60, 1999–2010. [Google Scholar] [CrossRef]
- Arnette, D.; Gibson, T.B.; Lawrence, M.C.; January, B.; Khoo, S.; McGlynn, K.; Vanderbilt, C.A.; Cobb, M.H. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J. Biol. Chem. 2003, 278, 32517–32525. [Google Scholar] [CrossRef] [PubMed]
- Trümper, J.; Ross, D.; Jahr, H.; Brendel, M.D.; Göke, R.; Hörsch, D. The Rap-B-Raf signaling pathway is activated by glucose and glucagon-like peptide-1 in human islet cells. Diabetologia 2005, 48, 1534–1540. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wijesekara, N.; Krishnamurthy, M.; Bhattacharjee, A.; Suhail, A.; Sweeney, G.; Wheeler, M.B. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J. Biol. Chem. 2010, 285, 33623–33631. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef]
- Goodyear, L.J.; Chang, P.Y.; Sherwood, D.J.; Dufresne, S.D.; Moller, D.E. Effects of exercise and insulin on mitogen-activated protein kinase signaling pathways in rat skeletal muscle. Am. J. Physiol. 1996, 271, E403–E408. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Steiler, T.L.; Zierath, J.R. Effects of insulin, contraction, and phorbol esters on mitogen-activated protein kinase signaling in skeletal muscle from lean and ob/ob mice. Diabetes 2004, 53, 1436–1444. [Google Scholar] [CrossRef]
- Seaberg, B.; Henslee, G.; Wang, S.; Paez-Colasante, X.; Landreth, G.E.; Rimer, M. Muscle-derived extracellular signal-regulated kinases 1 and 2 are required for the maintenance of adult myofibers and their neuromuscular junctions. Mol. Cell Biol. 2015, 35, 1238–1253. [Google Scholar] [CrossRef]
- Barreto, R.; Waning, D.L.; Gao, H.; Liu, Y.; Zimmers, T.A.; Bonetto, A. Chemotherapy-related cachexia is associated with mitochondrial depletion and the activation of ERK1/2 and p38 MAPKs. Oncotarget 2016, 7, 43442–43460. [Google Scholar] [CrossRef]
- Pu, J.; Peng, G.; Li, L.; Na, H.; Liu, Y.; Liu, P. Palmitic acid acutely stimulates glucose uptake via activation of Akt and ERK1/2 in skeletal muscle cells. J. Lipid Res. 2011, 52, 1319–1327. [Google Scholar] [CrossRef]
- Wu, W.; Sun, Y.; Zhao, C.; Chen, X.; Wang, G.; Pang, W.; Yang, G. Lipogenesis in myoblasts and its regulation of CTRP6 by AdipoR1/Erk/PPARγ signaling pathway. Acta Biochim. Biophys. Sin. (Shanghai) 2016, 48, 509–519. [Google Scholar] [CrossRef]
- Montori-Grau, M.; Tarrats, N.; Osorio-Conles, O.; Orozco, A.; Serrano-Marco, L.; Vázquez-Carrera, M.; Gómez-Foix, A.M. Glucose dependence of glycogen synthase activity regulation by GSK3 and MEK/ERK inhibitors and angiotensin-(1-7) action on these pathways in cultured human myotubes. Cell Signal. 2013, 25, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Behera, S.; Kapadia, B.; Kain, V.; Alamuru-Yellapragada, N.P.; Murunikkara, V.; Kumar, S.T.; Babu, P.P.; Seshadri, S.; Shivarudraiah, P.; Hiriyan, J.; et al. ERK1/2 activated PHLPP1 induces skeletal muscle ER stress through the inhibition of a novel substrate AMPK. Biochim Biophys Acta Mol. Basis Dis. 2018, 1864, 1702–1716. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.F.; Brüning, J.C. CNS leptin and insulin action in the control of energy homeostasis. Ann. N. Y. Acad. Sci. 2010, 1212, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Waterson, M.J.; Horvath, T.L. Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell Metab. 2015, 22, 962–970. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, Y.; Chen, C.; Yu, F.; Wang, Y.; Gu, J.; Ma, L.; Ho, G. ERK1/2 mediates glucose-regulated POMC gene expression in hypothalamic neurons. J. Mol. Endocrinol. 2015, 54, 125–135. [Google Scholar] [CrossRef]
- Morikawa, Y.; Ueyama, E.; Senba, E. Fasting-induced activation of mitogen-activated protein kinases (ERK/p38) in the mouse hypothalamus. J. Neuroendocrinol. 2004, 16, 105–112. [Google Scholar] [CrossRef]
- Ueyama, E.; Morikawa, Y.; Yasuda, T.; Senba, E. Attenuation of fasting-induced phosphorylation of mitogen-activated protein kinases (ERK/p38) in the mouse hypothalamus in response to refeeding. Neurosci Lett. 2004, 371, 40–44. [Google Scholar] [CrossRef]
- Rodrigues, B.A.; Muñoz, V.R.; Kuga, G.K.; Gaspar, R.C.; Nakandakari, S.C.B.R.; Crisol, B.M.; Botezelli, J.D.; Pauli, L.S.S.; da Silva, A.S.R.; de Moura, L.P.; et al. Obesity Increases Mitogen-Activated Protein Kinase Phosphatase-3 Levels in the Hypothalamus of Mice. Front. Cell Neurosci. 2017, 11, 313. [Google Scholar] [CrossRef]
- Rahmouni, K.; Sigmund, C.D.; Haynes, W.G.; Mark, A.L. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes 2009, 58, 536–542. [Google Scholar] [CrossRef]
- Myers, M.G. Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog. Horm. Res. 2004, 59, 287–304. [Google Scholar] [CrossRef]
- Gaspar, R.C.; Muñoz, V.R.; Kuga, G.K.; Nakandakari, S.C.B.R.; Minuzzi, L.G.; Botezelli, J.D.; da Silva, A.S.R.; Cintra, D.E.; de Moura, L.P.; Ropelle, E.R.; et al. Acute physical exercise increases leptin-induced hypothalamic extracellular signal-regulated kinase1/2 phosphorylation and thermogenesis of obese mice. J. Cell Biochem. 2019, 120, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Zhong, L.; Zhang, J.; Wang, Y.; Bornstein, S.R.; Triggle, C.R.; Ding, H.; Lam, K.S.; Xu, A. FGF21 maintains glucose homeostasis by mediating the cross talk between liver and brain during prolonged fasting. Diabetes 2014, 63, 4064–4075. [Google Scholar] [CrossRef] [PubMed]
