Molecular and Therapeutic Aspects of Hyperbaric Oxygen Therapy in Neurological Conditions


{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Hypoxia in Neurological Conditions
1.2. HBOT
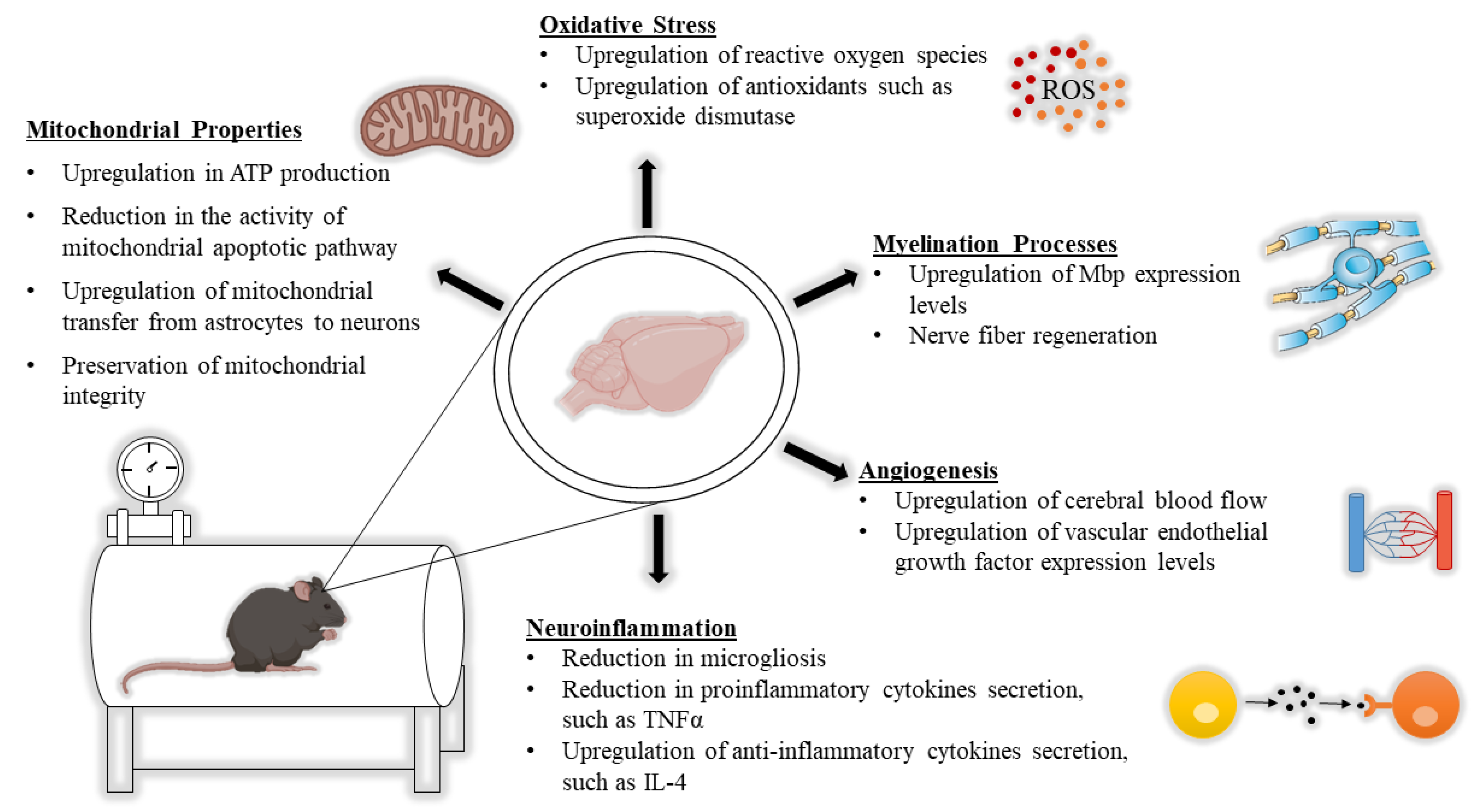
2. HBOT from a Neurobiological Point of View
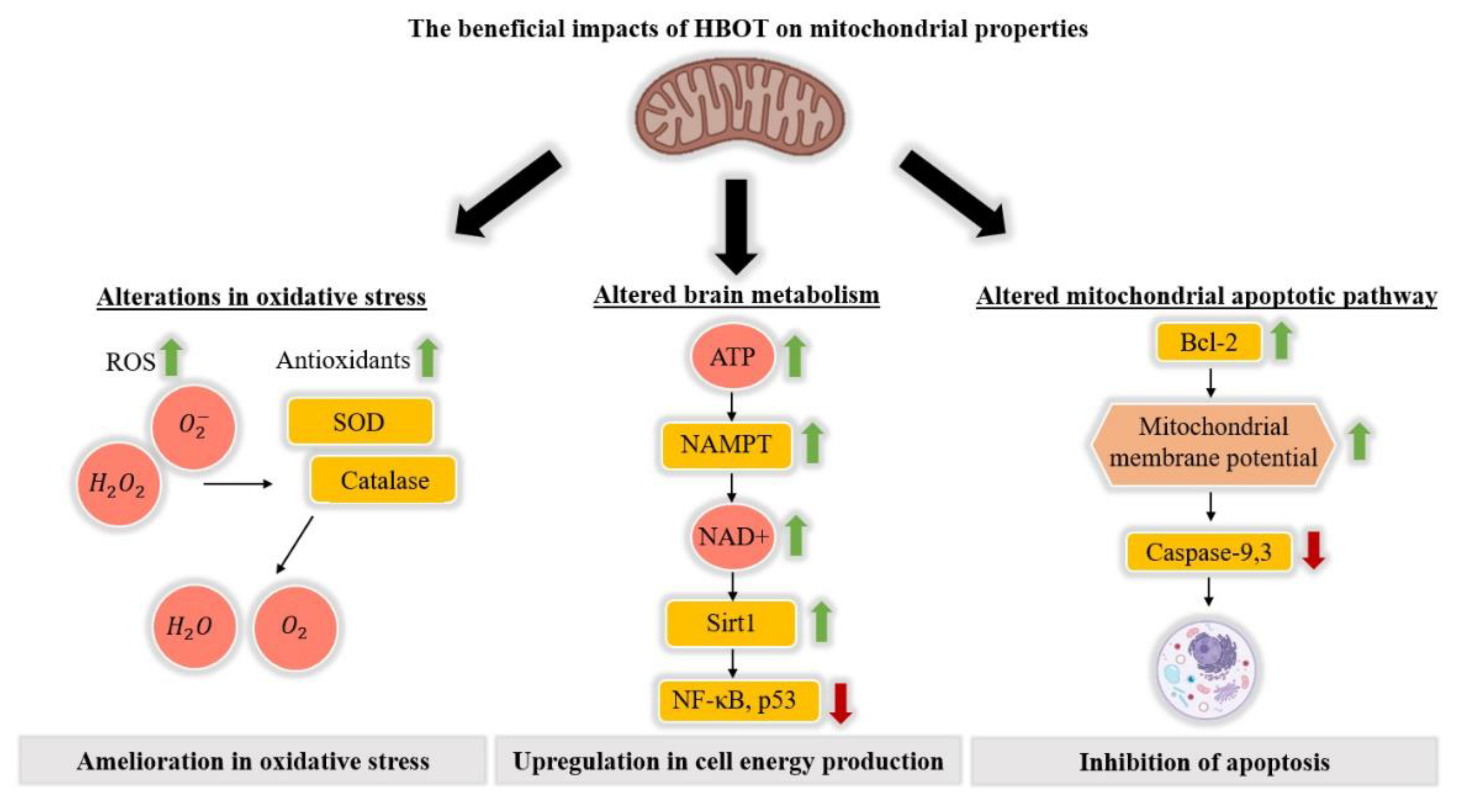
2.1. The Impact of HBOT on Mitochondrial Properties
2.2. The Impact of HBOT on Alterations in White Matter
2.3. The Impact of HBOT on Neuroinflammation
2.4. The Impact of HBOT on Oxidative Stress
2.5. The Impact of HBOT on Induction of Angiogenesis and Changes in CBF
3. HBOT from a Therapeutic Point of View
3.1. ASD
3.2. CP
3.3. TBI
4. Discussion and Future Research
Author Contributions
Funding
Conflicts of Interest
References
- Gibson, G.E.; Peterson, C.; Sansone, J. Decreases in Amino Acid and Acetylcholine Metabolism During Hypoxia. J. Neurochem. 1981, 37, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef]
- Solaini, G.; Baracca, A.; Lenaz, G.; Sgarbi, G. Hypoxia and mitochondrial oxidative metabolism. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 1171–1177. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Rossignol, L.W. Hyperbaric oxygen therapy may improve symptoms in autistic children. Med. Hypotheses 2006, 67, 216–228. [Google Scholar] [CrossRef]
- Van Tilborg, E.; Achterberg, E.J.M.; Van Kammen, C.M.; Van Der Toorn, A.; Groenendaal, F.; Dijkhuizen, R.M.; Heijnen, C.J.; Vanderschuren, L.J.M.J.; Benders, M.N.J.L.; Nijboer, C.H. Combined fetal inflammation and postnatal hypoxia causes myelin deficits and autism-like behavior in a rat model of diffuse white matter injury. Glia 2017, 66, 78–93. [Google Scholar] [CrossRef]
- Blomgren, K.; Hagberg, H. Free radicals, mitochondria, and hypoxia–ischemia in the developing brain. Free. Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef]
- Thompson, C.; Puterman, A.; Linley, L.; Hann, F.; Elst, C.; Molteno, C.; Malan, A. The value of a scoring system for hypoxic ischaemic encephalopathy in predicting neurodevelopmental outcome. Acta Paediatr. 1997, 86, 757–761. [Google Scholar] [CrossRef]
- Erecińska, M.; Silver, I.A. Tissue oxygen tension and brain sensitivity to hypoxia. Respir. Physiol. 2001, 128, 263–276. [Google Scholar] [CrossRef]
- Ghajar, J. Traumatic brain injury. Lancet 2000, 356, 923–929. [Google Scholar] [CrossRef]
- Sarrafzadeh, A.S.; Sakowitz, O.W.; Callsen, T.A.; Lanksch, W.R.; Unterberg, A.W. Bedside microdialysis for early detection of cerebral hypoxia in traumatic brain injury. Neurosurg. Focus 2000, 9, 1–6. [Google Scholar] [CrossRef]
- Zhang, X.; Le, W. Pathological role of hypoxia in Alzheimer’s disease. Exp. Neurol. 2010, 223, 299–303. [Google Scholar] [CrossRef]
- Caplan, L.R.; Hennerici, M. Impaired clearance of emboli (washout) is an important link between hypoperfusion, embolism, and ischemic stroke. Arch. Neurol. 1998, 55, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Grond, M.; Von Kummer, R.; Sobesky, J.; Rudolf, J.; Terstegge, K.; Heiss, W.-D.; Schmulling, S. Early X-Ray Hypoattenuation of Brain Parenchyma Indicates Extended Critical Hypoperfusion in Acute Stroke. Stroke 2000, 31, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Perlman, J.M. Intrapartum hypoxic-ischemic cerebral injury and subsequent cerebral palsy: Medicolegal issues. Pediatrics 1997, 99, 851–859. [Google Scholar] [CrossRef]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Sun, X.; He, G.; Qing, H.; Zhou, W.; Dobie, F.; Cai, F.; Staufenbiel, M.; Huang, L.E.; Song, W. Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 18727–18732. [Google Scholar] [CrossRef]
- Zürcher, N.R.; Bhanot, A.; McDougle, C.J.; Hooker, J.M. A systematic review of molecular imaging (PET and SPECT) in autism spectrum disorder: Current state and future research opportunities. Neurosci. Biobehav. Rev. 2015, 52, 56–73. [Google Scholar] [CrossRef]
