The Release of a Soluble Glycosylated Protein from Glycogen by Recombinant Lysosomal α-Glucosidase (rhGAA) In Vitro and Its Presence in Serum In Vivo

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Glycogen Substrates
2.2. Human Glycogen Samples
2.3. Source
2.4. Enzyme Assays
2.5. Carbohydrate Analysis
2.6. Protein Determination
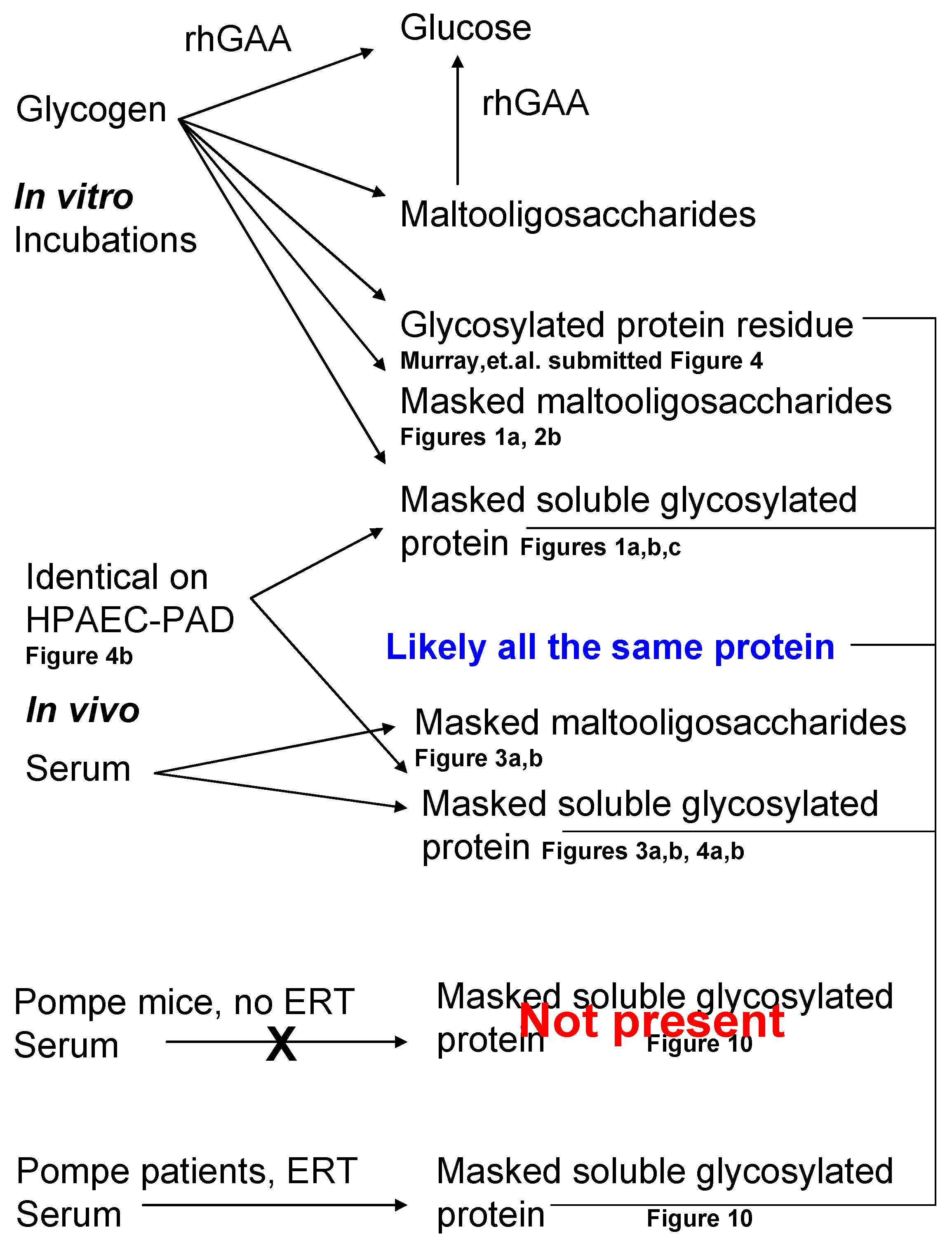
3. Results
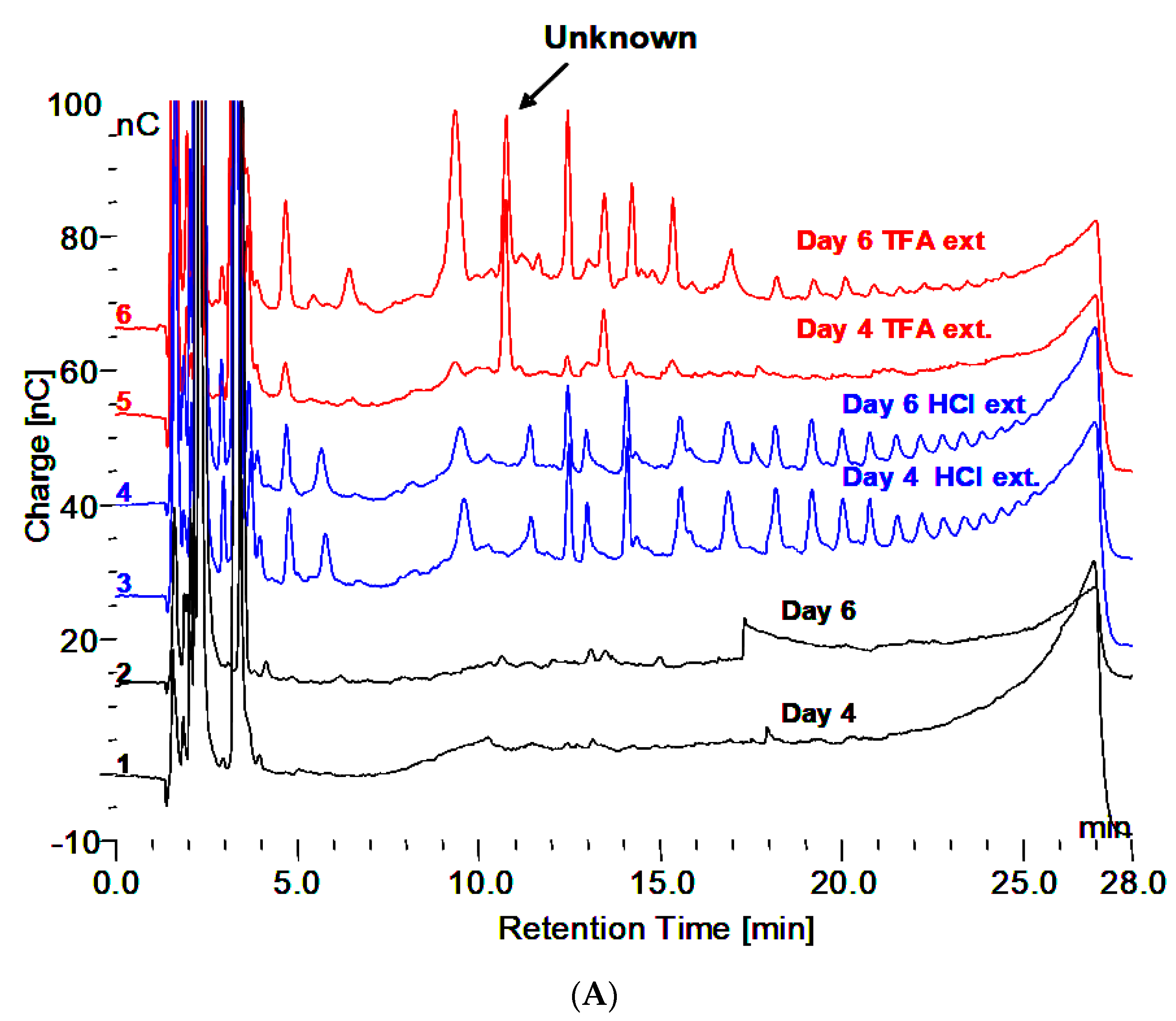
3.1. Incubation Medium Analysis