- Filippi, B.M.; Yang, C.S.; Tang, C.; Lam, T.K. Insulin activates Erk1/2 signaling in the dorsal vagal complex to inhibit glucose production. Cell Metab. 2012, 16, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Filippi, B.M.; Bassiri, A.; Abraham, M.A.; Duca, F.A.; Yue, J.T.; Lam, T.K. Insulin signals through the dorsal vagal complex to regulate energy balance. Diabetes 2014, 63, 892–899. [Google Scholar] [CrossRef]
- Subramaniam, S.; Ozdener, M.H.; Abdoul-Azize, S.; Saito, K.; Malik, B.; Maquart, G.; Hashimoto, T.; Marambaud, P.; Aribi, M.; Tordoff, M.G.; et al. ERK1/2 activation in human taste bud cells regulates fatty acid signaling and gustatory perception of fat in mice and humans. FASEB J. 2016, 30, 3489–3500. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Weston, C.R.; Davis, R.J. The JNK signal transduction pathway. Curr. Opin. Genet. Dev. 2002, 12, 14–21. [Google Scholar] [CrossRef]
- Li, L.; Feng, Z.; Porter, A.G. JNK-dependent phosphorylation of c-Jun on serine 63 mediates nitric oxide-induced apoptosis of neuroblastoma cells. J. Biol. Chem. 2004, 279, 4058–4065. [Google Scholar] [CrossRef]
- Aoki, H.; Kang, P.M.; Hampe, J.; Yoshimura, K.; Noma, T.; Matsuzaki, M.; Izumo, S. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J. Biol. Chem. 2002, 277, 10244–10250. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Ricci, R.; Sumara, G.; Sumara, I.; Rozenberg, I.; Kurrer, M.; Akhmedov, A.; Hersberger, M.; Eriksson, U.; Eberli, F.R.; Becher, B.; et al. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science 2004, 306, 1558–1561. [Google Scholar] [CrossRef] [PubMed]
- Sumara, G.; Belwal, M.; Ricci, R. “Jnking” atherosclerosis. Cell Mol. Life Sci. 2005, 62, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Nguyen, A.K.; Henstridge, D.C.; Holmes, A.G.; Chan, M.H.; Mesa, J.L.; Lancaster, G.I.; Southgate, R.J.; Bruce, C.R.; Duffy, S.J.; et al. HSP72 protects against obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Duan, X.; Homko, C.; Molina, E.J.; Song, W.; Perez, O.; Cheung, P.; Merali, S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 2008, 57, 2438–2444. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.M.; Oliveira, A.G.; Ueno, M.; Araújo, T.G.; Guadagnini, D.; Carvalho-Filho, M.A.; Geloneze, B.; Lima, M.M.; Pareja, J.C.; Carvalheira, J.B.; et al. Modulation of double-stranded RNA-activated protein kinase in insulin sensitive tissues of obese humans. Obesity (Silver Spring) 2013, 21, 2452–2457. [Google Scholar] [CrossRef]
- Tuncman, G.; Hirosumi, J.; Solinas, G.; Chang, L.; Karin, M.; Hotamisligil, G.S. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2006, 103, 10741–10746. [Google Scholar] [CrossRef]
- Yang, R.; Wilcox, D.M.; Haasch, D.L.; Jung, P.M.; Nguyen, P.T.; Voorbach, M.J.; Doktor, S.; Brodjian, S.; Bush, E.N.; Lin, E.; et al. Liver-specific knockdown of JNK1 up-regulates proliferator-activated receptor gamma coactivator 1 beta and increases plasma triglyceride despite reduced glucose and insulin levels in diet-induced obese mice. J. Biol. Chem. 2007, 282, 22765–22774. [Google Scholar] [CrossRef]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Hatazaki, M.; Miyatsuka, T.; Matsuoka, T.A.; Kajimoto, Y.; Matsuhisa, M.; Yamasaki, Y.; Hori, M. Modulation of the JNK pathway in liver affects insulin resistance status. J. Biol. Chem. 2004, 279, 45803–45809. [Google Scholar] [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Garcia-Haro, L.; Sabio, G.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Xu, J.; Shulha, H.P.; Garber, M.; et al. The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014, 20, 512–525. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; Hancer, N.J.; Opare-Ado, L.; Qiu, W.; Walsh, C.; White, M.F. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 2010, 11, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Cavanagh-Kyros, J.; Ko, H.J.; Jung, D.Y.; Gray, S.; Jun, J.Y.; Barrett, T.; Mora, A.; Kim, J.K.; Davis, R.J. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009, 10, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef]
- Galic, S.; Sachithanandan, N.; Kay, T.W.; Steinberg, G.R. Suppressor of cytokine signaling (SOCS) proteins as guardians of inflammatory responses critical for regulating insulin sensitivity. Biochem. J. 2014, 461, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Yamakuchi, M.; Biswas, K.K.; Aryal, B.; Yamada, S.; Hashiguchi, T.; Maruyama, I. HMGB1 is secreted by 3T3-L1 adipocytes through JNK signaling and the secretion is partially inhibited by adiponectin. Obesity (Silver Spring) 2016, 24, 1913–1921. [Google Scholar] [CrossRef]
- Jaeschke, A.; Czech, M.P.; Davis, R.J. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev. 2004, 18, 1976–1980. [Google Scholar] [CrossRef]
- Kou, Y.; Liu, Q.; Liu, W.; Sun, H.; Liang, M.; Kong, F.; Zhang, B.; Wei, Y.; Liu, Z.; Wang, Y. LIGHT/TNFSF14 signaling attenuates beige fat biogenesis. FASEB J. 2019, 33, 1595–1604. [Google Scholar] [CrossRef]