- Zilbovicius, M.; Boddaert, N.; Belin, P.; Poline, J.-B.; Rémy, P.; Mangin, J.-F.; Thivard, L.; Barthélémy, C.; Samson, Y. Temporal Lobe Dysfunction in Childhood Autism: A PET Study. Am. J. Psychiatry 2000, 157, 1988–1993. [Google Scholar] [CrossRef]
- Barak, B.; Feng, G. Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat. Neurosci. 2016, 19, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Omata, N.; Murata, T.; Takamatsu, S.; Maruoka, N.; Mitsuya, H.; Yonekura, Y.; Fujibayashi, Y.; Wada, Y. Neuroprotective effect of chronic lithium treatment against hypoxia in specific brain regions with upregulation of cAMP response element binding protein and brain-derived neurotrophic factor but not nerve growth factor: Comparison with acute lithium treatment. Bipolar Disord. 2008, 10, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Morán, J.; Stokowska, A.; Walker, F.R.; Mallard, C.; Hagberg, H.; Pekna, M. Intranasal C3a treatment ameliorates cognitive impairment in a mouse model of neonatal hypoxic–ischemic brain injury. Exp. Neurol. 2017, 290, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Tibbles, P.M.; Edelsberg, J.S. Hyperbaric-Oxygen Therapy. N. Engl. J. Med. 1996, 334, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.B. Hyperbaric Oxygen Therapy. J. Am. Coll. Certif. Wound Spec. 2010, 2, 9–13. [Google Scholar] [CrossRef]
- Hadanny, A.; Rittblat, M.; Bitterman, M.; May-Raz, I.; Suzin, G.; Boussi-Gross, R.; Zemel, Y.; Bechor, Y.; Catalogna, M.; Efrati, S. Hyperbaric oxygen therapy improves neurocognitive functions of post-stroke patients – a retrospective analysis. Restor. Neurol. Neurosci. 2020, 38, 93–107. [Google Scholar] [CrossRef]
- Efrati, S.; Fishlev, G.; Bechor, Y.; Volkov, O.; Bergan, J.; Kliakhandler, K.; Kamiager, I.; Gal, N.; Friedman, M.; Ben-Jacob, E.; et al. Hyperbaric Oxygen Induces Late Neuroplasticity in Post Stroke Patients—Randomized, Prospective Trial. PLoS ONE 2013, 8, e53716. [Google Scholar] [CrossRef]
- Shapira, R.; Solomon, B.; Efrati, S.; Frenkel, D.; Ashery, U. Hyperbaric oxygen therapy ameliorates pathophysiology of 3xTg-AD mouse model by attenuating neuroinflammation. Neurobiol. Aging 2018, 62, 105–119. [Google Scholar] [CrossRef]
- Gill, A.; Bell, C. Hyperbaric oxygen: Its uses, mechanisms of action and outcomes. QJM Int. J. Med. 2004, 97, 385–395. [Google Scholar] [CrossRef]
- Thackham, J.A.; McElwain, D.L.S.; Long, R.J. The use of hyperbaric oxygen therapy to treat chronic wounds: A review. Wound Repair. Regen. 2008, 16, 321–330. [Google Scholar] [CrossRef]
- Boerema, I.; Kroll, J.A.; Meyne, N.G.; Lokin, E.; Kroon, B.; Huiskes, J.W. High Atmospheric Pressure as an Aid to Cardiac Surgery. Surv. Anesthesiol. 1958, 2, 377–379. [Google Scholar] [CrossRef]
- Weaver, L.; Hopkins, R.O.; Chan, K.J.; Churchill, S.; Elliott, C.G.; Clemmer, T.P.; Orme, J.F.; Thomas, F.O.; Morris, A.H. Hyperbaric Oxygen for Acute Carbon Monoxide Poisoning. N. Engl. J. Med. 2002, 347, 1057–1067. [Google Scholar] [CrossRef]
- Golden, Z.L.; Neubauer, R.; Golden, C.J.; Greene, L.; Marsh, J.; Mleko, A. Improvement in Cerebral Metabolism in Chronic Brain Injury After Hyperbaric Oxygen Therapy. Int. J. Neurosci. 2002, 112, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Daugherty, W.P.; Sun, D.; Levasseur, J.E.; Altememi, N.; Hamm, R.J.; Rockswold, G.L.; Bullock, M.R. Protection of mitochondrial function and improvement in cognitive recovery in rats treated with hyperbaric oxygen following lateral fluid-percussion injury. J. Neurosurg. 2007, 106, 687–694. [Google Scholar] [CrossRef]
- Vlodavsky, E.; Palzur, E.; Soustiel, J.F. Hyperbaric oxygen therapy reduces neuroinflammation and expression of matrix metalloproteinase-9 in the rat model of traumatic brain injury. Neuropathol. Appl. Neurobiol. 2006, 32, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Boussi-Gross, R.; Golan, H.; Fishlev, G.; Bechor, Y.; Volkov, O.; Bergan, J.; Friedman, M.; Hoofien, D.; Shlamkovitch, N.; Ben-Jacob, E.; et al. Hyperbaric Oxygen Therapy Can Improve Post Concussion Syndrome Years after Mild Traumatic Brain Injury—Randomized Prospective Trial. PLoS ONE 2013, 8, e79995. [Google Scholar] [CrossRef] [PubMed]