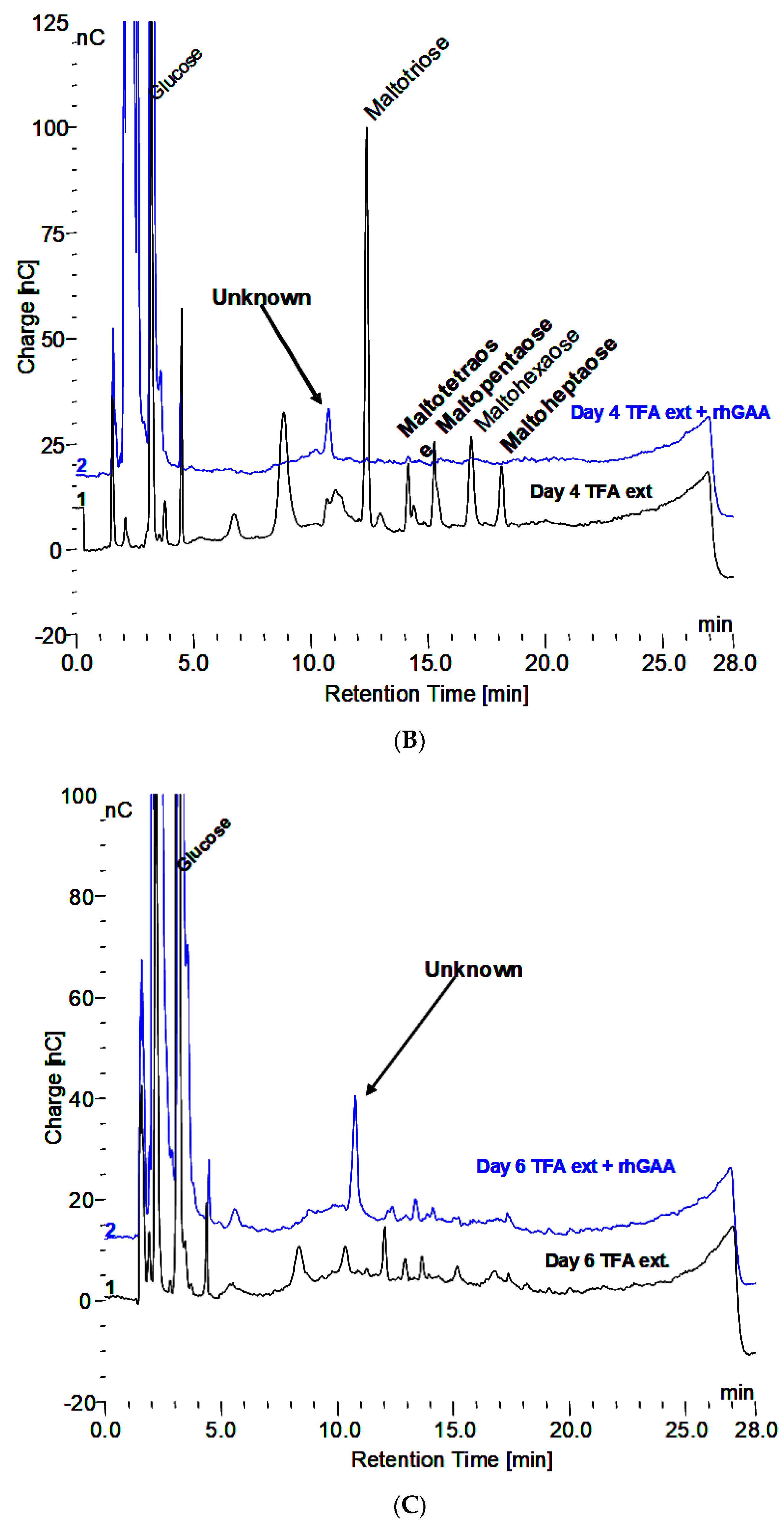
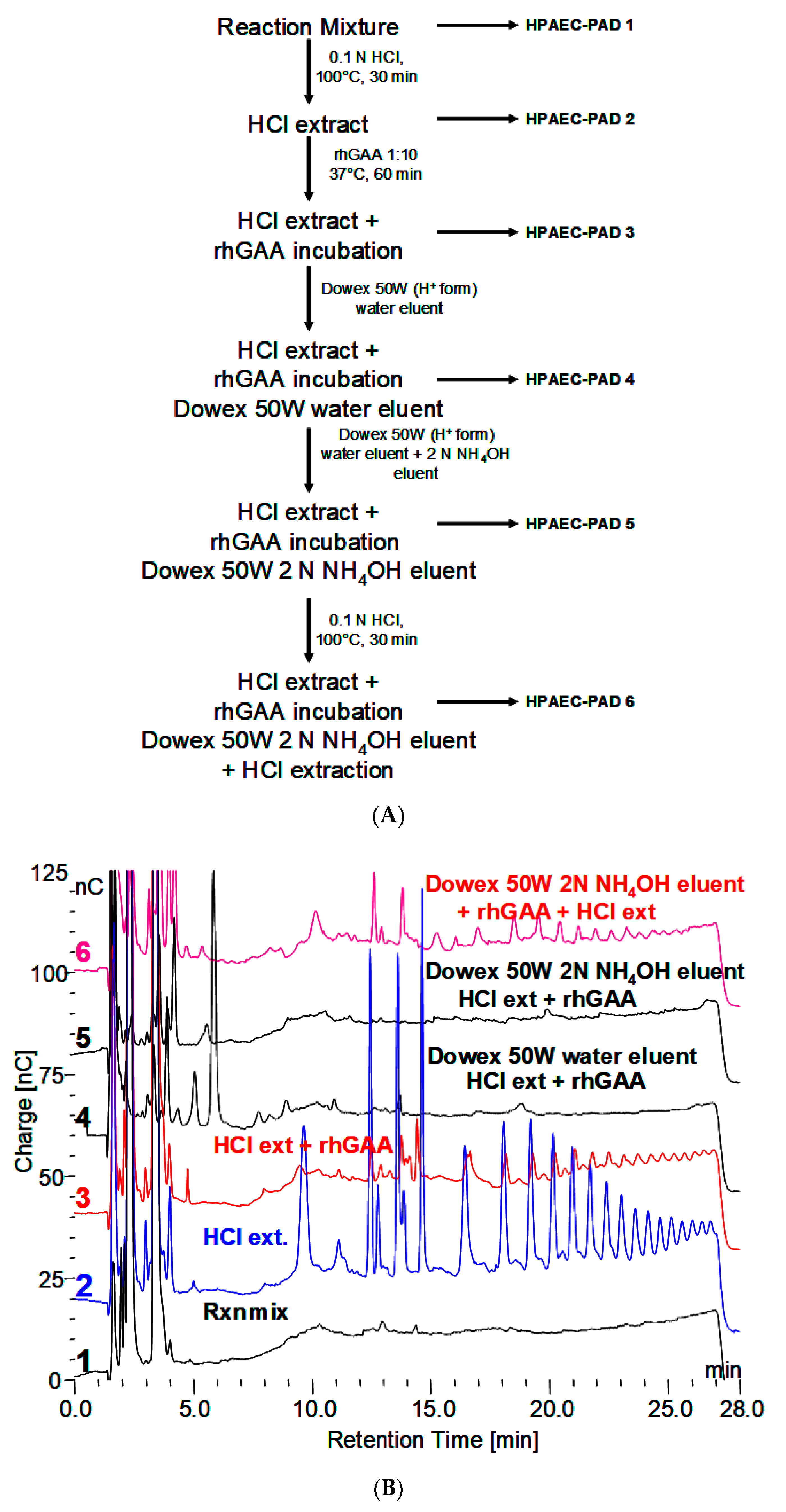
3.2. Characteristics of the Glycogen Fraction that is not Degraded by rhGAA
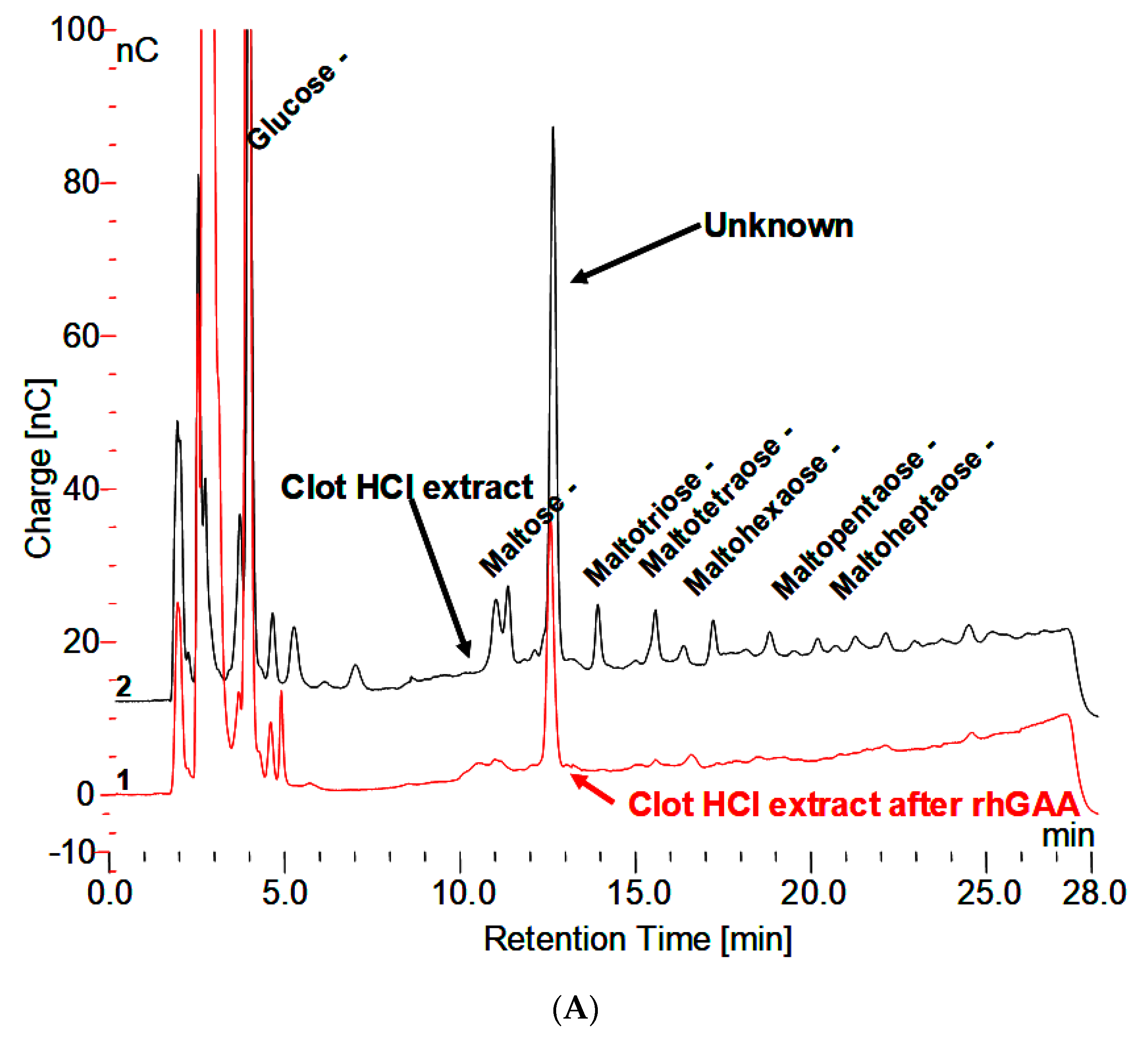
- HPAEC-PAD does not reveal any significant peaks that elute after monosaccharides. Which indicates no carbohydrates with ionizable hydroxyl groups are present?
- Extraction with 0.1 N HCl at 100 °C for 30 min reveals maltooligosaccharides from DP 2 to about 18 on HPAEC-PAD.