- Yuliana, A.; Daijo, A.; Jheng, H.F.; Kwon, J.; Nomura, W.; Takahashi, H.; Ara, T.; Kawada, T.; Goto, T. Endoplasmic Reticulum Stress Impaired Uncoupling Protein 1 Expression via the Suppression of Peroxisome Proliferator-Activated Receptor γ Binding Activity in Mice Beige Adipocytes. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Solinas, G.; Vilcu, C.; Neels, J.G.; Bandyopadhyay, G.K.; Luo, J.L.; Naugler, W.; Grivennikov, S.; Wynshaw-Boris, A.; Scadeng, M.; Olefsky, J.M.; et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007, 6, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Vallerie, S.N.; Furuhashi, M.; Fucho, R.; Hotamisligil, G.S. A predominant role for parenchymal c-Jun amino terminal kinase (JNK) in the regulation of systemic insulin sensitivity. PLoS ONE 2008, 3, e3151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, A.; Chung, S.K.; Cresser, J.H.; Sweeney, G.; Wong, R.L.; Lin, A.; Lam, K.S. Selective inactivation of c-Jun NH2-terminal kinase in adipose tissue protects against diet-induced obesity and improves insulin sensitivity in both liver and skeletal muscle in mice. Diabetes 2011, 60, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Camporez, J.G.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Jaeschke, A.; Rincón, M.; Doran, B.; Reilly, J.; Neuberg, D.; Greiner, D.L.; Shultz, L.D.; Rossini, A.A.; Flavell, R.A.; Davis, R.J. Disruption of the Jnk2 (Mapk9) gene reduces destructive insulitis and diabetes in a mouse model of type I diabetes. Proc. Natl. Acad. Sci. USA 2005, 102, 6931–6935. [Google Scholar] [CrossRef]
- Ammendrup, A.; Maillard, A.; Nielsen, K.; Aabenhus Andersen, N.; Serup, P.; Dragsbaek Madsen, O.; Mandrup-Poulsen, T.; Bonny, C. The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 2000, 49, 1468–1476. [Google Scholar] [CrossRef]
- Major, C.D.; Wolf, B.A. Interleukin-1beta stimulation of c-Jun NH(2)-terminal kinase activity in insulin-secreting cells: Evidence for cytoplasmic restriction. Diabetes 2001, 50, 2721–2728. [Google Scholar] [CrossRef][Green Version]
- Maedler, K.; Schulthess, F.T.; Bielman, C.; Berney, T.; Bonny, C.; Prentki, M.; Donath, M.Y.; Roduit, R. Glucose and leptin induce apoptosis in human beta-cells and impair glucose-stimulated insulin secretion through activation of c-Jun N-terminal kinases. FASEB J. 2008, 22, 1905–1913. [Google Scholar] [CrossRef]
- Subramanian, S.L.; Hull, R.L.; Zraika, S.; Aston-Mourney, K.; Udayasankar, J.; Kahn, S.E. cJUN N-terminal kinase (JNK) activation mediates islet amyloid-induced beta cell apoptosis in cultured human islet amyloid polypeptide transgenic mouse islets. Diabetologia 2012, 55, 166–174. [Google Scholar] [CrossRef]
- Tang, C.; Yeung, L.S.N.; Koulajian, K.; Zhang, L.; Tai, K.; Volchuk, A.; Giacca, A. Glucose-Induced β-Cell Dysfunction In Vivo: Evidence for a Causal Role of C-jun N-terminal Kinase Pathway. Endocrinology 2018, 159, 3643–3654. [Google Scholar] [CrossRef]
- Verma, G.; Bhatia, H.; Datta, M. JNK1/2 regulates ER-mitochondrial Ca2+ cross-talk during IL-1β-mediated cell death in RINm5F and human primary β-cells. Mol. Biol. Cell 2013, 24, 2058–2071. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, D.; Kaneto, H.; Nakatani, Y.; Matsuoka, T.A.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J. Biol. Chem. 2006, 281, 1091–1098. [Google Scholar] [CrossRef]
- Haefliger, J.A.; Tawadros, T.; Meylan, L.; Gurun, S.L.; Roehrich, M.E.; Martin, D.; Thorens, B.; Waeber, G. The scaffold protein IB1/JIP-1 is a critical mediator of cytokine-induced apoptosis in pancreatic beta cells. J. Cell Sci. 2003, 116, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Standen, C.L.; Kennedy, N.J.; Flavell, R.A.; Davis, R.J. Signal transduction cross talk mediated by Jun N-terminal kinase-interacting protein and insulin receptor substrate scaffold protein complexes. Mol. Cell Biol. 2009, 29, 4831–4840. [Google Scholar] [CrossRef] [PubMed]
- Abdelli, S.; Bonny, C. JNK3 maintains expression of the insulin receptor substrate 2 (IRS2) in insulin-secreting cells: Functional consequences for insulin signaling. PLoS ONE 2012, 7, e35997. [Google Scholar] [CrossRef] [PubMed]
- Prause, M.; Christensen, D.P.; Billestrup, N.; Mandrup-Poulsen, T. JNK1 protects against glucolipotoxicity-mediated beta-cell apoptosis. PLoS ONE 2014, 9, e87067. [Google Scholar] [CrossRef]
- Henstridge, D.C.; Bruce, C.R.; Pang, C.P.; Lancaster, G.I.; Allen, T.L.; Estevez, E.; Gardner, T.; Weir, J.M.; Meikle, P.J.; Lam, K.S.L.; et al. Skeletal muscle-specific overproduction of constitutively activated c-Jun N-terminal kinase (JNK) induces insulin resistance in mice. Diabetologia 2012, 55, 2769–2778. [Google Scholar] [CrossRef]
- Pal, M.; Wunderlich, C.M.; Spohn, G.; Brönneke, H.S.; Schmidt-Supprian, M.; Wunderlich, F.T. Alteration of JNK-1 signaling in skeletal muscle fails to affect glucose homeostasis and obesity-associated insulin resistance in mice. PLoS ONE 2013, 8, e54247. [Google Scholar] [CrossRef]
- Sabio, G.; Kennedy, N.J.; Cavanagh-Kyros, J.; Jung, D.Y.; Ko, H.J.; Ong, H.; Barrett, T.; Kim, J.K.; Davis, R.J. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol. Cell Biol. 2010, 30, 106–115. [Google Scholar] [CrossRef]
- Jung, T.W.; Chung, Y.H.; Kim, H.C.; Abd El-Aty, A.M.; Jeong, J.H. Hyperlipidemia-induced hepassocin in the liver contributes to insulin resistance in skeletal muscle. Mol. Cell Endocrinol. 2018, 470, 26–33. [Google Scholar] [CrossRef]