- Hadanny, A.; Golan, H.; Fishlev, G.; Bechor, Y.; Volkov, O.; Suzin, G.; Ben-Jacob, E.; Efrati, S. Hyperbaric oxygen can induce neuroplasticity and improve cognitive functions of patients suffering from anoxic brain damage. Restor. Neurol. Neurosci. 2015, 33, 471–486. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and Apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.C.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in Neuroplasticity and Neurological Disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovacs, R. Mitochondria and neuronal activity. Am. J. Physiol. Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef]
- Hu, Q.; Manaenko, A.; Bian, H.; Guo, Z.; Huang, J.-L.; Guo, Z.-N.; Yang, P.; Tang, J.; Zhang, J.H. Hyperbaric Oxygen Reduces Infarction Volume and Hemorrhagic Transformation Through ATP/NAD+/Sirt1 Pathway in Hyperglycemic Middle Cerebral Artery Occlusion Rats. Stroke 2017, 48, 1655–1664. [Google Scholar] [CrossRef]
- Palzur, E.; Zaaroor, M.; Vlodavsky, E.; Milman, F.; Soustiel, J.F. Neuroprotective effect of hyperbaric oxygen therapy in brain injury is mediated by preservation of mitochondrial membrane properties. Brain Res. 2008, 1221, 126–133. [Google Scholar] [CrossRef]
- Lippert, T.; Borlongan, C.V. Prophylactic treatment of hyperbaric oxygen treatment mitigates inflammatory response via mitochondria transfer. CNS Neurosci. Ther. 2019, 25, 815–823. [Google Scholar] [CrossRef]
- Sun, L.; Strelow, H.; Mies, G.; Veltkamp, R. Oxygen therapy improves energy metabolism in focal cerebral ischemia. Brain Res. 2011, 1415, 103–108. [Google Scholar] [CrossRef]
- Vlodavsky, E.; Palzur, E.; Feinsod, M.; Soustiel, J.F. Evaluation of the apoptosis-related proteins of the BCL-2 family In the traumatic penumbra area of the rat model of cerebral contusion, treated by hyperbaric oxygen therapy: A quantitative immunohistochemical study. Acta Neuropathol. 2005, 110, 120–126. [Google Scholar] [CrossRef]
- Ola, M.S.; Nawaz, M.I.; Ahsan, H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol. Cell. Biochem. 2011, 351, 41–58. [Google Scholar] [CrossRef]
- Fan, T.-J.; Han, L.-H.; Cong, R.-S.; Liang, J. Caspase Family Proteases and Apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef]
- Rossignol, D.A.; E Frye, R. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2011, 17, 290–314. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Rossignol, D.A. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr. Res. 2011, 69, 41R–47R. [Google Scholar] [CrossRef] [PubMed]
- Salzer, J.L.; Brophy, P.J.; Peles, E. Molecular domains of myelinated axons in the peripheral nervous system. Glia 2008, 56, 1532–1540. [Google Scholar] [CrossRef] [PubMed]
- Nave, K.-A.; Werner, H.B. Myelination of the Nervous System: Mechanisms and Functions. Annu. Rev. Cell Dev. Biol. 2014, 30, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, J.M. Physiological Basis of Conduction in Myelinated Nerve Fibers. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1984; pp. 117–145. [Google Scholar]
- Barak, B.; Zhang, Z.; Liu, Y.; Nir, A.; Trangle, S.S.; Ennis, M.; Levandowski, K.M.; Wang, N.; Quast, K.; Boulting, G.L.; et al. Neuronal deletion of Gtf2i, associated with Williams syndrome, causes behavioral and myelin alterations rescuable by a remyelinating drug. Nat. Neurosci. 2019, 22, 700–708. [Google Scholar] [CrossRef]
- Flygt, J.; Djupsjö, A.; Lenne, F.; Marklund, N. Myelin loss and oligodendrocyte pathology in white matter tracts following traumatic brain injury in the rat. Eur. J. Neurosci. 2013, 38, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.C.; Niu, J.; Hoi, K.K.; Zhao, C.; Caganap, S.D.; Henry, R.G.; Dao, D.; Zollinger, D.R.; Mei, F.; A Shen, Y.-A.; et al. Clemastine rescues myelination defects and promotes functional recovery in hypoxic brain injury. Brain 2018, 141, 85–98. [Google Scholar] [CrossRef]
