Interplay between Cell-Surface Receptors and Extracellular Matrix in Skin

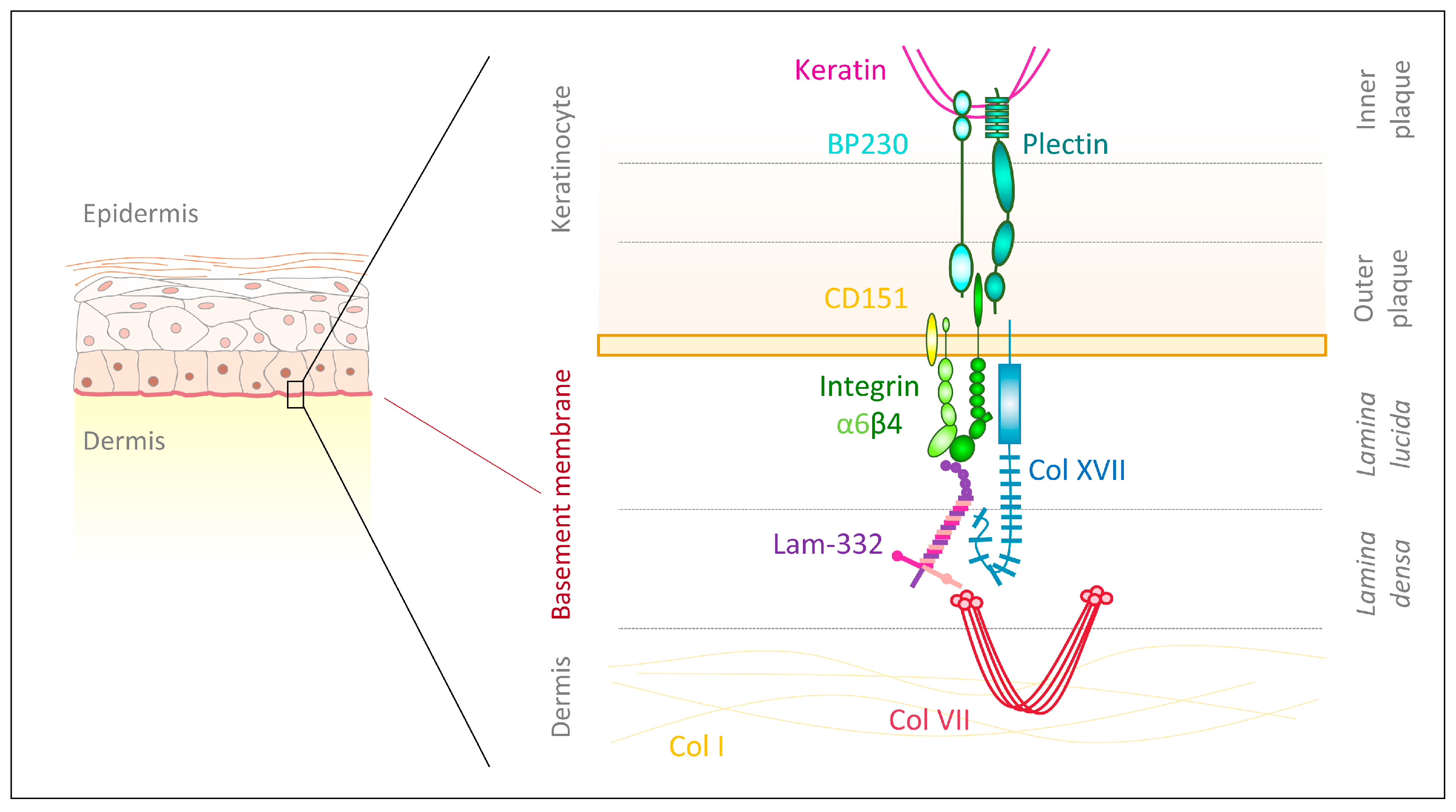{kind=link}
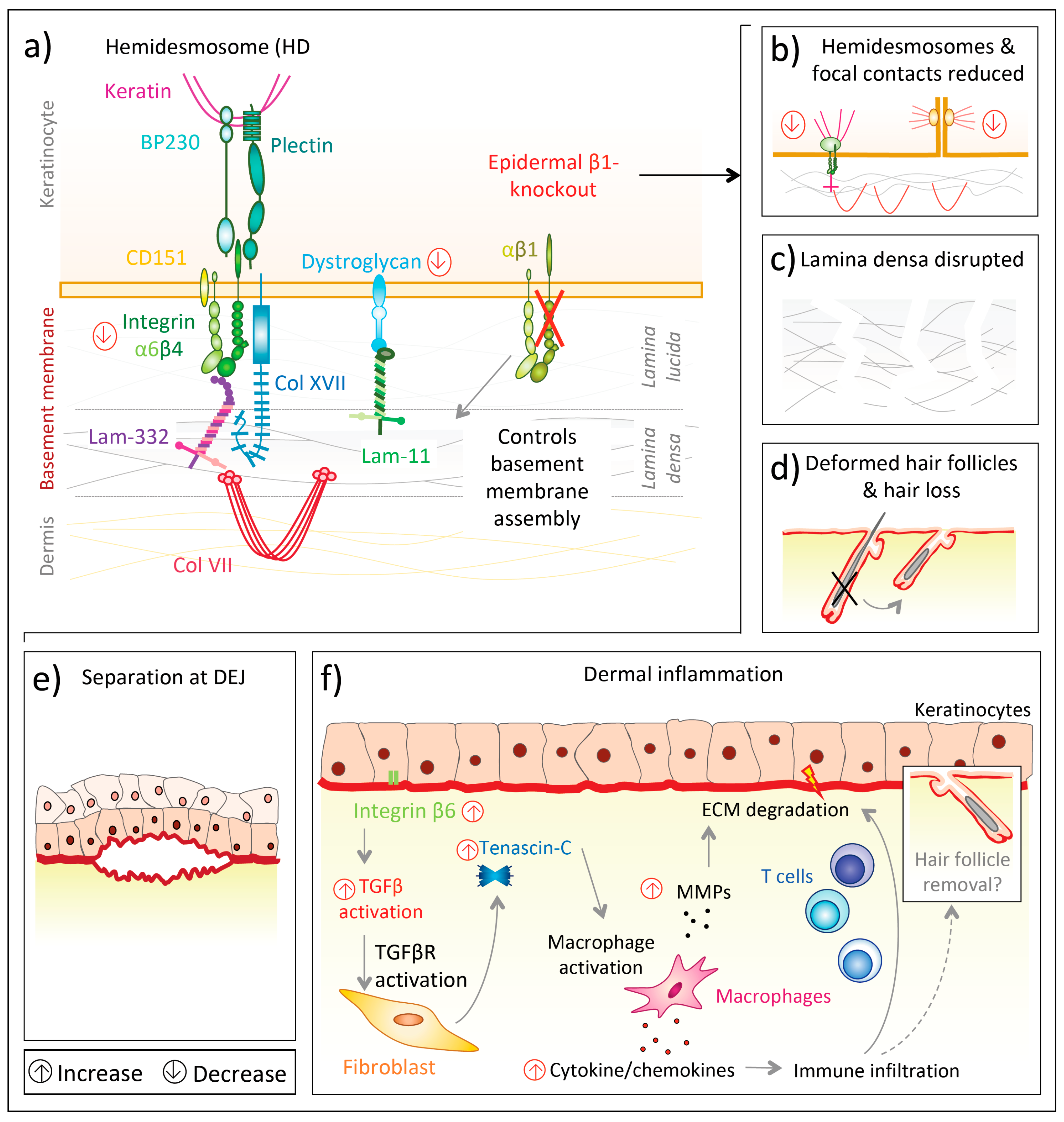{kind=link}
{kind=link}
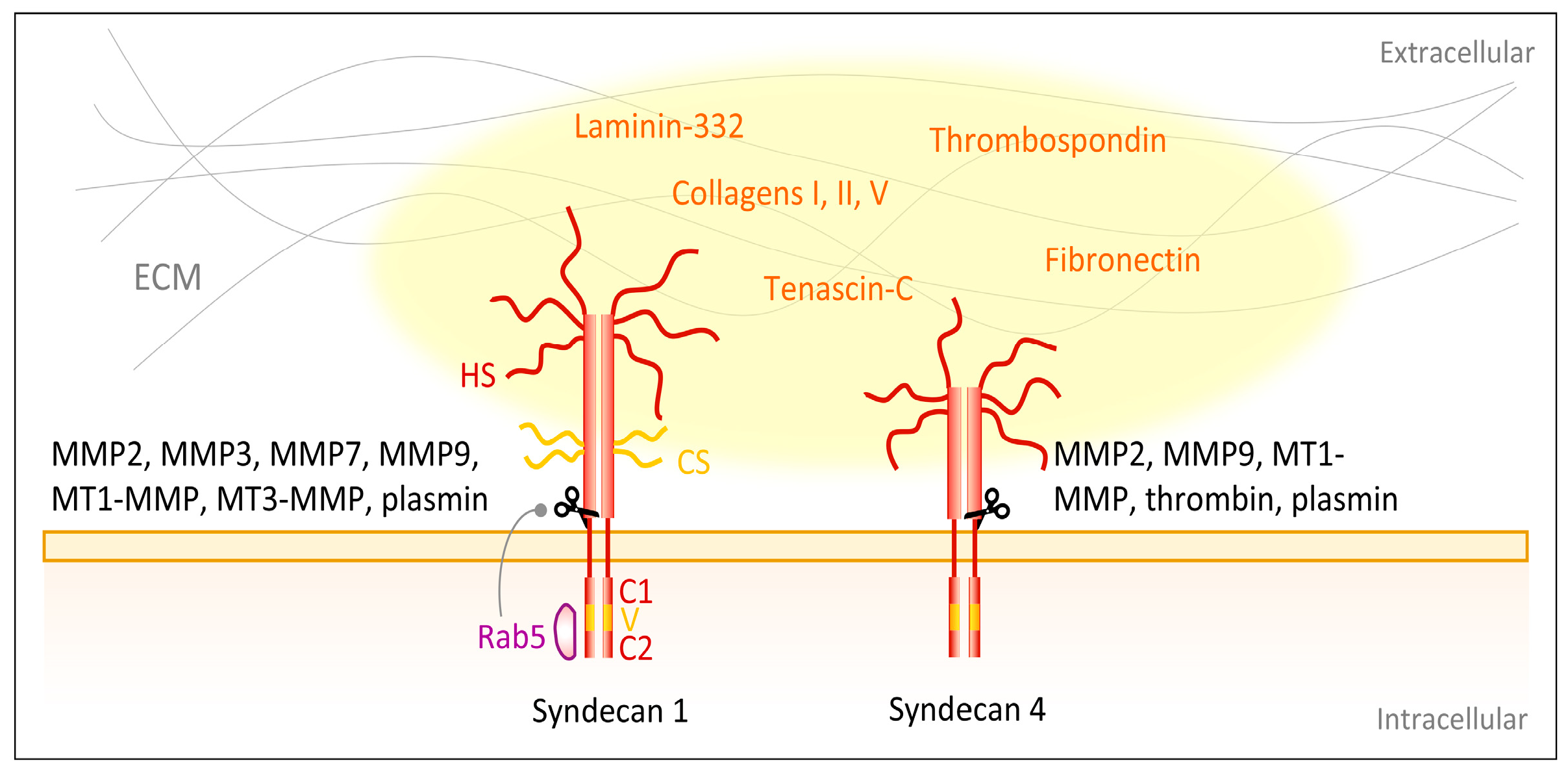{kind=link}
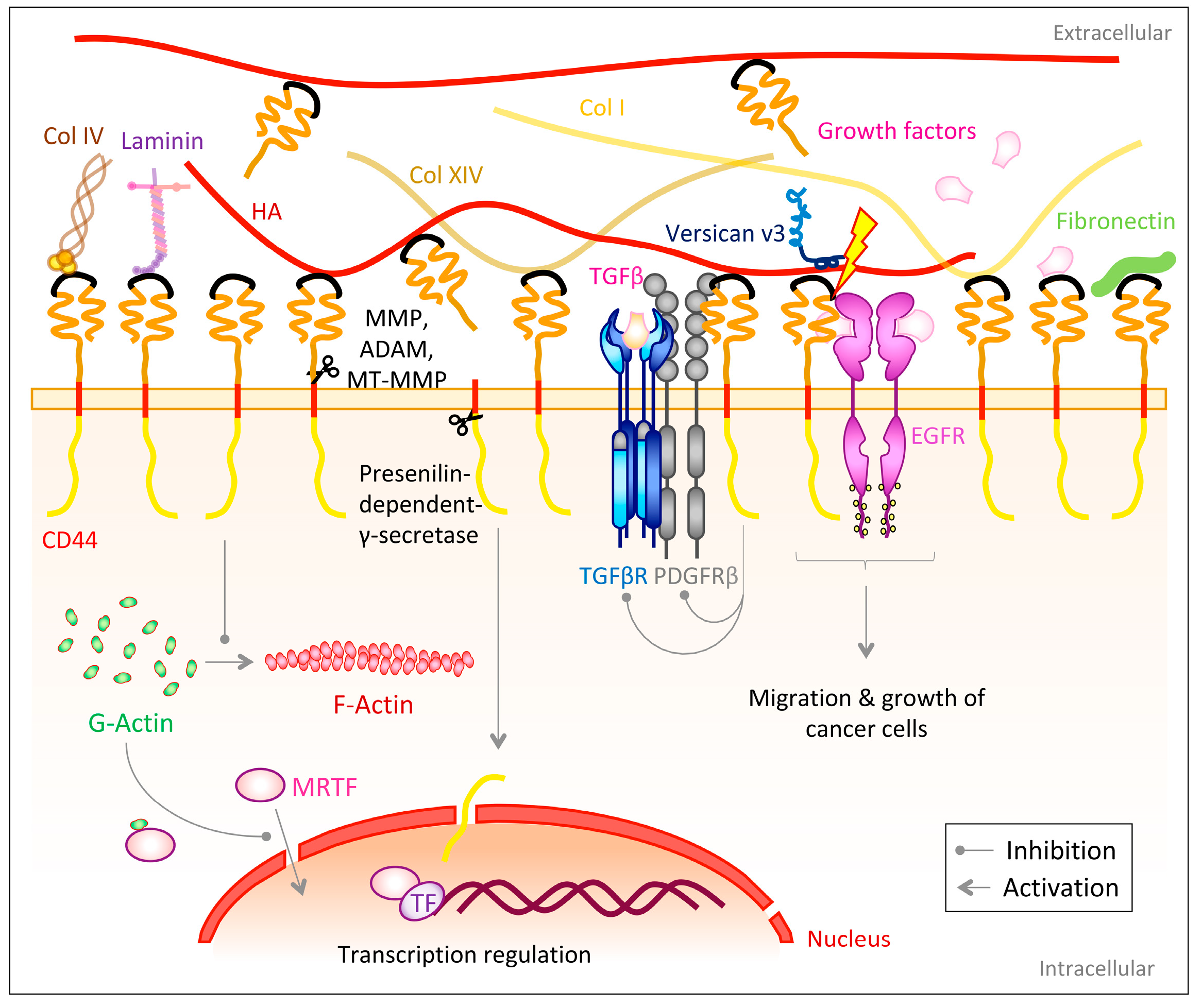{kind=link}
{kind=link}
Abstract
1. Introduction
2. Integrins
2.1. Integrin α6β4
2.2. Integrins Containing the β1 Subunit
2.3. Integrins Containing the αV Subunit
3. Proteoglycans
3.1. Syndecans

3.2. CD44
4. Growth Factor Receptors
4.1. TGFβR
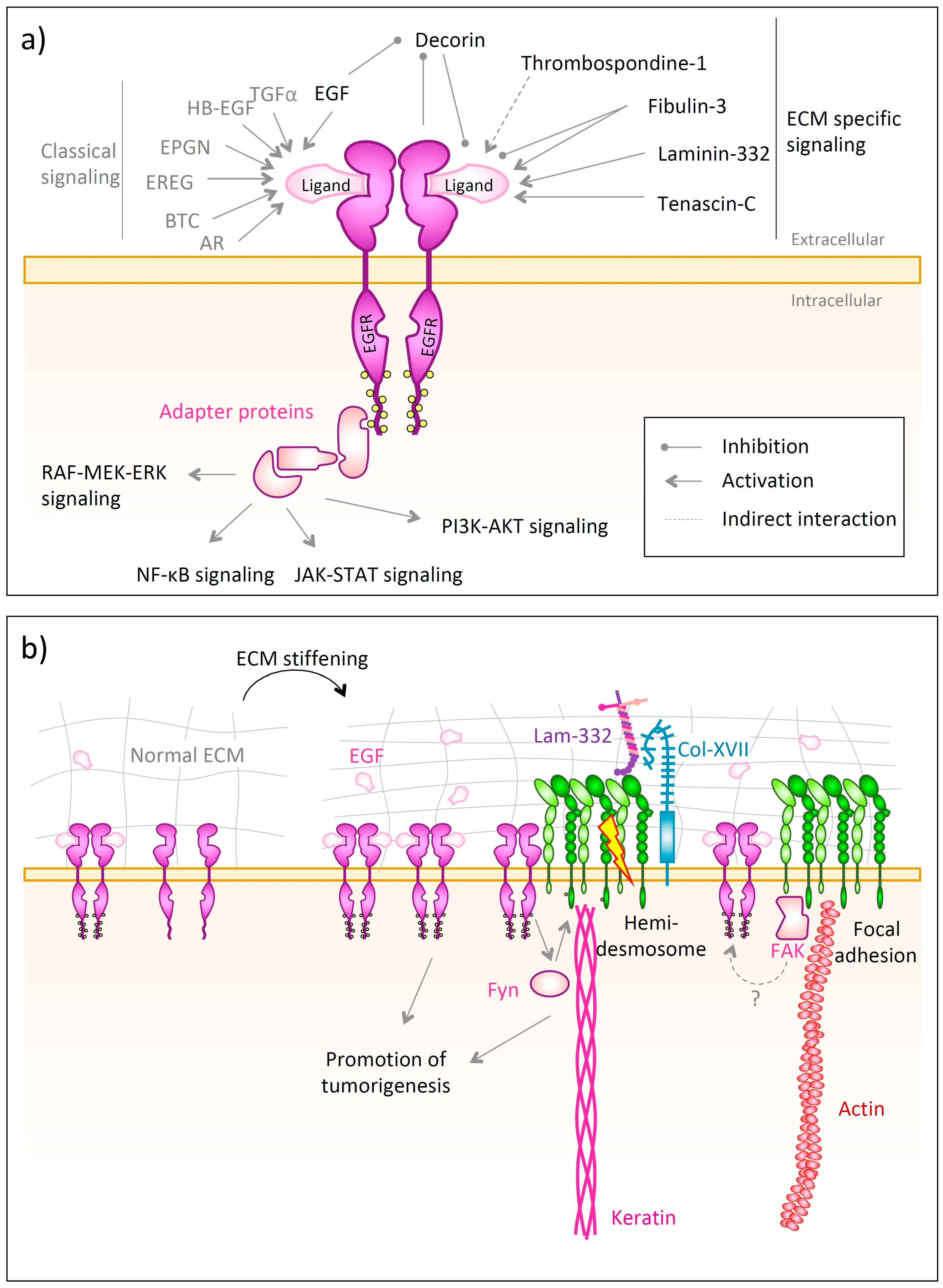
4.2. EGFR
5. Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Nestle, F.O.; Di Meglio, P.; Qin, J.-Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef]
- Yadav, N.; Parveen, S.; Chakravarty, S.; Banerjee, M. Skin anatomy and morphology. In Skin Aging & Cancer: Ambient UV-R Exposure; Dwivedi, A., Agarwal, N., Ray, L., Tripathi, A.K., Eds.; Springer: Singapore, 2019; pp. 1–10. ISBN 978-981-13-2541-0. [Google Scholar]
- Abdo, J.M.; Sopko, N.A.; Milner, S.M. The applied anatomy of human skin: A model for regeneration. Wound Med. 2020, 28, 100179. [Google Scholar] [CrossRef]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular matrix and dermal fibroblast function in the healing wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef]
- Nyström, A.; Bruckner-Tuderman, L. Matrix molecules and skin biology. Semin. Cell Dev. Biol. 2019, 89, 136–146. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Theocharis, A.D.; Neill, T.; Karamanos, N.K. Complexity of matrix phenotypes. Matrix Biol. Plus 2020, 6–7, 100038. [Google Scholar] [CrossRef]
- Uitto, J.; Has, C.; Vahidnezhad, H.; Youssefian, L.; Bruckner-Tuderman, L. Molecular pathology of the basement membrane zone in heritable blistering diseases: The paradigm of epidermolysis bullosa. Matrix Biol. 2017, 57–58, 76–85. [Google Scholar] [CrossRef]
- Has, C.; Nyström, A. Chapter four—Epidermal basement membrane in health and disease. In Current Topics in Membranes: Basement Membranes; Miner, J.H., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 117–170. ISBN 1063-5823. [Google Scholar]
- Kerr, B.A.; Byzova, T.V. Integrin alpha v (ITGAV). In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 2634–2645. ISBN 978-3-319-67199-4. [Google Scholar]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef]
- Hegde, S.; Raghavan, S. A skin-depth analysis of integrins: Role of the integrin network in health and disease. Cell Commun. Adhes. 2013, 20, 155–169. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Schiller, H.B.; Hermann, M.-R.; Polleux, J.; Vignaud, T.; Zanivan, S.; Friedel, C.C.; Sun, Z.; Raducanu, A.; Gottschalk, K.-E.; Théry, M.; et al. β1- and αv-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nat. Cell Biol. 2013, 15, 625–636. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef]
- Goodman, S.R. Chapter 6—Cell adhesion and the extracellular matrix. In Medical Cell Biology, 3rd ed.; Goodman, S.R., Ed.; Academic Press: Cambridge, MA, USA, 2008; pp. 191–225. ISBN 978-0-12-370458-0. [Google Scholar]
- Tiwari, S.; Askari, J.A.; Humphries, M.J.; Bulleid, N.J. Divalent cations regulate the folding and activation status of integrins during their intracellular trafficking. J. Cell Sci. 2011, 124, 1672–1680. [Google Scholar] [CrossRef]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Danen, E.H.J. Integrins: An overview of structural and functional aspects. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Tucker, R.P.; Chiquet-Ehrismann, R. Tenascin-C: Its functions as an integrin ligand. Int. J. Biochem. Cell Biol. 2015, 65, 165–168. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef]
- Knight, C.G.; Morton, L.F.; Onley, D.J.; Peachey, A.R.; Messent, A.J.; Smethurst, P.A.; Tuckwell, D.S.; Farndale, R.W.; Barnes, M.J. Identification in collagen type I of an integrin alpha2 beta1-binding site containing an essential GER sequence. J. Biol. Chem. 1998, 273, 33287–33294. [Google Scholar] [CrossRef]
- Rehn, M.; Veikkola, T.; Kukk-Valdre, E.; Nakamura, H.; Ilmonen, M.; Lombardo, C.; Pihlajaniemi, T.; Alitalo, K.; Vuori, K. Interaction of endostatin with integrins implicated in angiogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 1024–1029. [Google Scholar] [CrossRef]
- Woodall, B.P.; Nyström, A.; Iozzo, R.A.; Eble, J.A.; Niland, S.; Krieg, T.; Eckes, B.; Pozzi, A.; Iozzo, R.V. Integrin alpha2beta1 is the required receptor for endorepellin angiostatic activity. J. Biol. Chem. 2008, 283, 2335–2343. [Google Scholar] [CrossRef]
- Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc. Natl. Acad. Sci. USA 2003, 100, 4766–4771. [Google Scholar] [CrossRef]
- Luo, B.-H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef]
- Shimaoka, M.; Xiao, T.; Liu, J.-H.; Yang, Y.; Dong, Y.; Jun, C.-D.; McCormack, A.; Zhang, R.; Joachimiak, A.; Takagi, J.; et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 2003, 112, 99–111. [Google Scholar] [CrossRef]
- Sun, Z.; Costell, M.; Fässler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Schäfer, M.; Mykuliak, V.V.; Ripamonti, M.; Heiser, L.; Weißenbruch, K.; Krübel, S.; Franz, C.M.; Hytönen, V.P.; Wehrle-Haller, B.; et al. Induction of ligand promiscuity of αVβ3 integrin by mechanical force. J. Cell Sci. 2020. [Google Scholar] [CrossRef]
- Baker, E.L.; Zaman, M.H. The biomechanical integrin. J. Biomech. 2010, 43, 38–44. [Google Scholar] [CrossRef]
- Moore, S.W.; Roca-Cusachs, P.; Sheetz, M.P. Stretchy proteins on stretchy substrates: The important elements of integrin-mediated rigidity sensing. Dev. Cell 2010, 19, 194–206. [Google Scholar] [CrossRef]
- Puklin-Faucher, E.; Sheetz, M.P. The mechanical integrin cycle. J. Cell Sci. 2009, 122, 179–186. [Google Scholar] [CrossRef]
- Schwartz, M.A. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol. 2010, 2, a005066. [Google Scholar] [CrossRef]
- Duperret, E.K.; Dahal, A.; Ridky, T.W. Focal-adhesion-independent integrin-alpha v regulation of FAK and c-Myc is necessary for 3D skin formation and tumor invasion. J. Cell Sci. 2015, 128, 3997–4013. [Google Scholar] [CrossRef]
- Duperret, E.K.; Ridky, T.W. Focal adhesion complex proteins in epidermis and squamous cell carcinoma. Cell Cycle 2013, 12, 3272–3285. [Google Scholar] [CrossRef]
- Stutchbury, B.; Atherton, P.; Tsang, R.; Wang, D.-Y.; Ballestrem, C. Distinct focal adhesion protein modules control different aspects of mechanotransduction. J. Cell Sci. 2017, 130, 1612–1624. [Google Scholar] [CrossRef]
- Hu, Y.-L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci. Rep. 2014, 4, 6024. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Guan, J.-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M. Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 2002, 21, 3919–3926. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Increased expression of integrin αvβ3 contributes to the establishment of autocrine TGF-β signaling in scleroderma fibroblasts. J. Immunol. 2005, 175, 7708–7718. [Google Scholar] [CrossRef]
- Phillips, R.J.; Aplin, J.D.; Lake, B.D. Antigenic expression of integrin alpha 6 beta 4 in junctional epidermolysis bullosa. Histopathology 1994, 24, 571–576. [Google Scholar] [CrossRef]
- Brown, T.A.; Gil, S.G.; Sybert, V.P.; Lestringant, G.G.; Tadini, G.; Caputo, R.; Carter, W.G. Defective integrin alpha 6 beta 4 expression in the skin of patients with junctional epidermolysis bullosa and pyloric atresia. J. Investig. Dermatol. 1996, 107, 384–391. [Google Scholar] [CrossRef]
- Niessen, C.M.; van der Raaij-Helmer, M.H.; Hulsman, E.H.; van der Neut, R.; Jonkman, M.F.; Sonnenberg, A. Deficiency of the integrin beta 4 subunit in junctional epidermolysis bullosa with pyloric atresia: Consequences for hemidesmosome formation and adhesion properties. J. Cell Sci. 1996, 109 Pt 7, 1695–1706. [Google Scholar]
- Nishiuchi, R.; Takagi, J.; Hayashi, M.; Ido, H.; Yagi, Y.; Sanzen, N.; Tsuji, T.; Yamada, M.; Sekiguchi, K. Ligand-binding specificities of laminin-binding integrins: A comprehensive survey of laminin-integrin interactions using recombinant alpha3beta1, alpha6beta1, alpha7beta1 and alpha6beta4 integrins. Matrix Biol. 2006, 25, 189–197. [Google Scholar] [CrossRef]
- Spinardi, L.; Ren, Y.L.; Sanders, R.; Giancotti, F.G. The beta 4 subunit cytoplasmic domain mediates the interaction of alpha 6 beta 4 integrin with the cytoskeleton of hemidesmosomes. Mol. Biol. Cell 1993, 4, 871–884. [Google Scholar] [CrossRef] [PubMed]
- Kariya, Y.; Gu, J. N-glycosylation of β4 integrin controls the adhesion and motility of keratinocytes. PLoS ONE 2011, 6, e27084. [Google Scholar] [CrossRef] [PubMed]
- Dans, M.; Gagnoux-Palacios, L.; Blaikie, P.; Klein, S.; Mariotti, A.; Giancotti, F.G. Tyrosine phosphorylation of the beta 4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J. Biol. Chem. 2001, 276, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulos, S.N.; Blaikie, P.; Yoshioka, T.; Guo, W.; Puri, C.; Tacchetti, C.; Giancotti, F.G. Targeted deletion of the integrin beta4 signaling domain suppresses laminin-5-dependent nuclear entry of mitogen-activated protein kinases and NF-kappaB, causing defects in epidermal growth and migration. Mol. Cell Biol. 2005, 25, 6090–6102. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dong, Z.; Zhang, Y.; Miao, J. The roles of integrin β4 in vascular endothelial cells. J. Cell Physiol. 2012, 227, 474–478. [Google Scholar] [CrossRef]
- Walko, G.; Castanon, M.J.; Wiche, G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015, 360, 529–544. [Google Scholar] [CrossRef]
- Aho, S.; Uitto, J. Direct interaction between the intracellular domains of bullous pemphigoid antigen 2 (BP180) and beta 4 integrin, hemidesmosomal components of basal keratinocytes. Biochem. Biophys. Res. Commun. 1998, 243, 694–699. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef]
- Tidman, M.J.; Eady, R.A. Hemidesmosome heterogeneity in junctional epidermolysis bullosa revealed by morphometric analysis. J. Investig. Dermatol. 1986, 86, 51–56. [Google Scholar] [CrossRef]
- Eady, R.A.; McGrath, J.A.; McMillan, J.R. Ultrastructural clues to genetic disorders of skin: The dermal-epidermal junction. J. Investig. Dermatol. 1994, 103, 13S–18S. [Google Scholar] [CrossRef][Green Version]
- Falcioni, R.; Antonini, A.; Nistico, P.; Di Stefano, S.; Crescenzi, M.; Natali, P.G.; Sacchi, A. Alpha 6 beta 4 and alpha 6 beta 1 integrins associate with ErbB-2 in human carcinoma cell lines. Exp. Cell Res. 1997, 236, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, A.; Kedeshian, P.A.; Dans, M.; Curatola, A.M.; Gagnoux-Palacios, L.; Giancotti, F.G. EGF-R signaling through Fyn kinase disrupts the function of integrin α6β4 at hemidesmosomes: Role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 2001, 155, 447–458. [Google Scholar] [CrossRef]