- Tsaousidou, E.; Paeger, L.; Belgardt, B.F.; Pal, M.; Wunderlich, C.M.; Brönneke, H.; Collienne, U.; Hampel, B.; Wunderlich, F.T.; Schmidt-Supprian, M.; et al. Distinct Roles for JNK and IKK Activation in Agouti-Related Peptide Neurons in the Development of Obesity and Insulin Resistance. Cell Rep. 2014, 9, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Cavanagh-Kyros, J.; Barrett, T.; Jung, D.Y.; Ko, H.J.; Ong, H.; Morel, C.; Mora, A.; Reilly, J.; Kim, J.K.; et al. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev. 2010, 24, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Vernia, S.; Cavanagh-Kyros, J.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Davis, R.J. Diet-induced obesity mediated by the JNK/DIO2 signal transduction pathway. Genes Dev. 2013, 27, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Quiñones, M.; Al-Massadi, O.; Folgueira, C.; Bremser, S.; Gallego, R.; Torres-Leal, L.; Haddad-Tóvolli, R.; García-Caceres, C.; Hernandez-Bautista, R.; Lam, B.Y.H.; et al. p53 in AgRP neurons is required for protection against diet-induced obesity via JNK1. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sánchez, N.; Seoane-Collazo, P.; Contreras, C.; Varela, L.; Villarroya, J.; Rial-Pensado, E.; Buqué, X.; Aurrekoetxea, I.; Delgado, T.C.; Vázquez-Martínez, R.; et al. Hypothalamic AMPK-ER Stress-JNK1 Axis Mediates the Central Actions of Thyroid Hormones on Energy Balance. Cell Metab. 2017, 26, 212–229. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signaling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Lambert, P.F.; Kashanchi, F.; Radonovich, M.F.; Shiekhattar, R.; Brady, J.N. Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem. 1998, 273, 33048–33053. [Google Scholar] [CrossRef]
- Loughery, J.; Cox, M.; Smith, L.M.; Meek, D.W. Critical role for p53-serine 15 phosphorylation in stimulating transactivation at p53-responsive promoters. Nucleic Acids Res. 2014, 42, 7666–7680. [Google Scholar] [CrossRef]
- Teodoro, T.; Odisho, T.; Sidorova, E.; Volchuk, A. Pancreatic β-cells depend on basal expression of active ATF6α-p50 for cell survival even under nonstress conditions. Am. J. Physiol. Cell Physiol. 2012, 302, C992–C1003. [Google Scholar] [CrossRef]
- Luo, S.; Lee, A.S. Requirement of the p38 mitogen-activated protein kinase signaling pathway for the induction of the 78 kDa glucose-regulated protein/immunoglobulin heavy-chain binding protein by azetidine stress: Activating transcription factor 6 as a target for stress-induced phosphorylation. Biochem. J. 2002, 366, 787–795. [Google Scholar] [CrossRef]
- Schieven, G.L. The biology of p38 kinase: A central role in inflammation. Curr. Top. Med. Chem. 2005, 5, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed]
- Hemi, R.; Yochananov, Y.; Barhod, E.; Kasher-Meron, M.; Karasik, A.; Tirosh, A.; Kanety, H. p38 mitogen-activated protein kinase-dependent transactivation of ErbB receptor family: A novel common mechanism for stress-induced IRS-1 serine phosphorylation and insulin resistance. Diabetes 2011, 60, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Liu, W.; Cao, H.; Zhang, D.; Yao, X.; Zhang, S.; Xia, H.; Li, D.; Wang, Y.C.; Yan, J.; et al. Hepatic p38α regulates gluconeogenesis by suppressing AMPK. J. Hepatol. 2015, 62, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- González-Terán, B.; Matesanz, N.; Nikolic, I.; Verdugo, M.A.; Sreeramkumar, V.; Hernández-Cosido, L.; Mora, A.; Crainiciuc, G.; Sáiz, M.L.; Bernardo, E.; et al. p38γ and p38δ reprogram liver metabolism by modulating neutrophil infiltration. EMBO J. 2016, 35, 536–552. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Collins, Q.F.; Becker, T.C.; Robidoux, J.; Lupo, E.G.; Xiong, Y.; Daniel, K.W.; Floering, L.; Collins, S. p38 Mitogen-activated protein kinase plays a stimulatory role in hepatic gluconeogenesis. J. Biol. Chem. 2005, 280, 42731–42737. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Miller, A.L.; Webb, M.S.; Copik, A.J.; Wang, Y.; Johnson, B.H.; Kumar, R.; Thompson, E.B. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: Correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol. Endocrinol. 2005, 19, 1569–1583. [Google Scholar] [CrossRef]
- Itoh, M.; Adachi, M.; Yasui, H.; Takekawa, M.; Tanaka, H.; Imai, K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol. Endocrinol. 2002, 16, 2382–2392. [Google Scholar] [CrossRef]
- Krstic, M.D.; Rogatsky, I.; Yamamoto, K.R.; Garabedian, M.J. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol. Cell Biol. 1997, 17, 3947–3954. [Google Scholar] [CrossRef]
- Juge-Aubry, C.E.; Hammar, E.; Siegrist-Kaiser, C.; Pernin, A.; Takeshita, A.; Chin, W.W.; Burger, A.G.; Meier, C.A. Regulation of the transcriptional activity of the peroxisome proliferator-activated receptor alpha by phosphorylation of a ligand-independent trans-activating domain. J. Biol. Chem. 1999, 274, 10505–10510. [Google Scholar] [CrossRef] [PubMed]
- Deak, M.; Clifton, A.D.; Lucocq, L.M.; Alessi, D.R. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998, 17, 4426–4441. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; MacDougald, O.A.; Shao, J. CCAAT/enhancer-binding protein alpha mediates induction of hepatic phosphoenolpyruvate carboxykinase by p38 mitogen-activated protein kinase. J. Biol. Chem. 2006, 281, 24390–24397. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Croniger, C.M.; Lekstrom-Himes, J.; Zhang, P.; Fenyus, M.; Tenen, D.G.; Darlington, G.J.; Hanson, R.W. Metabolic response of mice to a postnatal ablation of CCAAT/enhancer-binding protein alpha. J. Biol. Chem. 2005, 280, 38689–38699. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Sun, C.; Zhou, Y.; Gokalp, D.; Herrema, H.; Park, S.W.; Davis, R.J.; Ozcan, U. p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 2011, 17, 1251–1260. [Google Scholar] [CrossRef]