- Moscarello, M.A.; Wood, D.D.; Ackerley, C.; Boulias, C. Myelin in multiple sclerosis is developmentally immature. J. Clin. Investig. 1994, 94, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Nir, A.; Barak, B. White matter alterations in Williams syndrome related to behavioral and motor impairments. Glia 2020. [Google Scholar] [CrossRef] [PubMed]
- Elroy-Stein, O. Mitochondrial malfunction in vanishing white matter disease: A disease of the cytosolic translation machinery. Neural Regen. Res. 2017, 12, 1610–1612. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Enein, F.; Rauschka, H.; Kornek, B.; Stadelmann, C.; Stefferl, A.; Brück, W.; Lucchinetti, C.; Schmidbauer, M.; Jellinger, K.; Lassmann, H. Preferential loss of myelin-associated glycoprotein reflects hypoxia-like white matter damage in stroke and inflammatory brain diseases. J. Neuropathol. Exp. Neurol. 2003, 62, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Back, S.A.; Han, B.H.; Luo, N.L.; Chricton, C.A.; Xanthoudakis, S.; Tam, J.; Arvin, K.L.; Holtzman, D.M. Selective Vulnerability of Late Oligodendrocyte Progenitors to Hypoxia–Ischemia. J. Neurosci. 2002, 22, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Tal, S.; Hadanny, A.; Sasson, E.; Suzin, G.; Efrati, S. Hyperbaric Oxygen Therapy Can Induce Angiogenesis and Regeneration of Nerve Fibers in Traumatic Brain Injury Patients. Front. Hum. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Assaf, Y.; Pasternak, O. Diffusion Tensor Imaging (DTI)-based White Matter Mapping in Brain Research: A Review. J. Mol. Neurosci. 2007, 34, 51–61. [Google Scholar] [CrossRef]
- Baratz-Goldstein, R.; Toussia-Cohen, S.; Elpaz, A.; Rubovitch, V.; Pick, C.G. Immediate and delayed hyperbaric oxygen therapy as a neuroprotective treatment for traumatic brain injury in mice. Mol. Cell. Neurosci. 2017, 83, 74–82. [Google Scholar] [CrossRef]
- Kraitsy, K.; Ueçal, M.; Grossauer, S.; Bruckmann, L.; Pfleger, F.; Ropele, S.; Fazekas, F.; Gruenbacher, G.; Patz, S.; Absenger, M.; et al. Repetitive Long-Term Hyperbaric Oxygen Treatment (HBOT) Administered after Experimental Traumatic Brain Injury in Rats Induces Significant Remyelination and a Recovery of Sensorimotor Function. PLoS ONE 2014, 9, e97750. [Google Scholar] [CrossRef]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef][Green Version]
- Bar, E.; Barak, B. Microglia roles in synaptic plasticity and myelination in homeostatic conditions and neurodevelopmental disorders. Glia 2019, 67, 2125–2141. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, L.; Pu, H.; Mao, L.; Hu, X.; Jiang, X.; Xu, N.; Stetler, R.A.; Zhang, J.H.; Liu, X.; et al. Rapid endothelial cytoskeletal reorganization enables early blood–brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 2016, 7, 10523. [Google Scholar] [CrossRef]
- Rao, J.S.; Kellom, M.; Kim, H.-W.; Rapoport, S.I.; Reese, E.A. Neuroinflammation and Synaptic Loss. Neurochem. Res. 2012, 37, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Chez, M.G.; Dowling, T.; Patel, P.B.; Khanna, P.; Kominsky, M. Elevation of Tumor Necrosis Factor-Alpha in Cerebrospinal Fluid of Autistic Children. Pediatr. Neurol. 2007, 36, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2004, 57, 67–81. [Google Scholar] [CrossRef]
- Pardo, C.A.; Vargas, D.L.; Zimmerman, A.W. Immunity, neuroglia and neuroinflammation in autism. Int. Rev. Psychiatry 2005, 17, 485–495. [Google Scholar] [CrossRef]
- Srivastava, I.N.; Shperdheja, J.; Baybis, M.; Ferguson, T.; Crino, P.B. mTOR pathway inhibition prevents neuroinflammation and neuronal death in a mouse model of cerebral palsy. Neurobiol. Dis. 2016, 85, 144–154. [Google Scholar] [CrossRef]
- Yoon, B.H.; Romero, R.; Park, J.S.; Kim, C.J.; Kim, S.H.; Choi, J.-H.; Han, T.R. Fetal exposure to an intra-amniotic inflammation and the development of cerebral palsy at the age of three years. Am. J. Obstet. Gynecol. 2000, 182, 675–681. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1α Is Essential for Myeloid Cell-Mediated Inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef]
