Investigation of Volatiles in Cork Samples Using Chromatographic Data and the Superposing Significant Interaction Rules (SSIR) Chemometric Tool



Abstract
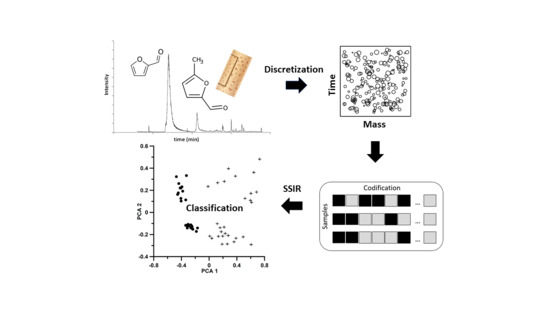
1. Introduction
2. Materials and Methods
2.1. Reagents and Solutions
2.2. Headspace Solid-Phase Microextraction Procedure
2.3. Equipment and Chromatographic Conditions
2.4. Cork Samples and Preparation of Cork Macerates
2.5. Dataset and Data Processing Algorithms
3. Results and Discussion
3.1. Targeted Analysis and First Attempt to Evaluate the Impact of Cork Treatment Using Principal Component Analysis
3.2. Data Processing Results Obtained with SSIR
3.3. Discrimination and Prediction
3.4. Identification of the Compounds Responsible for Group Separation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pereira, H. The Rationale behind Cork Properties: A Review of Structure and Chemistry. Bioresources 2015, 10, 1–23. [Google Scholar] [CrossRef]
- Silva, S.P.; Sabino, M.A.; Fernandes, E.M.; Correlo, V.M.; Boesel, L.F.; Reis, R.L. Cork: Properties, capabilities and applications. Int. Mater. Rev. 2005, 50, 345–365. [Google Scholar] [CrossRef]
- Fernandes, A.; Sousa, A.; Mateus, N.; Cabral, M.; de Freitas, V. Analysis of phenolic compounds in cork from Quercus suber L. by HPLC-DAD/ESI-MS. Food Chem. 2011, 125, 1398–1405. [Google Scholar] [CrossRef]
- Varea, S.; García-Vallejo, M.C.; Cadahía, E.; Fernández de Simon, B. Polyphenols susceptible to migrate from cork stoppers to wine. Eur. Food Res. Technol. 2001, 213, 56–61. [Google Scholar] [CrossRef]
- Pinto, J.; Oliveira, A.S.; Lopes, P.; Roseira, I.; Cabral, M.; Bastos, M.L.; Guedes de Pinho, P. Characterization of chemical compounds susceptible to be extracted from cork by the wine using GC-MS and 1H NMR metabolomics approaches. Food Chem. 2019, 271, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, J.; Fernades, I.L.; Lopes, P.; Roseira, I.; Cabral, M.; Mateus, N.; Freitas, V. Migration of phenolic compounds from different cork stoppers to wine model solutions: Antioxidant and biological relevance. Eur. Food Res. Technol. 2014, 239, 951–960. [Google Scholar] [CrossRef]
- Moreira, N.; Lopes, P.; Cabral, M.; Guedes de Pinho, P. HS-SPME/GC-MS methodologies for the analysis of volatile compounds in cork material. Eur. Food Res. Technol. 2016, 242, 457–466. [Google Scholar] [CrossRef]
- Barreto, M.C.; Vilas Boas, L.; Carneiro, L.C.; San Romao, M.V. Volatile compounds in samples of cork and also produced by selected fungi. J. Agric. Food Chem. 2011, 59, 6568–6574. [Google Scholar] [CrossRef]
- Prat, C.; Trias, R.; Culleré, L.; Escudero, A.; Anticó, E.; Bañeras, L. Off-odor compounds produced in cork by isolated bacteria and fungi: A gas chromatography-mass spectrometry and gas chromatography-olfactometry study. J. Agric. Food Chem. 2009, 57, 7473–7479. [Google Scholar] [CrossRef]
- Prat, C.; Besalú, E.; Bañeras, L.; Anticó, E. Multivariate analysis of volatile compounds detected by headspace solid-phase microextraction/gas chromatography: A tool for sensory classification of cork stoppers. Food Chem. 2011, 126, 1978–1984. [Google Scholar] [CrossRef]
- Rocha, S.; Delgadillo, I.; Ferrer Correia, A.J. GC-MS Study of Volatiles of Normal and Microbiologically Attacked Cork from Quercus suber L. J. Agric. Food Chem. 1996, 44, 865–871. [Google Scholar] [CrossRef]
- Belitz, H.-D.; Grosch, W.; Schieberle, P. Food Chemistry, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2004; ISBN 978-3-540-69933-0. [Google Scholar]
- Rocha, S.; Delgadillo, I.; Ferrer Correia, A.J.; Barros, A.; Wells, P. Application of an electronic aroma sensing system to cork stopper quality control. J. Agric. Food Chem. 1998, 46, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Culleré, L.; Cacho, J.; Ferreira, V. Comparative study of the aromatic profile of different kinds of wine cork stoppers. Food Chem. 2009, 112, 381–387. [Google Scholar] [CrossRef]