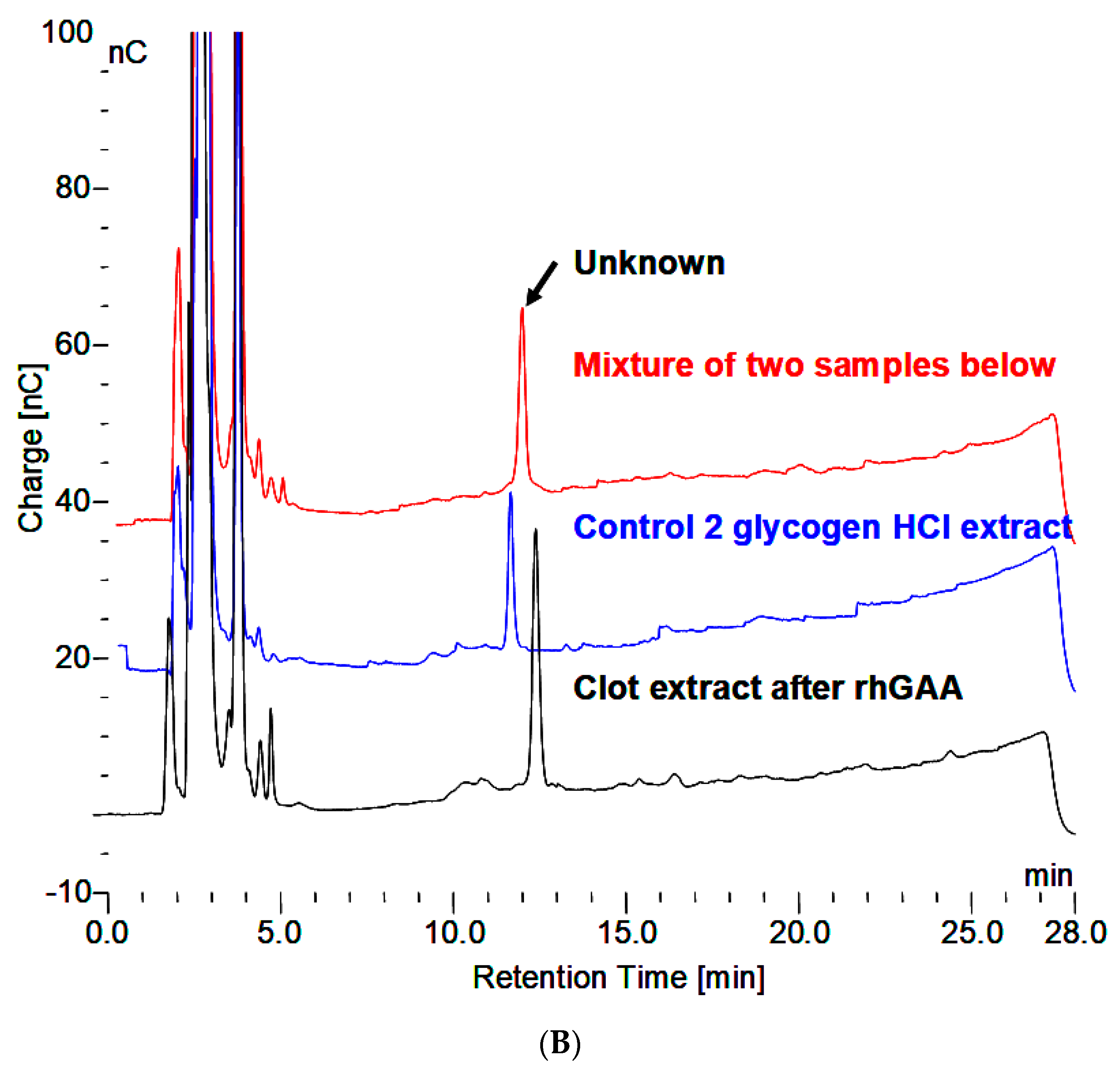
- The material in the incubation medium binds to a Dowex 50W ion exchange column and elutes in 2.0 N NH4OH. This is indicative of binding by a charged species such as protein or amino acids. After taken to dryness, it can be extracted with 0.1 N HCl at 100 °C to reveal the maltooligosaccharides.
- Incubation with amyloglucosidase does not do anything to the samples.