- Lewis, J.S.; Lee, J.A.; Underwood, J.C.E.; Harris, A.L.; Lewis, C.E. Macrophage responses to hypoxia: Relevance to disease mechanisms. J. Leukoc. Biol. 1999, 66, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lu, J.; Sivakumar, V.; Ling, E.A.; Kaur, C. Amoeboid Microglia in the Periventricular White Matter Induce Oligodendrocyte Damage through Expression of Proinflammatory Cytokines via MAP Kinase Signaling Pathway in Hypoxic Neonatal Rats. Brain Pathol. 2008, 18, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A. Hyperbaric oxygen therapy might improve certain pathophysiological findings in autism. Med. Hypotheses 2007, 68, 1208–1227. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Landreth, G.E. Microglial interaction with β-amyloid: Implications for the pathogenesis of Alzheimer’s disease. Microsc. Res. Tech. 2001, 54, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Sheng, J.G.; Griffin, W.S.T. Glial Cytokines in Alzheimer’s Disease. Hum. Pathol. 1995, 26, 816–823. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Bertram, L. Twenty Years of the Alzheimer’s Disease Amyloid Hypothesis: A Genetic Perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Chen, X.; Duan, X.-S.; Xu, L.-J.; Zhao, J.-J.; She, Z.-F.; Chen, W.-W.; Zheng, Z.-J.; Jiang, G.-D. Interleukin-10 mediates the neuroprotection of hyperbaric oxygen therapy against traumatic brain injury in mice. Neuroscience 2014, 266, 235–243. [Google Scholar] [CrossRef]
- Rosario, E.R.; Kaplan, S.E.; Khonsari, S.; Vazquez, G.; Solanki, N.; Lane, M.; Brownell, H.; Rosenberg, S.S. The Effect of Hyperbaric Oxygen Therapy on Functional Impairments Caused by Ischemic Stroke. Neurol. Res. Int. 2018, 2018, 1–12. [Google Scholar] [CrossRef]
- Jamieson, D.; Chance, B.; Cadenas, E.; Boveris, A. The Relation of Free Radical Production to Hyperoxia. Annu. Rev. Physiol. 1986, 48, 703–719. [Google Scholar] [CrossRef]
- Yusa, T.; Beckman, J.S.; Crapo, J.D.; Freeman, B.A. Hyperoxia increases H2O2 production by brain in vivo. J. Appl. Physiol. 1987, 63, 353–358. [Google Scholar] [CrossRef]
- Massaad, C.A.; Klann, E. Reactive Oxygen Species in the Regulation of Synaptic Plasticity and Memory. Antioxidants Redox Signal. 2011, 14, 2013–2054. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.; Balin, A.K. Oxidative influence on development and differentiation: An overview of a free radical theory of development. Free. Radic. Biol. Med. 1989, 6, 631–661. [Google Scholar] [CrossRef]
- Yu, B.P. Cellular defenses against damage from reactive oxygen species. Physiol. Rev. 1994, 74, 139–162. [Google Scholar] [CrossRef] [PubMed]
- Matchett, G.A.; Martin, R.D.; Zhang, J.H. Hyperbaric oxygen therapy and cerebral ischemia: Neuroprotective mechanisms. Neurol. Res. 2009, 31, 114–121. [Google Scholar] [CrossRef]
- Wada, K.; Miyazawa, T.; Nomura, N.; Tsuzuki, N.; Nawashiro, H.; Shima, K. Preferential Conditions for and Possible Mechanisms of Induction of Ischemic Tolerance by Repeated Hyperbaric Oxygenation in Gerbil Hippocampus. Neurosurgery 2001, 49, 160–167. [Google Scholar] [CrossRef]
- Pablos, M.I.; Reiter, R.J.; Chuang, J.-I.; Ortiz, G.G.; Guerrero, J.M.; Sewerynek, E.; Agapito, M.T.; Melchiorri, D.; Lawrence, R.; Deneke, S.M. Acutely administered melatonin reduces oxidative damage in lung and brain induced by hyperbaric oxygen. J. Appl. Physiol. 1997, 83, 354–358. [Google Scholar] [CrossRef]
- Oter, S.; Korkmaz, A.; Topal, T.; Özcan, Ö.; Sadir, S.; Ozler, M.; Ogur, R.; Bilgic, H. Correlation between hyperbaric oxygen exposure pressures and oxidative parameters in rat lung, brain, and erythrocytes. Clin. Biochem. 2005, 38, 706–711. [Google Scholar] [CrossRef]
