Meridianins and Lignarenone B as Potential GSK3β Inhibitors and Inductors of Structural Neuronal Plasticity

 ,
,  , and
, and
Abstract
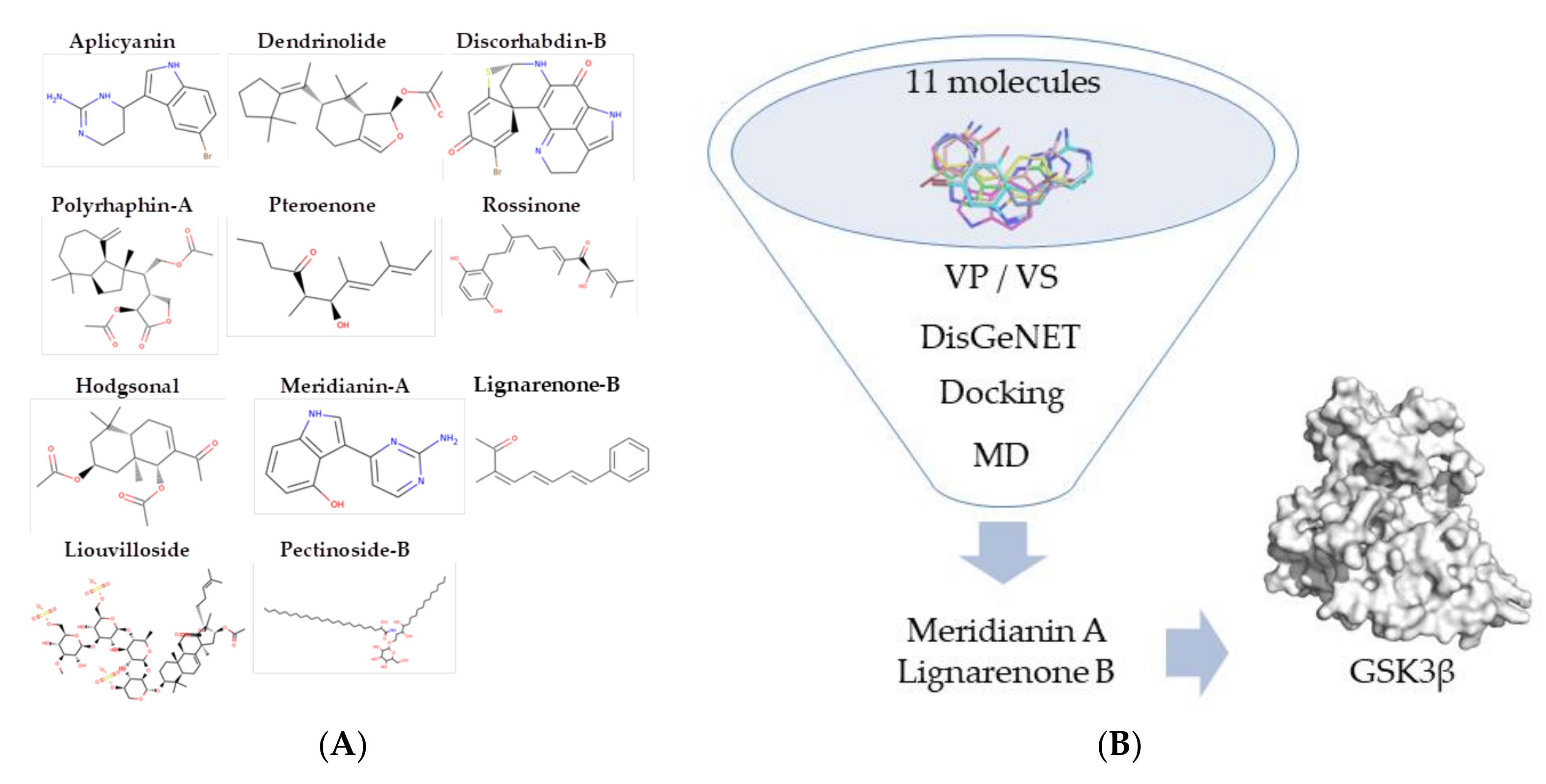
1. Introduction
2. Materials and Methods
2.1. Computational Analysis
2.1.1. Target Selection and Modelling
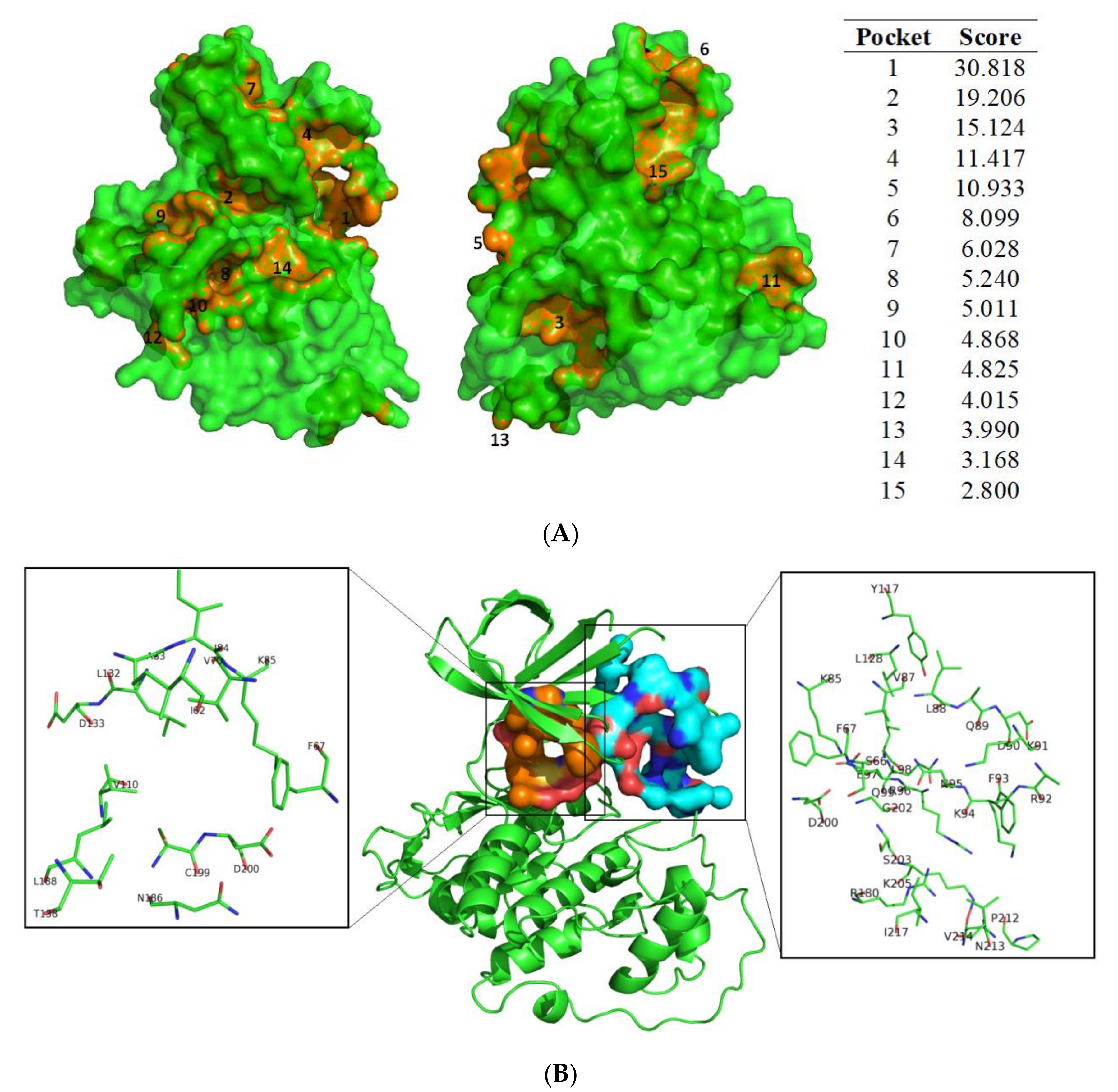
2.1.2. Cavity Search
2.1.3. Docking Calculations
2.1.4. Molecular Dynamics Simulation
2.1.5. Molecular Dynamics Analysis
2.1.6. Molecular Mechanics Generalized Born Surface Area
2.1.7. ADMET Prediction
2.1.8. Graphical Representations
2.2. In Vitro Analysis
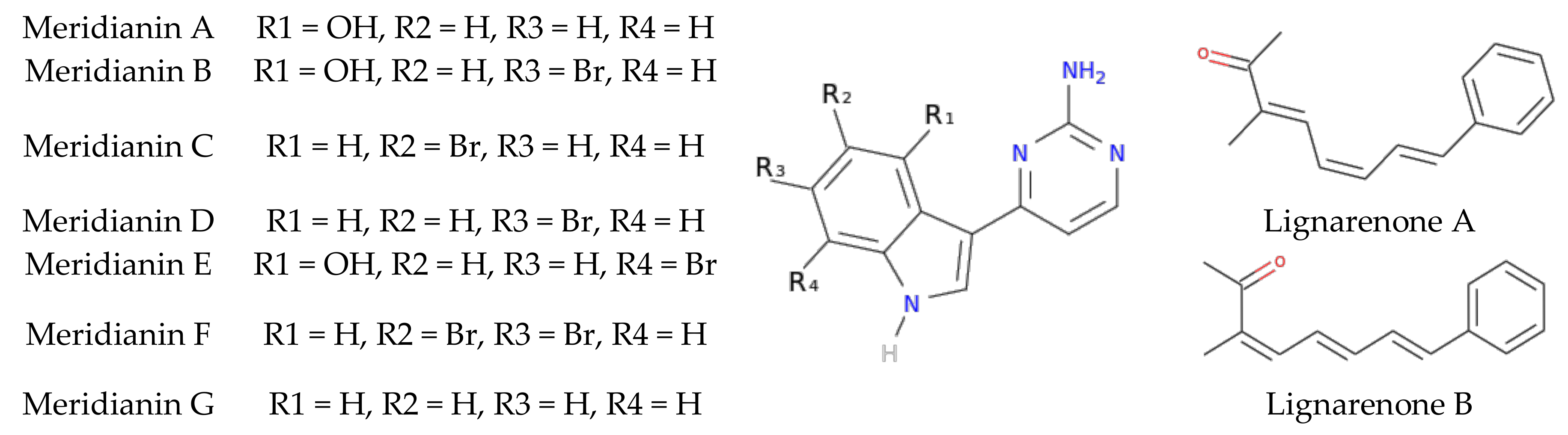
2.2.1. Marine Molecules
2.2.2. Primary Cortical Neuron Cultures
2.2.3. Immunoblot Analysis
2.2.4. Immunocytochemical Staining
2.2.5. Imaging and Analysis
2.3. Statistical Analysis
3. Results
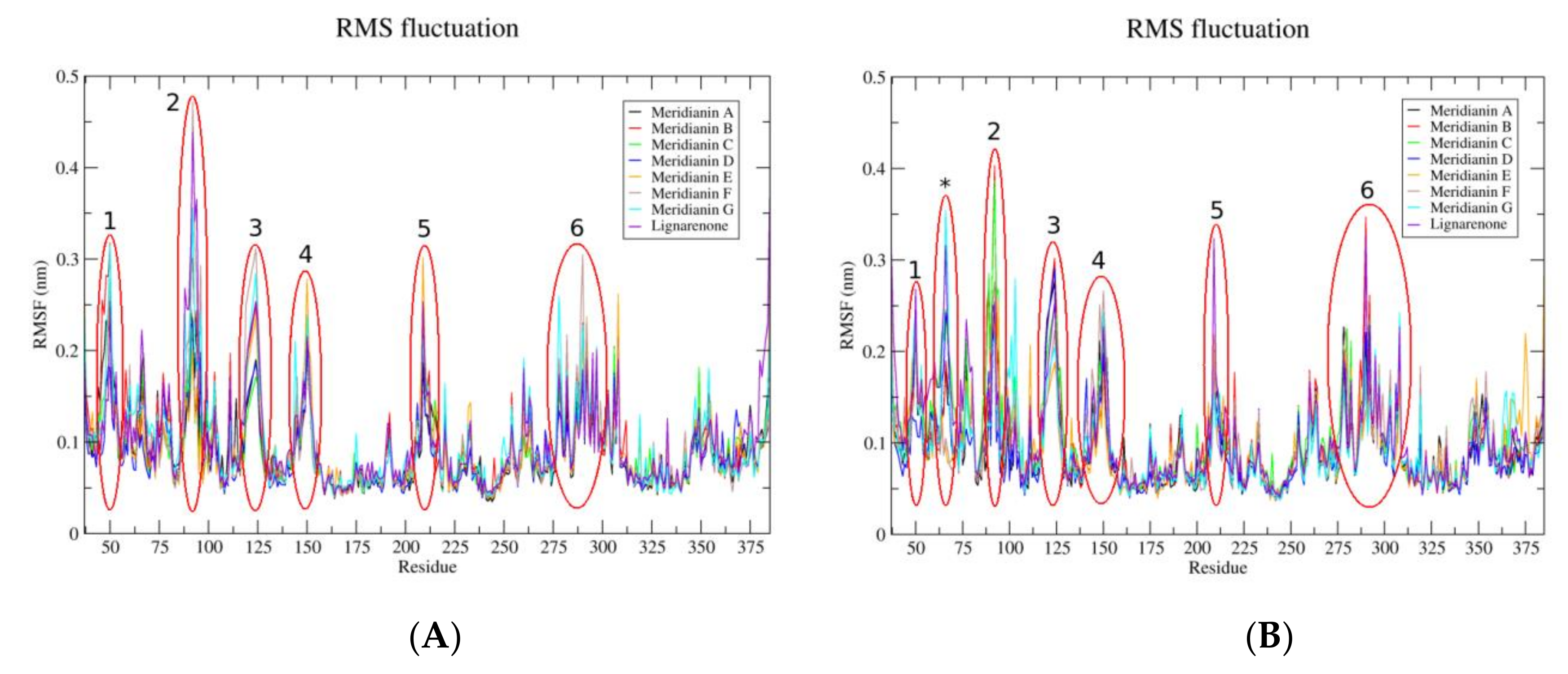
3.1. Exploring Druggable Binding Sites on GSK3β
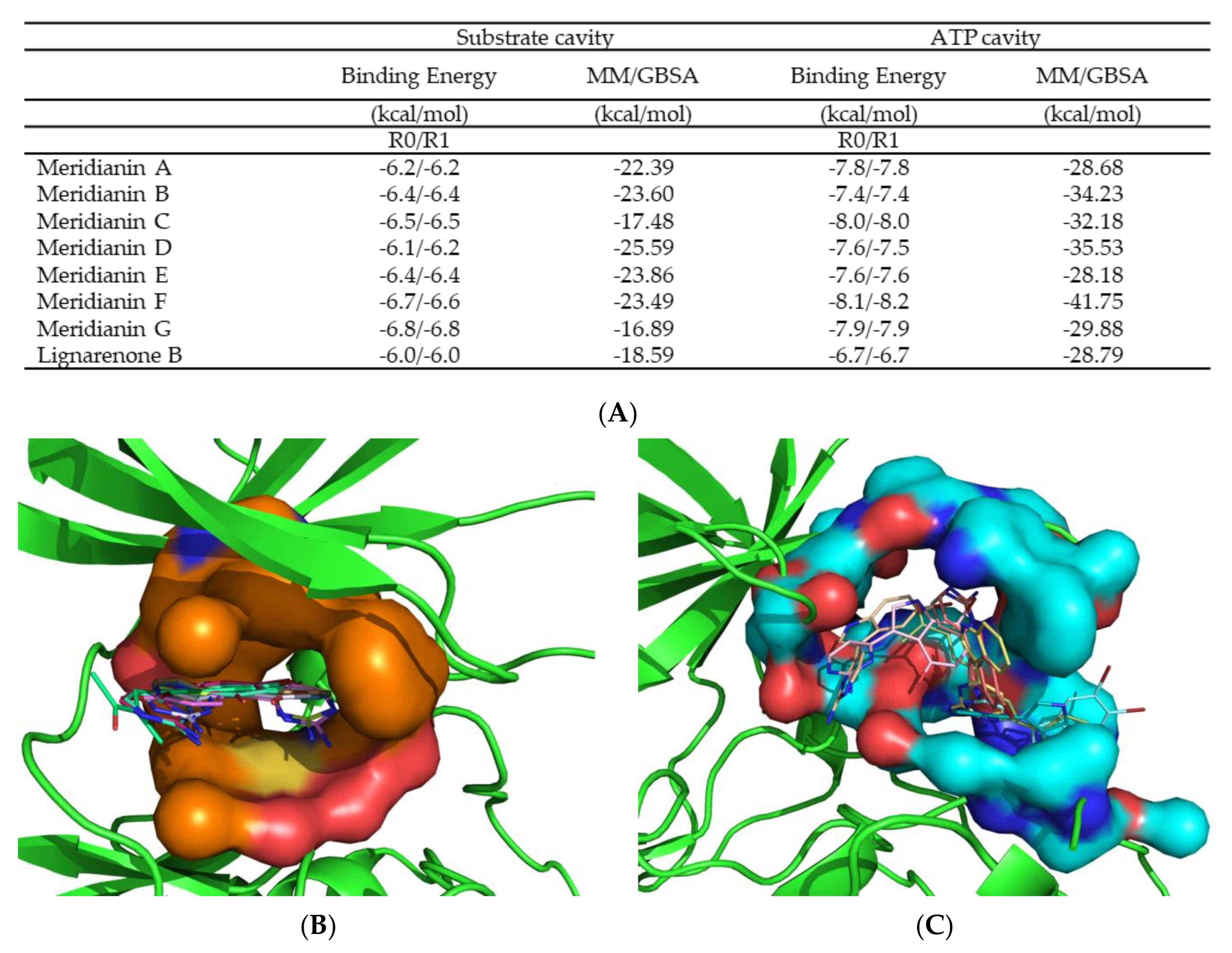
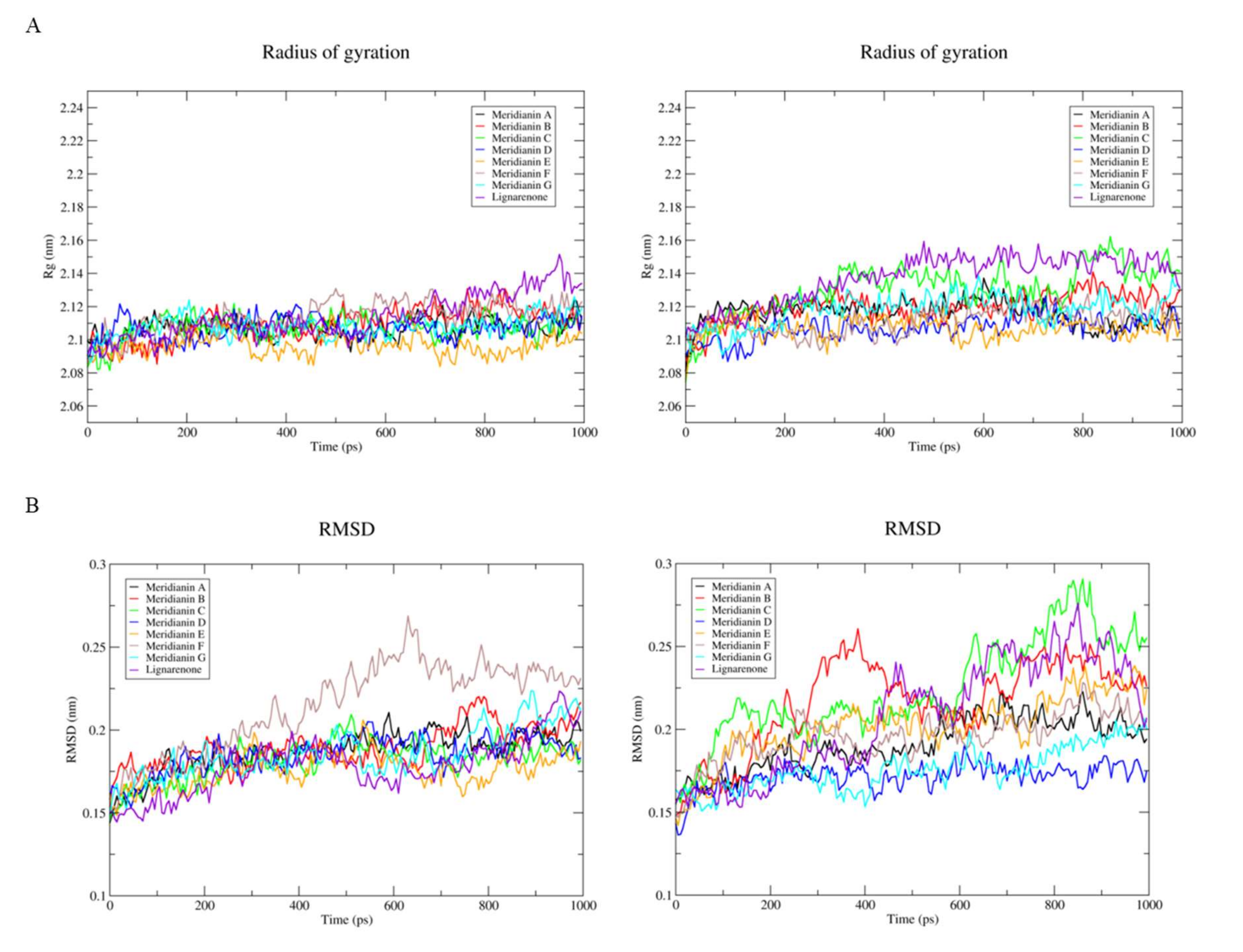
3.2. Binding of Meridianins and Lignarenone B to the ATP and Substrate Cavities
3.3. Pharmacokinetic Properties Evaluation
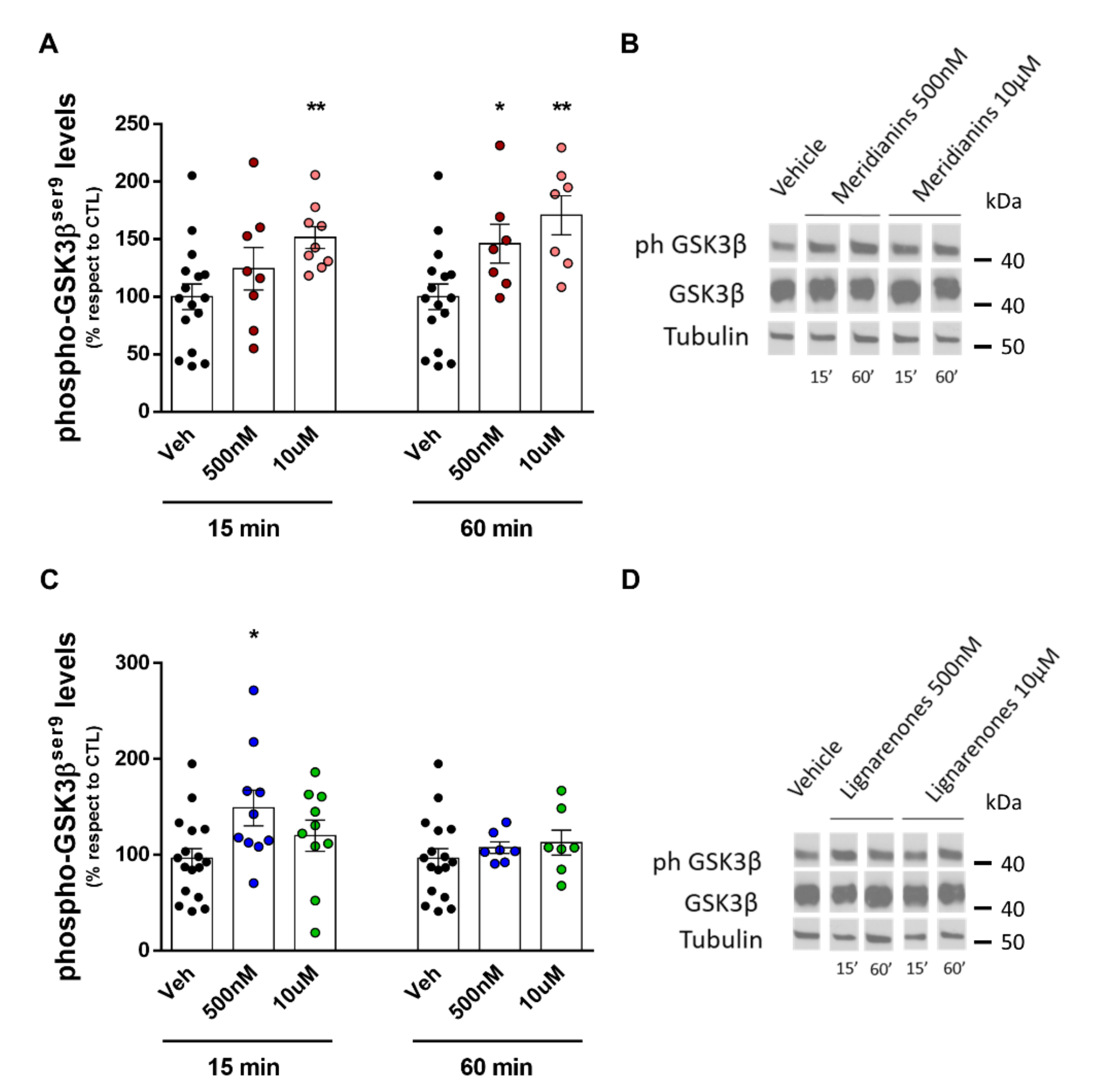
3.4. Meridianins and Lignarenones Differentially Increased pGSK3β Ser9, but Not Total GSK3β Levels In Vitro
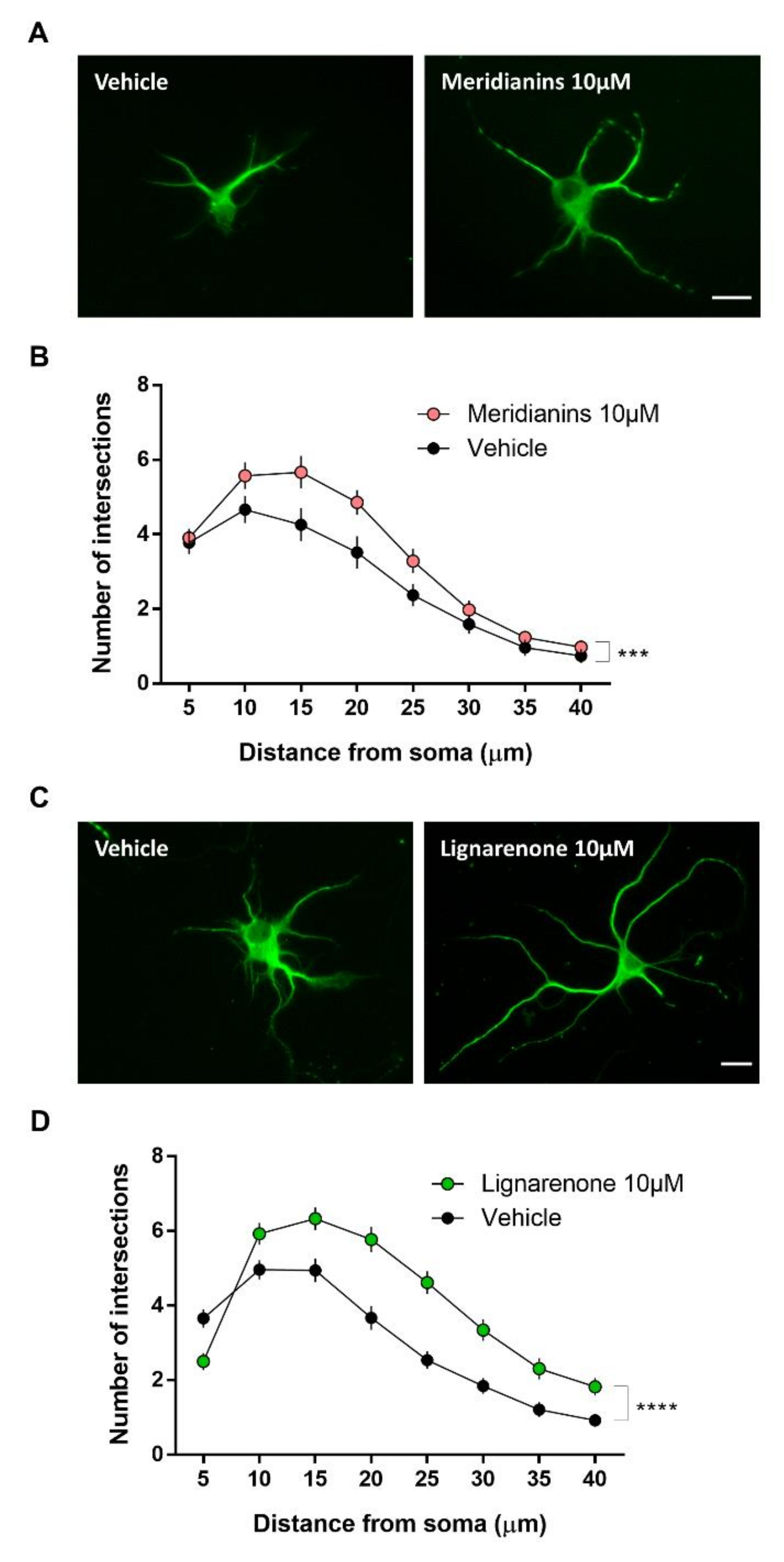
3.5. Meridianins and Lignarenones Regulate Neurite Complexity in Vitro
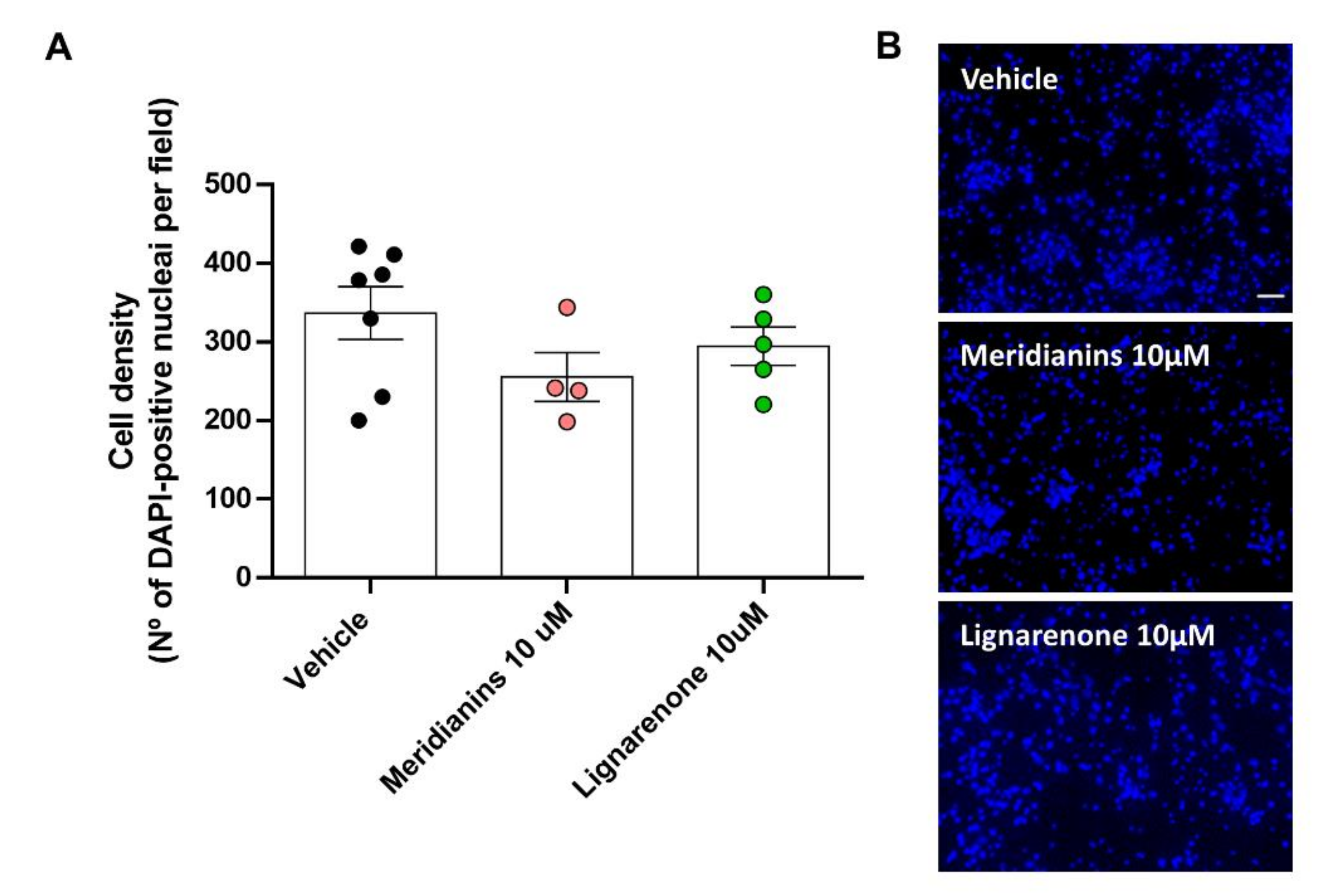
3.6. Effect of Meridianins and Lignarenones on Neuronal Viability
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.-P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Mandelkow, E.-M.; Drewes, G.; Biernat, J.; Gustke, N.; Van Lint, J.; Vandenheede, J.R.; Mandelkow, E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992, 314, 315–321. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Ding, V.W.; Chen, R.H.; McCormick, F. Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J. Biol. Chem. 2000, 275, 32475–32481. [Google Scholar] [CrossRef] [PubMed]