- Aroso, I.M.; Araújo, A.R.; Fernandes, J.P.; Santos, T.; Batista, M.T.; Pires, R.A.; Mano, J.F.; Reis, R.L. Hydroalcoholic extracts form the bark of Quercus suber L. (cork): Optimization of extraction conditions, chemical composition and antioxidant potential. Wood Sci. Technol. 2017, 51, 855–872. [Google Scholar] [CrossRef]
- Macku, C.; Gonzalez, L.; Schleussner, C.; Mesquita, A.C.; Herwatt, J.W.; Kirch, L.C.; Schwartz, R.J. Sensory screening for large format natural cork by “dry soak” testing and its correlation to headspace solid-phase microextraction (SPME) gas chromatography/mass spectrometry (GC/MS) releasable trichloroanisole (TCA) analysis. J. Agric. Food Chem. 2009, 57, 7962–7968. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Rauhut, D.; Junga, R. “Cork taint” responsible compounds. Determination of haloanisoles and halophenols in cork matrix: A review. Talanta 2017, 175, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Soleas, G.J.; Yan, J.; Seaver, T.; Goldberg, D.M. Method for gas chromatographic assay with mass selective detection of trichloro compounds in corks and wines applied to elucidate the potential cause of cork taint. J. Agric. Food Chem. 2002, 50, 1032–1039. [Google Scholar] [CrossRef]
- Castro, R.; Natera, R.; Duran, E.; García-Barroso, C. Application of solid phase extraction techniques to analyse volatile compounds in wines and other enological products. Eur. Food Res. Technol. 2008, 228, 1–18. [Google Scholar] [CrossRef]
- Pawliszyn, J. Solid Phase Microextraction Theory and Practice; Wiley-VCH: New York, NY, USA, 1997; ISBN 0-471-19034-9. [Google Scholar]
- Amon, J.M.; Vandepeer, J.M.; Simpson, R.F. Compounds responsible for cork taint in wine. Wine Ind. J. 1989, 4, 62–69. [Google Scholar]
- Chatonnet, P.; Bonnet, S.; Boutou, S.; Labadie, M.-D. Identification and Responsibility of 2,4,6-Tribromoanisole in Musty, Corked Odors in Wine. J. Agric. Food Chem. 2004, 52, 1255–1262. [Google Scholar] [CrossRef]
- Alvárez-Rodríguez, M.L.; López-Ocaña, L.; López-Coronado, J.M.; Rodríguez, E.; Martínez, M.J.; Larriba, G.; Coque, J.-J.R. Cork taint of wines: Role of filamentous fungi isolated from cork in the formation of 2,4,6-trichloroanisole by O methylation of 2,4,6-trichlorophenol. Appl. Environ. Microbiol. 2002, 68, 5860–5869. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Byrd, N.; Williams, J. An assessment of the effect of the ROSA treatment on the level of TCA in naturally-contaminated cork granules. Aust. N. Z. Grapegrow. Winemak. 2004, 484, 57–59. [Google Scholar]
- Sefton, M.A.; Simpson, R.F. Compounds causing cork taint and the factors affecting their transfer from natural cork closures to wine—A review. Aust. J. Grape Wine Res. 2005, 11, 226–240. [Google Scholar] [CrossRef]
- Corsi, A.; Robles Hernandez, F.C.; Cordúa Cruz, G.; Neal, J.A. The effectiveness of electron beam irradiation to reduce or eliminate mould in cork stoppers. Int. J. Food Sci. Technol. 2016, 51, 389–395. [Google Scholar] [CrossRef]
- Recio, E.; Álvarez-Rodríguez, M.L.; Rumbero, A.; Garzón, E.; Coque, J.-J.R. Destruction of Chloroanisoles by Using a Hydrogen Peroxide Activated Method and Its Application To Remove Chloroanisoles from Cork Stoppers. J. Agric. Food Chem. 2011, 59, 12589–12597. [Google Scholar] [CrossRef]
- Vlachos, P.; Kampioti, A.; Kornaros, M.; Lyberatos, G. Development and evaluation of alternative processes for sterilization and deodorization of cork barks and natural cork stoppers. Eur. Food Res. Technol. 2007, 225, 653–663. [Google Scholar] [CrossRef]
- Martí, M.P.; Boqué, R.; Busto, O.; Guasch, J. Electronic noses in the quality control of alcoholic beverages. Trends Anal. Chem. 2005, 24, 57–66. [Google Scholar] [CrossRef]
- Boudaoud, N.; Eveleigh, L. A New Approach to the Characterization of Volatile Signatures of Cork Wine Stoppers. J. Agric. Food Chem. 2003, 51, 1530–1533. [Google Scholar] [CrossRef]
- Besalú, E. Fast Modeling of Binding Affinities by means of Superposing Significant Interaction Rules (SSIR) method. Int. J. Mol. Sci. 2016, 17, 827. [Google Scholar] [CrossRef]
- MINITAB Version 14 for Windows; Minitab Inc.: State College, PA, USA, 2004.
