Structural Determinants Responsible for the Preferential Insertion of Ribonucleotides by Bacterial NHEJ PolDom


Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Oligonucleotides
2.2. Expression and Purification of Recombinant Pa-PolDom
2.3. Site-Directed Mutagenesis
2.4. Nucleotide Insertion Assays on Defined DNA Molecules
2.5. Steady-State Primer Extension Assays
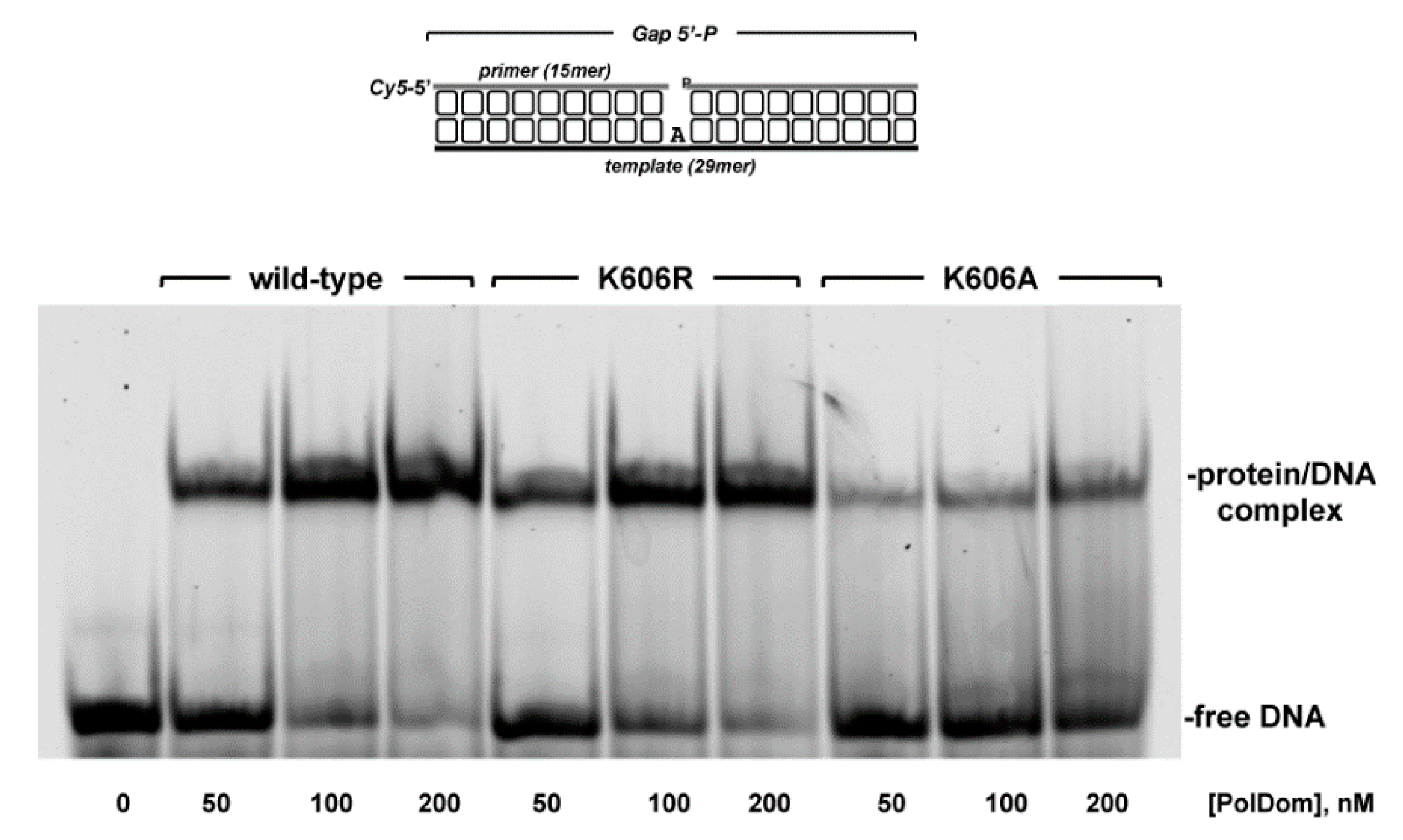
2.6. Electrophoretic Mobility Shift Assays (EMSAs)
2.7. Kinetic Measurement of 8oxoGMP Incorporation by PaPolDom
3. Results and Discussion
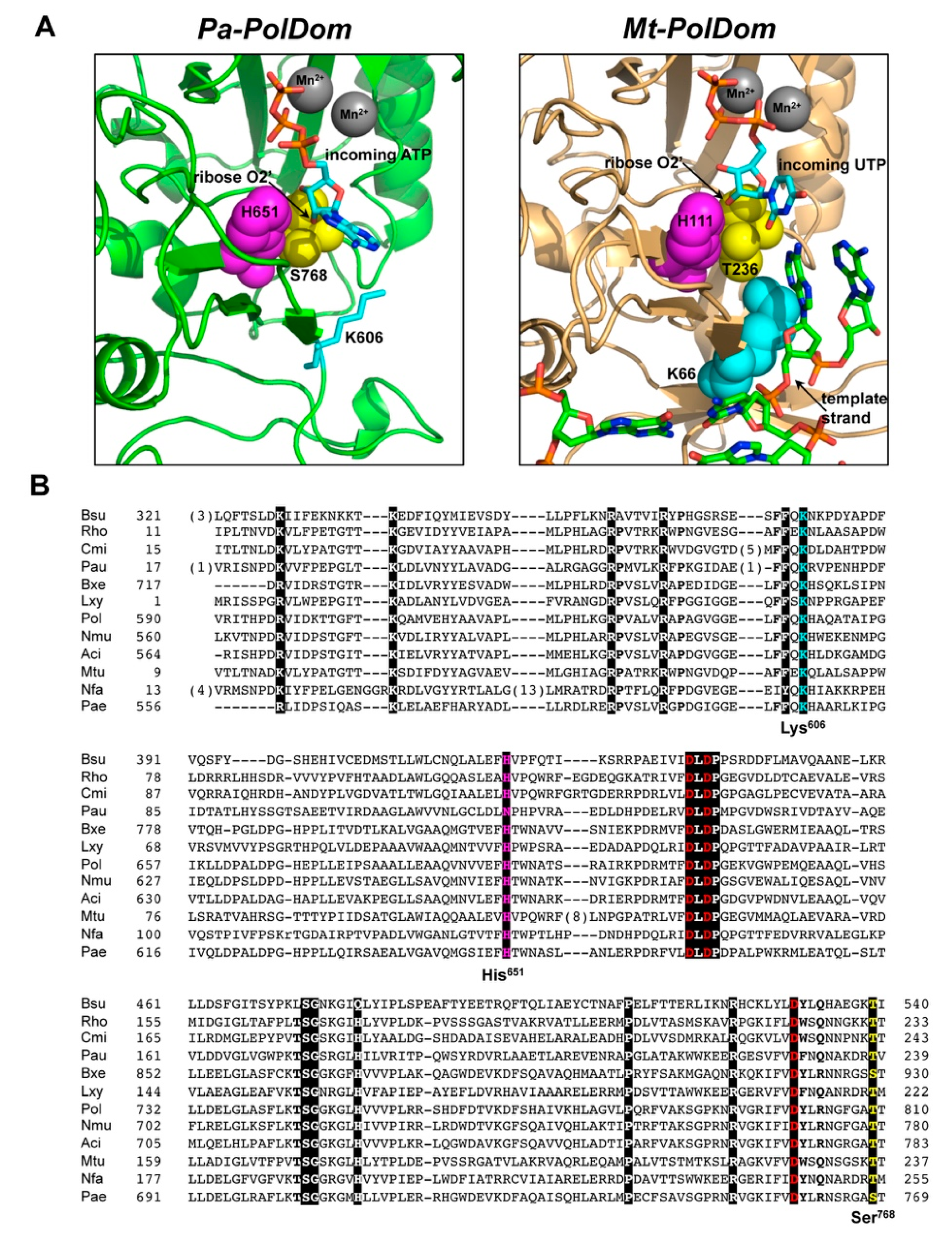
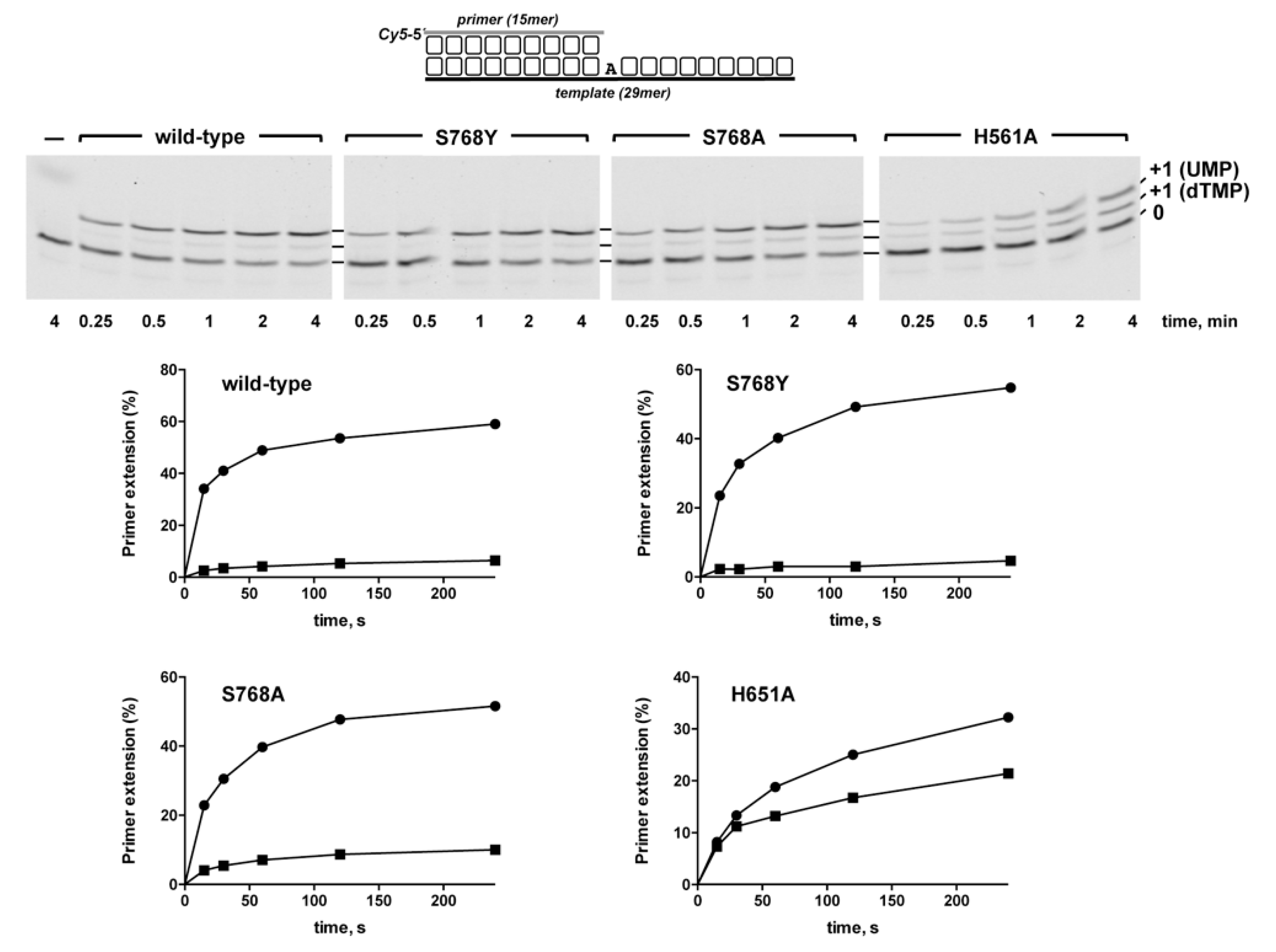
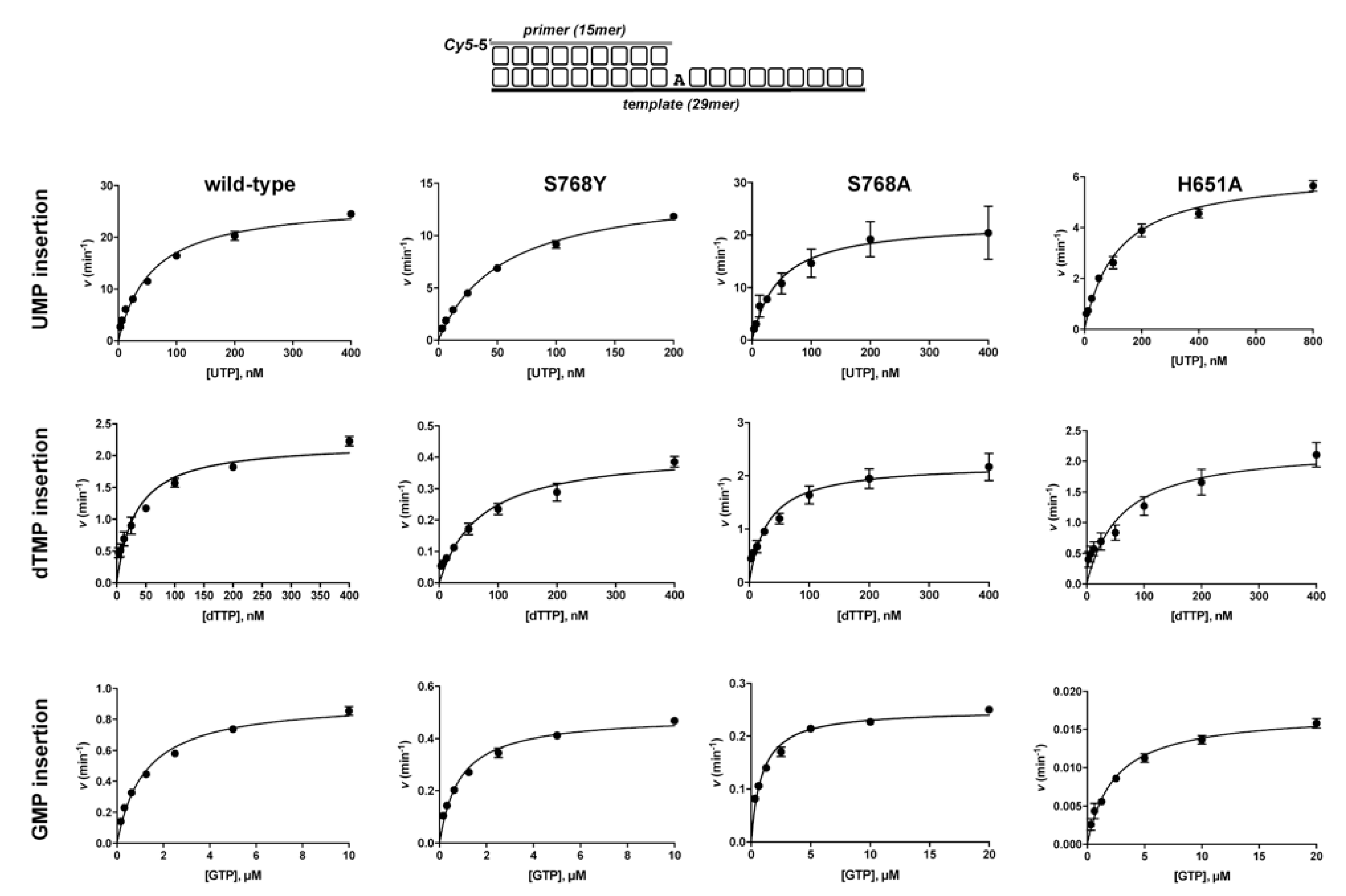
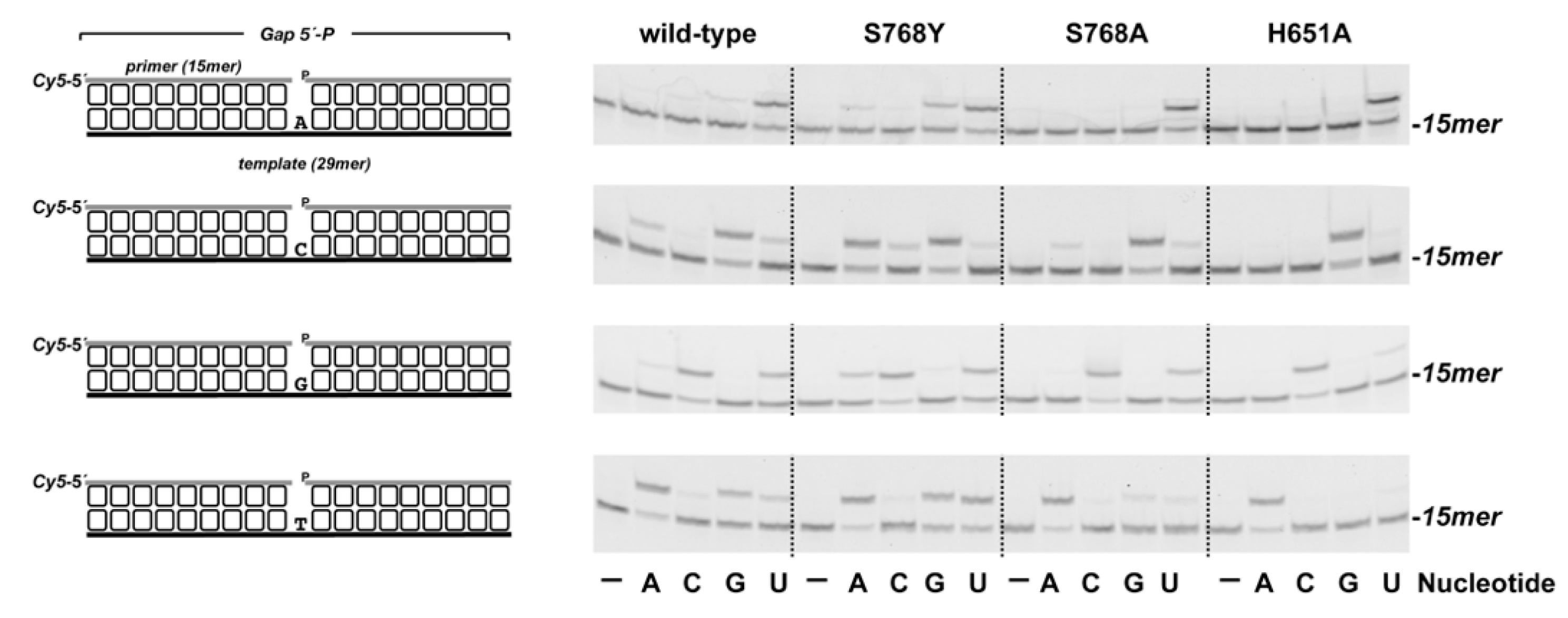
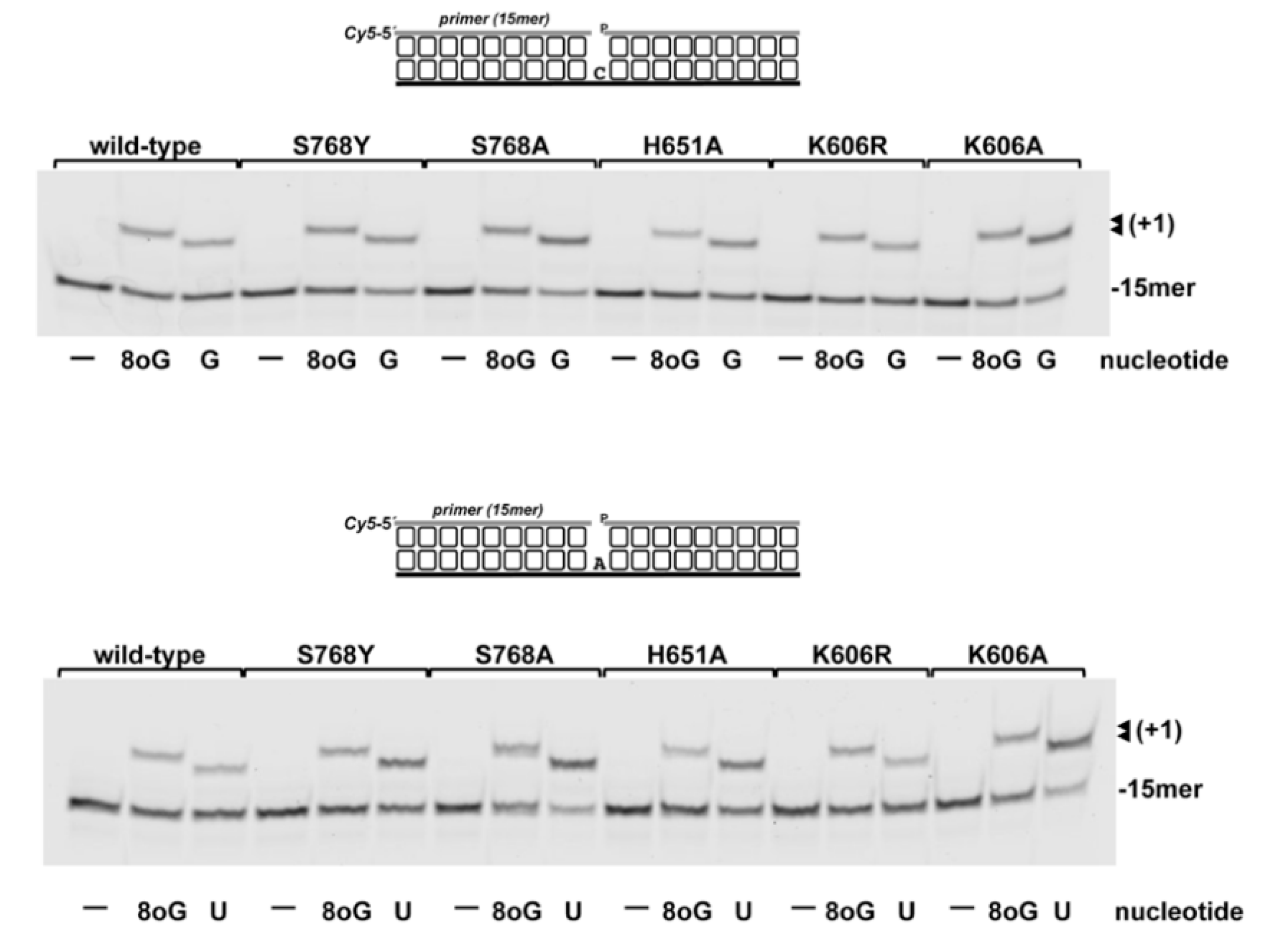
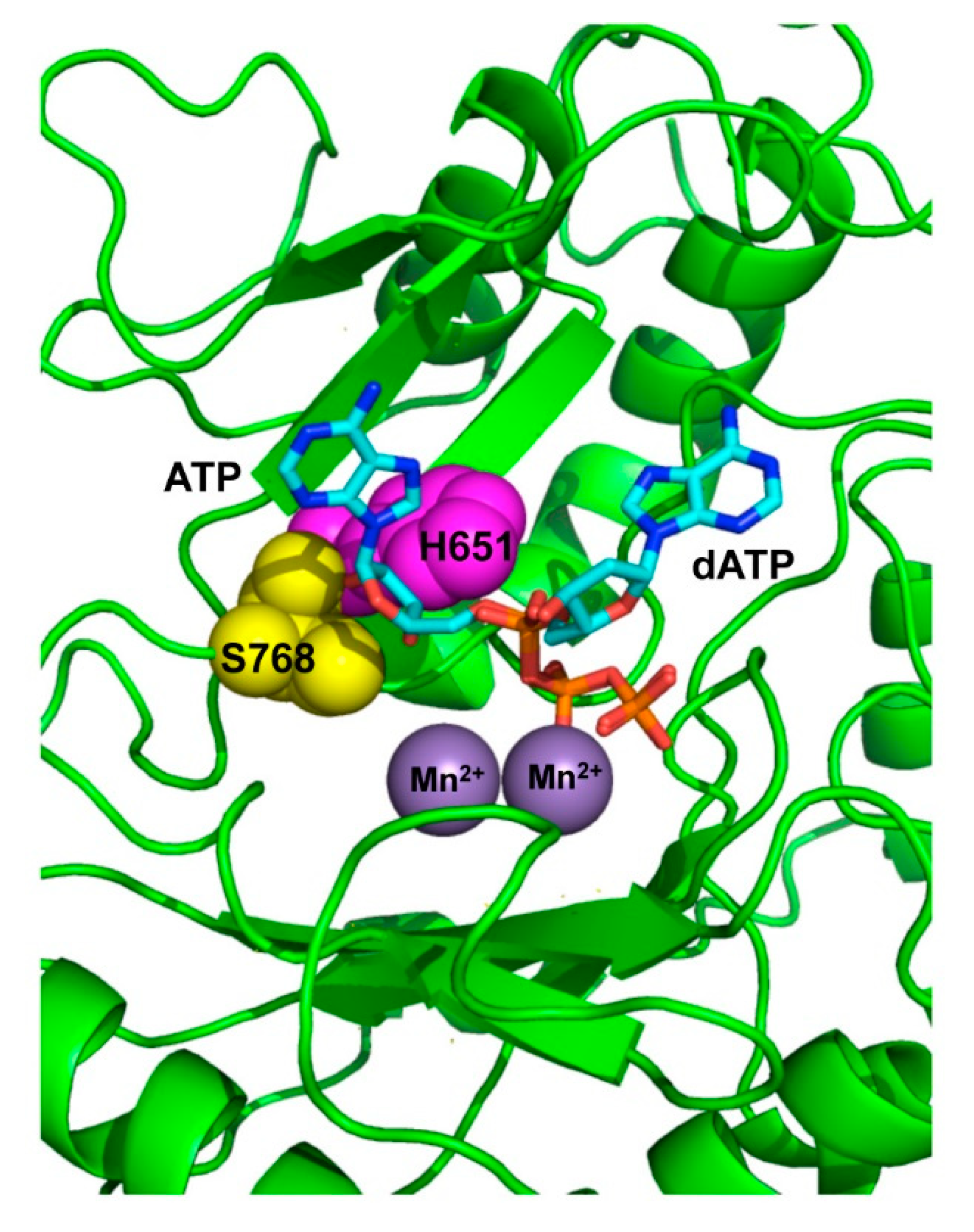
3.1. Role of Pa-LigD Residues His651 and Ser768 in Preferential Insertion of Ribonucleotides and Fidelity
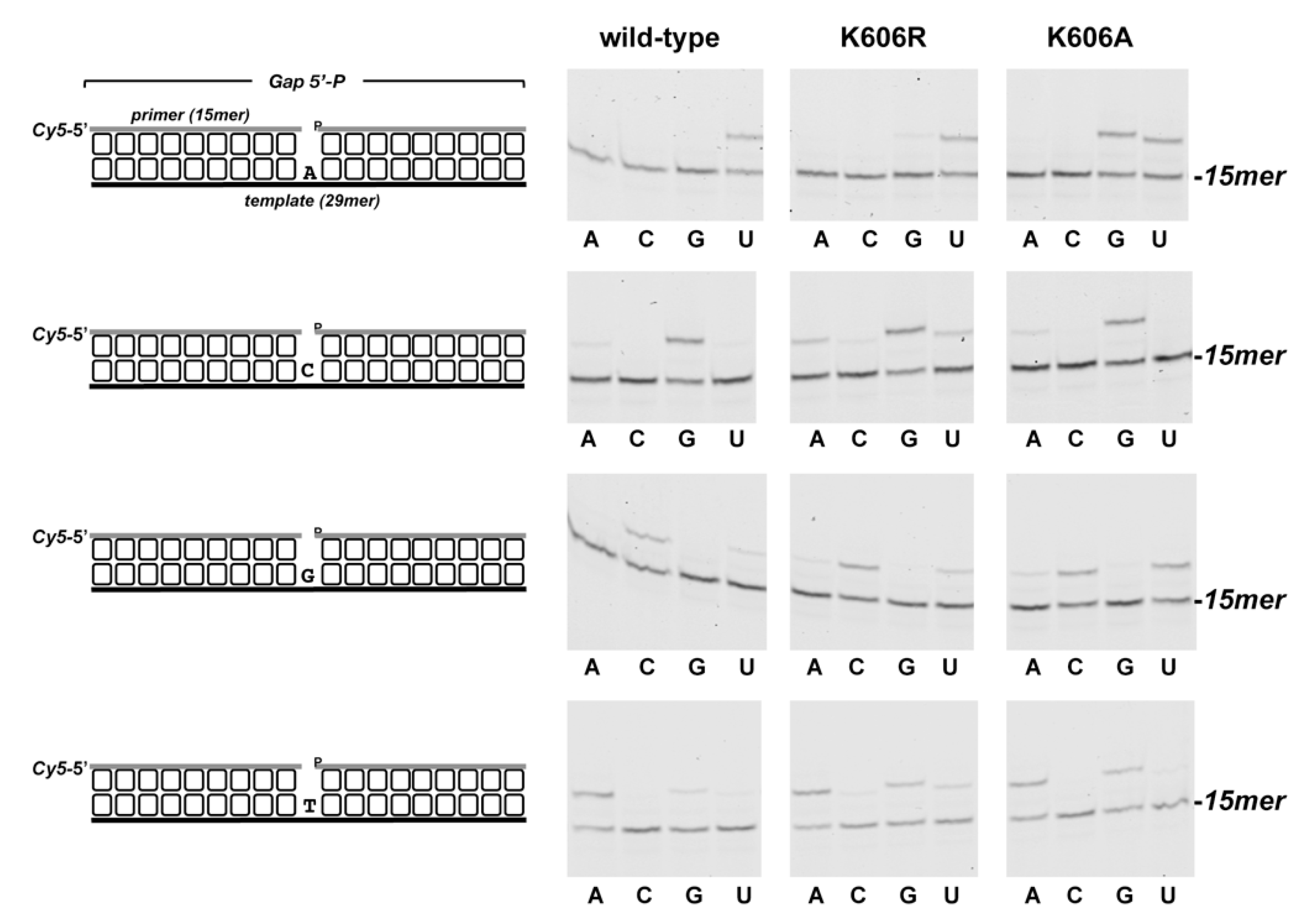
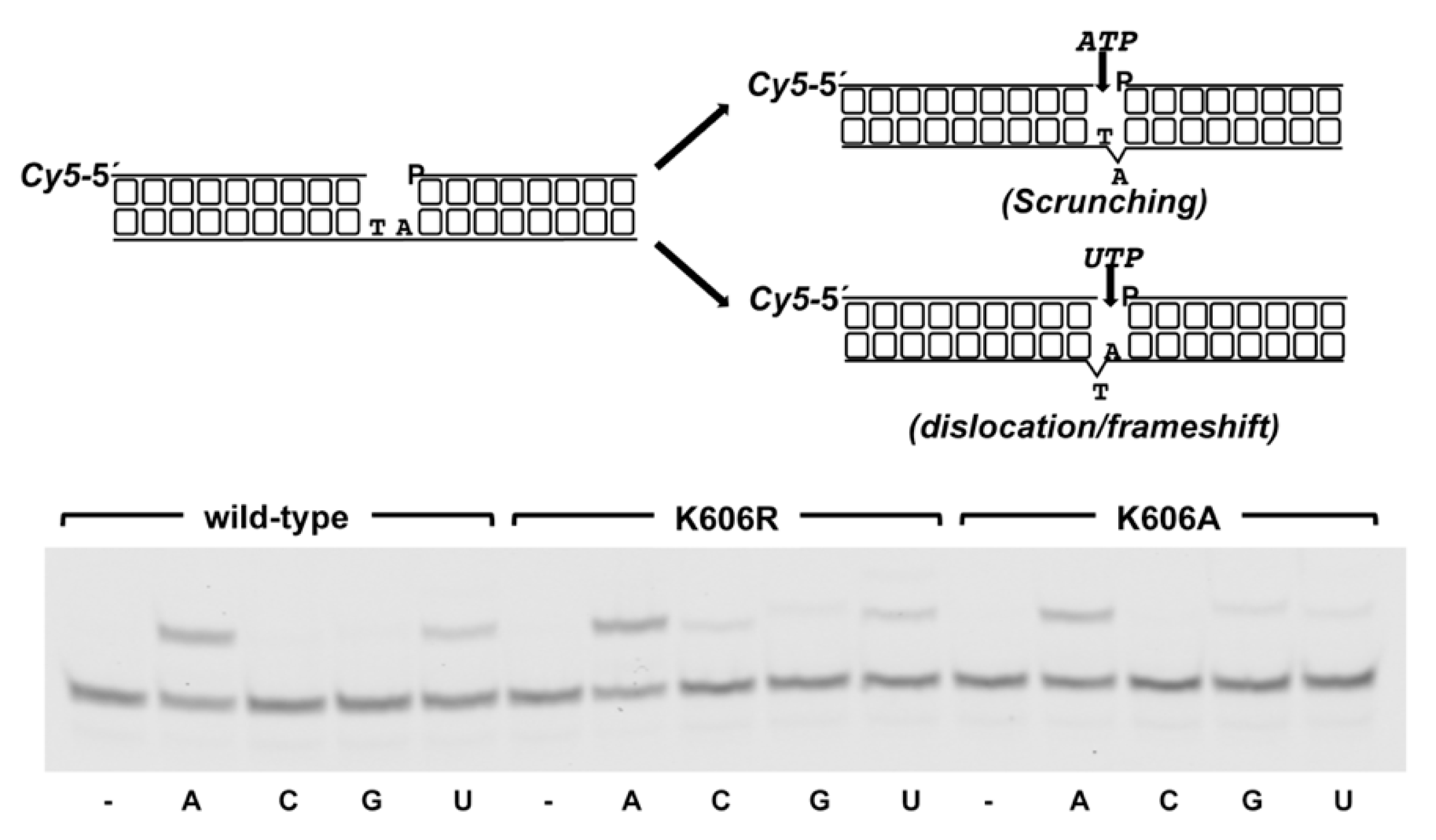
3.2. Role of Pa-LigD Lys606 in Dislocation of Proximal Templating Nucleotides
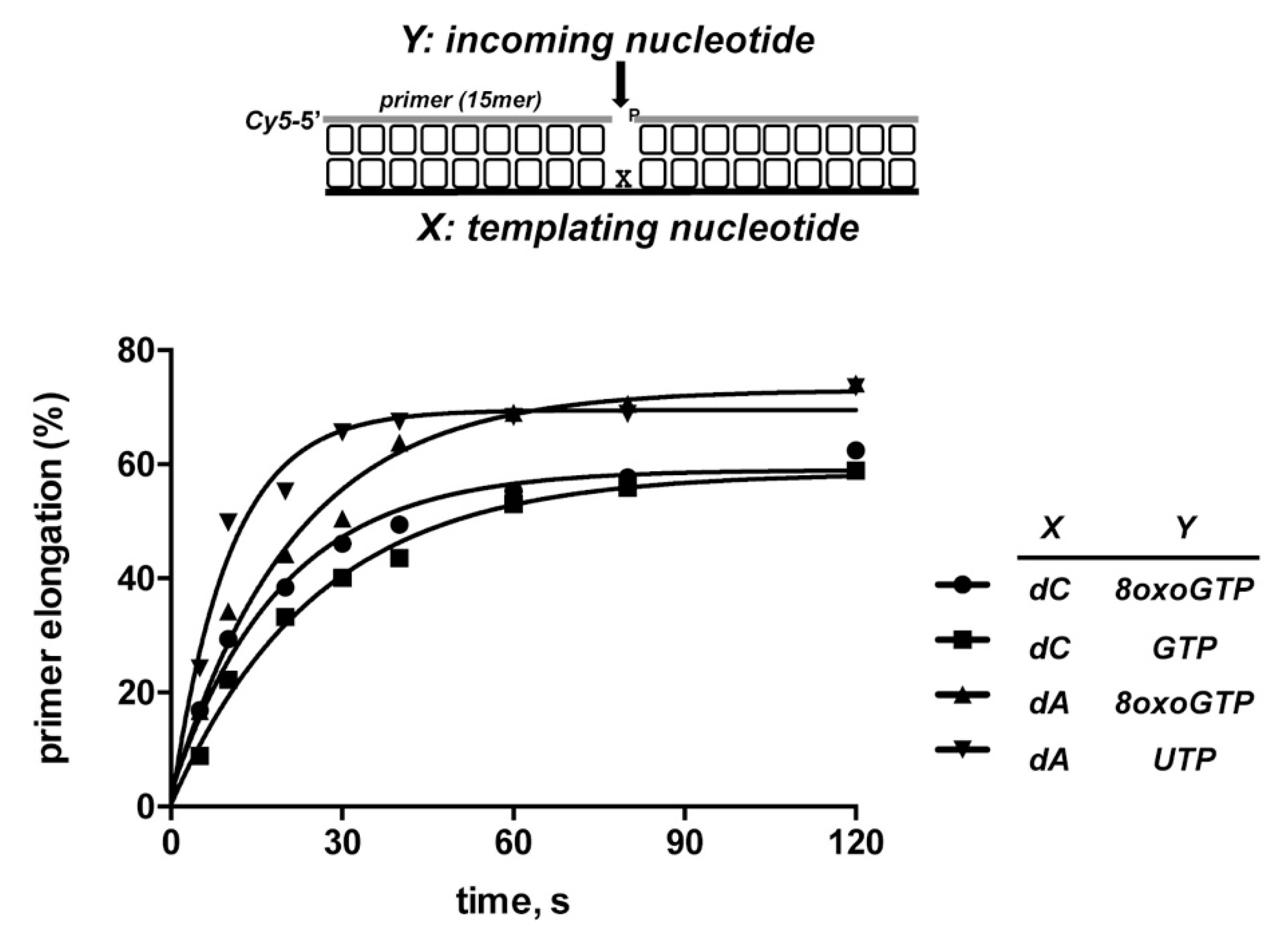
3.3. 8oxoGMP Is Efficiently Inserted During In Vitro Polymerization by PolDom
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double−strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end−joining for repair of DNA double−strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.G.; Cooper, J.P. Two modes of DNA double−strand break repair are reciprocally regulated through the fission yeast cell cycle. Genes Dev. 2004, 18, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Morrison, C.; Hashimoto, M.; Utsumi, H.; Yamaguchi−Iwai, Y.; Shinohara, A.; Takeda, S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998, 17, 5497–5508. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Palmbos, P.L.; Wu, D.; Wilson, T.E. Nonhomologous end joining in yeast. Annu. Rev. Genet. 2005, 39, 431–451. [Google Scholar] [CrossRef]
- Mahaney, B.L.; Meek, K.; Lees−Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009, 417, 639–650. [Google Scholar] [CrossRef]
- Gottlieb, T.M.; Jackson, S.P. The DNA−dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 1993, 72, 131–142. [Google Scholar] [CrossRef]
- Lees-Miller, S.P.; Meek, K. Repair of DNA double strand breaks by non−homologous end joining. Biochimie 2003, 85, 1161–1173. [Google Scholar]