- Xiong, Y.; Collins, Q.F.; An, J.; Lupo, E.; Liu, H.Y.; Liu, D.; Robidoux, J.; Liu, Z.; Cao, W. p38 mitogen-activated protein kinase plays an inhibitory role in hepatic lipogenesis. J. Biol. Chem. 2007, 282, 4975–4982. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, L.M.; Zhang, J.L.; Sabri, A.; Wang, S.J.; Qin, G.J.; Guo, C.Q.; Wen, H.T.; Du, B.B.; Zhang, D.H.; et al. Dual Specificity Phosphatase 12 Regulates Hepatic Lipid Metabolism Through Inhibition of the Lipogenesis and Apoptosis Signal-Regulating Kinase 1 Pathways. Hepatology 2019, 70, 1099–1118. [Google Scholar] [CrossRef]
- Wang, S.; Yan, Z.Z.; Yang, X.; An, S.; Zhang, K.; Qi, Y.; Zheng, J.; Ji, Y.X.; Wang, P.X.; Fang, C.; et al. Hepatocyte DUSP14 maintains metabolic homeostasis and suppresses inflammation in the liver. Hepatology 2018, 67, 1320–1338. [Google Scholar] [CrossRef]
- Ye, P.; Liu, J.; Xu, W.; Liu, D.; Ding, X.; Le, S.; Zhang, H.; Chen, S.; Chen, M.; Xia, J. Dual-Specificity Phosphatase 26 Protects Against Nonalcoholic Fatty Liver Disease in Mice Through Transforming Growth Factor Beta-Activated Kinase 1 Suppression. Hepatology 2019, 69, 1946–1964. [Google Scholar] [CrossRef]
- Tang, P.; Low, H.B.; Png, C.W.; Torta, F.; Kumar, J.K.; Lim, H.Y.; Zhou, Y.; Yang, H.; Angeli, V.; Shabbir, A.; et al. Protective Function of Mitogen-Activated Protein Kinase Phosphatase 5 in Aging- and Diet-Induced Hepatic Steatosis and Steatohepatitis. Hepatol. Commun. 2019, 3, 748–762. [Google Scholar] [CrossRef]
- Wang, Y.; Yan, S.; Xiao, B.; Zuo, S.; Zhang, Q.; Chen, G.; Yu, Y.; Chen, D.; Liu, Q.; Liu, Y.; et al. Prostaglandin F. Diabetes 2018, 67, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [PubMed]
- Matesanz, N.; Bernardo, E.; Acín-Pérez, R.; Manieri, E.; Pérez-Sieira, S.; Hernández-Cosido, L.; Montalvo-Romeral, V.; Mora, A.; Rodríguez, E.; Leiva-Vega, L.; et al. MKK6 controls T3-mediated browning of white adipose tissue. Nat. Commun. 2017, 8, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.; Wang, X.; Cui, T.; et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cao, H.; Li, Y.; Jing, Y.; Liu, S.; Ye, C.; Wang, H.; Yu, S.; Peng, C.; Hui, L.; et al. Metabolic benefits of inhibition of p38α in white adipose tissue in obesity. PLoS Biol. 2018, 16, e2004225. [Google Scholar] [CrossRef] [PubMed]
- Matesanz, N.; Nikolic, I.; Leiva, M.; Pulgarín-Alfaro, M.; Santamans, A.M.; Bernardo, E.; Mora, A.; Herrera-Melle, L.; Rodríguez, E.; Beiroa, D.; et al. p38α blocks brown adipose tissue thermogenesis through p38δ inhibition. PLoS Biol. 2018, 16, e2004455. [Google Scholar] [CrossRef]
- Mottillo, E.P.; Shen, X.J.; Granneman, J.G. beta3-adrenergic receptor induction of adipocyte inflammation requires lipolytic activation of stress kinases p38 and JNK. Biochim. Biophys. Acta 2010, 1801, 1048–1055. [Google Scholar] [CrossRef]
- Engelman, J.A.; Lisanti, M.P.; Scherer, P.E. Specific inhibitors of p38 mitogen-activated protein kinase block 3T3-L1 adipogenesis. J. Biol. Chem. 1998, 273, 32111–32120. [Google Scholar] [CrossRef]
- Tanaka, T.; Yoshida, N.; Kishimoto, T.; Akira, S. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 1997, 16, 7432–7443. [Google Scholar] [CrossRef]
- Aouadi, M.; Laurent, K.; Prot, M.; Le Marchand-Brustel, Y.; Binétruy, B.; Bost, F. Inhibition of p38MAPK increases adipogenesis from embryonic to adult stages. Diabetes 2006, 55, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Takeda, K.; Kadowaki, H.; Ueda, I.; Namba, Y.; Ouchi, Y.; Nishitoh, H.; Ichijo, H. Involvement of ASK1-p38 pathway in the pathogenesis of diabetes triggered by pancreatic ß cell exhaustion. Biochim. Biophys. Acta 2013, 1830, 3656–3663. [Google Scholar] [CrossRef]
- Wei, X.; Gu, N.; Feng, N.; Guo, X.; Ma, X. Inhibition of p38 mitogen-activated protein kinase exerts a hypoglycemic effect by improving β cell function via inhibition of β cell apoptosis in db/db mice. J. Enzyme Inhib. Med. Chem. 2018, 33, 1494–1500. [Google Scholar] [CrossRef]
- Balakrishnan, S.; Sadasivam, M.; Kannan, A.; Panneerselvam, A.; Prahalathan, C. Glucose modulates Pax6 expression through the JNK/p38 MAP kinase pathway in pancreatic beta-cells. Life Sci. 2014, 109, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wohltmann, M.; Tan, M.; Bao, S.; Ladenson, J.H.; Turk, J. Group VIA PLA2 (iPLA2β) is activated upstream of p38 mitogen-activated protein kinase (MAPK) in pancreatic islet β-cell signaling. J. Biol. Chem. 2012, 287, 5528–5541. [Google Scholar] [CrossRef]
- Sumara, G.; Formentini, I.; Collins, S.; Sumara, I.; Windak, R.; Bodenmiller, B.; Ramracheya, R.; Caille, D.; Jiang, H.; Platt, K.A.; et al. Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell 2009, 136, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Goginashvili, A.; Zhang, Z.; Erbs, E.; Spiegelhalter, C.; Kessler, P.; Mihlan, M.; Pasquier, A.; Krupina, K.; Schieber, N.; Cinque, L.; et al. Insulin granules. Insulin secretory granules control autophagy in pancreatic β cells. Science 2015, 347, 878–882. [Google Scholar] [CrossRef]
- Lyu, J.; Imachi, H.; Yoshimoto, T.; Fukunaga, K.; Sato, S.; Ibata, T.; Kobayashi, T.; Dong, T.; Yonezaki, K.; Yamaji, N.; et al. Thyroid stimulating hormone stimulates the expression of glucose transporter 2 via its receptor in pancreatic β cell line, INS-1 cells. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Sato, S.; Imachi, H.; Lyu, J.; Miyai, Y.; Fukunaga, K.; Dong, T.; Ibata, T.; Kobayashi, T.; Yoshimoto, T.; Kikuchi, F.; et al. Effect of TNF-α on the expression of ABCA1 in pancreatic β-cells. J. Mol. Endocrinol. 2018, 61, 185–193. [Google Scholar] [CrossRef]