- Benedetti, S.; Lamorgese, A.; Piersantelli, M.; Pagliarani, S.; Benvenuti, F.; Canestrari, F. Oxidative stress and antioxidant status in patients undergoing prolonged exposure to hyperbaric oxygen. Clin. Biochem. 2004, 37, 312–317. [Google Scholar] [CrossRef]
- Narkowicz, C.K.; Vial, J.H.; McCartney, P.W. Hyperbaric Oxygen Therapy Increases Free Radical Levels in the Blood of Humans. Free. Radic. Res. Commun. 1993, 19, 71–80. [Google Scholar] [CrossRef]
- Şimşek, K.; Ozler, M.; Yildirim, A.O.; Sadir, S.; Demirbas, S.; Oztosun, M.; Korkmaz, A.; Ay, H.; Oter, S.; Yildiz, S. Evaluation of the Oxidative Effect of Long-Term Repetitive Hyperbaric Oxygen Exposures on Different Brain Regions of Rats. Sci. World J. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Dennog, C.; Hartmann, A.; Frey, G.; Speit, G. Detection of DNA damage after hyperbaric oxygen (HBO) therapy. Mutagenesis 1996, 11, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Xiong, L.; Lao, N.; Chen, S.; Xu, N.; Zhu, Z. Hyperbaric Oxygen Preconditioning Induces Tolerance against Spinal Cord Ischemia by Upregulation of Antioxidant Enzymes in Rabbits. Br. J. Pharmacol. 2005, 26, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Y.; Gibson, J.J.; Rollins, M.D.; Hopf, H.W.; Hussain, Z.; Hunt, T.K. Effect of hyperoxia on vascular endothelial growth factor levels in a wound model. Arch. Surg. 2000, 135, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Thom, S.R. Oxidative stress is fundamental to hyperbaric oxygen therapy. J. Appl. Physiol. 2009, 106, 988–995. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling: Role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef]
- Hadanny, A.; Meir, O.; Bechor, Y.; Fishlev, G.; Bergan, J.; Efrati, S. Seizures during hyperbaric oxygen therapy: Retrospective analysis of 62,614 treatment sessions. Undersea Hyperb. Med. 2016, 43, 21–28. [Google Scholar]
- Critchley, H.D.; Daly, E.; Bullmore, E.T.; Williams, S.; Van Amelsvoort, T.; Robertson, D.; Rowe, A.; Phillips, M.; McAlonan, G.; Howlin, P.; et al. The functional neuroanatomy of social behaviour. Brain 2000, 123, 2203–2212. [Google Scholar] [CrossRef]
- Harch, P.G.; Fogarty, E.F. Hyperbaric oxygen therapy for Alzheimer’s dementia with positron emission tomography imaging: A case report. Med. Gas Res. 2019, 8, 181–184. [Google Scholar] [CrossRef]
- Cardenas, D.P.; Muir, E.R.; Huang, S.; Boley, A.; Lodge, D.; Duong, T.Q. Functional MRI during hyperbaric oxygen: Effects of oxygen on neurovascular coupling and BOLD fMRI signals. NeuroImage 2015, 119, 382–389. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, Y.; Wang, Z.; Yang, J.; Gao, C.; Su, Q. Effect of VEGF and CX43 on the promotion of neurological recovery by hyperbaric oxygen treatment in spinal cord–injured rats. Spine J. 2014, 14, 119–127. [Google Scholar] [CrossRef]
- Peng, Z.-R.; Yang, A.-L.; Yang, Q.-D. The effect of hyperbaric oxygen on intracephalic angiogenesis in rats with intracerebral hemorrhage. J. Neurol. Sci. 2014, 342, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Knopf, A. Autism prevalence increases from 1 in 60 to 1 in 54: CDC. Brown Univ. Child Adolesc. Behav. Lett. 2020, 36, 4. [Google Scholar] [CrossRef]
- Barnea-Goraly, N.; Kwon, H.; Menon, V.; Eliez, S.; Lotspeich, L.; Reiss, A.L. White matter structure in autism: Preliminary evidence from diffusion tensor imaging. Biol. Psychiatry 2004, 55, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Chou, K.-H.; Chen, I.-Y.; Fan, Y.-T.; Decety, J.; Lin, C.-P. Atypical development of white matter microstructure in adolescents with autism spectrum disorders. NeuroImage 2010, 50, 873–882. [Google Scholar] [CrossRef]
- Ohnishi, T.; Matsuda, H.; Hashimoto, T.; Kunihiro, T.; Nishikawa, M.; Uema, T.; Sasaki, M. Abnormal regional cerebral blood flow in childhood autism. Brain 2000, 123, 1838–1844. [Google Scholar] [CrossRef]