- Aravind, L.; Koonin, E.V. Prokaryotic homologs of the eukaryotic DNA−end−binding protein Ku, novel domains in the Ku protein and prediction of a prokaryotic double−strand break repair system. Genome Res. 2001, 11, 1365–1374. [Google Scholar] [CrossRef]
- Doherty, A.J.; Jackson, S.P.; Weller, G.R. Identification of bacterial homologues of the Ku DNA repair proteins. FEBS Lett. 2001, 500, 186–188. [Google Scholar] [CrossRef]
- Pitcher, R.S.; Brissett, N.C.; Doherty, A.J. Nonhomologous end−joining in bacteria: A microbial perspective. Annu. Rev. Microbiol. 2007, 61, 259–282. [Google Scholar] [CrossRef] [PubMed]
- Weller, G.R.; Kysela, B.; Roy, R.; Tonkin, L.M.; Scanlan, E.; Della, M.; Devine, S.K.; Day, J.P.; Wilkinson, A.; d’Adda di Fagagna, F.; et al. Identification of a DNA nonhomologous end−joining comple× in bacteria. Science 2002, 297, 1686–1689. [Google Scholar] [CrossRef] [PubMed]
- Della, M.; Palmbos, P.L.; Tseng, H.M.; Tonkin, L.M.; Daley, J.M.; Topper, L.M.; Pitcher, R.S.; Tomkinson, A.E.; Wilson, T.E.; Doherty, A.J. Mycobacterial Ku and ligase proteins constitute a two−component NHEJ repair machine. Science 2004, 306, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Bongiorno, P.; Martins, A.; Stephanou, N.C.; Zhu, H.; Shuman, S.; Glickman, M.S. Mechanism of nonhomologous end−joining in mycobacteria: A low−fidelity repair system driven by Ku, ligase D and ligase C. Nat. Struct. Mol. Biol. 2005, 12, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Martins, A.; Bongiorno, P.; Glickman, M.; Shuman, S. Biochemical and genetic analysis of the four DNA ligases of mycobacteria. J. Biol. Chem. 2004, 279, 20594–20606. [Google Scholar] [CrossRef]
- Koonin, E.V.; Wolf, Y.I.; Kondrashov, A.S.; Aravind, L. Bacterial homologs of the small subunit of eukaryotic DNA primase. J. Mol. Microbiol. Biotechnol. 2000, 2, 509–512. [Google Scholar]
- Weller, G.R.; Doherty, A.J. A family of DNA repair ligases in bacteria? FEBS Lett. 2001, 505, 340–342. [Google Scholar] [CrossRef]
- Moeller, R.; Stackebrandt, E.; Reitz, G.; Berger, T.; Rettberg, P.; Doherty, A.J.; Horneck, G.; Nicholson, W.L. Role of DNA repair by nonhomologous−end joining in Bacillus subtilis spore resistance to e×treme dryness, mono− and polychromatic UV, and ionizing radiation. J. Bacteriol. 2007, 189, 3306–3311. [Google Scholar] [CrossRef]
- Pitcher, R.S.; Green, A.J.; Brzostek, A.; Korycka−Machala, M.; Dziadek, J.; Doherty, A.J. NHEJ protects mycobacteria in stationary phase against the harmful effects of desiccation. DNA Repair (Amst) 2007, 6, 1271–1276. [Google Scholar] [CrossRef][Green Version]