- Palsgaard, J.; Emanuelli, B.; Winnay, J.N.; Sumara, G.; Karsenty, G.; Kahn, C.R. Cross-talk between insulin and Wnt signaling in preadipocytes: Role of Wnt co-receptor low density lipoprotein receptor-related protein-5 (LRP5). J. Biol. Chem. 2012, 287, 12016–12026. [Google Scholar] [CrossRef]
- Li, D.-W.; Liu, Z.-Q.; Chen, W.; Yao, M.; Li, G.-R. Association of glycogen synthase kinase-3β with Parkinson’s disease. Mol. Med. Rep. 2014, 9, 2043–2050. [Google Scholar] [CrossRef]
- Llorens-Martín, M.; Jurado, J.; Hernández, F.; Avila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Lim, N.K.H.; Hung, L.W.; Pang, T.Y.; Mclean, C.A.; Liddell, J.R.; Hilton, J.B.; Li, Q.-X.; White, A.R.; Hannan, A.J.; Crouch, P.J. Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington’s disease affected brain. Hum. Mol. Genet. 2014, 23, 4051–4063. [Google Scholar] [CrossRef]
- Fernández-Nogales, M.; Hernández, F.; Miguez, A.; Alberch, J.; Ginés, S.; Pérez-Navarro, E.; Lucas, J. Decreased glycogen synthase kinase-3 levels and activity contribute to Huntington’s disease.Abstract—Europe PMC. Hum. Mol. Genet. 2015, 24, 5040–5052. [Google Scholar] [CrossRef]
- Kremer, A.; Louis, J.V.; Jaworski, T.; Van Leuven, F. GSK3 and Alzheimer’s Disease: Facts and Fiction…. Front. Mol. Neurosci. 2011, 4, 17. [Google Scholar] [CrossRef]
- Goedert, M.; Klug, A.; Crowther, R.A. Tau protein, the paired helical filament and Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D.; Ripova, D. Structure and Pathology of Tau Protein in Alzheimer Disease. Int. J. Alzheimers Dis. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Soteras, I.; Perez, D.I.; Perez, C.; Gil, C.; Eugenia Campillo, N.; Martinez, A. Exploring the Binding Sites of Glycogen Synthase Kinase 3. Identification and Characterization of Allosteric Modulation Cavities. J. Med. Chem. 2011, 54, 8461–8470. [Google Scholar] [CrossRef]