- Xiong, T.; Chen, H.; Luo, R.; Mu, D. Hyperbaric oxygen therapy for autism spectrum disorder (ASD) in children and adults. Cochrane Database Syst. Rev. 2014, 1. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Rossignol, L.W.; Smith, S.; Schneider, C.; Logerquist, S.; Usman, A.; Neubrander, J.; Madren, E.M.; Hintz, G.; Grushkin, B.; et al. Hyperbaric treatment for children with autism: A multicenter, randomized, double-blind, controlled trial. BMC Pediatr. 2009, 9, 21. [Google Scholar] [CrossRef]
- Granpeesheh, R.; Tarbox, J.; Dixon, D.R.; Wilke, A.E.; Allen, M.S.; Bradstreet, J.J. Randomized trial of hyperbaric oxygen therapy for children with autism. Res. Autism Spectr. Disord. 2010, 4, 268–275. [Google Scholar] [CrossRef]
- Jepson, B.; Granpeesheh, R.; Tarbox, J.; Olive, M.L.; Stott, C.; Braud, S.; Yoo, J.H.; Wakefield, A.; Allen, M.S. Controlled Evaluation of the Effects of Hyperbaric Oxygen Therapy on the Behavior of 16 Children with Autism Spectrum Disorders. J. Autism Dev. Disord. 2010, 41, 575–588. [Google Scholar] [CrossRef]
- Lerman, D.C.; Sansbury, T.; Hovanetz, A.; Wolever, E.; Garcia, A.; O’Brien, E.; Adedipe, H. Using Behavior Analysis to Examine the Outcomes of Unproven Therapies: An Evaluation of Hyperbaric Oxygen Therapy for Children with Autism. Behav. Anal. Pr. 2008, 1, 50–58. [Google Scholar] [CrossRef]
- Luo, C.-W.; Deng, X.-Y.; Cheng, J.-L.; Xiao, D.-X.; Zhang, C.-Y.; Feng, J.-X.; Chen, S.-Q.; Hu, N. Altered anxiety and social behaviors in a mouse model of Fragile X syndrome treated with hyperbaric oxygen therapy. J. Clin. Neurosci. 2020, 73, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Rossignol, L.W.; James, S.J.; Melnyk, S.; Mumper, E. The effects of hyperbaric oxygen therapy on oxidative stress, inflammation, and symptoms in children with autism: An open-label pilot study. BMC Pediatr. 2007, 7, 36. [Google Scholar] [CrossRef] [PubMed]
- El-Baz, F.; Elhossiny, R.M.; Azeem, Y.A.; Girgis, M. Study the effect of hyperbaric oxygen therapy in Egyptian autistic children: A clinical trial. Egypt. J. Med Hum. Genet. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- Chungpaibulpatana, J.; Sumpatanarax, T.; Thadakul, N.; Chantharatreerat, C.; Konkaew, M.; Aroonlimsawas, M. Hyperbaric oxygen therapy in Thai autistic children. J. Med Assoc. Thail. 2008, 91, 1232–1238. [Google Scholar]
- Bent, S.; Bertoglio, K.; Ashwood, P.; Nemeth, E.; Hendren, R.L. Brief Report: Hyperbaric Oxygen Therapy (HBOT) in Children with Autism Spectrum Disorder: A Clinical Trial. J. Autism Dev. Disord. 2011, 42, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Sampanthavivat, M.; Singkhwa, W.; Chaiyakul, T.; Karoonyawanich, S.; Ajpru, H. Hyperbaric oxygen in the treatment of childhood autism: A randomised controlled trial. Diving Hyperb. Med. J. 2012, 42, 128–133. [Google Scholar]
- Mukherjee, A.; Raison, M.; Sahni, T.; Arya, A.; Lambert, J.; Marois, P.; James, P.B.; Parent, A.; Ballaz, L. Intensive rehabilitation combined with HBO2 therapy in children with cerebral palsy: A controlled longitudinal study. Undersea Hyperb. Med. 2014, 41, 77–85. [Google Scholar]
- Rossignol, D.A.; Bradstreet, J.J. Evidence of Mitochondrial Dysfunction in Autism and Implications for Treatment. Am. J. Biochem. Biotechnol. 2008, 4, 208–217. [Google Scholar] [CrossRef]
- Fukuda, S.; Mizuno, K.; Kawai, S.; Kakita, H.; Goto, T.; Hussein, M.H.; Daoud, G.A.; Ito, T.; Kato, I.; Suzuki, S.; et al. Reduction in cerebral blood flow volume in infants complicated with hypoxic ischemic encephalopathy resulting in cerebral palsy. Brain Dev. 2008, 30, 246–253. [Google Scholar] [CrossRef]
- Lou, H.C.; Lassen, N.A.; Friis-Hansen, B. Low Cerebral Blood Flow in Hypotensive Perinatal Distress. Acta Neurol. Scand. 1977, 56, 343–352. [Google Scholar] [CrossRef]
- McDonagh, M.S.; Morgan, D.; Carson, S.; Russman, B.S. Systematic review of hyperbaric oxygen therapy for cerebral palsy: The state of the evidence. Dev. Med. Child Neurol. 2007, 49, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.P.; Vanasse, M.; Marois, P.; Amar, M.; Goldberg, J.; Lambert, J.; Lassonde, M.; Hardy, P.; Fortin, J.; Tremblay, S.D.; et al. Hyperbaric oxygen for children with cerebral palsy: A randomised multicentre trial. Lancet 2001, 357, 582–586. [Google Scholar] [CrossRef]