- Bertrand, C.; Thibessard, A.; Bruand, C.; Lecointe, F.; Leblond, P. Bacterial NHEJ: A never ending story. Mol. Microbiol. 2019, 111, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Brissett, N.C.; Martin, M.J.; Bartlett, E.J.; Bianchi, J.; Blanco, L.; Doherty, A.J. Molecular basis for DNA double−strand break annealing and primer e×tension by an NHEJ DNA polymerase. Cell Rep. 2013, 5, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Brissett, N.C.; Martin, M.J.; Pitcher, R.S.; Bianchi, J.; Juarez, R.; Green, A.J.; Fox, G.C.; Blanco, L.; Doherty, A.J. Structure of a preternary comple× involving a prokaryotic NHEJ DNA polymerase. Mol. Cell 2011, 41, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Brissett, N.C.; Pitcher, R.S.; Juarez, R.; Picher, A.J.; Green, A.J.; Dafforn, T.R.; Fox, G.C.; Blanco, L.; Doherty, A.J. Structure of a NHEJ polymerase−mediated DNA synaptic comple×. Science 2007, 318, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wen, J.; Lin, Y.; Wang, S.; Xue, P.; Zhang, Z.; Zhou, Y.; Wang, X.; Sui, L.; Bi, L.J.; et al. A Sir2−like protein participates in mycobacterial NHEJ. PLoS ONE 2011, 6, e20045. [Google Scholar] [CrossRef]
- Sinha, K.M.; Stephanou, N.C.; Gao, F.; Glickman, M.S.; Shuman, S. Mycobacterial UvrD1 is a Ku−dependent DNA helicase that plays a role in multiple DNA repair events, including double−strand break repair. J. Biol. Chem. 2007, 282, 15114–15125. [Google Scholar] [CrossRef]
- Bartlett, E.J.; Brissett, N.C.; Doherty, A.J. Ribonucleolytic resection is required for repair of strand displaced nonhomologous end-joining intermediates. Proc. Natl. Acad. Sci. USA 2013, 110, E1984–E1991. [Google Scholar] [CrossRef]
- Iyer, L.M.; Koonin, E.V.; Leipe, D.D.; Aravind, L. Origin and evolution of the archaeo−eukaryotic primase superfamily and related palm−domain proteins: Structural insights and new members. Nucleic Acids Res. 2005, 33, 3875–3896. [Google Scholar] [CrossRef]
- Guilliam, T.A.; Keen, B.A.; Brissett, N.C.; Doherty, A.J. Primase−polymerases are a functionally diverse superfamily of replication and repair enzymes. Nucleic Acids Res. 2015, 43, 6651–6664. [Google Scholar] [CrossRef]
- Lipps, G.; Weinzierl, A.O.; von Scheven, G.; Buchen, C.; Cramer, P. Structure of a bifunctional DNA primase−polymerase. Nat. Struct. Mol. Biol. 2004, 11, 157–162. [Google Scholar] [CrossRef]