- Walz, A.; Ugolkov, A.; Chandra, S.; Kozikowski, A.; Carneiro, B.A.; O’Halloran, T.V.; Giles, F.J.; Billadeau, D.D.; Mazar, A.P. Molecular Pathways: Revisiting Glycogen Synthase Kinase-3β as a Target for the Treatment of Cancer. Clin. Cancer Res. 2017, 23, 1891–1897. [Google Scholar] [CrossRef]
- Pandey, M.K.; DeGrado, T.R. Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics 2016, 6, 571–593. [Google Scholar] [CrossRef]
- Martinez, A.; Gil, C.; Perez, D.I. Glycogen Synthase Kinase 3 Inhibitors in the Next Horizon for Alzheimer’s Disease Treatment. Int. J. Alzheimers Dis. 2011, 2011, 1–7. [Google Scholar] [CrossRef]
- Bhat, R.V.; Andersson, U.; Andersson, S.; Knerr, L.; Bauer, U.; Sundgren-Andersson, A.K. The Conundrum of GSK3 Inhibitors: Is it the Dawn of a New Beginning? J. Alzheimers Dis. 2018, 64, S547–S554. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Mohutsky, M.A.; Rehmel, J.L.F.; Ke, A.B. Investigational small-molecule drug selectively suppresses constitutive cyp2b6 activity at the gene transcription level: Physiologically based pharmacokinetic model assessment of clinical drug interaction risk. Drug Metab. Dispos. 2014, 42, 1008–1015. [Google Scholar] [CrossRef]
- Del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andrés, M.V.; Gómez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; León, T. Treatment of Alzheimer’s Disease with the GSK-3 Inhibitor Tideglusib: A Pilot Study. J. Alzheimers Dis. 2012, 33, 205–215. [Google Scholar] [CrossRef]
- Tolosa, E.; Litvan, I.; Höglinger, G.U.; Burn, D.; Lees, A.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; del Ser, T. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 2014, 29, 470–478. [Google Scholar] [CrossRef]
- Nwankwo, N.; Zhang, Z.; Wang, T.; Collins, C.; Resta, L.; Ermisch, S.; Day, J.; Decker, R.; Kornberg, L.; Nicol, S.; et al. Phase i study of enzastaurin and bevacizumab in patients with advanced cancer: Safety, efficacy and pharmacokinetics. Investig. New Drugs 2013, 31, 653–660. [Google Scholar] [CrossRef]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Kotliarova, S.; Walling, J.; Musib, L.; Thornton, D.; Albert, P.S.; Fine, H.A. A phase I trial of enzastaurin in patients with recurrent gliomas. Clin. Cancer Res. 2009, 15, 3617–3623. [Google Scholar] [CrossRef]