- Gagnoux-Palacios, L.; Dans, M.; van’t Hof, W.; Mariotti, A.; Pepe, A.; Meneguzzi, G.; Resh, M.D.; Giancotti, F.G. Compartmentalization of integrin alpha6beta4 signaling in lipid rafts. J. Cell Biol. 2003, 162, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Tsubota, Y.; Hashimoto, J.; Kariya, Y.; Miyazaki, K. The short arm of laminin gamma2 chain of laminin-5 (laminin-332) binds syndecan-1 and regulates cellular adhesion and migration by suppressing phosphorylation of integrin beta4 chain. Mol. Biol. Cell 2007, 18, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Mainiero, F.; Murgia, C.; Wary, K.K.; Curatola, A.M.; Pepe, A.; Blumemberg, M.; Westwick, J.K.; Der, C.J.; Giancotti, F.G. The coupling of alpha6beta4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO J. 1997, 16, 2365–2375. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell 2001, 107, 643–654. [Google Scholar] [CrossRef]
- Shaw, L.M.; Rabinovitz, I.; Wang, H.H.; Toker, A.; Mercurio, A.M. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell 1997, 91, 949–960. [Google Scholar] [CrossRef]
- Kligys, K.R.; Wu, Y.; Hopkinson, S.B.; Kaur, S.; Platanias, L.C.; Jones, J.C.R. α6β4 integrin, a master regulator of expression of integrins in human keratinocytes. J. Biol. Chem. 2012, 287, 17975–17984. [Google Scholar] [CrossRef]
- Chung, J.; Bachelder, R.E.; Lipscomb, E.A.; Shaw, L.M.; Mercurio, A.M. Integrin (alpha 6 beta 4) regulation of eIF-4E activity and VEGF translation: A survival mechanism for carcinoma cells. J. Cell Biol. 2002, 158, 165–174. [Google Scholar] [CrossRef]
- Tani, T.; Karttunen, T.; Kiviluoto, T.; Kivilaakso, E.; Burgeson, R.E.; Sipponen, P.; Virtanen, I. Alpha 6 beta 4 integrin and newly deposited laminin-1 and laminin-5 form the adhesion mechanism of gastric carcinoma. Continuous expression of laminins but not that of collagen VII is preserved in invasive parts of the carcinomas: Implications for acquisition of the invading phenotype. Am. J. Pathol. 1996, 149, 781–793. [Google Scholar]
- Kariya, Y.; Oyama, M.; Hashimoto, Y.; Gu, J.; Kariya, Y. beta4-integrin/PI3K signaling promotes tumor progression through the galectin-3-N-glycan complex. Mol. Cancer Res. 2018, 16, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitz, I.; Mercurio, A.M. The integrin alpha 6 beta 4 and the biology of carcinoma. Biochem. Cell Biol. 1996, 74, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, S.W.; Theivakumar, S.; Owens, D.M. Epidermal alpha6beta4 integrin stimulates the influx of immunosuppressive cells during skin tumor promotion. J. Dermatol. Sci. 2012, 66, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.L.; O’Connor, K.L. Clinical significance of the integrin α6β4 in human malignancies. Lab. Investig. 2015, 95, 976–986. [Google Scholar] [CrossRef]
- Kimmel, K.A.; Carey, T.E. Altered expression in squamous carcinoma cells of an orientation restricted epithelial antigen detected by monoclonal antibody A9. Cancer Res. 1986, 46, 3614–3623. [Google Scholar]
- Van Waes, C.; Surh, D.M.; Chen, Z.; Kirby, M.; Rhim, J.S.; Brager, R.; Sessions, R.B.; Poore, J.; Wolf, G.T.; Carey, T.E. Increase in suprabasilar integrin adhesion molecule expression in human epidermal neoplasms accompanies increased proliferation occurring with immortalization and tumor progression. Cancer Res. 1995, 55, 5434–5444. [Google Scholar]
- Wolf, G.T.; Carey, T.E.; Schmaltz, S.P.; McClatchey, K.D.; Poore, J.; Glaser, L.; Hayashida, D.J.; Hsu, S. Altered antigen expression predicts outcome in squamous cell carcinoma of the head and neck. J. Natl. Cancer Inst. 1990, 82, 1566–1572. [Google Scholar] [CrossRef]
- Kariya, Y.; Kariya, Y.; Gu, J. Roles of integrin α6β4 glycosylation in cancer. Cancers 2017, 9, 79. [Google Scholar] [CrossRef]
- Michael, M.; Begum, R.; Chan, G.K.; Whitewood, A.J.; Matthews, D.R.; Goult, B.T.; McGrath, J.A.; Parsons, M. Kindlin-1 regulates epidermal growth factor receptor signaling. J. Investig. Dermatol. 2019, 139, 369–379. [Google Scholar] [CrossRef]
- Brakebusch, C.; Fässler, R. The integrin-actin connection, an eternal love affair. EMBO J. 2003, 22, 2324–2333. [Google Scholar] [CrossRef]
- Rippa, A.L.; Vorotelyak, E.A.; Vasiliev, A.V.; Terskikh, V.V. The role of integrins in the development and homeostasis of the epidermis and skin appendages. Acta Nat. 2013, 5, 22–33. [Google Scholar] [CrossRef]
- Moser, M.; Legate, K.R.; Zent, R.; Fässler, R. The tail of integrins, talin, and kindlins. Science 2009, 324, 895. [Google Scholar] [CrossRef] [PubMed]
- Bouaouina, M.; Lad, Y.; Calderwood, D.A. The N-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate beta1 and beta3 integrins. J. Biol. Chem. 2008, 283, 6118–6125. [Google Scholar] [CrossRef] [PubMed]
- Meves, A.; Geiger, T.; Zanivan, S.; DiGiovanni, J.; Mann, M.; Faessler, R. Beta 1 integrin cytoplasmic tyrosines promote skin tumorigenesis independent of their phosphorylation. Proc. Natl. Acad. Sci. USA 2011, 108, 15213–15218. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Jove, R.; Fässler, R.; Mosher, D.F. Role of the cytoplasmic tyrosines of β1A integrins in transformation by v-src. Proc. Natl. Acad. Sci. USA 2001, 98, 3808. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva, Y.; Giancotti, F.G. Development requires activation but not phosphorylation of β1 integrins. Genes Dev. 2006, 20, 1057–1060. [Google Scholar] [CrossRef]
- Plantefaber, L.C.; Hynes, R.O. Changes in integrin receptors on oncogenically transformed cells. Cell 1989, 56, 281–290. [Google Scholar] [CrossRef]
- Morgner, J.; Ghatak, S.; Jakobi, T.; Dieterich, C.; Aumailley, M.; Wickström, S.A. Integrin-linked kinase regulates the niche of quiescent epidermal stem cells. Nat. Commun. 2015, 6, 8198. [Google Scholar] [CrossRef]
- Fujiwara, H.; Ferreira, M.; Donati, G.; Marciano, D.K.; Linton, J.M.; Sato, Y.; Hartner, A.; Sekiguchi, K.; Reichardt, L.F.; Watt, F.M. The basement membrane of hair follicle stem cells is a muscle cell niche. Cell 2011, 144, 577–589. [Google Scholar] [CrossRef]
- Symington, B.; Takada, Y.; Carter, W. Interaction of integrins α3β1 and α2β1: Potential role in keratinocyte intercellular adhesion. J. Cell Biol. 1993, 120, 523–535. [Google Scholar] [CrossRef]
- Woltersdorf, C.; Bonk, M.; Leitinger, B.; Huhtala, M.; Käpylä, J.; Heino, J.; Gil Girol, C.; Niland, S.; Eble, J.A.; Bruckner, P.; et al. The binding capacity of α1β1-, α2β1- and α10β1-integrins depends on non-collagenous surface macromolecules rather than the collagens in cartilage fibrils. Matrix Biol. 2017, 63, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Zwolanek, D.; Eckes, B.; Niland, S.; Käpylä, J.; Zweers, M.C.; Ishada-Yamamoto, A.; Krieg, T.; Heino, J.; Eble, J.A.; et al. Collagen XXIII, novel ligand for integrin alpha2beta1 in the epidermis. J. Biol. Chem. 2011, 286, 27804–27813. [Google Scholar] [CrossRef] [PubMed]
- Riikonen, T.; Westermarck, J.; Koivisto, L.; Broberg, A.; Kähäri, V.-M.; Heino, J. Integrin α2β1 is a positive regulator of collagenase (MMP-1) and collagen α1(I) gene expression. J. Biol. Chem. 1995, 270, 13548–13552. [Google Scholar] [CrossRef] [PubMed]
- Langholz, O.; Röckel, D.; Mauch, C.; Kozlowska, E.; Bank, I.; Krieg, T.; Eckes, B. Collagen and collagenase gene expression in three-dimensional collagen lattices are differentially regulated by alpha 1 beta 1 and alpha 2 beta 1 integrins. J. Cell Biol. 1995, 131, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Ravanti, L.; Heino, J.; Lopez-Otin, C.; Kahari, V.M. Induction of collagenase-3 (MMP-13) expression in human skin fibroblasts by three-dimensional collagen is mediated by p38 mitogen-activated protein kinase. J. Biol. Chem. 1999, 274, 2446–2455. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Reunanen, H.; Westermarck, J.; Koivisto, L.; Kähäri, V.-M.; Heino, J. Integrin α2β1 mediates isoform-specific activation of p38 and upregulation of collagen gene transcription by a mechanism involving the α2 cytoplasmic tail. J. Cell Biol. 1999, 147, 401–416. [Google Scholar] [CrossRef]
- Ojalill, M.; Parikainen, M.; Rappu, P.; Aalto, E.; Jokinen, J.; Virtanen, N.; Siljamäki, E.; Heino, J. Integrin α2β1 decelerates proliferation, but promotes survival and invasion of prostate cancer cells. Oncotarget 2018, 9, 32435–32447. [Google Scholar] [CrossRef]
- Parks, W.C. What is the α2β1 integrin doing in the epidermis? J. Investig. Dermatol. 2007, 127, 264–266. [Google Scholar] [CrossRef][Green Version]
- Zweers, M.C.; Davidson, J.M.; Pozzi, A.; Hallinger, R.; Janz, K.; Quondamatteo, F.; Leutgeb, B.; Krieg, T.; Eckes, B. Integrin alpha2beta1 is required for regulation of murine wound angiogenesis but is dispensable for reepithelialization. J. Investig. Dermatol. 2007, 127, 467–478. [Google Scholar] [CrossRef]
- DiPersio, C.M.; Hodivala-Dilke, K.M.; Jaenisch, R.; Kreidberg, J.A.; Hynes, R.O. Alpha3beta1 integrin is required for normal development of the epidermal basement membrane. J. Cell Biol. 1997, 137, 729–742. [Google Scholar] [CrossRef]
- Delwel, G.O.; de Melker, A.A.; Hogervorst, F.; Jaspars, L.H.; Fles, D.L.; Kuikman, I.; Lindblom, A.; Paulsson, M.; Timpl, R.; Sonnenberg, A. Distinct and overlapping ligand specificities of the alpha 3A beta 1 and alpha 6A beta 1 integrins: Recognition of laminin isoforms. Mol. Biol. Cell 1994, 5, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, J.; Miosge, N. Basement membrane components are key players in specialized extracellular matrices. Cell Mol. Life Sci. 2010, 67, 2879–2895. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Spartà, G.; Kiritsi, D.; Weibel, L.; Moeller, A.; Vega-Warner, V.; Waters, A.; He, Y.; Anikster, Y.; Esser, P.; et al. Integrin α3 mutations with kidney, lung, and skin disease. N. Engl. J. Med. 2012, 366, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Raymond, K.; Kreft, M.; Sachs, N.; Janssen, H.; Sonnenberg, A. Integrin α3β1 inhibits directional migration and wound re-epithelialization in the skin. J. Cell Sci. 2009, 122, 278. [Google Scholar] [CrossRef]
- Hodivala-Dilke, K.M.; DiPersio, C.M.; Kreidberg, J.A.; Hynes, R.O. Novel roles for alpha3beta1 integrin as a regulator of cytoskeletal assembly and as a trans-dominant inhibitor of integrin receptor function in mouse keratinocytes. J. Cell Biol. 1998, 142, 1357–1369. [Google Scholar] [CrossRef]
- Singh, P.; Chen, C.; Pal-Ghosh, S.; Stepp, M.A.; Sheppard, D.; van de Water, L. Loss of integrin alpha9beta1 results in defects in proliferation, causing poor re-epithelialization during cutaneous wound healing. J. Investig. Dermatol. 2009, 129, 217–228. [Google Scholar] [CrossRef]