- Zhu, H.; Nandakumar, J.; Aniukwu, J.; Wang, L.K.; Glickman, M.S.; Lima, C.D.; Shuman, S. Atomic structure and nonhomologous end−joining function of the polymerase component of bacterial DNA ligase D. Proc. Natl. Acad. Sci. USA 2006, 103, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- García−Gómez, S.; Reyes, A.; Martínez−Jiménez, M.I.; Chocrón, E.S.; Mouron, S.; Terrados, G.; Powell, C.; Salido, E.; Méndez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Guilliam, T.A.; Tsuda, M.; Yamamoto, J.; Bailey, L.J.; Iwai, S.; Takeda, S.; Doherty, A.J.; Hirota, K. Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain−terminating nucleosides. Cell Cycle 2016, 15, 1997–2008. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, M.I.; Garcia-Gomez, S.; Bebenek, K.; Sastre-Moreno, G.; Calvo, P.A.; Diaz-Talavera, A.; Kunkel, T.A.; Blanco, L. Alternative solutions and new scenarios for translesion DNA synthesis by human PrimPol. DNA Repair (Amst) 2015, 29, 127–138. [Google Scholar] [CrossRef]
- Mourón, S.; Rodríguez-Acebes, S.; Martínez-Jiménez, M.I.; García-Gómez, S.; Chocrón, S.; Blanco, L.; Méndez, J. Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat. Struct Mol. Biol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef]
- Schiavone, D.; Jozwiakowski, S.K.; Romanello, M.; Guilbaud, G.; Guilliam, T.A.; Bailey, L.J.; Sale, J.E.; Doherty, A.J. PrimPol Is Required for Replicative Tolerance of G Quadruple×es in Vertebrate Cells. Mol. Cell 2016, 61, 161–169. [Google Scholar] [CrossRef]
- Svikovic, S.; Crisp, A.; Tan−Wong, S.M.; Guilliam, T.A.; Doherty, A.J.; Proudfoot, N.J.; Guilbaud, G.; Sale, J.E. R−loop formation during S phase is restricted by PrimPol−mediated repriming. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Wan, L.; Lou, J.; Xia, Y.; Su, B.; Liu, T.; Cui, J.; Sun, Y.; Lou, H.; Huang, J. hPrimpol1/CCDC111 is a human DNA primase−polymerase required for the maintenance of genome integrity. EMBO Rep. 2013, 14, 1104–1112. [Google Scholar] [CrossRef]
- Shuman, S.; Glickman, M.S. Bacterial DNA repair by non−homologous end joining. Nat. Rev. Microbiol. 2007, 5, 852–861. [Google Scholar] [CrossRef]
- de Vega, M. The minimal Bacillus subtilis nonhomologous end joining repair machinery. PLoS ONE 2013, 8, e64232. [Google Scholar] [CrossRef]
- Traut, T.W. Physiological concentrations of purines and pyrimidines. Mol. Cell Biochem 1994, 140, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shuman, S. Bacterial nonhomologous end joining ligases preferentially seal breaks with a 3′−OH monoribonucleotide. J. Biol. Chem. 2008, 283, 8331–8339. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shuman, S. A primer−dependent polymerase function of pseudomonas aeruginosa ATP−dependent DNA ligase (LigD). J. Biol. Chem. 2005, 280, 418–427. [Google Scholar] [CrossRef] [PubMed]