- Hampel, H.; Ewers, M.; Bürger, K.; Annas, P.; Mörtberg, A.; Bogstedt, A.; Frölich, L.; Schröder, J.; Schönknecht, P.; Riepe, M.W.; et al. Lithium trial in Alzheimer’s disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 2009, 70, 922–931. [Google Scholar] [CrossRef]
- Macdonald, A.; Briggs, K.; Poppe, M.; Higgins, A.; Velayudhan, L.; Lovestone, S. A feasibility and tolerability study of lithium in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2008, 23, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Kisialiou, A.; Lamonaca, P.; Moroni, R.; Prinzi, G.; Fini, M. New Drugs from Marine Organisms in Alzheimer’s Disease. Mar. Drugs 2015, 14, 5. [Google Scholar] [CrossRef]
- Ansari, N.; Khodagholi, F. Natural Products as Promising Drug Candidates for the Treatment of Alzheimer’s Disease: Molecular Mechanism Aspect. Curr. Neuropharmacol. 2013, 11, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Bhattacharya, R.; Mukherjee, A.; Pandey, D.K. Natural products against Alzheimer’s disease: Pharmaco-therapeutics and biotechnological interventions. Biotechnol. Adv. 2017, 35, 178–216. [Google Scholar] [CrossRef]
- Llorach-Pares, L.; Nonell-Canals, A.; Sanchez-Martinez, M.; Avila, C. Computer-Aided Drug Design Applied to Marine Drug Discovery: Meridianins as Alzheimer’s Disease Therapeutic Agents. Mar. Drugs 2017, 15, 366. [Google Scholar] [CrossRef] [PubMed]
- Llorach-Pares, L.; Nonell-Canals, A.; Avila, C.; Sanchez-Martinez, M. Kororamides, Convolutamines, and Indole Derivatives as Possible Tau and Dual-Specificity Kinase Inhibitors for Alzheimer’s Disease: A Computational Study. Mar. Drugs 2018, 16, 386. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [PubMed]