- Zhang, S.; Kaplan, M.H. The p38 mitogen-activated protein kinase is required for IL-12-induced IFN-gamma expression. J. Immunol. 2000, 165, 1374–1380. [Google Scholar] [CrossRef]
- Rincón, M.; Enslen, H.; Raingeaud, J.; Recht, M.; Zapton, T.; Su, M.S.; Penix, L.A.; Davis, R.J.; Flavell, R.A. Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. 1998, 17, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Park, Y.C.; Kim, J.H.; Kim, H.R.; Yu, T.; Byeon, S.E.; Unsworth, L.D.; Lee, J.; Cho, J.Y. Nanostructured, self-assembling peptide K5 blocks TNF-α and PGE2 production by suppression of the AP-1/p38 pathway. Mediators Inflamm. 2012, 2012. [Google Scholar] [CrossRef]
- Kang, Y.J.; Chen, J.; Otsuka, M.; Mols, J.; Ren, S.; Wang, Y.; Han, J. Macrophage deletion of p38alpha partially impairs lipopolysaccharide-induced cellular activation. J. Immunol. 2008, 180, 5075–5082. [Google Scholar] [CrossRef] [PubMed]
- Risco, A.; del Fresno, C.; Mambol, A.; Alsina-Beauchamp, D.; MacKenzie, K.F.; Yang, H.T.; Barber, D.F.; Morcelle, C.; Arthur, J.S.; Ley, S.C.; et al. p38γ and p38δ kinases regulate the Toll-like receptor 4 (TLR4)-induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc. Natl. Acad. Sci. USA 2012, 109, 11200–11205. [Google Scholar] [CrossRef]
- González-Terán, B.; Cortés, J.R.; Manieri, E.; Matesanz, N.; Verdugo, Á.; Rodríguez, M.E.; González-Rodríguez, Á.; Valverde, Á.; Valverde, Á.; Martín, P.; et al. Eukaryotic elongation factor 2 controls TNF-α translation in LPS-induced hepatitis. J. Clin. Investig. 2013, 123, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Ittner, A.; Block, H.; Reichel, C.A.; Varjosalo, M.; Gehart, H.; Sumara, G.; Gstaiger, M.; Krombach, F.; Zarbock, A.; Ricci, R. Regulation of PTEN activity by p38δ-PKD1 signaling in neutrophils confers inflammatory responses in the lung. J. Exp. Med. 2012, 209, 2229–2246. [Google Scholar] [CrossRef]
- Bertola, A.; Park, O.; Gao, B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: A critical role for E-selectin. Hepatology 2013, 58, 1814–1823. [Google Scholar] [CrossRef]
- Koistinen, H.A.; Chibalin, A.V.; Zierath, J.R. Aberrant p38 mitogen-activated protein kinase signaling in skeletal muscle from Type 2 diabetic patients. Diabetologia 2003, 46, 1324–1328. [Google Scholar] [CrossRef]
- Brown, A.E.; Palsgaard, J.; Borup, R.; Avery, P.; Gunn, D.A.; De Meyts, P.; Yeaman, S.J.; Walker, M. p38 MAPK activation upregulates proinflammatory pathways in skeletal muscle cells from insulin-resistant type 2 diabetic patients. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E63–E70. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, D.; Cui, Y.; Lu, S.; Gao, D.; Liu, J. Proinflammatory macrophages impair skeletal muscle differentiation in obesity through secretion of tumor necrosis factor-α via sustained activation of p38 mitogen-activated protein kinase. J. Cell Physiol. 2019, 234, 2566–2580. [Google Scholar] [CrossRef]
- Fukawa, T.; Yan-Jiang, B.C.; Min-Wen, J.C.; Jun-Hao, E.T.; Huang, D.; Qian, C.N.; Ong, P.; Li, Z.; Chen, S.; Mak, S.Y.; et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat. Med. 2016, 22, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Bosquet, A.; Girona, J.; Guaita-Esteruelas, S.; Heras, M.; Saavedra-García, P.; Martínez-Micaelo, N.; Masana, L.; Rodríguez-Calvo, R. FABP4 inhibitor BMS309403 decreases saturated-fatty-acid-induced endoplasmic reticulum stress-associated inflammation in skeletal muscle by reducing p38 MAPK activation. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2018, 1863, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Ohki, K.; Wakui, H.; Kishio, N.; Azushima, K.; Uneda, K.; Haku, S.; Kobayashi, R.; Haruhara, K.; Kinguchi, S.; Yamaji, T.; et al. Angiotensin II Type 1 Receptor-associated Protein Inhibits Angiotensin II-induced Insulin Resistance with Suppression of Oxidative Stress in Skeletal Muscle Tissue. Sci. Rep. 2018, 8, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Min, K.; Zhang, L.; Canfran-Duque, A.; Jurczak, M.J.; Camporez, J.P.G.; Nie, Y.; Gavin, T.P.; Shulman, G.I.; Fernandez-Hernando, C.; et al. Skeletal Muscle-Specific Deletion of MKP-1 Reveals a p38 MAPK/JNK/Akt Signaling Node That Regulates Obesity-Induced Insulin Resistance. Diabetes 2018, 67, 624–635. [Google Scholar] [CrossRef]
- Yuasa, K.; Okubo, K.; Yoda, M.; Otsu, K.; Ishii, Y.; Nakamura, M.; Itoh, Y.; Horiuchi, K. Targeted ablation of p38α MAPK suppresses denervation-induced muscle atrophy. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Pogozelski, A.R.; Geng, T.; Li, P.; Yin, X.; Lira, V.A.; Zhang, M.; Chi, J.T.; Yan, Z. p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS ONE 2009, 4, e7934. [Google Scholar] [CrossRef]
- Ho, R.C.; Alcazar, O.; Fujii, N.; Hirshman, M.F.; Goodyear, L.J. p38gamma MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R342–R349. [Google Scholar] [CrossRef]
- Zhang, G.; Li, Y.P. p38β MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPβ. Skelet Muscle 2012, 2, 1–9. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, G.; Sin, K.W.; Liu, Z.; Lin, R.K.; Li, M.; Li, Y.P. Activin A induces skeletal muscle catabolism via p38β mitogen-activated protein kinase. J. Cachexia. Sarcopenia. 2017, 8, 202–212. [Google Scholar] [CrossRef]