- de Ory, A.; Nagler, K.; Carrasco, B.; Raguse, M.; Zafra, O.; Moeller, R.; de Vega, M. Identification of a conserved 5′−dRP lyase activity in bacterial DNA repair ligase D and its potential role in base e×cision repair. Nucleic Acids Res. 2016, 44, 1833–1844. [Google Scholar] [CrossRef]
- O’Flaherty, D.K.; Guengerich, F.P. Steady−state kinetic analysis of DNA polymerase single−nucleotide incorporation products. Curr. Protoc. Nucleic Acid Chem. 2014, 59, 7–21. [Google Scholar] [CrossRef]
- González-Barrera, S.; Sánchez, A.; Ruiz, J.F.; Juárez, R.; Picher, A.J.; Terrados, G.; Andrade, P.; Blanco, L. Characterization of SpPol4, a unique X−family DNA polymerase in Schizosaccharomyces pombe. Nucleic Acids Res. 2005, 33, 4762–4774. [Google Scholar] [CrossRef]
- Ruiz, J.F.; Juárez, R.; García-Díaz, M.; Terrados, G.; Picher, A.J.; González-Barrera, S.; Fernández de Henestrosa, A.R.; Blanco, L. Lack of sugar discrimination by human Pol mu requires a single glycine residue. Nucleic Acids Res. 2003, 31, 4441–4449. [Google Scholar] [CrossRef]
- Pitcher, R.S.; Brissett, N.C.; Picher, A.J.; Andrade, P.; Juárez, R.; Thompson, D.; Fox, G.C.; Blanco, L.; Doherty, A.J. Structure and function of a mycobacterial NHEJ DNA repair polymerase. J. Mol. Biol. 2007, 366, 391–405. [Google Scholar] [CrossRef]
- Liu, L.; Komori, K.; Ishino, S.; Bocquier, A.A.; Cann, I.K.; Kohda, D.; Ishino, Y. The archaeal DNA primase: Biochemical characterization of the p41−p46 comple× from Pyrococcus furiosus. J. Biol. Chem. 2001, 276, 45484–45490. [Google Scholar] [CrossRef]
- Díaz-Talavera, A.; Calvo, P.A.; Gonzalez-Acosta, D.; Diaz, M.; Sastre-Moreno, G.; Blanco-Franco, L.; Guerra, S.; Martinez-Jimenez, M.I.; Mendez, J.; Blanco, L. A cancer-associated point mutation disables the steric gate of human PrimPol. Sci Rep. 2019, 9, 1121. [Google Scholar] [CrossRef]
- Yakovleva, L.; Shuman, S. Nucleotide misincorporation, 3′−mismatch e×tension, and responses to abasic sites and DNA adducts by the polymerase component of bacterial DNA ligase D. J. Biol. Chem. 2006, 281, 25026–25040. [Google Scholar] [CrossRef] [PubMed]