- Llorach-Pares, L. Computer Aided Drug Design Applied to Marine Drug Discovery. Ph.D. Thesis, University of Barcelona, Barcelona, Catalonia, Spain, 2019. [Google Scholar]
- Llorach-Pares, L.; Nonell-Canals, A.; Sanchez-Martinez, M.; Avila, C. In silico studies to find new therapeutic indications for marine molecules. In press.
- Núñez-Pons, L.; Forestieri, R.; Nieto, R.M.; Varela, M.; Nappo, M.; Rodríguez, J.; Jiménez, C.; Castelluccio, F.; Carbone, M.; Ramos-Espla, A.; et al. Chemical defenses of tunicates of the genus Aplidium from the Weddell Sea (Antarctica). Polar Biol. 2010, 33, 1319–1329. [Google Scholar] [CrossRef]
- Núñez-Pons, L.; Carbone, M.; Vázquez, J.; Rodríguez, J.; Nieto, R.M.; Varela, M.M.; Gavagnin, M.; Avila, C. Natural Products from Antarctic Colonial Ascidians of the Genera Aplidium and Synoicum: Variability and Defensive Role. Mar. Drugs 2012, 10, 1741–1764. [Google Scholar] [CrossRef] [PubMed]
- Cutignano, A.; Avila, C.; Rosica, A.; Romano, G.; Laratta, B.; Domenech-Coll, A.; Cimino, G.; Mollo, E.; Fontana, A. Biosynthesis and Cellular Localization of Functional Polyketides in the Gastropod Mollusc Scaphander lignarius. ChemBioChem 2012, 13, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Cutignano, A.; Avila, C.; Domenech-Coll, A.; d’Ippolito, G.; Cimino, G.; Fontana, A. First Biosynthetic Evidence on the Phenyl-Containing Polyketides of the Marine Mollusc Scaphander lignarius. Org. Lett. 2008, 10, 2963–2966. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Alonso, M.; Castro, A.; Concepción Pérez, A.; Moreno, F.J. First Non-ATP Competitive Glycogen Synthase Kinase 3 β (GSK-3β) Inhibitors: Thiadiazolidinones (TDZD) as Potential Drugs for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef]
- Palomo, V.; Perez, D.I.; Roca, C.; Anderson, C.; Rodríguez-Muela, N.; Perez, C.; Morales-Garcia, J.A.; Reyes, J.A.; Campillo, N.E.; Perez-Castillo, A.M.; et al. Subtly Modulating Glycogen Synthase Kinase 3 β: Allosteric Inhibitor Development and Their Potential for the Treatment of Chronic Diseases. J. Med. Chem. 2017, 60, 4983–5001. [Google Scholar] [CrossRef] [PubMed]