- Geller, S.; Arribat, Y.; Netzahualcoyotzi, C.; Lagarrigue, S.; Carneiro, L.; Zhang, L.; Amati, F.; Lopez-Mejia, I.C.; Pellerin, L. Tanycytes Regulate Lipid Homeostasis by Sensing Free Fatty Acids and Signaling to Key Hypothalamic Neuronal Populations via FGF21 Secretion. Cell Metab. 2019, 30, 833–844. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Kempuraj, D.; Prabhakaran, K.; Jayakumar, A.R.; Devi, R.S.; Suthanthirarajan, N.; Namasivayam, A. A short-term diabetes induced changes of catecholamines and p38-MAPK in discrete areas of rat brain. Life Sci. 2005, 77, 1825–1835. [Google Scholar] [CrossRef]
- Liao, M.J.; Lin, H.; He, Y.W.; Zou, C. NFATc3 deficiency protects against high fat diet (HFD)-induced hypothalamus inflammation and apoptosis via p38 and JNK suppression. Biochem. Biophys. Res. Commun. 2018, 499, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Benomar, Y.; Gertler, A.; De Lacy, P.; Crépin, D.; Ould Hamouda, H.; Riffault, L.; Taouis, M. Central resistin overexposure induces insulin resistance through Toll-like receptor 4. Diabetes 2013, 62, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.; Müller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell Signal. 2012, 24, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Regan, C.P.; Li, W.; Boucher, D.M.; Spatz, S.; Su, M.S.; Kuida, K. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. USA 2002, 99, 9248–9253. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Lee, J.D. Role of the BMK1/ERK5 signaling pathway: Lessons from knockout mice. J. Mol. Med. (Berl) 2004, 82, 800–808. [Google Scholar] [CrossRef]
- Horie, T.; Park, G.; Inaba, Y.; Hashiuchi, E.; Iezaki, T.; Tokumura, K.; Fukasawa, K.; Yamada, T.; Hiraiwa, M.; Kitaguchi, Y.; et al. MAPK Erk5 in Leptin Receptor‒Expressing Neurons Controls Body Weight and Systemic Energy Homeostasis in Female Mice. Endocrinology 2019, 160, 2837–2848. [Google Scholar] [CrossRef]
- Zhu, H.; Guariglia, S.; Li, W.; Brancho, D.; Wang, Z.V.; Scherer, P.E.; Chow, C.W. Role of extracellular signal-regulated kinase 5 in adipocyte signaling. J. Biol. Chem. 2014, 289, 6311–6322. [Google Scholar] [CrossRef]
- Nam, D.H.; Han, J.H.; Lim, J.H.; Park, K.M.; Woo, C.H. CHOP Deficiency Ameliorates ERK5 Inhibition-Mediated Exacerbation of Streptozotocin-Induced Hyperglycemia and Pancreatic β-Cell Apoptosis. Mol. Cells 2017, 40, 457–465. [Google Scholar] [CrossRef]
- Chen, C.; Wu, S.; Lin, X.; Wu, D.; Fischbach, S.; Xiao, X. ERK5 plays an essential role in gestational beta-cell proliferation. Cell Prolif. 2018, 51, e12410. [Google Scholar] [CrossRef]
- Coulombe, P.; Rodier, G.; Pelletier, S.; Pellerin, J.; Meloche, S. Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol. Cell Biol. 2003, 23, 4542–4558. [Google Scholar] [CrossRef] [PubMed]
- Sauma, S.; Friedman, E. Increased expression of protein kinase C beta activates ERK3. J. Biol. Chem. 1996, 271, 11422–11426. [Google Scholar] [CrossRef] [PubMed]
- Seternes, O.M.; Mikalsen, T.; Johansen, B.; Michaelsen, E.; Armstrong, C.G.; Morrice, N.A.; Turgeon, B.; Meloche, S.; Moens, U.; Keyse, S.M. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 2004, 23, 4780–4791. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, S.; Laass, K.; Kant, S.; Shi, Y.; Visel, A.; Gruber, A.D.; Kotlyarov, A.; Gaestel, M. Scaffolding by ERK3 regulates MK5 in development. EMBO J. 2004, 23, 4770–4779. [Google Scholar] [CrossRef] [PubMed]
- El-Merahbi, R.; Viera, J.T.; Valdes, A.L.; Kolczynska, K.; Reuter, S.; Löffler, M.C.; Erk, M.; Ade, C.P.; Karwen, T.; Mayer, A.E.; et al. The adrenergic-induced ERK3 pathway drives lipolysis and suppresses energy dissipation. Genes Dev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Anhê, G.F.; Torrão, A.S.; Nogueira, T.C.; Caperuto, L.C.; Amaral, M.E.; Medina, M.C.; Azevedo-Martins, A.K.; Carpinelli, A.R.; Carvalho, C.R.; Curi, R.; et al. ERK3 associates with MAP2 and is involved in glucose-induced insulin secretion. Mol. Cell Endocrinol. 2006, 251, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Bogucka, K.; Pompaiah, M.; Marini, F.; Binder, H.; Harms, G.; Kaulich, M.; Klein, M.; Michel, C.; Radsak, M.P.; Rosigkeit, S. ERK3/MAPK6 controls IL-8 production and chemotaxis. Elife 2020, 9, e52511. [Google Scholar] [CrossRef]
- Okamura, M.; Inagaki, T.; Tanaka, T.; Sakai, J. Role of histone methylation and demethylation in adipogenesis and obesity. Organogenesis 2010, 6, 24–32. [Google Scholar] [CrossRef]
- Takada, I.; Kouzmenko, A.P.; Kato, S. PPAR-gamma Signaling Crosstalk in Mesenchymal Stem Cells. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef]
- Ross, S.E.; Hemati, N.; Longo, K.A.; Bennett, C.N.; Lucas, P.C.; Erickson, R.L.; MacDougald, O.A. Inhibition of adipogenesis by Wnt signaling. Science 2000, 289, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.F.; Zhang, L.; Liu, Y.; Li, J.; Shen, H.; Liu, Y.Z.; Tian, Q.; He, H.; Wu, S.; Ran, S.; et al. Meta-analysis of genome-wide association data identifies novel susceptibility loci for obesity. Hum. Mol. Genet. 2014, 23, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Kortenjann, M.; Nehls, M.; Smith, A.J.; Carsetti, R.; Schüler, J.; Köhler, G.; Boehm, T. Abnormal bone marrow stroma in mice deficient for nemo-like kinase, Nlk. Eur. J. Immunol. 2001, 31, 3580–3587. [Google Scholar] [CrossRef]
- Supakankul, P.; Mekchay, S. Association of NLK polymorphisms with intramuscular fat content and fatty acid composition traits in pigs. Meat. Sci. 2016, 118, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Hasygar, K.; Hietakangas, V. p53- and ERK7-dependent ribosome surveillance response regulates Drosophila insulin-like peptide secretion. PLoS Genet. 2014, 10, e1004764. [Google Scholar] [CrossRef]