- Besalú, E.; Pogliani, L.; De Julián-Ortiz, J.V. The Superposing Significant Interaction Rules (SSIR) Method in Applied Chemistry and Chemical Engineering; Volume 4 (Experimental Techniques and Methodical Developments); Haghi, A.K., Pogliani, L., Castro, E.A., Balköse, D., Mukbaniani, O.V., Chia, C.H., Eds.; Apple Academic Press (AAP): Waretown, NJ, USA, 2017; ISBN 9781771885874. [Google Scholar]
- Besalú, E.; Pogliani, L.; De Julián-Ortiz, J.V. Superposing Significant Interaction Rules (SSIR) method: A simple procedure for rapid ranking of congeneric compounds. Croat. Chem. Acta 2016, 89, 481–492. [Google Scholar] [CrossRef]
- Besalú, E.; Pogliani, L.; De Julián-Ortiz, J.V. Fast Qualitative Inspection of Designed Experiments by Means of the Superposing Significant Interaction Rules (SSIR) Method. In Physical Chemistry for Chemists and Chemical Engineers. Multidisciplinary Research Perspectives; Vakhrushev, A.V., Haghi, R., De Julián-Ortiz, J.V., Eds.; Series: Innovations in Physical Chemistry: Monograph Series (Volume 9); Apple Academic Press (AAP): Waretown, NJ, USA, 2019; ISBN 9781771886550. [Google Scholar]
- Rocha, S.M.; Coimbra, M.A.; Delgadillo, I. Occurrence of furfuraldehydes during the processing volatile of Quercus suber L. cork. Simultaneous determination of furfural, 5-hydroxymethylfurfural and 5-methylfurfural and their relation with cork polysaccharides. Carbohydr. Polym. 2004, 56, 287–293. [Google Scholar] [CrossRef]
- Horn, W.; Ullrich, D.; Seifert, B. VOC emissions from cork products for indoor use. Indoor Air 1998, 8, 39–46. [Google Scholar] [CrossRef]
- Brandão, P.F.; Ramos, R.M.; Almeida, P.J.; Rodrigues, J.A. Determination of carbonyl compounds in cork agglomerates by GDME-HPLC-UV: Identification of the extracted compounds by HPLC-MS/MS. J. Agric. Food Chem. 2017, 65, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Heras, M.; González-Sanjosé, M.L.; González-Huerta, C. Consideration of the influence of aging process, type of wine and oenological classic parameters on the levels of wood volatile compounds present in red wines. Food Chem. 2007, 103, 1434–1448. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Retention Time (min) | m/z 1 | Concentration (µg L−1) |
|---|---|---|---|
| 1,8-cineol (eucalyptol) | 12.33 | 81,108,139,154 | 3.95 |
| MDMP 2 | 12.66 | 137 | 0.46 |
| IPMP 2 | 13.55 | 137 | 0.38 |
| Guaiacol | 13.51 | 81,109,124 | 19.58 |
| (±)-linalool | 13.88 | 71,93,121 | 3.33 |
| (+)-fenchol | 14.33 | 81 | 5.26 |
| 1,2-dimethoxy benzene (veratrol) | 14.84 | 95,123,138 | 15.62 |
| Camphor | 15.00 | 95 | 3.66 |
| Sec-IBMP 2 | 15.42 | 124,138 | 0.68 |
| (-)-borneol | 15.6 | 95 | 4.25 |
| IBMP 2 | 15.65 | 124 | 21.12 |