- Griebel, G.; Stemmelin, J.; Lopez-Grancha, M.; Boulay, D.; Boquet, G.; Slowinski, F.; Pichat, P.; Beeské, S.; Tanaka, S.; Mori, A.; et al. The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Borer, B.C.; Taylor, R.J.K. ChemInform Abstract: The Synthesis of Lignarenone A and Lignarenone B, Metabolites of the Mediterranean Opisthobranch Mollusc Scaphander lignarius. J. Chem. Res. 1990, 5, 162–163. [Google Scholar] [CrossRef]
- Bell, P.T.; Donaldson, W.A. Synthesis of Lignarenone B. J. Nat. Prod. 1992, 55, 1669–1671. [Google Scholar] [CrossRef] [PubMed]
- Tibiletti, F.; Simonetti, M.; Nicholas, K.M.; Palmisano, G.; Parravicini, M.; Imbesi, F.; Tollari, S.; Penoni, A. One-pot synthesis of meridianins and meridianin analogues via indolization of nitrosoarenes. Tetrahedron 2010, 66, 1280–1288. [Google Scholar] [CrossRef]
- Walker, S.R.; Czyz, M.L.; Morris, J.C. Concise syntheses of meridianins and meriolins using a catalytic domino amino-palladation reaction. Org. Lett. 2014, 16, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Wagman, A.S.; Boyce, R.S.; Brown, S.P.; Fang, E.; Goff, D.; Jansen, J.M.; Le, V.P.; Levine, B.H.; Ng, S.C.; Ni, Z.-J.; et al. Synthesis, Binding Mode, and Antihyperglycemic Activity of Potent and Selective (5-Imidazol-2-yl-4-phenylpyrimidin-2-yl)[2-(2-pyridylamino)ethyl]amine Inhibitors of Glycogen Synthase Kinase 3. J. Med. Chem. 2017, 60, 8482–8514. [Google Scholar] [CrossRef] [PubMed]
- Desaphy, J.; Bret, G.; Rognan, D.; Kellenberger, E. sc-PDB: A 3D-database of ligandable binding sites—10 years on. Nucleic Acids Res. 2014, 43, D399–D404. [Google Scholar] [CrossRef]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffeery, P. Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Felix, E.; Santamaría-Navarro, E.; Sanchez-Martinez, M.; Nonell-Canals, A. Itzamna. Available online: https://www.mindthebyte.com/ (accessed on 25 May 2018).