- Brakebusch, C.; Grose, R.; Quondamatteo, F.; Ramirez, A.; Jorcano, J.L.; Pirro, A.; Svensson, M.; Herken, R.; Sasaki, T.; Timpl, R.; et al. Skin and hair follicle integrity is crucially dependent on beta 1 integrin expression on keratinocytes. EMBO J. 2000, 19, 3990–4003. [Google Scholar] [CrossRef]
- Henry, M.D.; Campbell, K.P. A role for dystroglycan in basement membrane assembly. Cell 1998, 95, 859–870. [Google Scholar] [CrossRef]
- Raghavan, S.; Bauer, C.; Mundschau, G.; Li, Q.; Fuchs, E. Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J. Cell Biol. 2000, 150, 1149–1160. [Google Scholar] [CrossRef]
- Kurbet, A.S.; Hegde, S.; Bhattacharjee, O.; Marepally, S.; Vemula, P.K.; Raghavan, S. Sterile inflammation enhances ECM degradation in integrin beta1 KO embryonic skin. Cell Rep. 2016, 16, 3334–3347. [Google Scholar] [CrossRef]
- Liu, S.; Leask, A. Integrin β1 is required for dermal homeostasis. J. Investig. Dermatol. 2013, 133, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xu, S.-w.; Blumbach, K.; Eastwood, M.; Denton, C.P.; Eckes, B.; Krieg, T.; Abraham, D.J.; Leask, A. Expression of integrin beta1 by fibroblasts is required for tissue repair in vivo. J. Cell Sci. 2010, 123, 3674–3682. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kapoor, M.; Denton, C.P.; Abraham, D.J.; Leask, A. Loss of beta1 integrin in mouse fibroblasts results in resistance to skin scleroderma in a mouse model. Arthritis Rheum. 2009, 60, 2817–2821. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.-N.; Zeltz, C.; Sørensen, I.W.; Barczyk, M.; Carracedo, S.; Hallinger, R.; Niehoff, A.; Eckes, B.; Gullberg, D. Reduced granulation tissue and wound strength in the absence of α11β1 integrin. J. Investig. Dermatol. 2015, 135, 1435–1444. [Google Scholar] [CrossRef]
- Schulz, J.-N.; Plomann, M.; Sengle, G.; Gullberg, D.; Krieg, T.; Eckes, B. New developments on skin fibrosis—Essential signals emanating from the extracellular matrix for the control of myofibroblasts. Matrix Biol. 2018, 68–69, 522–532. [Google Scholar] [CrossRef]
- Schulz, J.-N.; Blumbach, K.; Brunner, G.; Niehoff, A.; Gullberg, D.; Krieg, T.; Eckes, B. Disruption of the b1-integrin-ILK axis attenuates fibrotic reactions. Wound Repair Regen 2014, 22, A96. [Google Scholar]
- Brockbank, E.C.; Bridges, J.; Marshall, C.J.; Sahai, E. Integrin β1 is required for the invasive behaviour but not proliferation of squamous cell carcinoma cells in vivo. Br. J. Cancer 2005, 92, 102–112. [Google Scholar] [CrossRef]
- Martins, V.L.; Caley, M.P.; Moore, K.; Szentpetery, Z.; Marsh, S.T.; Murrell, D.F.; Kim, M.H.; Avari, M.; McGrath, J.A.; Cerio, R.; et al. Suppression of TGFbeta and angiogenesis by type VII collagen in cutaneous SCC. J. Natl. Cancer Inst. 2016. [Google Scholar] [CrossRef]
- Sachs, N.; Secades, P.; van Hulst, L.; Kreft, M.; Song, J.-Y.; Sonnenberg, A. Loss of integrin α3 prevents skin tumor formation by promoting epidermal turnover and depletion of slow-cycling cells. Proc. Natl. Acad. Sci. USA 2012, 109, 21468. [Google Scholar] [CrossRef]
- Xu, Z.; Zou, L.; Ma, G.; Wu, X.; Huang, F.; Feng, T.; Li, S.; Lin, Q.; He, X.; Liu, Z.; et al. Integrin β1 is a critical effector in promoting metastasis and chemo-resistance of esophageal squamous cell carcinoma. Am. J. Cancer Res. 2017, 7, 531–542. [Google Scholar]
- Wang, D.; Muller, S.; Amin, A.R.M.; Huang, D.; Su, L.; Hu, Z.; Rahman, M.A.; Nannapaneni, S.; Koenig, L.; Chen, Z.; et al. The pivotal role of integrin beta1 in metastasis of head and neck squamous cell carcinoma. Clin. Cancer Res. 2012, 18, 4589–4599. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Nakamura, S.; Sasaki, M.; Kurahara, S.; Ikebe, T.; Harada, T.; Shirasuna, K. Expression of integrins in squamous cell carcinoma of the oral cavity. Correlations with tumor invasion and metastasis. Am. J. Clin. Pathol. 1999, 111, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, H.; Mueller, S.C.; Nomizu, M.; Yamada, Y.; Yeh, Y.; Chen, W.-T. Activation of β1 integrin signaling stimulates tyrosine phosphorylation of p190 RhoGAP and membrane-protrusive activities at invadopodia. J. Biol. Chem. 1998, 273, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.C.; Coppolino, M.G. SNARE-dependent interaction of Src, EGFR and β1 integrin regulates invadopodia formation and tumor cell invasion. J. Cell Sci. 2014, 127, 1712. [Google Scholar] [CrossRef]
- Kean, M.J.; Williams, K.C.; Skalski, M.; Myers, D.; Burtnik, A.; Foster, D.; Coppolino, M.G. VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J. Cell Sci. 2009, 122, 4089. [Google Scholar] [CrossRef]
- Yu, X.; Miyamoto, S.; Mekada, E. Integrin alpha 2 beta 1-dependent EGF receptor activation at cell-cell contact sites. J. Cell Sci. 2000, 113 Pt 12, 2139–2147. [Google Scholar]
- Reginato, M.J.; Mills, K.R.; Paulus, J.K.; Lynch, D.K.; Sgroi, D.C.; Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 2003, 5, 733–740. [Google Scholar] [CrossRef]
- Wang, F.; Weaver, V.M.; Petersen, O.W.; Larabell, C.A.; Dedhar, S.; Briand, P.; Lupu, R.; Bissell, M.J. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: A different perspective in epithelial biology. Proc. Natl. Acad. Sci. USA 1998, 95, 14821–14826. [Google Scholar] [CrossRef]
- Eke, I.; Zscheppang, K.; Dickreuter, E.; Hickmann, L.; Mazzeo, E.; Unger, K.; Krause, M.; Cordes, N. Simultaneous β1 integrin-EGFR targeting and radiosensitization of human head and neck cancer. J. Natl. Cancer Inst. 2015. [Google Scholar] [CrossRef]
- Koistinen, P.; Heino, J. The selective regulation of alpha v beta 1 integrin expression is based on the hierarchical formation of alpha v-containing heterodimers. J. Biol. Chem. 2002, 277, 24835–24841. [Google Scholar] [CrossRef]
- Janes, S.M.; Watt, F.M. Switch from alpha v beta 5 to alpha v beta 6 integrin expression protects squamous cell carcinomas from anoikis. J. Cell Biol. 2004, 166, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, R.; Bodorova, J.; Ye, S.; Hemler, M.E. A study of the structure, function and distribution of beta 5 integrins using novel anti-beta 5 monoclonal antibodies. J. Cell Sci. 1993, 105 Pt 1, 101–111. [Google Scholar]
- Longmate, W.M.; DiPersio, C.M. Integrin regulation of epidermal functions in wounds. Adv. Wound Care 2014, 3, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef]
- Wipff, P.-J.; Hinz, B. Integrins and the activation of latent transforming growth factor beta1—An intimate relationship. Eur. J. Cell Biol. 2008, 87, 601–615. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, J.; Dong, X.; Shi, M.; Lu, C.; Springer, T.A. GARP regulates the bioavailability and activation of TGFβ. Mol. Biol. Cell 2012, 23, 1129–1139. [Google Scholar] [CrossRef]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural biology and evolution of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2016. [Google Scholar] [CrossRef]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP–dependent activation of TGF-β1. J. Cell Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef]
- Scaffidi, A.K.; Petrovic, N.; Moodley, Y.P.; Fogel-Petrovic, M.; Kroeger, K.M.; Seeber, R.M.; Eidne, K.A.; Thompson, P.J.; Knight, D.A. Alpha(v)beta(3) integrin interacts with the transforming growth factor beta (TGFbeta) type II receptor to potentiate the proliferative effects of TGFbeta1 in living human lung fibroblasts. J. Biol. Chem. 2004, 279, 37726–37733. [Google Scholar] [CrossRef] [PubMed]
- Galliher, A.J.; Schiemann, W.P. Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006, 8, R42. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Tamaki, K. Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am. J. Pathol. 2006, 168, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Barlati, S.; Colombi, M. FAK-independent alphavbeta3 integrin-EGFR complexes rescue from anoikis matrix-defective fibroblasts. Biochim. Biophys. Acta 2008, 1783, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Häkkinen, L.; Koivisto, L.; Gardner, H.; Saarialho-Kere, U.; Carroll, J.M.; Lakso, M.; Rauvala, H.; Laato, M.; Heino, J.; Larjava, H. Increased expression of beta6-integrin in skin leads to spontaneous development of chronic wounds. Am. J. Pathol. 2004, 164, 229–242. [Google Scholar] [CrossRef]
- AlDahlawi, S.; Eslami, A.; Hakkinen, L.; Larjava, H.S. The alphavbeta6 integrin plays a role in compromised epidermal wound healing. Wound Repair Regen 2006, 14, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Duperret, E.K.; Natale, C.A.; Monteleon, C.; Dahal, A.; Ridky, T.W. The integrin αv-TGFβ signaling axis is necessary for epidermal proliferation during cutaneous wound healing. Cell Cycle 2016, 15, 2077–2086. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Storm, G.; Prakash, J. Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev. 2018, 129, 37–53. [Google Scholar] [CrossRef]
- Jakhu, H.; Gill, G.; Singh, A. Role of integrins in wound repair and its periodontal implications. J. Oral Biol. Craniofac. Res. 2018, 8, 122–125. [Google Scholar] [CrossRef]
- Thomas, G.J.; Lewis, M.P.; Whawell, S.A.; Russell, A.; Sheppard, D.; Hart, I.R.; Speight, P.M.; Marshall, J.F. Expression of the αvβ6 integrin promotes migration and invasion in squamous carcinoma cells. J. Investig. Dermatol. 2001, 117, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Ramos, D.M.; But, M.; Regezi, J.; Schmidt, B.L.; Atakilit, A.; Dang, D.; Ellis, D.; Jordan, R.; Li, X. Expression of integrin beta 6 enhances invasive behavior in oral squamous cell carcinoma. Matrix Biol. 2002, 21, 297–307. [Google Scholar] [CrossRef]
- Ahmed, N.; Niu, J.; Dorahy, D.J.; Gu, X.; Andrews, S.; Meldrum, C.J.; Scott, R.J.; Baker, M.S.; Macreadie, I.G.; Agrez, M.V. Direct integrin alphavbeta6-ERK binding: Implications for tumour growth. Oncogene 2002, 21, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Pomin, V.H.; Mulloy, B. Glycosaminoglycans and proteoglycans. Pharmaceuticals 2018, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Bartlett, A.H.; Hayashida, K.; Park, P.W. Molecular and cellular mechanisms of syndecans in tissue injury and inflammation. Mol. Cells 2007, 24, 153–166. [Google Scholar]
- Deepa, S.S.; Yamada, S.; Zako, M.; Goldberger, O.; Sugahara, K. Chondroitin sulfate chains on syndecan-1 and syndecan-4 from normal murine mammary gland epithelial cells are structurally and functionally distinct and cooperate with heparan sulfate chains to bind growth factors. A novel function to control binding of midkine, pleiotrophin, and basic fibroblast growth factor. J. Biol. Chem. 2004, 279, 37368–37376. [Google Scholar] [CrossRef]
- Rapraeger, A.C. Molecular interactions of syndecans during development. Semin. Cell Dev. Biol. 2001, 12, 107–116. [Google Scholar] [CrossRef]