| Menthol | 15.71 | 95 | 3.82 |
| Methylisoborneol | 16.02 | 95,108 | 0.57 |
| α-terpineol | 16.13 | 93,121,136 | 5.54 |
| Benzothiazole | 16.86 | 108,135 | 21.89 |
| TCA | 19.30 | 210,212 | 0.01 |
| d5-TCA | 19.22 | 215,217 | 0.06 |
| Geosmin | 21.50 | 112,125 | 0.6 |
| Variable # | Time Interval (min) | m/z Interval (amu) 1 | 1st PCA Loading in Figure 3 | 2nd PCA Loading in Figure 3 |
|---|---|---|---|---|
| 1 | [7.3, 7.7) | 95 | 0.335 | −0.106 |
| 2 | [6.7, 7.0) | 95 | 0.225 | 0.286 |
| 3 | [10.3, 10.7) | 110 | 0.341 | 0.194 |
| 4 | [10.3, 10.7) | 109 | 0.332 | 0.221 |
| 5 | [7.0, 7.3) | 97 | 0.315 | −0.058 |
| 6 | [7.0, 7.3) | 67 | 0.294 | 0.166 |
| 7 | [6.7, 7.0) | 96 | 0.249 | 0.198 |
| 8 | [7.3, 7.7) | 96 | 0.299 | −0.243 |
| 9 | [10.7, 11.0) | 109 | 0.285 | −0.217 |
| 10 | [10.7, 11.0) | 110 | 0.272 | −0.286 |
| 11 | [7.0, 7.3) | 51 | 0.308 | 0.237 |
| 12 | [8.7, 9.0) | 51 | −0.106 | 0.497 |
| 13 | [9.0, 9.3) | 78 | −0.111 | 0.509 |
| Variable # | High Level | Low Level | p-Value 1 | ||
|---|---|---|---|---|---|
| Non-Treated Samples | Treated Samples | Non-Treated Samples | Treated Samples | ||
| 1 | 2 | 27 | 26 | 1 | 1.4·10−12 |
| 2 | 5 | 28 | 23 | 0 | 3.1·10−11 |
| 3 | 0 | 23 | 28 | 5 | 3.1·10−11 |
| 4 | 0 | 23 | 28 | 5 | 3.1·10−11 |
| 5 | 4 | 25 | 24 | 3 | 9.3·10−9 |
| 6 | 0 | 16 | 28 | 12 | 7.3·10−7 |
| 7 | 15 | 28 | 13 | 0 | 2.0·10−5 |
| 8 | 14 | 27 | 14 | 1 | 7.1·10−5 |
| 9 | 0 | 11 | 28 | 17 | 1.4·10−4 |
| 10 | 0 | 11 | 28 | 17 | 1.4·10−4 |
| 11 | 0 | 10 | 28 | 18 | 3.7·10−4 |
| 12 | 10 | 0 | 18 | 28 | 3.7·10−4 |
| 13 | 12 | 1 | 16 | 27 | 4.7·10−4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Besalú, E.; Prat, C.; Anticó, E. Investigation of Volatiles in Cork Samples Using Chromatographic Data and the Superposing Significant Interaction Rules (SSIR) Chemometric Tool. Biomolecules 2020, 10, 896. https://doi.org/10.3390/biom10060896
Besalú E, Prat C, Anticó E. Investigation of Volatiles in Cork Samples Using Chromatographic Data and the Superposing Significant Interaction Rules (SSIR) Chemometric Tool. Biomolecules. 2020; 10(6):896. https://doi.org/10.3390/biom10060896
Chicago/Turabian StyleBesalú, Emili, Chantal Prat, and Enriqueta Anticó. 2020. "Investigation of Volatiles in Cork Samples Using Chromatographic Data and the Superposing Significant Interaction Rules (SSIR) Chemometric Tool" Biomolecules 10, no. 6: 896. https://doi.org/10.3390/biom10060896
APA StyleBesalú, E., Prat, C., & Anticó, E. (2020). Investigation of Volatiles in Cork Samples Using Chromatographic Data and the Superposing Significant Interaction Rules (SSIR) Chemometric Tool. Biomolecules, 10(6), 896. https://doi.org/10.3390/biom10060896