- Yong, H.Y.; Koh, M.S.; Moon, A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert. Opin. Investig. Drugs 2009, 18, 1893–1905. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Nelson, L.J.; Ávila, M.A.; Cubero, F.J. Mitogen-Activated Protein Kinases (MAPKs) and Cholangiocarcinoma: The Missing Link. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Elkhawad, M.; Rudd, J.H.; Sarov-Blat, L.; Cai, G.; Wells, R.; Davies, L.C.; Collier, D.J.; Marber, M.S.; Choudhury, R.P.; Fayad, Z.A.; et al. Effects of p38 mitogen-activated protein kinase inhibition on vascular and systemic inflammation in patients with atherosclerosis. JACC Cardiovasc. Imaging 2012, 5, 911–922. [Google Scholar] [CrossRef]
- Emami, H.; Vucic, E.; Subramanian, S.; Abdelbaky, A.; Fayad, Z.A.; Du, S.; Roth, E.; Ballantyne, C.M.; Mohler, E.R.; Farkouh, M.E.; et al. The effect of BMS-582949, a P38 mitogen-activated protein kinase (P38 MAPK) inhibitor on arterial inflammation: A multicenter FDG-PET trial. Atherosclerosis 2015, 240, 490–496. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stimulus | MAPKKK | MAPKK | MAPK | Substrates and Biological Functions |
|---|---|---|---|---|
| Growth factors, ligand for GPCRs, cytokines, osmotic stress, microtubule disorganization, and insulin. | RAF family (ARAF, BRAF, CRAF) | MEK 1/2 | ERK1/2 | RSK family (gene transcription, cell proliferation, growth, and survival) MSK1/2 (gene transcription, nucleosome dynamics) MNK1/2 (mRNA translation) Elk-1 (transcription of c-Fos) c-Fos (transcription) Synapsin I, focal adhesion kinase [FAK] and myosin light-chain kinase (actin polymerization) Neurofilaments and paxillin (cytoskeleton organization) CD120a, spleen tyrosine kinase [SYK], and calnexin (plasma membrane dynamics) Death-associated protein kinase [DAPK] (cell death) Tuberous sclerosis complex 2 [TSC2] (nutrient sensing) Nuclear factor of activated T-cells [NF-AT], myocyte enhancer factor 2 [MEF2] and c-Myc (transcription) Signal transducer and activator of transcription 3 [STAT3] (signaling) |
| Stress (hypoxia, UV, and ionizing radiation), cytokines, growth factors, pathogens, toxins, drugs, metabolic changes (obesity and hyperlipidaemia). | MEKK1 to –4 Mixed lineage kinase 1/2/3 [MLK1/2/3] Tumor progression locus 2 [Tpl-2] Delta-like non-canonical Notch ligand [DLK] TAO1/2 TGF-β-activated kinase 1 [TAK1] Apoptosis signal-regulating kinase 1/2 [ASK1/2] | MKK4 MKK7 | JNKs | c-jun, (transcription, cell cycle and apoptosis) BH3-only family of Bcl2 proteins (apoptosis) p53 (apoptosis) Activating transcription factor 2 [ATF-2], nuclear factor of activated T-cells, cytoplasmic 1 [NF-ATc1], Elk-1, Heat shock factor protein 1 [HSF-1], STAT3, c-Myc, JunB (transcription) |
| Oxidative stress, UV irradiation, hypoxia, ischemia, inflammatory cytokines, ligand for GPCRs, and Rho family GTPases. | MEKK1 to -3 MLK2/3 ASK1 Tpl-2 TAK1 TAO1/2 | MKK3 MKK6 TAK1 binding protein 1 [TAB1] ZAP70 LCK | p38s | MSK1/2 (gene transcription, nucleosome response) MNK1/2 (mRNA translation) p53 (preventing p53 proteasomal degradation) ATF (regulation of ER stress response) ATF-2 and Nuclear factor NF-kappa-B [NF-κB] (expression of inflammatory cytokines) Elk-1 (transcription) protein kinase D1 [PKD1] (Trans-Golgi dynamics, signaling) |
| Growth factors, inflammatory cytokines, oxidative and osmotic stresses, ischaemia, and hypoxia. | MEKK2 MEKK3 | MEK5 | ERK5 | SGK (kinase) MEF2, Elk-1 and Sap1a (transcription) |
| Liver | Adipose Tissue | Pancreatic β-Cells | Skeletal Muscle | CNS | |
|---|---|---|---|---|---|
| ERK1/2 | (-) insulin sensitivity | (-) insulin sensitivity (+) adiposity | (+) glucose-stimulated insulin secretion (+) β-cell survival | ?? | (+) energy expenditure (-) adiposity (-) food intake |
| JNKs | contradictory results | (-) hepatic insulin sensitivity (+) hepatic steatosis | β-cell dysfunction | no effect on adiposity contradictory results on insulin sensitivity | (+) adiposity (-) glucose tolerance (-) insulin sensitivity (+) hepatic steatosis |
| p38s | p38α: (+) luconeogenesis (+) fasting hyperglycemia p38α/β (-) lipogenesis p38γ/p38δ (+) hepatic steatosis | Contradictory results | p38δ: (-) insulin secretion p38α: (+) glucose uptake | p38γ: (+) glucose uptake | ?? |
| ERK5 | no obvious phenotype | (-) adiposity (+) leptin/insulin sensitivity | (-) hyperglycaemia | ?? | (-) adiposity |
| ERK3 | ?? | (-) insulin sensitivity (+) adiposity | ?? | ?? | ?? |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kassouf, T.; Sumara, G. Impact of Conventional and Atypical MAPKs on the Development of Metabolic Diseases. Biomolecules 2020, 10, 1256. https://doi.org/10.3390/biom10091256
Kassouf T, Sumara G. Impact of Conventional and Atypical MAPKs on the Development of Metabolic Diseases. Biomolecules. 2020; 10(9):1256. https://doi.org/10.3390/biom10091256
Chicago/Turabian StyleKassouf, Toufic, and Grzegorz Sumara. 2020. "Impact of Conventional and Atypical MAPKs on the Development of Metabolic Diseases" Biomolecules 10, no. 9: 1256. https://doi.org/10.3390/biom10091256
APA StyleKassouf, T., & Sumara, G. (2020). Impact of Conventional and Atypical MAPKs on the Development of Metabolic Diseases. Biomolecules, 10(9), 1256. https://doi.org/10.3390/biom10091256