- Kasza, I.; Suh, Y.; Wollny, D.; Clark, R.J.; Roopra, A.; Colman, R.J.; MacDougald, O.A.; Shedd, T.A.; Nelson, D.W.; Yen, M.-I.; et al. Syndecan-1 is required to maintain intradermal fat and prevent cold stress. PLoS Genet. 2014, 10, e1004514. [Google Scholar] [CrossRef]
- Dews, I.C.; Mackenzie, K.R. Transmembrane domains of the syndecan family of growth factor coreceptors display a hierarchy of homotypic and heterotypic interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20782–20787. [Google Scholar] [CrossRef]
- Choi, Y.; Chung, H.; Jung, H.; Couchman, J.R.; Oh, E.-S. Syndecans as cell surface receptors: Unique structure equates with functional diversity. Matrix Biol. 2011, 30, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Lee, E.; Kwon, S.; Park, H.; Yi, J.Y.; Kim, S.; Han, I.-O.; Yun, Y.; Oh, E.-S. Transmembrane domain-induced oligomerization is crucial for the functions of syndecan-2 and syndecan-4. J. Biol. Chem. 2005, 280, 42573–42579. [Google Scholar] [CrossRef] [PubMed]
- Carey, D.J.; Bendt, K.M.; Stahl, R.C. The cytoplasmic domain of syndecan-1 is required for cytoskeleton association but not detergent insolubility. Identification of essential cytoplasmic domain residues. J. Biol. Chem. 1996, 271, 15253–15260. [Google Scholar] [CrossRef]
- Elfenbein, A.; Simons, M. Syndecan-4 signaling at a glance. J. Cell Sci. 2013, 126, 3799. [Google Scholar] [CrossRef] [PubMed]
- Ott, V.L.; Rapraeger, A.C. Tyrosine phosphorylation of syndecan-1 and -4 cytoplasmic domains in adherent B82 fibroblasts. J. Biol. Chem. 1998, 273, 35291–35298. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, P.; Colles, S.M.; Fox, P.L.; Graham, L.M. Protein kinase Cδ—Dependent phosphorylation of syndecan-4 regulates cell migration. Circ. Res. 2005, 97, 674–681. [Google Scholar] [CrossRef]
- Woods, A. Syndecans: Transmembrane modulators of adhesion and matrix assembly. J. Clin. Investig. 2001, 107, 935–941. [Google Scholar] [CrossRef]
- Couchman, J.R. Syndecans: Proteoglycan regulators of cell-surface microdomains? Nat. Rev. Mol. Cell Biol. 2003, 4, 926–937. [Google Scholar] [CrossRef]
- Sulka, B.; Lortat-Jacob, H.; Terreux, R.; Letourneur, F.; Rousselle, P. Tyrosine dephosphorylation of the syndecan-1 PDZ binding domain regulates syntenin-1 recruitment. J. Biol. Chem. 2009, 284, 10659–10671. [Google Scholar] [CrossRef]
- Choi, Y.; Yun, J.-H.; Yoo, J.; Lee, I.; Kim, H.; Son, H.-N.; Kim, I.-S.; Yoon, H.S.; Zimmermann, P.; Couchman, J.R.; et al. New structural insight of C-terminal region of syntenin-1, enhancing the molecular dimerization and inhibitory function related on syndecan-4 signaling. Sci. Rep. 2016, 6, 36818. [Google Scholar] [CrossRef]
- Friand, V.; David, G.; Zimmermann, P. Syntenin and syndecan in the biogenesis of exosomes. Biol. Cell 2015, 107, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.; Zhang, Z.; Degeest, G.; Mortier, E.; Leenaerts, I.; Coomans, C.; Schulz, J.; N’Kuli, F.; Courtoy, P.J.; David, G. Syndecan recyling is controlled by syntenin-PIP2 interaction and Arf6. Dev. Cell 2005, 9, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Rapraeger, A.; Jalkanen, M.; Endo, E.; Koda, J.; Bernfield, M. The cell surface proteoglycan from mouse mammary epithelial cells bears chondroitin sulfate and heparan sulfate glycosaminoglycans. J. Biol. Chem. 1985, 260, 11046–11052. [Google Scholar] [PubMed]
- Zhang, Y.; McKown, R.L.; Raab, R.W.; Rapraeger, A.C.; Laurie, G.W. Focus on molecules: Syndecan-1. Exp. Eye Res. 2011, 93, 329–330. [Google Scholar] [CrossRef]
- Shworak, N.W.; Shirakawa, M.; Mulligan, R.C.; Rosenberg, R.D. Characterization of ryudocan glycosaminoglycan acceptor sites. J. Biol. Chem. 1994, 269, 21204–21214. [Google Scholar]
- Gallo, R.L. Proteoglycans and cutaneous vascular defense and repair. J. Investig. Dermatol. Symp. Proc. 2000, 5, 55–60. [Google Scholar] [CrossRef]
- Fears, C.Y.; Gladson, C.L.; Woods, A. Syndecan-2 is expressed in the microvasculature of gliomas and regulates angiogenic processes in microvascular endothelial cells. J. Biol. Chem. 2006, 281, 14533–14536. [Google Scholar] [CrossRef]
- Manon-Jensen, T.; Multhaupt, H.A.B.; Couchman, J.R. Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. FEBS J. 2013, 280, 2320–2331. [Google Scholar] [CrossRef]
- Endo, K.; Takino, T.; Miyamori, H.; Kinsen, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 2003, 278, 40764–40770. [Google Scholar] [CrossRef]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef]
- Hayashida, K.; Stahl, P.D.; Park, P.W. Syndecan-1 ectodomain shedding is regulated by the small GTPase Rab5. J. Biol. Chem. 2008, 283, 35435–35444. [Google Scholar] [CrossRef]
- Fitzgerald, M.L.; Wang, Z.; Park, P.W.; Murphy, G.; Bernfield, M. Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell Biol. 2000, 148, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Elenius, V.; Gotte, M.; Reizes, O.; Elenius, K.; Bernfield, M. Inhibition by the soluble syndecan-1 ectodomains delays wound repair in mice overexpressing syndecan-1. J. Biol. Chem. 2004, 279, 41928–41935. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.V.; Fitzgerald, M.L.; Bernfield, M. Regulated shedding of syndecan-1 and -4 ectodomains by thrombin and growth factor receptor activation. J. Biol. Chem. 1997, 272, 14713–14720. [Google Scholar] [CrossRef] [PubMed]
- Park, P.W.; Pier, G.B.; Preston, M.J.; Goldberger, O.; Fitzgerald, M.L.; Bernfield, M. Syndecan-1 shedding is enhanced by LasA, a secreted virulence factor of Pseudomonas aeruginosa. J. Biol. Chem. 2000, 275, 3057–3064. [Google Scholar] [CrossRef]
- Park, P.W.; Pier, G.B.; Hinkes, M.T.; Bernfield, M. Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature 2001, 411, 98–102. [Google Scholar] [CrossRef]
- Hadigal, S.; Koganti, R.; Yadavalli, T.; Agelidis, A.; Suryawanshi, R.; Shukla, D. Heparanase-regulated syndecan-1 shedding facilitates herpes simplex virus 1 egress. J. Virol. 2020. [Google Scholar] [CrossRef]
- Yang, Y.; Yaccoby, S.; Liu, W.; Langford, J.K.; Pumphrey, C.Y.; Theus, A.; Epstein, J.; Sanderson, R.D. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood 2002, 100, 610–617. [Google Scholar] [CrossRef]
- Mahtouk, K.; Hose, D.; Raynaud, P.; Hundemer, M.; Jourdan, M.; Jourdan, E.; Pantesco, V.; Baudard, M.; de Vos, J.; Larroque, M.; et al. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood 2007, 109, 4914–4923. [Google Scholar] [CrossRef]
- Ramani, V.C.; Pruett, P.S.; Thompson, C.A.; DeLucas, L.D.; Sanderson, R.D. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J. Biol. Chem. 2012, 287, 9952–9961. [Google Scholar] [CrossRef]
- Gallo, R.; Kim, C.; Kokenyesi, R.; Scott Adzick, N.; Bernfield, M. Syndecans-1 and -4 are induced during wound repair of neonatal but not fetal skin. J. Investig. Dermatol. 1996, 107, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Oksala, O.; Salo, T.; Tammi, R.; Hakkinen, L.; Jalkanen, M.; Inki, P.; Larjava, H. Expression of proteoglycans and hyaluronan during wound healing. J. Histochem. Cytochem. 1995, 43, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, P.; Beck, K. Laminin 332 processing impacts cellular behavior. Cell Adh. Migr. 2013, 7, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Stepp, M.A.; Pal-Ghosh, S.; Tadvalkar, G.; Rajjoub, L.; Jurjus, R.A.; Gerdes, M.; Ryscavage, A.; Cataisson, C.; Shukla, A.; Yuspa, S.H. Loss of syndecan-1 is associated with malignant conversion in skin carcinogenesis. Mol. Carcinog. 2010, 49, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, H.M.; Edwards, S.N.; Jalkanen, M. Analysis of transport and targeting of syndecan-1: Effect of cytoplasmic tail deletions. Mol. Biol. Cell 1994, 5, 1325–1339. [Google Scholar] [CrossRef]
- Haubeck, H.-D. Syndecane. In Lexikon der Medizinischen Laboratoriumsdiagnostik; Gressner, A.M., Arndt, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 2246–2247. ISBN 978-3-662-48986-4. [Google Scholar]
- Jalkanen, M.; Rapraeger, A.; Bernfield, M. Mouse mammary epithelial cells produce basement membrane and cell surface heparan sulfate proteoglycans containing distinct core proteins. J. Cell Biol. 1988, 106, 953–962. [Google Scholar] [CrossRef]
- Stepp, M.A.; Gibson, H.E.; Gala, P.H.; Iglesia, D.D.S.; Pajoohesh-Ganji, A.; Pal-Ghosh, S.; Brown, M.; Aquino, C.; Schwartz, A.M.; Goldberger, O.; et al. Defects in keratinocyte activation during wound healing in the syndecan-1-deficient mouse. J. Cell Sci. 2002, 115, 4517–4531. [Google Scholar] [CrossRef]
- Lee, J.-h.; Park, H.; Chung, H.; Choi, S.; Kim, Y.; Yoo, H.; Kim, T.-Y.; Hann, H.-J.; Seong, I.; Kim, J.; et al. Syndecan-2 regulates the migratory potential of melanoma cells. J. Biol. Chem. 2009, 284, 27167–27175. [Google Scholar] [CrossRef]
- Ruiz, X.D.; Mlakar, L.R.; Yamaguchi, Y.; Su, Y.; Larregina, A.T.; Pilewski, J.M.; Feghali-Bostwick, C.A. Syndecan-2 is a novel target of insulin-like growth factor binding protein-3 and is over-expressed in fibrosis. PLoS ONE 2012, 7, e43049. [Google Scholar] [CrossRef]
- Bagabir, R.A.; Syed, F.; Shenjere, P.; Paus, R.; Bayat, A. Identification of a potential molecular diagnostic biomarker in keloid disease: Syndecan-1 (CD138) is overexpressed in keloid scar tissue. J. Investig. Dermatol. 2016, 136, 2319–2323. [Google Scholar] [CrossRef]
- Bayer-Garner, I.; Dilday, B.; Sanderson, R.; Smoller, B. Acantholysis and spongiosis are associated with loss of syndecan-1 expression. J. Cutan. Pathol. 2001, 28, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Søgaard, P.; Multhaupt, H.A.B.; Pataki, C.; Okina, E.; Xian, X.; Pedersen, M.E.; Stevens, T.; Griesbeck, O.; Park, P.W.; et al. Transmembrane proteoglycans control stretch-activated channels to set cytosolic calcium levels. J. Cell Biol. 2015, 210, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S. Syndecans in inflammation at a glance. Front. Immunol. 2020, 11, 227. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K.; Sadasivam, M.; Archer, N.K.; Miller, R.J.; Dillen, C.A.; Ravipati, A.; Park, P.W.; Chakravarti, S.; Miller, L.S.; Hamad, A.R.A. Syndecan-1 regulates psoriasiform dermatitis by controlling homeostasis of IL-17-producing γδ T cells. J. Immunol. 2018, 201, 1651–1661. [Google Scholar] [CrossRef]
- Elenius, K.; Vainio, S.; Laato, M.; Salmivirta, M.; Thesleff, I.; Jalkanen, M. Induced expression of syndecan in healing wounds. J. Cell Biol. 1991, 114, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Johnston, D.R.; Goldberger, O.; Park, P.W. Syndecan-1 expression in epithelial cells is induced by transforming growth factor beta through a PKA-dependent pathway. J. Biol. Chem. 2006, 281, 24365–24374. [Google Scholar] [CrossRef]