- Hu, Q.; Manaenko, A.; Xu, T.; Guo, Z.; Tang, J.; Zhang, J.H. Hyperbaric oxygen therapy for traumatic brain injury: Bench-to-bedside. Med. Gas Res. 2016, 6, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Obenaus, A. Hyperbaric oxygen therapy for traumatic brain injury. Med. Gas Res. 2011, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- Rockswold, S.B.; Rockswold, G.L.; Defillo, A. Hyperbaric oxygen in traumatic brain injury. Neurol. Res. 2007, 29, 162–172. [Google Scholar] [CrossRef]
- Harch, P.G.; Kriedt, C.; Van Meter, K.W.; Sutherland, R.J. Hyperbaric oxygen therapy improves spatial learning and memory in a rat model of chronic traumatic brain injury. Brain Res. 2007, 1174, 120–129. [Google Scholar] [CrossRef]
- Rockswold, S.B.; Rockswold, G.L.; Vargo, J.M.; Erickson, C.A.; Sutton, R.L.; Bergman, T.A.; Biros, M.H. Effects of hyperbaric oxygenation therapy on cerebral metabolism and intracranial pressure in severely brain injured patients. J. Neurosurg. 2001, 94, 403–411. [Google Scholar] [CrossRef]
- Rockswold, G.L.; Ford, S.E.; Anderson, D.; Bergman, T.A.; Sherman, R.E. Results of a prospective randomized trial for treatment of severely brain-injured patients with hyperbaric oxygen. J. Neurosurg. 1992, 76, 929–934. [Google Scholar] [CrossRef]
- Tal, S.; Hadanny, A.; Berkovitz, N.; Sasson, E.; Ben-Jacob, E.; Efrati, S. Hyperbaric oxygen may induce angiogenesis in patients suffering from prolonged post-concussion syndrome due to traumatic brain injury. Restor. Neurol. Neurosci. 2015, 33, 943–951. [Google Scholar] [CrossRef]
- Eve, D.J.; Steele, M.R.; Sanberg, P.R.; Borlongan, C.V. Hyperbaric oxygen therapy as a potential treatment for post-traumatic stress disorder associated with traumatic brain injury. Neuropsychiatr. Dis. Treat. 2016, 12, 2689–2705. [Google Scholar] [CrossRef]
- Rockswold, S.B.; Rockswold, G.L.; Zaun, D.A.; Liu, J. A prospective, randomized Phase II clinical trial to evaluate the effect of combined hyperbaric and normobaric hyperoxia on cerebral metabolism, intracranial pressure, oxygen toxicity, and clinical outcome in severe traumatic brain injury. J. Neurosurg. 2013, 118, 1317–1328. [Google Scholar] [CrossRef] [PubMed]
- Ziabreva, I.; Campbell, G.R.; Rist, J.; Zambonin, J.L.; Rorbach, J.; Wydro, M.M.; Lassmann, H.; Franklin, R.J.M.; Mahad, D. Injury and differentiation following inhibition of mitochondrial respiratory chain complex IV in rat oligodendrocytes. Glia 2010, 58, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, R.; Wong, A.; Silva, J.; Li, M.; Itoh, A.; Horiuchi, M.; Itoh, T.; Pleasure, D.; Cortopassi, G. Oligodendroglial differentiation induces mitochondrial genes and inhibition of mitochondrial function represses oligodendroglial differentiation. Mitochondrion 2010, 10, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Plafki, C.; Peters, P.; Almeling, M.; Welslau, W.; Busch, R. Complications and side effects of hyperbaric oxygen therapy. Aviat. Space Environ. Med. 2000, 71, 119–124. [Google Scholar] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, I.; Barak, B. Molecular and Therapeutic Aspects of Hyperbaric Oxygen Therapy in Neurological Conditions. Biomolecules 2020, 10, 1247. https://doi.org/10.3390/biom10091247
Fischer I, Barak B. Molecular and Therapeutic Aspects of Hyperbaric Oxygen Therapy in Neurological Conditions. Biomolecules. 2020; 10(9):1247. https://doi.org/10.3390/biom10091247
Chicago/Turabian StyleFischer, Inbar, and Boaz Barak. 2020. "Molecular and Therapeutic Aspects of Hyperbaric Oxygen Therapy in Neurological Conditions" Biomolecules 10, no. 9: 1247. https://doi.org/10.3390/biom10091247
APA StyleFischer, I., & Barak, B. (2020). Molecular and Therapeutic Aspects of Hyperbaric Oxygen Therapy in Neurological Conditions. Biomolecules, 10(9), 1247. https://doi.org/10.3390/biom10091247