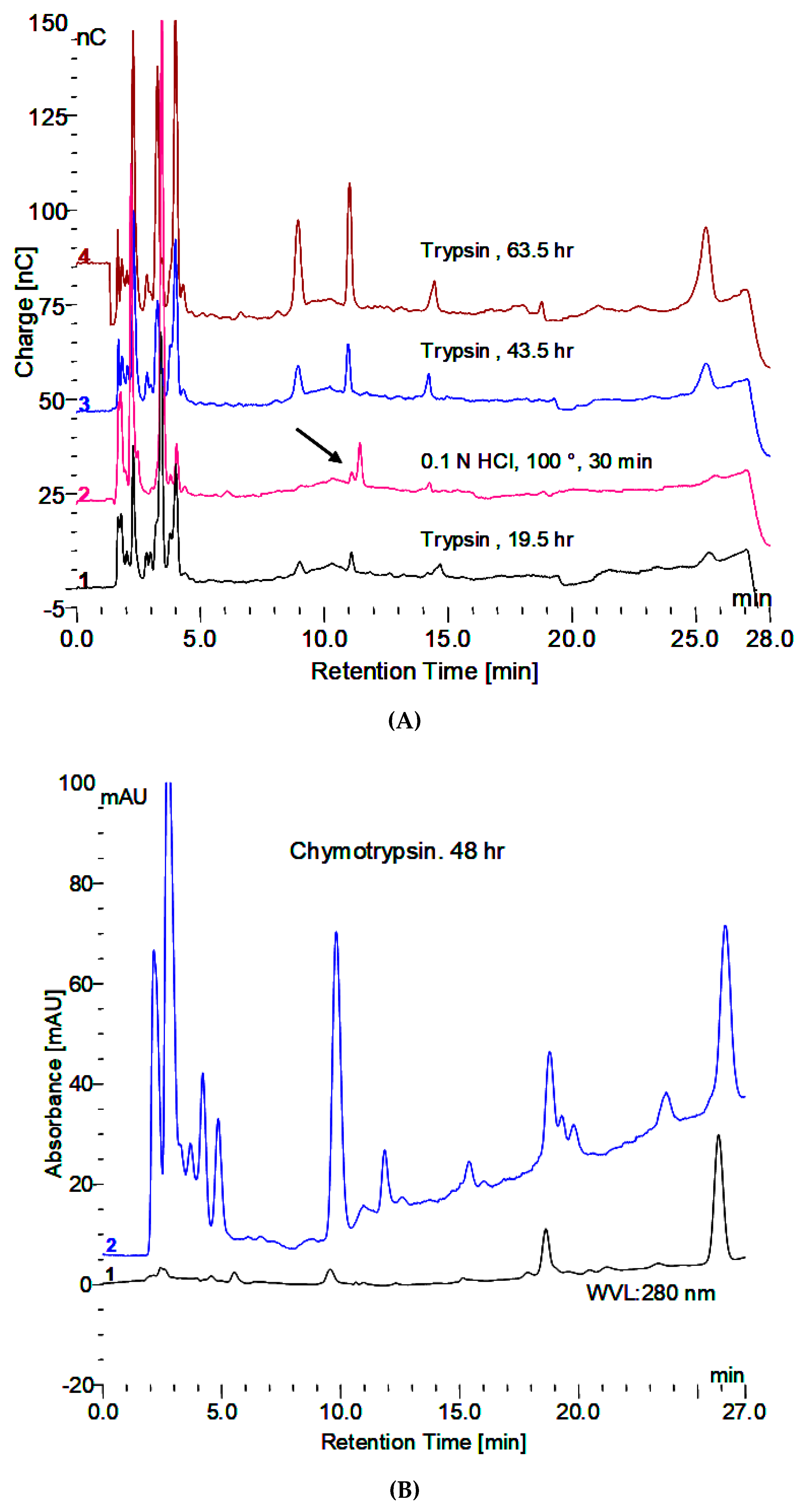
- Incubation with trypsin reveals some smaller oligosaccharides that elute in the region of up to about DP 4 and one at about DP 7 or 8. Additionally, trypsin treatment before HCl extraction appears to facilitate the appearance of more larger oligosaccharides. This is indicative of oligosaccharides being released or their appearance facilitated by the removal of protein.