- Stepp, M.A.; Liu, Y.; Pal-Ghosh, S.; Jurjus, R.A.; Tadvalkar, G.; Sekaran, A.; Losicco, K.; Jiang, L.; Larsen, M.; Li, L.; et al. Reduced migration, altered matrix and enhanced TGFbeta1 signaling are signatures of mouse keratinocytes lacking Sdc1. J. Cell Sci. 2007, 120, 2851–2863. [Google Scholar] [CrossRef] [PubMed]
- Echtermeyer, F.; Streit, M.; Wilcox-Adelman, S.; Saoncella, S.; Denhez, F.; Detmar, M.; Goetinck, P. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J. Clin. Investig. 2001, 107, R9–R14. [Google Scholar] [CrossRef]
- Carulli, S.; Beck, K.; Dayan, G.; Boulesteix, S.; Lortat-Jacob, H.; Rousselle, P. Cell surface proteoglycans syndecan-1 and -4 bind overlapping but distinct sites in laminin α3 LG45 protein domain. J. Biol. Chem. 2012, 287, 12204–12216. [Google Scholar] [CrossRef]
- Okamoto, O.; Bachy, S.; Odenthal, U.; Bernaud, J.; Rigal, D.; Lortat-Jacob, H.; Smyth, N.; Rousselle, P. Normal human keratinocytes bind to the alpha3LG4/5 domain of unprocessed laminin-5 through the receptor syndecan-1. J. Biol. Chem. 2003, 278, 44168–44177. [Google Scholar] [CrossRef]
- Tsubota, Y.; Yasuda, C.; Kariya, Y.; Ogawa, T.; Hirosaki, T.; Mizushima, H.; Miyazaki, K. Regulation of biological activity and matrix assembly of laminin-5 by COOH-terminal, LG4-5 domain of alpha3 chain. J. Biol. Chem. 2005, 280, 14370–14377. [Google Scholar] [CrossRef]
- Pulido, D.; Hussain, S.-A.; Hohenester, E. Crystal structure of the heterotrimeric integrin-binding region of laminin-111. Structure 2017, 25, 530–535. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bachy, S.; Letourneur, F.; Rousselle, P. Syndecan-1 interaction with the LG4/5 domain in laminin-332 is essential for keratinocyte migration. J. Cell. Physiol. 2008, 214, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.E.; Shen, X.-N.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B. MDA-9/Syntenin (SDCBP) modulates small GTPases RhoA and Cdc42 via transforming growth factor β1 to enhance epithelial-mesenchymal transition in breast cancer. Oncotarget 2016, 7, 80175–80189. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Leavitt, L.; Ramaswamy, R.; Rapraeger, A.C. Interaction of syndecan and alpha6beta4 integrin cytoplasmic domains: Regulation of ErbB2-mediated integrin activation. J. Biol. Chem. 2010, 285, 13569–13579. [Google Scholar] [CrossRef]
- Wang, H.; Jin, H.; Beauvais, D.M.; Rapraeger, A.C. Cytoplasmic domain interactions of syndecan-1 and syndecan-4 with α6β4 integrin mediate human epidermal growth factor receptor (HER1 and HER2)-dependent motility and survival. J. Biol. Chem. 2014, 289, 30318–30332. [Google Scholar] [CrossRef]
- Wang, H.; Jin, H.; Rapraeger, A.C. Syndecan-1 and syndecan-4 capture epidermal growth factor receptor family members and the alpha3beta1 integrin via binding sites in their ectodomains: Novel synstatins prevent kinase capture and inhibit alpha6beta4-integrin-dependent epithelial cell motility. J. Biol. Chem. 2015, 290, 26103–26113. [Google Scholar] [CrossRef]
- Koda, J.E.; Rapraeger, A.; Bernfield, M. Heparan sulfate proteoglycans from mouse mammary epithelial cells. Cell surface proteoglycan as a receptor for interstitial collagens. J. Biol. Chem. 1985, 260, 8157–8162. [Google Scholar]
- Sun, X.; Mosher, D.F.; Rapraeger, A. Heparan sulfate-mediated binding of epithelial cell surface proteoglycan to thrombospondin. J. Biol. Chem. 1989, 264, 2885–2889. [Google Scholar]
- Huang, W.; Chiquet-Ehrismann, R.; Moyano, J.V.; Garcia-Pardo, A.; Orend, G. Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res. 2001, 61, 8586–8594. [Google Scholar]
- Greene, D.K.; Tumova, S.; Couchman, J.R.; Woods, A. Syndecan-4 associates with alpha-actinin. J. Biol. Chem. 2003, 278, 7617–7623. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Longley, R.L.; Tumova, S.; Couchman, J.R. Syndecan-4 binding to the high affinity heparin-binding domain of fibronectin drives focal adhesion formation in fibroblasts. Arch. Biochem. Biophys. 2000, 374, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Wilcox-Adelman, S.A.; Denhez, F.; Goetinck, P.F. Syndecan-4 modulates focal adhesion kinase phosphorylation. J. Biol. Chem. 2002, 277, 32970–32977. [Google Scholar] [CrossRef] [PubMed]
- Saoncella, S.; Echtermeyer, F.; Denhez, F.; Nowlen, J.K.; Mosher, D.F.; Robinson, S.D.; Hynes, R.O.; Goetinck, P.F. Syndecan-4 signals cooperatively with integrins in a Rhodependent manner in the assembly of focal adhesions and actin stress fibers. Proc. Natl. Acad. Sci. USA 1999, 96, 2805. [Google Scholar] [CrossRef]
- Ishiguro, K.; Kadomatsu, K.; Kojima, T.; Muramatsu, H.; Tsuzuki, S.; Nakamura, E.; Kusugami, K.; Saito, H.; Muramatsu, T. Syndecan-4 deficiency impairs focal adhesion formation only under restricted conditions. J. Biol. Chem. 2000, 275, 5249–5252. [Google Scholar] [CrossRef]
- Echtermeyer, F.; Baciu, P.C.; Saoncella, S.; Ge, Y.; Goetinck, P.F. Syndecan-4 core protein is sufficient for the assembly of focal adhesions and actin stress fibers. J. Cell Sci. 1999, 112 Pt 20, 3433–3441. [Google Scholar]
- Mostafavi-Pour, Z.; Askari, J.A.; Parkinson, S.J.; Parker, P.J.; Ng, T.T.C.; Humphries, M.J. Integrin-specific signaling pathways controlling focal adhesion formation and cell migration. J. Cell Biol. 2003, 161, 155–167. [Google Scholar] [CrossRef]
- Baciu, P.C.; Goetinck, P.F. Protein kinase C regulates the recruitment of syndecan-4 into focal contacts. Mol. Biol. Cell 1995, 6, 1503–1513. [Google Scholar] [CrossRef]
- Midwood, K.S.; Valenick, L.V.; Hsia, H.C.; Schwarzbauer, J.E. Coregulation of fibronectin signaling and matrix contraction by tenascin-C and syndecan-4. Mol. Biol. Cell 2004, 15, 5670–5677. [Google Scholar] [CrossRef]
- Chronopoulos, A.; Thorpe, S.D.; Cortes, E.; Lachowski, D.; Rice, A.J.; Mykuliak, V.V.; Róg, T.; Lee, D.A.; Hytönen, V.P.; del Río Hernández, A.E. Syndecan-4 tunes cell mechanics by activating the kindlin-integrin-RhoA pathway. Nat. Mater. 2020, 19, 669–678. [Google Scholar] [CrossRef]
- Huang, C.-P.; Cheng, C.-M.; Su, H.-L.; Lin, Y.-W. Syndecan-4 promotes epithelial tumor cells spreading and regulates the turnover of PKCα activity under mechanical stimulation on the elastomeric substrates. Cell Physiol. Biochem. 2015, 36, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Bellin, R.M.; Kubicek, J.D.; Frigault, M.J.; Kamien, A.J.; Steward, R.L.; Barnes, H.M.; DiGiacomo, M.B.; Duncan, L.J.; Edgerly, C.K.; Morse, E.M.; et al. Defining the role of syndecan-4 in mechanotransduction using surface-modification approaches. Proc. Natl. Acad. Sci. USA 2009, 106, 22102. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chaikof, E.L. Mechanical stress regulates syndecan-4 expression and redistribution in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 61–68. [Google Scholar] [CrossRef]
- Nardone, G.; La Oliver-De Cruz, J.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skladal, P.; Pesl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Saunders, S.; Nguyen, H.; Bernfield, M. Loss of cell surface syndecan-1 causes epithelia to transform into anchorage-independent mesenchyme-like cells. Mol. Biol. Cell 1995, 6, 559–576. [Google Scholar] [CrossRef] [PubMed]
- Szatmári, T.; Ötvös, R.; Hjerpe, A.; Dobra, K. Syndecan-1 in cancer: Implications for cell signaling, differentiation, and prognostication. Dis. Markers 2015. [Google Scholar] [CrossRef]
- Kivisaari, A.K.; Kallajoki, M.; Mirtti, T.; McGrath, J.A.; Bauer, J.W.; Weber, F.; Konigova, R.; Sawamura, D.; Sato-Matsumura, K.C.; Shimizu, H.; et al. Transformation-specific matrix metalloproteinases (MMP)-7 and MMP-13 are expressed by tumour cells in epidermolysis bullosa-associated squamous cell carcinomas. Br. J. Dermatol. 2008, 158, 778–785. [Google Scholar] [CrossRef]
- Chen, P.; Abacherli, L.E.; Nadler, S.T.; Wang, Y.; Li, Q.; Parks, W.C. MMP7 shedding of syndecan-1 facilitates re-epithelialization by affecting alpha(2)beta(1) integrin activation. PLoS ONE 2009, 4, e6565. [Google Scholar] [CrossRef]
- Beauvais, D.M.; Burbach, B.J.; Rapraeger, A.C. The syndecan-1 ectodomain regulates alphavbeta3 integrin activity in human mammary carcinoma cells. J. Cell Biol. 2004, 167, 171–181. [Google Scholar] [CrossRef]
- Orian-Rousseau, V. CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 2010, 46, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Govindaraju, P.; Todd, L.; Shetye, S.; Monslow, J.; Pure, E. CD44-dependent inflammation, fibrogenesis, and collagenolysis regulates extracellular matrix remodeling and tensile strength during cutaneous wound healing. Matrix Biol. 2019, 75–76, 314–330. [Google Scholar] [CrossRef] [PubMed]
- Basakran, N.S. CD44 as a potential diagnostic tumor marker. Saudi. Med. J. 2015, 36, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Anderegg, U.; Simon, J.C.; Averbeck, M. More than just a filler—The role of hyaluronan for skin homeostasis. Exp. Dermatol. 2014, 23, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, S.; Jalkanen, M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J. Cell Biol. 1992, 116, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Kawashima, H.; Tanaka, T.; Hirose, M.; Toyama-Sorimachi, N.; Matsuzawa, Y.; Miyasaka, M. CD44 binds a chondroitin sulfate proteoglycan, aggrecan. Int. Immunol. 2001, 13, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Toyama-Sorimachi, N.; Sorimachi, H.; Tobita, Y.; Kitamura, F.; Yagita, H.; Suzuki, K.; Miyasaka, M. A novel ligand for CD44 is serglycin, a hematopoietic cell lineage-specific proteoglycan. Possible involvement in lymphoid cell adherence and activation. J. Biol. Chem. 1995, 270, 7437–7444. [Google Scholar] [CrossRef]
- Kawashima, H.; Hirose, M.; Hirose, J.; Nagakubo, D.; Plaas, A.H.; Miyasaka, M. Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J. Biol. Chem. 2000, 275, 35448–35456. [Google Scholar] [CrossRef]
- Bennett, K.L.; Jackson, D.G.; Simon, J.C.; Tanczos, E.; Peach, R.; Modrell, B.; Stamenkovic, I.; Plowman, G.; Aruffo, A. CD44 isoforms containing exon V3 are responsible for the presentation of heparin-binding growth factor. J. Cell Biol. 1995, 128, 687–698. [Google Scholar] [CrossRef]
- Ishii, S.; Ford, R.; Thomas, P.; Nachman, A.; Steele, G., Jr.; Jessup, J.M. CD44 participates in the adhesion of human colorectal carcinoma cells to laminin and type IV collagen. Surg. Oncol. 1993, 2, 255–264. [Google Scholar] [CrossRef]