- Paris, U.; Mikkel, K.; Tavita, K.; Saumaa, S.; Teras, R.; Kivisaar, M. NHEJ enzymes LigD and Ku participate in stationary−phase mutagenesis in Pseudomonas putida. DNA Repair (Amst) 2015, 31, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Stephanou, N.C.; Gao, F.; Bongiorno, P.; Ehrt, S.; Schnappinger, D.; Shuman, S.; Glickman, M.S. Mycobacterial nonhomologous end joining mediates mutagenic repair of chromosomal double−strand DNA breaks. J. Bacteriol 2007, 189, 5237–5246. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A.; Yin, Y.W. Accuracy, lesion bypass, strand displacement and translocation by DNA polymerases. Philos Trans. R. Soc. Lond B Biol. Sci. 2004, 359, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shuman, S. Novel 3′−ribonuclease and 3′−phosphatase activities of the bacterial non−homologous end−joining protein, DNA ligase D. J. Biol. Chem. 2005, 280, 25973–25981. [Google Scholar] [CrossRef] [PubMed]
- García-Díaz, M.; Bebenek, K.; Larrea, A.A.; Havener, J.M.; Perera, L.; Krahn, J.M.; Pedersen, L.C.; Ramsden, D.A.; Kunkel, T.A. Template strand scrunching during DNA gap repair synthesis by human polymerase lambda. Nat. Struct. Mol. Biol. 2009, 16, 967–972. [Google Scholar] [CrossRef]
- Bjelland, S.; Seeberg, E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 2003, 531, 37–80. [Google Scholar] [CrossRef]
- Imlay, J.A. Pathways of oxidative damage. Annu Rev. Microbiol 2003, 57, 395–418. [Google Scholar] [CrossRef]
- Sekiguchi, T.; Ito, R.; Hayakawa, H.; Sekiguchi, M. Elimination and utilization of oxidized guanine nucleotides in the synthesis of RNA and its precursors. J. Biol Chem 2013, 288, 8128–8135. [Google Scholar] [CrossRef]
- Nakabeppu, Y. Cellular levels of 8−oxoguanine in either DNA or the nucleotide pool play pivotal roles in carcinogenesis and survival of cancer cells. Int. J. Mol. Sci. 2014, 15, 12543–12557. [Google Scholar] [CrossRef]
- Batra, V.K.; Shock, D.D.; Beard, W.A.; McKenna, C.E.; Wilson, S.H. Binary comple× crystal structure of DNA polymerase beta reveals multiple conformations of the templating 8−oxoguanine lesion. Proc. Natl. Acad. Sci. USA 2012, 109, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Krahn, J.M.; Beard, W.A.; Miller, H.; Grollman, A.P.; Wilson, S.H. Structure of DNA polymerase beta with the mutagenic DNA lesion 8−oxodeoxyguanine reveals structural insights into its coding potential. Structure 2003, 11, 121–127. [Google Scholar] [CrossRef]