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Jenson, C. Temperature dependence of TIP3P, SPC, and TIP4P water from NPT Monte Carlo simulations: Seeking temperatures of maximum density. J. Comput. Chem. 1998, 19, 1179–1186. [Google Scholar] [CrossRef]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber, An Accessory Software Package For Molecular Mechanical Calculations. Natl. Inst. Health 2000, 21, 1049–1074. [Google Scholar]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. The Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Rastelli, G.; Degliesposti, G.; Del Rio, A.; Sgobba, M. Binding Estimation after Refinement, a New Automated Procedure for the Refinement and Rescoring of Docked Ligands in Virtual Screening. Chem. Biol. Drug Des. 2009, 73, 283–286. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Vidal, D.; Nonell-Canals, A. ADMET Models. Available online: https://www.mindthebyte.com/ (accessed on 19 July 2018).
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Grace User’s Guide (for Grace-5.1.22). Available online: http://plasma-gate.weizmann.ac.il/Grace/doc/UsersGuide.html (accessed on 26 April 2019).
- RDKit: Open-Source Cheminformatics; GitHub: San Francisco, CA, USA, 2019.
- Anglada-Huguet, M.; Vidal-Sancho, L.; Giralt, A.; García-Díaz Barriga, G.; Xifró, X.; Alberch, J. Prostaglandin E2 EP2 activation reduces memory decline in R6/1 mouse model of Huntington’s disease by the induction of BDNF-dependent synaptic plasticity. Neurobiol. Dis. 2016, 95, 22–34. [Google Scholar] [CrossRef]
- Xifró, X.; García-Martínez, J.M.; del Toro, D.; Alberch, J.; Pérez-Navarro, E. Calcineurin is involved in the early activation of NMDA-mediated cell death in mutant huntingtin knock-in striatal cells. J. Neurochem. 2008, 105, 1596–1612. [Google Scholar] [CrossRef]
- Osolodkin, D.I.; Palyulin, V.A.; Zefirov, N.S. Glycogen Synthase Kinase 3 as an Anticancer Drug Target: Novel Experimental Findings and Trends in the Design of Inhibitors. Curr. Pharm. Des. 2013, 19, 665–679. [Google Scholar] [CrossRef]
- Bidon-Chanal, A.; Fuertes, A.; Alonso, D.; Pérez, D.I.; Martínez, A.; Luque, F.J.; Medina, M. Evidence for a new binding mode to GSK-3: Allosteric regulation by the marine compound palinurin. Eur. J. Med. Chem. 2013, 60, 479–489. [Google Scholar] [CrossRef]
- Alqahtani, S. In silico ADME-Tox modeling: Progress and prospects. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1147–1158. [Google Scholar] [CrossRef]
- Ruiz-Garcia, A.; Bermejo, M.; Moss, A.; Casabo, V.G. Pharmacokinetics in Drug Discovery. J. Pharm. Sci. 2008, 97, 654–690. [Google Scholar] [CrossRef]
- Meijer, L.; Flajolet, M.; Greengard, P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 2004, 25, 471–480. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef] [PubMed]
- Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal structure of glycogen synthase kinase 3β: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 2001, 105, 721–732. [Google Scholar] [CrossRef]
- Zhan, F.; Phiel, C.J.; Spece, L.; Gurvich, N.; Klein, P.S. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium: Evidence for autoregulation of GSK-3. J. Biol. Chem. 2003, 278, 33067–33077. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- García-Díaz Barriga, G.; Giralt, A.; Anglada-Huguet, M.; Gaja-Capdevila, N.; Orlandi, J.G.; Soriano, J.; Canals, J.-M.; Alberch, J. 7,8-dihydroxyflavone ameliorates cognitive and motor deficits in a Huntington’s disease mouse model through specific activation of the PLCγ1 pathway. Hum. Mol. Genet. 2017, 26, 3144–3160. [Google Scholar] [CrossRef]
- Cabezas-Llobet, N.; Vidal-Sancho, L.; Masana, M.; Fournier, A.; Alberch, J.; Vaudry, D.; Xifró, X. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Enhances Hippocampal Synaptic Plasticity and Improves Memory Performance in Huntington’s Disease. Mol. Neurobiol. 2018, 55, 8263–8277. [Google Scholar] [CrossRef]
- Badrinarayan, P.; Sastry, G. Rational Approaches Towards Lead Optimization of Kinase Inhibitors: The Issue of Specificity. Curr. Pharm. Des. 2013, 19, 4714–4738. [Google Scholar] [CrossRef]
- Hamann, M.; Alonso, D.; Martín-Aparicio, E.; Fuertes, A.; Pérez-Puerto, M.J.; Castro, A.; Morales, S.; Navarro, M.L.; del Monte-Millán, M.; Medina, M.; et al. Glycogen Synthase Kinase-3 (GSK-3) Inhibitory Activity and Structure–Activity Relationship (SAR) Studies of the Manzamine Alkaloids. Potential for Alzheimer’s Disease. J. Nat. Prod. 2007, 70, 1397–1405. [Google Scholar] [CrossRef]
- Osolodkin, D.I.; Shulga, D.A.; Palyulin, V.A.; Zefirov, N.S. Interaction of manzamine A with glycogen synthase kinase 3β: A molecular dynamics study. Russ. Chem. Bull. 2010, 59, 1983–1993. [Google Scholar] [CrossRef]
- Eldar-Finkelman, H.; Licht-Murava, A.; Pietrokovski, S.; Eisenstein, M. Substrate Competitive GSK-3 Inhibitors {single bond} strategy and Implications. Biochim. Biophys. ActaProteins Proteom. 2010, 1804, 598–603. [Google Scholar] [CrossRef]
- Nussinov, R.; Ma, B.; Tsai, C.J. Multiple conformational selection and induced fit events take place in allosteric propagation. Biophys. Chem. 2014, 186, 22–30. [Google Scholar] [CrossRef]
- Kempner, E.S. Movable lobes and flexible loops in proteins Structural deformations that control biochemical activity. FEBS Lett. 1993, 326, 4–10. [Google Scholar] [CrossRef]
- Ilouz, R.; Kowalsman, N.; Eisenstein, M.; Eldar-Finkelman, H. Identification of novel glycogen synthase kinase-3beta substrate-interacting residues suggests a common mechanism for substrate recognition. J. Biol. Chem. 2006, 281, 30621–30630. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Yadav, R.; Battula, S.; Vishwakarma, R. Meridianins: Marine-Derived Potent Kinase Inhibitors. Mini Rev. Med. Chem. 2012, 12, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Alonso, D.; Martnez, A. Marine Compounds as a New Source for Glycogen Synthase Kinase 3 Inhibitors. In Glycogen Synthase Kinase 3 (GSK-3) and Its Inhibitors; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 307–331. [Google Scholar]