- Ehnis, T.; Dieterich, W.; Bauer, M.; Lampe, B.; Schuppan, D. A chondroitin/dermatan sulfate form of CD44 is a receptor for collagen XIV (undulin). Exp. Cell Res. 1996, 229, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Porsch, H.; Mehic, M.; Olofsson, B.; Heldin, P.; Heldin, C.-H. Platelet-derived growth factor beta-receptor, transforming growth factor beta type I receptor, and CD44 protein modulate each other’s signaling and stability. J. Biol. Chem. 2014, 289, 19747–19757. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Heldin, C.-H.; Heldin, P. Inhibition of platelet-derived growth factor-BB-induced receptor activation and fibroblast migration by hyaluronan activation of CD44. J. Biol. Chem. 2006, 281, 26512–26519. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, Y.-S.; Choe, J.; Lee, H.; Kim, Y.-M.; Jeoung, D. CD44-epidermal growth factor receptor interaction mediates hyaluronic acid-promoted cell motility by activating protein kinase C signaling involving Akt, Rac1, Phox, reactive oxygen species, focal adhesion kinase, and MMP-2. J. Biol. Chem. 2008, 283, 22513–22528. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Misra, S.; Toole, B.P. Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 2005, 280, 8875–8883. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Zhu, H.; Zhou, B.; Diedrich, F.; Singleton, P.A.; Hung, M.C. Hyaluronan promotes CD44v3-Vav2 interaction with Grb2-p185(HER2) and induces Rac1 and Ras signaling during ovarian tumor cell migration and growth. J. Biol. Chem. 2001, 276, 48679–48692. [Google Scholar] [CrossRef]
- Nagano, O.; Murakami, D.; Hartmann, D.; de Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca(2+) influx and PKC activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef]
- Suenaga, N.; Mori, H.; Itoh, Y.; Seiki, M. CD44 binding through the hemopexin-like domain is critical for its shedding by membrane-type 1 matrix metalloproteinase. Oncogene 2005, 24, 859–868. [Google Scholar] [CrossRef]
- Kajita, M.; Itoh, Y.; Chiba, T.; Mori, H.; Okada, A.; Kinoh, H.; Seiki, M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 2001, 153, 893–904. [Google Scholar] [CrossRef]
- Ahrens, T.; Sleeman, J.P.; Schempp, C.M.; Howells, N.; Hofmann, M.; Ponta, H.; Herrlich, P.; Simon, J.C. Soluble CD44 inhibits melanoma tumor growth by blocking cell surface CD44 binding to hyaluronic acid. Oncogene 2001, 20, 3399–3408. [Google Scholar] [CrossRef]
- Murakami, D.; Okamoto, I.; Nagano, O.; Kawano, Y.; Tomita, T.; Iwatsubo, T.; de Strooper, B.; Yumoto, E.; Saya, H. Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of CD44. Oncogene 2003, 22, 1511–1516. [Google Scholar] [CrossRef]
- Lammich, S.; Okochi, M.; Takeda, M.; Kaether, C.; Capell, A.; Zimmer, A.-K.; Edbauer, D.; Walter, J.; Steiner, H.; Haass, C. Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an Aβ-like peptide. J. Biol. Chem. 2002, 277, 44754–44759. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mack, J.A.; Maytin, E.V. CD44 inhibits alpha-SMA gene expression via a novel G-actin/MRTF-mediated pathway that intersects with TGFbetaR/p38MAPK signaling in murine skin fibroblasts. J. Biol. Chem. 2019, 294, 12779–12794. [Google Scholar] [CrossRef]
- Hernández, D.; Miquel-Serra, L.; Docampo, M.-J.; Marco-Ramell, A.; Cabrera, J.; Fabra, A.; Bassols, A. V3 versican isoform alters the behavior of human melanoma cells by interfering with CD44/ErbB-dependent signaling. J. Biol. Chem. 2011, 286, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Thorne, R.F.; Legg, J.W.; Isacke, C.M. The role of the CD44 transmembrane and cytoplasmic domains in co-ordinating adhesive and signalling events. J. Cell Sci. 2004, 117, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Zöller, M. CD44, hyaluronan, the hematopoietic stem cell, and leukemia-initiating cells. Front. Immunol. 2015, 6, 235. [Google Scholar] [CrossRef]
- Dzwonek, J.; Wilczynski, G.M. CD44: Molecular interactions, signaling and functions in the nervous system. Front. Cell Neurosci. 2015. [Google Scholar] [CrossRef]
- Skelton, T.P.; Zeng, C.; Nocks, A.; Stamenkovic, I. Glycosylation provides both stimulatory and inhibitory effects on cell surface and soluble CD44 binding to hyaluronan. J. Cell Biol. 1998, 140, 431–446. [Google Scholar] [CrossRef]
- Hurt-Camejo, E.; Rosengren, B.; Sartipy, P.; Elfsberg, K.; Camejo, G.; Svensson, L. CD44, a cell surface chondroitin sulfate proteoglycan, mediates binding of interferon-gamma and some of its biological effects on human vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 18957–18964. [Google Scholar] [CrossRef]
- Chetty, C.; Vanamala, S.K.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. MMP-9 induces CD44 cleavage and CD44 mediated cell migration in glioblastoma xenograft cells. Cell Signal. 2012, 24, 549–559. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999, 13, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Kawano, Y.; Murakami, D.; Sasayama, T.; Araki, N.; Miki, T.; Wong, A.J.; Saya, H. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J. Cell Biol. 2001, 155, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Teye, K.; Numata, S.; Ishii, N.; Krol, R.P.; Tsuchisaka, A.; Hamada, T.; Koga, H.; Karashima, T.; Ohata, C.; Tsuruta, D.; et al. Isolation of all CD44 transcripts in human epidermis and regulation of their expression by various agents. PLoS ONE 2016, 11, e0160952. [Google Scholar] [CrossRef]
- Rajarajan, A.; Bloor, B.; Desai, H.; Stokes, A.; Odell, E. Variant CD44 expression by human fibroblasts. Biomarkers 2008, 13, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tammi, M.; Tammi, R. Distribution of hyaluronan and its CD44 receptor in the epithelia of human skin appendages. Histochemistry 1992, 98, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Seelentag, W.K.; Günthert, U.; Saremaslani, P.; Futo, E.; Pfaltz, M.; Heitz, P.U.; Roth, J. CD44 standard and variant isoform expression in normal human skin appendages and epidermis. Histochem. Cell Biol. 1996, 106, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Penneys, N.S. CD44 expression in normal and inflamed skin. J. Cutan. Pathol. 1993, 20, 250–253. [Google Scholar] [CrossRef]
- Man, M.; Elias, P.M.; Man, W.; Wu, Y.; Bourguignon, L.Y.W.; Feingold, K.R.; Man, M.-Q. The role of CD44 in cutaneous inflammation. Exp. Dermatol. 2009, 18, 962–968. [Google Scholar] [CrossRef]
- Lugović-Mihić, L.; Novak-Bilić, G.; Vučić, M.; Japundžić, I.; Bukvić, I. CD44 expression in human skin: High expression in irritant and allergic contact dermatitis and moderate expression in psoriasis lesions in comparison with healthy controls. Contact Dermat. 2020, 82, 297–306. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W.; Ramez, M.; Gilad, E.; Singleton, P.A.; Man, M.-Q.; Crumrine, D.A.; Elias, P.M.; Feingold, K.R. Hyaluronan-CD44 interaction stimulates keratinocyte differentiation, lamellar body formation/secretion, and permeability barrier homeostasis. J. Investig. Dermatol. 2006, 126, 1356–1365. [Google Scholar] [CrossRef]
- Kirschner, N.; Haftek, M.; Niessen, C.M.; Behne, M.J.; Furuse, M.; Moll, I.; Brandner, J.M. CD44 regulates tight-junction assembly and barrier function. J. Investig. Dermatol. 2011, 131, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Shatirishvili, M.; Burk, A.S.; Franz, C.M.; Pace, G.; Kastilan, T.; Breuhahn, K.; Hinterseer, E.; Dierich, A.; Bakiri, L.; Wagner, E.F.; et al. Epidermal-specific deletion of CD44 reveals a function in keratinocytes in response to mechanical stress. Cell Death Dis. 2016, 7, e2461. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.M.; Wells, A.; Thomas, D.; Stephens, P.; Steadman, R.; Phillips, A. Aging fibroblasts resist phenotypic maturation because of impaired hyaluronan-dependent CD44/epidermal growth factor receptor signaling. Am. J. Pathol. 2010, 176, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Ansorge, H.L.; Beredjiklian, P.K.; Soslowsky, L.J. CD44 deficiency improves healing tendon mechanics and increases matrix and cytokine expression in a mouse patellar tendon injury model. J. Orthop. Res. 2009, 27, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Velasco, J.; Li, J.; DiPietro, L.; Stepp, M.A.; Sandy, J.D.; Plaas, A. Adamts5 deletion blocks murine dermal repair through CD44-mediated aggrecan accumulation and modulation of transforming growth factor β1 (TGFβ1) signaling. J. Biol. Chem. 2011, 286, 26016–26027. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.A. Transforming growth factor-beta: A general review. Eur. Cytokine Netw. 1996, 7, 363–374. [Google Scholar]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016. [Google Scholar] [CrossRef]
- Hinck, A.P. Structural studies of the TGF-βs and their receptors – insights into evolution of the TGF-β superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef]
- Chen, R.H.; Derynck, R. Homomeric interactions between type II transforming growth factor-beta receptors. J. Biol. Chem. 1994, 269, 22868–22874. [Google Scholar] [PubMed]
- Gilboa, L.; Wells, R.G.; Lodish, H.F.; Henis, Y.I. Oligomeric structure of type I and type II transforming growth factor beta receptors: Homodimers form in the ER and persist at the plasma membrane. J. Cell Biol. 1998, 140, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Gutman, O.; Knaus, P.; Henis, Y.I. Oligomeric interactions of TGF-beta and BMP receptors. FEBS Lett. 2012, 586, 1885–1896. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, Y.; Wang, Q.; Ma, X.; Xiao, Z.; Zuo, W.; Fang, X.; Chen, Y.-G. Single-molecule imaging reveals transforming growth factor-beta-induced type II receptor dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 15679–15683. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Laiho, M.; Weis, F.M.; Boyd, F.T.; Ignotz, R.A.; Massague, J. Responsiveness to transforming growth factor-beta (TGF-beta) restored by genetic complementation between cells defective in TGF-beta receptors I and II. J. Biol. Chem. 1991, 266, 9108–9112. [Google Scholar]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef]
- Massagué, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Huse, M.; Chen, Y.G.; Massague, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.G.; Kuriyan, J.; Massague, J. The TGF beta receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Mulder, K.M.; Morris, S.L. Activation of p21ras by transforming growth factor beta in epithelial cells. J. Biol. Chem. 1992, 267, 5029–5031. [Google Scholar] [PubMed]
- Yan, Z.; Winawer, S.; Friedman, E. Two different signal transduction pathways can be activated by transforming growth factor beta 1 in epithelial cells. J. Biol. Chem. 1994, 269, 13231–13237. [Google Scholar] [PubMed]
- Mucsi, I.; Skorecki, K.L.; Goldberg, H.J. Extracellular signal-regulated kinase and the small GTP-binding protein, Rac, contribute to the effects of transforming growth factor-beta1 on gene expression. J. Biol. Chem. 1996, 271, 16567–16572. [Google Scholar] [CrossRef] [PubMed]
- Hanafusa, H.; Ninomiya-Tsuji, J.; Masuyama, N.; Nishita, M.; Fujisawa, J.; Shibuya, H.; Matsumoto, K.; Nishida, E. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J. Biol. Chem. 1999, 274, 27161–27167. [Google Scholar] [CrossRef]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef]