- Katafuchi, A.; Nohmi, T. DNA polymerases involved in the incorporation of oxidized nucleotides into DNA: Their efficiency and template base preference. Mutat Res. 2010, 703, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Beard, W.A.; Batra, V.K.; Wilson, S.H. DNA polymerase structure−based insight on the mutagenic properties of 8−oxoguanine. Mutat. Res. 2010, 703, 18–23. [Google Scholar] [CrossRef]
- Burak, M.J.; Guja, K.E.; Garcia−Diaz, M. Nucleotide binding interactions modulate dNTP selectivity and facilitate 8−oxo−dGTP incorporation by DNA polymerase lambda. Nucleic Acids Res. 2015, 43, 8089–8099. [Google Scholar] [CrossRef]
- Zafra, O.; Pérez de Ayala, L.; de Vega, M. The anti/syn conformation of 8−oxo−7,8−dihydro−2′−deoxyguanosine is modulated by Bacillus subtilis PolX active site residues His255 and Asn263. Efficient processing of damaged 3′−ends. DNA Repair (Amst) 2017, 52, 59–69. [Google Scholar] [CrossRef]
- de Vega, M.; Salas, M. A highly conserved Tyrosine residue of family B DNA polymerases contributes to dictate translesion synthesis past 8−oxo−7,8−dihydro−2′−deoxyguanosine. Nucleic Acids Res. 2007, 35, 5096–5107. [Google Scholar] [CrossRef]
- Dalgaard, J.Z. Causes and consequences of ribonucleotide incorporation into nuclear DNA. Trends Genet. 2012, 28, 592–597. [Google Scholar] [CrossRef]
- Sparks, J.L.; Chon, H.; Cerritelli, S.M.; Kunkel, T.A.; Johansson, E.; Crouch, R.J.; Burgers, P.M. RNase H2−initiated ribonucleotide e×cision repair. Mol. Cell 2012, 47, 980–986. [Google Scholar] [CrossRef]
- Schroeder, J.W.; Randall, J.R.; Hirst, W.G.; O’Donnell, M.E.; Simmons, L.A. Mutagenic cost of ribonucleotides in bacterial DNA. Proc. Natl. Acad. Sci. USA 2017, 114, 11733–11738. [Google Scholar] [CrossRef]
- Cilli, P.; Minoprio, A.; Bossa, C.; Bignami, M.; Mazzei, F. Formation and Repair of Mismatches Containing Ribonucleotides and O×idized Bases at Repeated DNA Sequences. J. Biol. Chem. 2015, 290, 26259–26269. [Google Scholar] [CrossRef] [PubMed]
- Crespan, E.; Furrer, A.; Rosinger, M.; Bertoletti, F.; Mentegari, E.; Chiapparini, G.; Imhof, R.; Ziegler, N.; Sturla, S.J.; Hubscher, U.; et al. Impact of ribonucleotide incorporation by DNA polymerases beta and lambda on oxidative base e×cision repair. Nat. Commun. 2016, 7, 10805. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, H.; Shuman, S. Mycobacterium smegmatis DinB2 misincorporates deoxyribonucleotides and ribonucleotides during templated synthesis and lesion bypass. Nucleic Acids Res. 2014, 42, 12722–12734. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Moreno, G.; Sanchez, A.; Esteban, V.; Blanco, L. ATP insertion opposite 8-oxo-deoxyguanosine by Pol4 mediates error-free tolerance in Schizosaccharomyces pombe. Nucleic Acids Res. 2014, 42, 9821–9837. [Google Scholar] [CrossRef]