- Hansch, C.; Björkroth, J.P.; Leo, A. Hydrophobicity and central nervous system agents: On the principle of minimal hydrophobicity in drug design. J. Pharm. Sci. 1987, 76, 663–687. [Google Scholar] [CrossRef]
- Wang, W.; Bodles-Brakhop, A.M.; Barger, S.W. A Role for P-Glycoprotein in Clearance of Alzheimer Amyloid β -Peptide from the Brain. Curr. Alzheimer Res. 2016, 13, 615–620. [Google Scholar] [CrossRef]
- Miller, D.S.; Bauer, B.; Hartz, A.M.S. Modulation of P-glycoprotein at the blood-brain barrier: Opportunities to improve central nervous system pharmacotherapy. Pharmacol. Rev. 2008, 60, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.L.; Pee, H.N.; Yang, S.; Ho, P.C. Influence of drug transporters and stereoselectivity on the brain penetration of pioglitazone as a potential medicine against Alzheimer’s disease. Sci. Rep. 2015, 5, 9000. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Deane, R.; Fagan, A.M.; Spinner, M.L.; Parsadanian, M.; Finn, M.B.; Jiang, H.; Prior, J.L.; Sagare, A.; Bales, K.R.; et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2005, 115, 3285–3290. [Google Scholar] [CrossRef]
- Roden, D.M.; Viswanathan, P.C. Genetics of acquired long QT syndrome. J. Clin. Investig. 2005, 115, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Swaab, D.F.; Hofman, M.A.; Lucassen, P.J.; Salehi, A.; Uylings, H.B.M. Neuronal atrophy, not cell death, is the main hallmark of Alzheimer’s disease. Neurobiol. Aging 1994, 15, 369–371. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. ActaMol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. J. Chem. Res. 2019, 5, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Garrido, J.J.; Wandosell, F.G. Modulation of GSK-3 as a therapeutic strategy on tau pathologies. Front. Mol. Neurosci. 2011, 4, 24. [Google Scholar] [CrossRef]
- Kingwell, K. Drug delivery: New targets for drug delivery across the BBB. Nat. Rev. Drug Discov. 2016, 15, 84–85. [Google Scholar] [CrossRef]
- Banks, W.A. From blood–brain barrier to blood–brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-mediated brain drug delivery: Overcoming blood–brain barrier to treat neurodegenerative diseases. J. Med. Chem. 2016, 235, 34–47. [Google Scholar] [CrossRef]
- Liu, B.; Xu, R.; He, X. Kinetic Energy-Based Temperature Computation in Non-Equilibrium Molecular Dynamics Simulation. J. Comput. Theor. Nanosci. 2009, 9, 428–433. [Google Scholar]
- Merz, P.T.; Shirts, M.R. Testing for physical validity in molecular simulations. PLoS ONE 2018, 13, e0202764. [Google Scholar] [CrossRef]
- Jepps, A.; Ayton, J.; Evans, E. Microscopic expressions for the thermodynamic temperature. Phys. Rev. E. Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 2000, 62, 4757–4763. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [PubMed]
- Maiorov, V.N.; Crippen, G.M. Significance of Root-Mean-Square Deviation in Comparing Three-dimensional Structures of Globular Proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Mackerell, A.D. Computer-Aided Drug Design Methods. Methods Mol. Biol. 2017, 1520, 93–94. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent/Chemical/Antibody | Manufacturer | Reference |
|---|---|---|
| Neurobasal medium | Gibco | 21103049 |
| B27 | Gibco | 17504-044 |
| GlutaMAX | Gibco | 35050-038 |
| Tripanblue solution | Sigma | T8154 |
| Chemilluminescence ECL kit | Santa Cruz Biotechnology | sc-2048 |
| Normal donkey serum | Jackson Immunores. Labs. | 017-000-121 |
| 4% paraformaldehyde | Pierce | 28908 |
| Anti-GSK3β | Cell Signalling | #9315 |
| Anti-phosphoGSK3βat Ser9 | Cell Signalling | #9336 |
| Anti-MAP2 | Sigma-Aldrich | M1406 |
| Anti-α-Tubulin | Sigma-Aldrich | T9026 |
| Horseradish peroxidase-conjugated secondary antibody | Promega | W4021 |
| Alexa fluor 488-conjugatedanti-mouse | Jackson Immunores. Labs. | 715-545-150 |
| DAPI Fluoromont | Southern Biotech | 010020 |
| Absorption | Distribution | Toxicity | ||||||
|---|---|---|---|---|---|---|---|---|
| Mol Weight | logS | Pgp | Caco2 | logP | BBB | PPB | hERG | |
| Meridianin A | 226.2 | −4.2 | inactive | High | 1.5 | NO | High | <4.0 |
| Meridianin B | 305.1 | −5.0 | inactive | High | 2.4 | NO | High | <4.0 |
| Meridianin C | 289.1 | −5.6 | inactive | High | 3.1 | NO | High | <4.0 |
| Meridianin D | 289.1 | −5.6 | inactive | High | 3.1 | NO | High | <4.0 |
| Meridianin E | 305.1 | −5.0 | inactive | High | 2.4 | NO | High | <4.0 |
| Meridianin F | 368.0 | −6.2 | inactive | High | 3.6 | NO | High | <4.0 |
| Meridianin G | 210.2 | −4.5 | inactive | High | 2.4 | NO | High | <4.0 |
| Lignarenone B | 212.3 | −3.2 | inactive | Low | 3.6 | NO | High | <4.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Llorach-Pares, L.; Rodriguez-Urgelles, E.; Nonell-Canals, A.; Alberch, J.; Avila, C.; Sanchez-Martinez, M.; Giralt, A. Meridianins and Lignarenone B as Potential GSK3β Inhibitors and Inductors of Structural Neuronal Plasticity. Biomolecules 2020, 10, 639. https://doi.org/10.3390/biom10040639
Llorach-Pares L, Rodriguez-Urgelles E, Nonell-Canals A, Alberch J, Avila C, Sanchez-Martinez M, Giralt A. Meridianins and Lignarenone B as Potential GSK3β Inhibitors and Inductors of Structural Neuronal Plasticity. Biomolecules. 2020; 10(4):639. https://doi.org/10.3390/biom10040639
Chicago/Turabian StyleLlorach-Pares, Laura, Ened Rodriguez-Urgelles, Alfons Nonell-Canals, Jordi Alberch, Conxita Avila, Melchor Sanchez-Martinez, and Albert Giralt. 2020. "Meridianins and Lignarenone B as Potential GSK3β Inhibitors and Inductors of Structural Neuronal Plasticity" Biomolecules 10, no. 4: 639. https://doi.org/10.3390/biom10040639
APA StyleLlorach-Pares, L., Rodriguez-Urgelles, E., Nonell-Canals, A., Alberch, J., Avila, C., Sanchez-Martinez, M., & Giralt, A. (2020). Meridianins and Lignarenone B as Potential GSK3β Inhibitors and Inductors of Structural Neuronal Plasticity. Biomolecules, 10(4), 639. https://doi.org/10.3390/biom10040639