- Frey, R.S.; Mulder, K.M. Involvement of extracellular signal-regulated kinase 2 and stress-activated protein kinase/Jun N-terminal kinase activation by transforming growth factor beta in the negative growth control of breast cancer cells. Cancer Res. 1997, 57, 628–633. [Google Scholar]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef]
- Wilkes, M.C.; Mitchell, H.; Penheiter, S.G.; Dore, J.J.; Suzuki, K.; Edens, M.; Sharma, D.K.; Pagano, R.E.; Leof, E.B. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 2005, 65, 10431–10440. [Google Scholar] [CrossRef]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef]
- Freudlsperger, C.; Bian, Y.; Contag Wise, S.; Burnett, J.; Coupar, J.; Yang, X.; Chen, Z.; van Waes, C. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene 2013, 32, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Gingery, A.; Bradley, E.; Pederson, L.; Ruan, M.; Horwood, N.; Oursler, M. TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp. Cell Res. 2008, 314, 2725–2738. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Edlund, S.; Landstrom, M.; Heldin, C.-H.; Aspenstrom, P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, M.C.; Murphy, S.J.; Garamszegi, N.; Leof, E.B. Cell-type-specific activation of PAK2 by transforming growth factor beta independent of Smad2 and Smad3. Mol. Cell. Biol 2003, 23, 8878–8889. [Google Scholar] [CrossRef]
- Mythreye, K.; Blobe, G.C. The type III TGF-beta receptor regulates epithelial and cancer cell migration through beta-arrestin2-mediated activation of Cdc42. Proc. Natl. Acad. Sci. USA 2009, 106, 8221–8226. [Google Scholar] [CrossRef]
- You, H.J.; How, T.; Blobe, G.C. The type III transforming growth factor-beta receptor negatively regulates nuclear factor kappa B signaling through its interaction with beta-arrestin2. Carcinogenesis 2009, 30, 1281–1287. [Google Scholar] [CrossRef]
- López-Casillas, F.; Wrana, J.L.; Massagué, J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 1993, 73, 1435–1444. [Google Scholar] [CrossRef]
- Moustakas, A.; Lin, H.Y.; Henis, Y.I.; Plamondon, J.; O’Connor-McCourt, M.D.; Lodish, H.F. The transforming growth factor beta receptors types I, II, and III form hetero-oligomeric complexes in the presence of ligand. J. Biol. Chem. 1993, 268, 22215–22218. [Google Scholar]
- Varadaraj, A.; Jenkins, L.M.; Singh, P.; Chanda, A.; Snider, J.; Lee, N.Y.; Amsalem-Zafran, A.R.; Ehrlich, M.; Henis, Y.I.; Mythreye, K. TGF-β triggers rapid fibrillogenesis via a novel TβRII-dependent fibronectin-trafficking mechanism. Mol. Biol. Cell 2017, 28, 1195–1207. [Google Scholar] [CrossRef]
- Garamszegi, N.; Garamszegi, S.P.; Samavarchi-Tehrani, P.; Walford, E.; Schneiderbauer, M.M.; Wrana, J.L.; Scully, S.P. Extracellular matrix-induced transforming growth factor-beta receptor signaling dynamics. Oncogene 2010, 29, 2368–2380. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Itin, P.; Rufli, T. In situ analysis of transforming growth factors-beta (TGF-beta 1, TGF-beta 2, TGF-beta 3) and TGF-beta type II receptor expression in basal cell carcinomas. Br. J. Dermatol. 1996, 134, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Myokai, F.; Arata, J.; Noji, S.; Taniguchi, S. Expression of type II transforming growth factor-beta receptor mRNA in human skin, as revealed by in situ hybridization. J. Dermatol. Sci. 1994, 8, 25–32. [Google Scholar] [CrossRef]
- Kudo, H.; Jinnin, M.; Asano, Y.; Trojanowska, M.; Nakayama, W.; Inoue, K.; Honda, N.; Kajihara, I.; Makino, K.; Fukushima, S.; et al. Decreased interleukin-20 expression in scleroderma skin contributes to cutaneous fibrosis. Arthritis Rheumatol. 2014, 66, 1636–1647. [Google Scholar] [CrossRef]
- Sowden, H.M.; Karoo, R.O.S.; Tobin, D.J. Transforming growth factor-beta receptor II is preferentially expressed in the companion layer of the human anagen hair follicle. Br. J. Dermatol. 2007, 157, 161–164. [Google Scholar] [CrossRef]
- Gold, L.I.; Sung, J.J.; Siebert, J.W.; Longaker, M.T. Type I (RI) and type II (RII) receptors for transforming growth factor-beta isoforms are expressed subsequent to transforming growth factor-beta ligands during excisional wound repair. Am. J. Pathol. 1997, 150, 209–222. [Google Scholar]
- Qin, Z.; Fisher, G.J.; Voorhees, J.J.; Quan, T. Actin cytoskeleton assembly regulates collagen production via TGF-beta type II receptor in human skin fibroblasts. J. Cell Mol. Med. 2018, 22, 4085–4096. [Google Scholar] [CrossRef]
- Fisher, G.J.; Shao, Y.; He, T.; Qin, Z.; Perry, D.; Voorhees, J.J.; Quan, T. Reduction of fibroblast size/mechanical force down-regulates TGF-β type II receptor: Implications for human skin aging. Aging Cell 2016, 15, 67–76. [Google Scholar] [CrossRef]
- Quan, T.; Fisher, G.J. Role of age-associated alterations of the dermal extracellular matrix microenvironment in human skin aging: A mini-review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef]
- March, J.T.; Golshirazi, G.; Cernisova, V.; Carr, H.; Leong, Y.; Lu-Nguyen, N.; Popplewell, L.J. Targeting TGFβ signaling to address fibrosis using antisense oligonucleotides. Biomedicines 2018, 6, 74. [Google Scholar] [CrossRef]
- Pohlers, D.; Brenmoehl, J.; Löffler, I.; Müller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-β and fibrosis in different organs—Molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The role of the TGF-β family in wound healing, burns and scarring: A review. Int. J. Burns Trauma 2012, 2, 18–28. [Google Scholar] [PubMed]
- Pakyari, M.; Farrokhi, A.; Maharlooei, M.K.; Ghahary, A. Critical role of transforming growth factor beta in different phases of wound healing. Adv. Wound Care 2013, 2, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Liarte, S.; Bernabé-García, Á.; Nicolás, F.J. Role of TGF—In skin chronic wounds: A keratinocyte perspective. Cells 2020, 9, 306. [Google Scholar] [CrossRef] [PubMed]
- Kiritsi, D.; Nystrom, A. The role of TGFbeta in wound healing pathologies. Mech. Ageing Dev. 2018, 172, 51–58. [Google Scholar] [CrossRef]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (TGF-beta) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef]
- Liu, J.; Johnson, K.; Li, J.; Piamonte, V.; Steffy, B.M.; Hsieh, M.H.; Ng, N.; Zhang, J.; Walker, J.R.; Ding, S.; et al. Regenerative phenotype in mice with a point mutation in transforming growth factor beta type I receptor (TGFBR1). Proc. Natl. Acad. Sci. USA 2011, 108, 14560. [Google Scholar] [CrossRef]
- Wang, X.J.; Greenhalgh, D.A.; Bickenbach, J.R.; Jiang, A.; Bundman, D.S.; Krieg, T.; Derynck, R.; Roop, D.R. Expression of a dominant-negative type II transforming growth factor beta (TGF-beta) receptor in the epidermis of transgenic mice blocks TGF-beta-mediated growth inhibition. Proc. Natl. Acad. Sci. USA 1997, 94, 2386–2391. [Google Scholar] [CrossRef]
- Amendt, C.; Mann, A.; Schirmacher, P.; Blessing, M. Resistance of keratinocytes to TGFbeta-mediated growth restriction and apoptosis induction accelerates re-epithelialization in skin wounds. J. Cell Sci. 2002, 115, 2189–2198. [Google Scholar] [PubMed]
- Martinez-Ferrer, M.; Afshar-Sherif, A.-R.; Uwamariya, C.; de Crombrugghe, B.; Davidson, J.M.; Bhowmick, N.A. Dermal transforming growth factor-beta responsiveness mediates wound contraction and epithelial closure. Am. J. Pathol. 2010, 176, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Khan, K.; Hoyles, R.K.; Shiwen, X.; Leoni, P.; Chen, Y.; Eastwood, M.; Abraham, D.J. Inducible lineage-specific deletion of TbetaRII in fibroblasts defines a pivotal regulatory role during adult skin wound healing. J. Investig. Dermatol. 2009, 129, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Zheng, B.; Evans, L.A.; Shi-wen, X.; Ong, V.H.; Fisher, I.; Lazaridis, K.; Abraham, D.J.; Black, C.M.; de Crombrugghe, B. Fibroblast-specific expression of a kinase-deficient type II transforming growth factor beta (TGFbeta) receptor leads to paradoxical activation of TGFbeta signaling pathways with fibrosis in transgenic mice. J. Biol. Chem. 2003, 278, 25109–25119. [Google Scholar] [CrossRef]
- Oshima, M.; Oshima, H.; Taketo, M.M. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996, 179, 297–302. [Google Scholar] [CrossRef]
- Sonnylal, S.; Denton, C.P.; Zheng, B.; Keene, D.R.; He, R.; Adams, H.P.; Vanpelt, C.S.; Geng, Y.J.; Deng, J.M.; Behringer, R.R.; et al. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007, 56, 334–344. [Google Scholar] [CrossRef]
- Dudas, M.; Kim, J.; Li, W.-Y.; Nagy, A.; Larsson, J.; Karlsson, S.; Chai, Y.; Kaartinen, V. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev. Biol. 2006, 296, 298–314. [Google Scholar] [CrossRef]
- Guerra, L.; Odorisio, T.; Zambruno, G.; Castiglia, D. Stromal microenvironment in type VII collagen-deficient skin: The ground for squamous cell carcinoma development. Matrix Biol. 2017, 63, 1–10. [Google Scholar] [CrossRef]
- Knaup, J.; Gruber, C.; Krammer, B.; Ziegler, V.; Bauer, J.; Verwanger, T. TGFbeta-signaling in squamous cell carcinoma occurring in recessive dystrophic epidermolysis bullosa. Anal. Cell Pathol. (Amst.) 2011, 34, 339–353. [Google Scholar] [CrossRef]
- Cammareri, P.; Rose, A.M.; Vincent, D.F.; Wang, J.; Nagano, A.; Libertini, S.; Ridgway, R.A.; Athineos, D.; Coates, P.J.; McHugh, A.; et al. Inactivation of TGFβ receptors in stem cells drives cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12493. [Google Scholar] [CrossRef]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Piepkorn, M.; Predd, H.; Underwood, R.; Cook, P. Proliferation-differentiation relationships in the expression of heparin-binding epidermal growth factor-related factors and erbB receptors by normal and psoriatic human keratinocytes. Arch. Dermatol. Res. 2003, 295, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, E.; Mahmoudi, M.; Rahmani, F.; Yousefi, B.; Sarafnejad, A.; Kavosi, H.; Karimizadeh, E.; Jamshidi, A.; Gharibdoost, F. Attenuation of aquaporin-3 and epidermal growth factor receptor expression and activation in systemic sclerosis dermal fibroblasts. J. Cell Physiol. 2019, 234, 12876–12883. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Ferguson, K.M. A structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef]
- Normanno, N.; de Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; de Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Sun, L.; Carpenter, G. Epidermal growth factor activation of NF-kappaB is mediated through IkappaBalpha degradation and intracellular free calcium. Oncogene 1998, 16, 2095–2102. [Google Scholar] [CrossRef]
- Andl, C.D.; Mizushima, T.; Oyama, K.; Bowser, M.; Nakagawa, H.; Rustgi, A.K. EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1227–G1237. [Google Scholar] [CrossRef]