- Malfatti, M.C.; Henneke, G.; Balachander, S.; Koh, K.D.; Newnam, G.; Uehara, R.; Crouch, R.J.; Storici, F.; Tell, G. Unlike the Escherichia coli counterpart, archaeal RNase HII cannot process ribose monophosphate abasic sites and oxidized ribonucleotides embedded in DNA. J. Biol. Chem. 2019, 294, 13061–13072. [Google Scholar] [CrossRef] [PubMed]
- Sassa, A.; Caglayan, M.; Rodriguez, Y.; Beard, W.A.; Wilson, S.H.; Nohmi, T.; Honma, M.; Yasui, M. Impact of Ribonucleotide Backbone on Translesion Synthesis and Repair of 7,8-Dihydro−8-oxoguanine. J. Biol. Chem. 2016, 291, 24314–24323. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Nucleotide | kcat (s−1) | Km (nM) | Cat.eff. (s−1·nM−1) | f | Fins |
|---|---|---|---|---|---|---|
| wild−type | UTP dTTP GTP | 26.9 ± 0.8 2.2 ± 0.1 0.92 ± 0.02 | 58.4 ± 5.1 33.3 ± 4.9 1211 ± 90 | 0.46 ± 0.02 0.07 ± 0.01 8 × 10−4 ± 3 × 10−5 | 7 | 625 |
| S768Y | UTP dTTP GTP | 14.8 ± 0.4 0.42 ± 0.02 0.49 ± 0.01 | 55.5 ± 3.5 70.4 ± 9.9 865 ± 70 | 0.27 ± 0.01 6 × 10−3 ± 9 × 10−4 6 × 10−4 ± 3 × 10−5 | 44 | 473 |
| S768A | UTP dTTP GTP | 22.6 ± 1.5 2.2 ± 0.1 0.25 ± 4 ×10−3 | 46 ± 9 31.2± 4.6 890 ± 66 | 0.49 ± 0.15 0.07 ± 3 × 10−3 3 × 10−4 ± 6 × 10−6 | 7 | 1749 |
| H651A | UTP dTTP GTP | 6.2 ± 0.2 2.2 ± 0.2 0.02 ± 4 × 10−4 | 117 ± 10 60 ± 13 2385 ± 200 | 0.05 ± 4 × 10−3 0.04 ± 6 × 10−3 7 × 10−6 ± 7 ×1 0−7 | 1.3 | 7361 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Salvador, A.; de Vega, M. Structural Determinants Responsible for the Preferential Insertion of Ribonucleotides by Bacterial NHEJ PolDom. Biomolecules 2020, 10, 203. https://doi.org/10.3390/biom10020203
Sánchez-Salvador A, de Vega M. Structural Determinants Responsible for the Preferential Insertion of Ribonucleotides by Bacterial NHEJ PolDom. Biomolecules. 2020; 10(2):203. https://doi.org/10.3390/biom10020203
Chicago/Turabian StyleSánchez-Salvador, Alejandro, and Miguel de Vega. 2020. "Structural Determinants Responsible for the Preferential Insertion of Ribonucleotides by Bacterial NHEJ PolDom" Biomolecules 10, no. 2: 203. https://doi.org/10.3390/biom10020203
APA StyleSánchez-Salvador, A., & de Vega, M. (2020). Structural Determinants Responsible for the Preferential Insertion of Ribonucleotides by Bacterial NHEJ PolDom. Biomolecules, 10(2), 203. https://doi.org/10.3390/biom10020203