- Li, Y.; Macdonald-Obermann, J.; Westfall, C.; Piwnica-Worms, D.; Pike, L.J. Quantitation of the effect of ErbB2 on epidermal growth factor receptor binding and dimerization. J. Biol. Chem. 2012, 287, 31116–31125. [Google Scholar] [CrossRef]
- Duan, L.; Miura, Y.; Dimri, M.; Majumder, B.; Dodge, I.L.; Reddi, A.L.; Ghosh, A.; Fernandes, N.; Zhou, P.; Mullane-Robinson, K.; et al. Cbl-mediated ubiquitinylation is required for lysosomal sorting of epidermal growth factor receptor but is dispensable for endocytosis. J. Biol. Chem. 2003, 278, 28950–28960. [Google Scholar] [CrossRef] [PubMed]
- Umebayashi, K.; Stenmark, H.; Yoshimori, T. Ubc4/5 and c-Cbl continue to ubiquitinate EGF receptor after internalization to facilitate polyubiquitination and degradation. Mol. Biol. Cell 2008, 19, 3454–3462. [Google Scholar] [CrossRef] [PubMed]
- Kenny, F.N.; Drymoussi, Z.; Delaine-Smith, R.; Kao, A.P.; Laly, A.C.; Knight, M.M.; Philpott, M.P.; Connelly, J.T. Tissue stiffening promotes keratinocyte proliferation through activation of epidermal growth factor signaling. J. Cell Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Liu, S.; Yang, B.; Hajal, C.; Changede, R.; Hu, J.; Wolfenson, H.; Hone, J.; Sheetz, M.P. EGFR and HER2 activate rigidity sensing only on rigid matrices. Nat. Mater. 2017, 16, 775–781. [Google Scholar] [CrossRef]
- Kim, J.-H.; Asthagiri, A.R. Matrix stiffening sensitizes epithelial cells to EGF and enables the loss of contact inhibition of proliferation. J. Cell Sci. 2011, 124, 1280–1287. [Google Scholar] [CrossRef]
- Mainiero, F.; Pepe, A.; Yeon, M.; Ren, Y.; Giancotti, F.G. The intracellular functions of alpha6beta4 integrin are regulated by EGF. J. Cell Biol. 1996, 134, 241–253. [Google Scholar] [CrossRef]
- Grassian, A.R.; Schafer, Z.T.; Brugge, J.S. ErbB2 stabilizes epidermal growth factor receptor (EGFR) expression via Erk and Sprouty2 in extracellular matrix-detached cells. J. Biol. Chem. 2011, 286, 79–90. [Google Scholar] [CrossRef]
- Yarwood, S.J.; Woodgett, J.R. Extracellular matrix composition determines the transcriptional response to epidermal growth factor receptor activation. Proc. Natl. Acad. Sci. USA 2001, 98, 4472. [Google Scholar] [CrossRef]
- Kim, S.; Jeon, M.; Lee, J.; Han, J.; Oh, S.-J.; Jung, T.; Nam, S.J.; Kil, W.H.; Lee, J.E. Induction of fibronectin in response to epidermal growth factor is suppressed by silibinin through the inhibition of STAT3 in triple negative breast cancer cells. Oncol. Rep. 2014, 32, 2230–2236. [Google Scholar] [CrossRef]
- Hsu, J.-Y.; Chang, J.-Y.; Chang, K.-Y.; Chang, W.-C.; Chen, B.-K. Epidermal growth factor-induced pyruvate dehydrogenase kinase 1 expression enhances head and neck squamous cell carcinoma metastasis via up-regulation of fibronectin. FASEB J. 2017, 31, 4265–4276. [Google Scholar] [CrossRef]
- Santra, M.; Reed, C.C.; Iozzo, R.V. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope. J. Biol. Chem. 2002, 277, 35671–35681. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Santra, M.; Reed, C.C.; Eichstetter, I.; McQuillan, D.J.; Gross, D.; Nugent, M.A.; Hajnoczky, G.; Iozzo, R.V. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J. Biol. Chem. 2000, 275, 32879–32887. [Google Scholar] [CrossRef] [PubMed]
- Gubbiotti, M.A.; Vallet, S.D.; Ricard-Blum, S.; Iozzo, R.V. Decorin interacting network: A comprehensive analysis of decorin-binding partners and their versatile functions. Matrix Biol. 2016, 55, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.K.V.; Tran, K.T.; Borysenko, C.W.; Cascio, M.; Camacho, C.J.; Blair, H.C.; Bahar, I.; Wells, A. Tenascin cytotactin epidermal growth factor-like repeat binds epidermal growth factor receptor with low affinity. J. Cell Physiol. 2007, 211, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Hintermann, E.; Bilban, M.; Koshikawa, N.; Hojilla, C.; Khokha, R.; Quaranta, V. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J. Cell Biol. 2003, 161, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Garg, P.; Yang, S.; Gong, P.; Pallero, M.A.; Annis, D.S.; Liu, Y.; Passaniti, A.; Mann, D.; Mosher, D.F.; et al. Epidermal growth factor-like repeats of thrombospondins activate phospholipase Cgamma and increase epithelial cell migration through indirect epidermal growth factor receptor activation. J. Biol. Chem. 2009, 284, 6389–6402. [Google Scholar] [CrossRef]
- Zhang, Y.; Marmorstein, L.Y. Focus on molecules: Fibulin-3 (EFEMP1). Exp. Eye Res. 2010, 90, 374–375. [Google Scholar] [CrossRef]
- Wolf, C.; Qian, Y.; Brooke, M.A.; Kelsell, D.P.; Franzke, C.-W. ADAM17/EGFR axis promotes transglutaminase-dependent skin barrier formation through phospholipase C γ1 and protein kinase C pathways. Sci. Rep. 2016, 6, 39780. [Google Scholar] [CrossRef]
- Schneider, M.R.; Werner, S.; Paus, R.; Wolf, E. Beyond wavy hairs: The epidermal growth factor receptor and its ligands in skin biology and pathology. Am. J. Pathol. 2008, 173, 14–24. [Google Scholar] [CrossRef]
- Wakita, H.; Takigawa, M. Activation of epidermal growth factor receptor promotes late terminal differentiation of cell-matrix interaction-disrupted keratinocytes. J. Biol. Chem. 1999, 274, 37285–37291. [Google Scholar] [CrossRef]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Klarlund, J.K.; Block, E.R. Free edges in epithelia as cues for motility. Cell Adh. Migr. 2011, 5, 106–110. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mazie, A.R.; Spix, J.K.; Block, E.R.; Achebe, H.B.; Klarlund, J.K. Epithelial cell motility is triggered by activation of the EGF receptor through phosphatidic acid signaling. J. Cell Sci. 2006, 119, 1645. [Google Scholar] [CrossRef]
- Klarlund, J.K. Dual modes of motility at the leading edge of migrating epithelial cell sheets. Proc. Natl. Acad. Sci. USA 2012, 109, 15799. [Google Scholar] [CrossRef] [PubMed]
- Bill, H.M.; Knudsen, B.; Moores, S.L.; Muthuswamy, S.K.; Rao, V.R.; Brugge, J.S.; Miranti, C.K. Epidermal growth factor receptor-dependent regulation of integrin-mediated signaling and cell cycle entry in epithelial cells. Mol. Cell Biol. 2004, 24, 8586–8599. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.; Morton, P.E.; Takeichi, T.; Salam, A.; Roberts, N.; Proudfoot, L.E.; Mellerio, J.E.; Aminu, K.; Wellington, C.; Patil, S.N.; et al. Epithelial inflammation resulting from an inherited loss-of-function mutation in EGFR. J. Investig. Dermatol. 2014, 134, 2570–2578. [Google Scholar] [CrossRef]
- Gaudry, C.A.; Palka, H.L.; Dusek, R.L.; Huen, A.C.; Khandekar, M.J.; Hudson, L.G.; Green, K.J. Tyrosine-phosphorylated plakoglobin is associated with desmogleins but not desmoplakin after epidermal growth factor receptor activation. J. Biol. Chem. 2001, 276, 24871–24880. [Google Scholar] [CrossRef]
- Klessner, J.L.; Desai, B.V.; Amargo, E.V.; Getsios, S.; Green, K.J. EGFR and ADAMs cooperate to regulate shedding and endocytic trafficking of the desmosomal cadherin desmoglein 2. Mol. Biol. Cell 2009, 20, 328–337. [Google Scholar] [CrossRef]
- Yin, T.; Getsios, S.; Caldelari, R.; Godsel, L.M.; Kowalczyk, A.P.; Muller, E.J.; Green, K.J. Mechanisms of plakoglobin-dependent adhesion: Desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J. Biol. Chem. 2005, 280, 40355–40363. [Google Scholar] [CrossRef]
- Lorch, J.H.; Klessner, J.; Park, J.K.; Getsios, S.; Wu, Y.L.; Stack, M.S.; Green, K.J. Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J. Biol. Chem. 2004, 279, 37191–37200. [Google Scholar] [CrossRef]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.-M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef]
- Franzke, C.-W.; Cobzaru, C.; Triantafyllopoulou, A.; Löffek, S.; Horiuchi, K.; Threadgill, D.W.; Kurz, T.; van Rooijen, N.; Bruckner-Tuderman, L.; Blobel, C.P. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Exp. Med. 2012, 209, 1105–1119. [Google Scholar] [CrossRef]
- Hitomi, K. Transglutaminases in skin epidermis. Eur. J. Dermatol. 2005, 15, 313–319. [Google Scholar]
- Blaydon, D.C.; Biancheri, P.; Di, W.-L.; Plagnol, V.; Cabral, R.M.; Brooke, M.A.; van Heel, D.A.; Ruschendorf, F.; Toynbee, M.; Walne, A.; et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 2011, 365, 1502–1508. [Google Scholar] [CrossRef]
- Brooke, M.A.; O’Toole, E.A.; Kelsell, D.P. Exoming into rare skin disease: EGFR deficiency. J. Investig. Dermatol. 2014, 134, 2486–2488. [Google Scholar] [CrossRef]
- Grasset, E.M.; Bertero, T.; Bozec, A.; Friard, J.; Bourget, I.; Pisano, S.; Lecacheur, M.; Maiel, M.; Bailleux, C.; Emelyanov, A.; et al. Matrix stiffening and EGFR cooperate to promote the collective invasion of cancer cells. Cancer Res. 2018, 78, 5229. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Commandeur, S.; van Drongelen, V.; de Gruijl, F.R.; El Ghalbzouri, A. Epidermal growth factor receptor activation and inhibition in 3D in vitro models of normal skin and human cutaneous squamous cell carcinoma. Cancer Sci. 2012, 103, 2120–2126. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Diociaiuti, A.; Steinke, H.; Nyström, A.; Schwieger-Briel, A.; Meiss, F.; Pfannenberg, C.; Bruckner-Tuderman, L.; Ruf, J.; Vito, R.D.; Hachem, M.E.; et al. EGFR inhibition for metastasized cutaneous squamous cell carcinoma in dystrophic epidermolysis bullosa. Orphanet. J. Rare Dis. 2019, 14, 1–6. [Google Scholar] [CrossRef]
- Kim, M.; Li, M.; Mather-Hillon, J.; Melbourne, W.; Tran, K.; Daniel, B.; de Souza, P.; Mallesara, G.; Murrell, D. Increased expression of EGFR in squamous cell carcinoma in recessive dystrophic epidermolysis bullosa and response to Cetuximab. Australas J. Dermatol. 2011, 52, 32. [Google Scholar]
- Filoni, A.; Cicco, G.; Lospalluti, L.; Maglietta, A.; Foti, C.; Annichiarico, G.; Resta, L.; Bonamonte, D. Morphological and morphometric analysis of cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa: A retrospective study. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 1707–1714. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleiser, S.; Nyström, A. Interplay between Cell-Surface Receptors and Extracellular Matrix in Skin. Biomolecules 2020, 10, 1170. https://doi.org/10.3390/biom10081170
Kleiser S, Nyström A. Interplay between Cell-Surface Receptors and Extracellular Matrix in Skin. Biomolecules. 2020; 10(8):1170. https://doi.org/10.3390/biom10081170
Chicago/Turabian StyleKleiser, Svenja, and Alexander Nyström. 2020. "Interplay between Cell-Surface Receptors and Extracellular Matrix in Skin" Biomolecules 10, no. 8: 1170. https://doi.org/10.3390/biom10081170
APA StyleKleiser, S., & Nyström, A. (2020). Interplay between Cell-Surface Receptors and Extracellular Matrix in Skin. Biomolecules, 10(8), 1170. https://doi.org/10.3390/biom10081170