- Incubation with concanavalin A does not appear to bind the material. This indicates the absence of exposed glucose or mannose residues, including glycogen, which would be bound by the concanavalin A protein [14].
3.3. Summary of Characteristics
- Lack of chemical detection of ionizable hydroxyls of carbohydrate.
- Lack of biological recognition of carbohydrate by rhGAA, concanavalin A, or amyloglucosidase.
- Binding to Dowex 50W indicative of a charged species.
- Exposure of carbohydrate by incubation with a protease (trypsin).
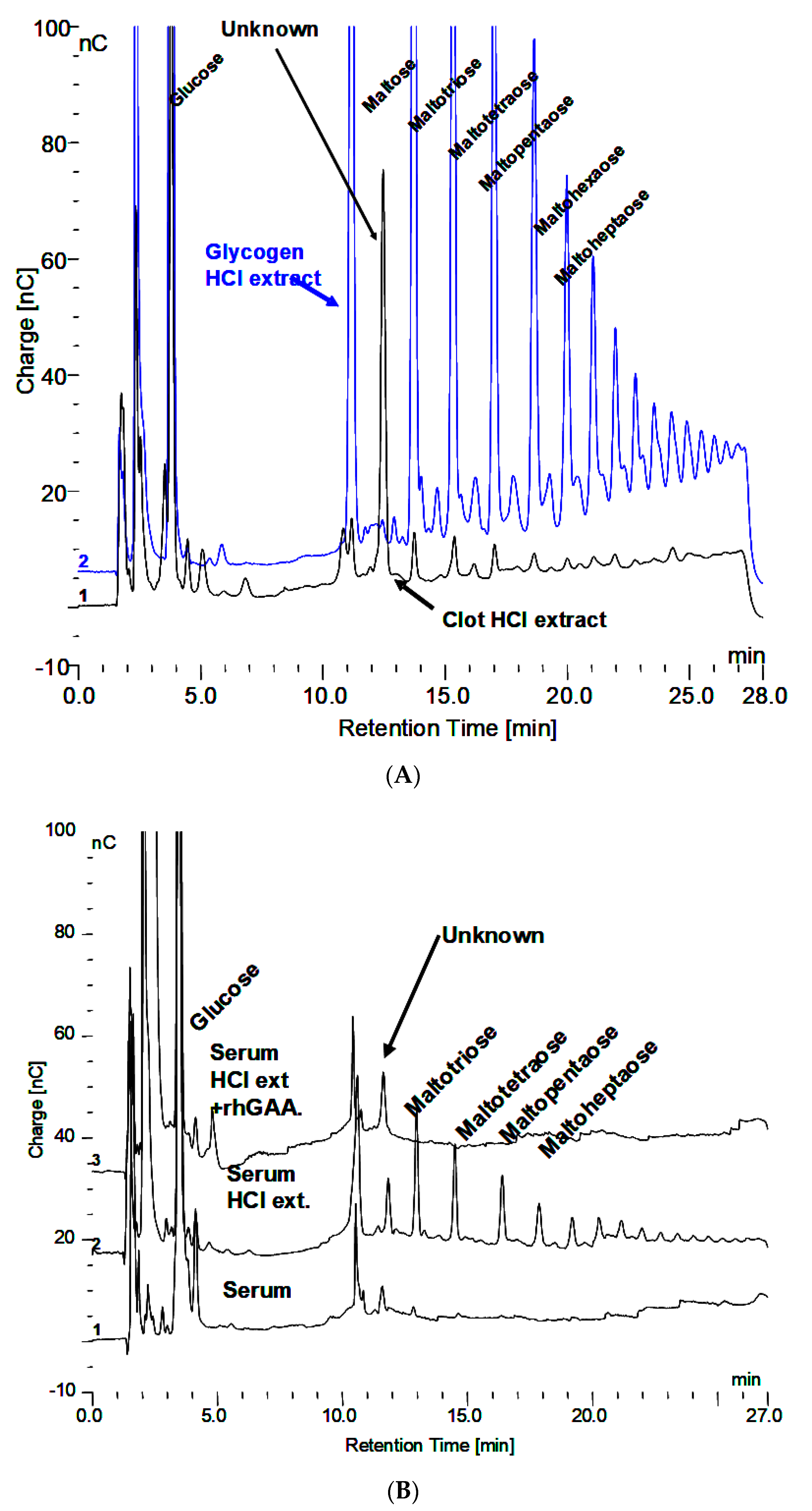
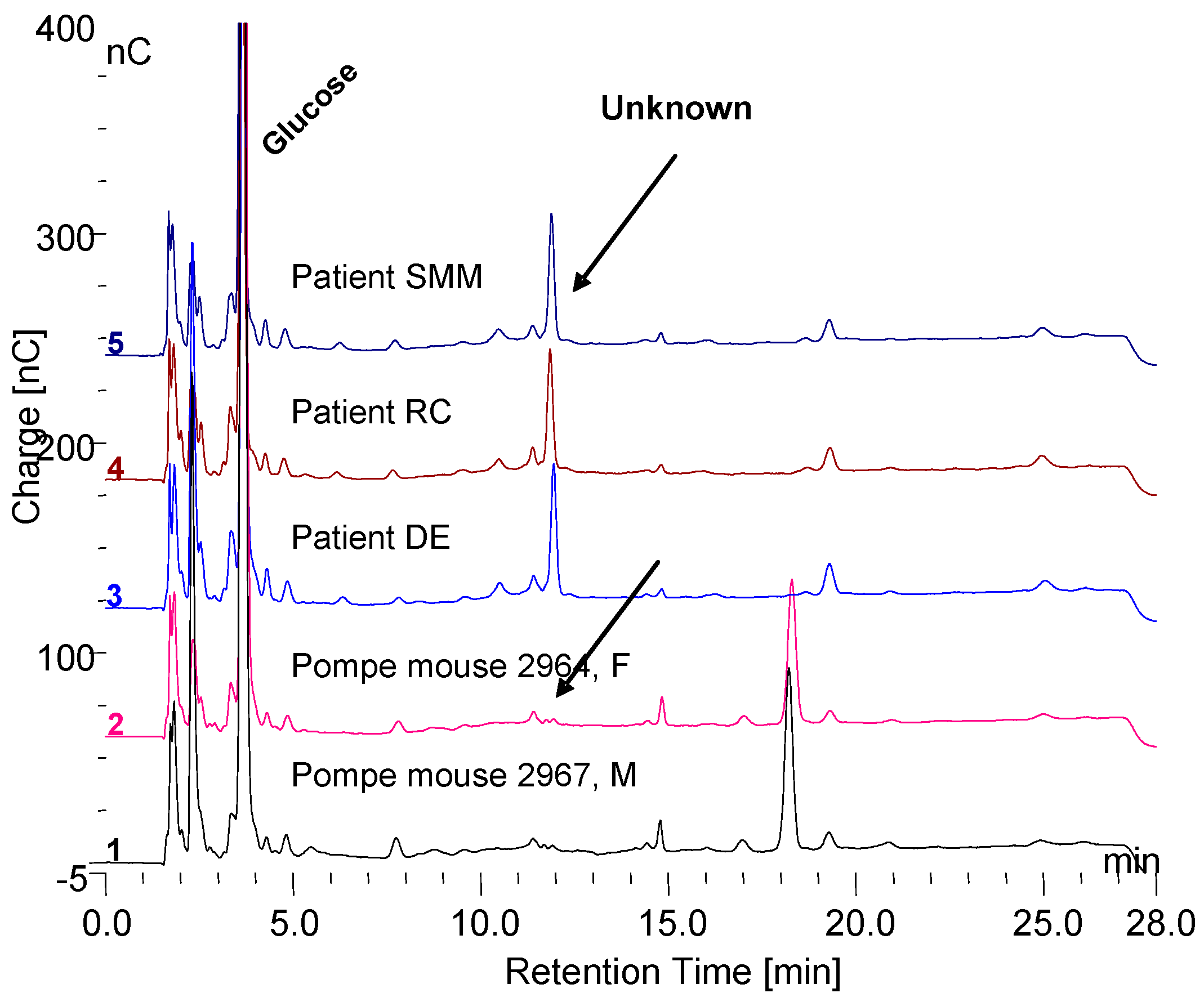
3.4. Serum Investigation
3.5. Masking of Carbohydrate by Protein
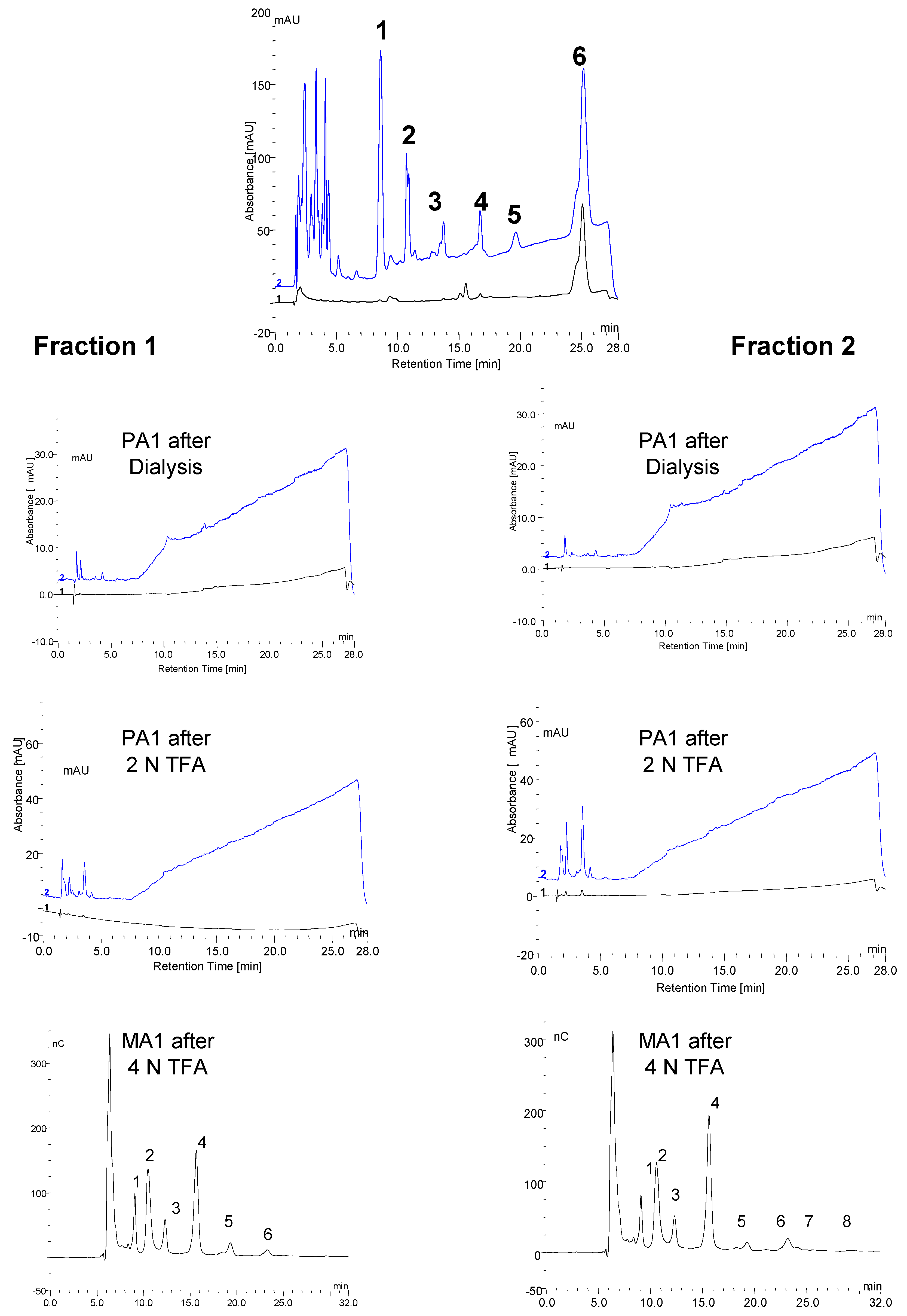
3.6. Fraction Collection and Evidence of Protein Masking by Carbohydrate in Serum
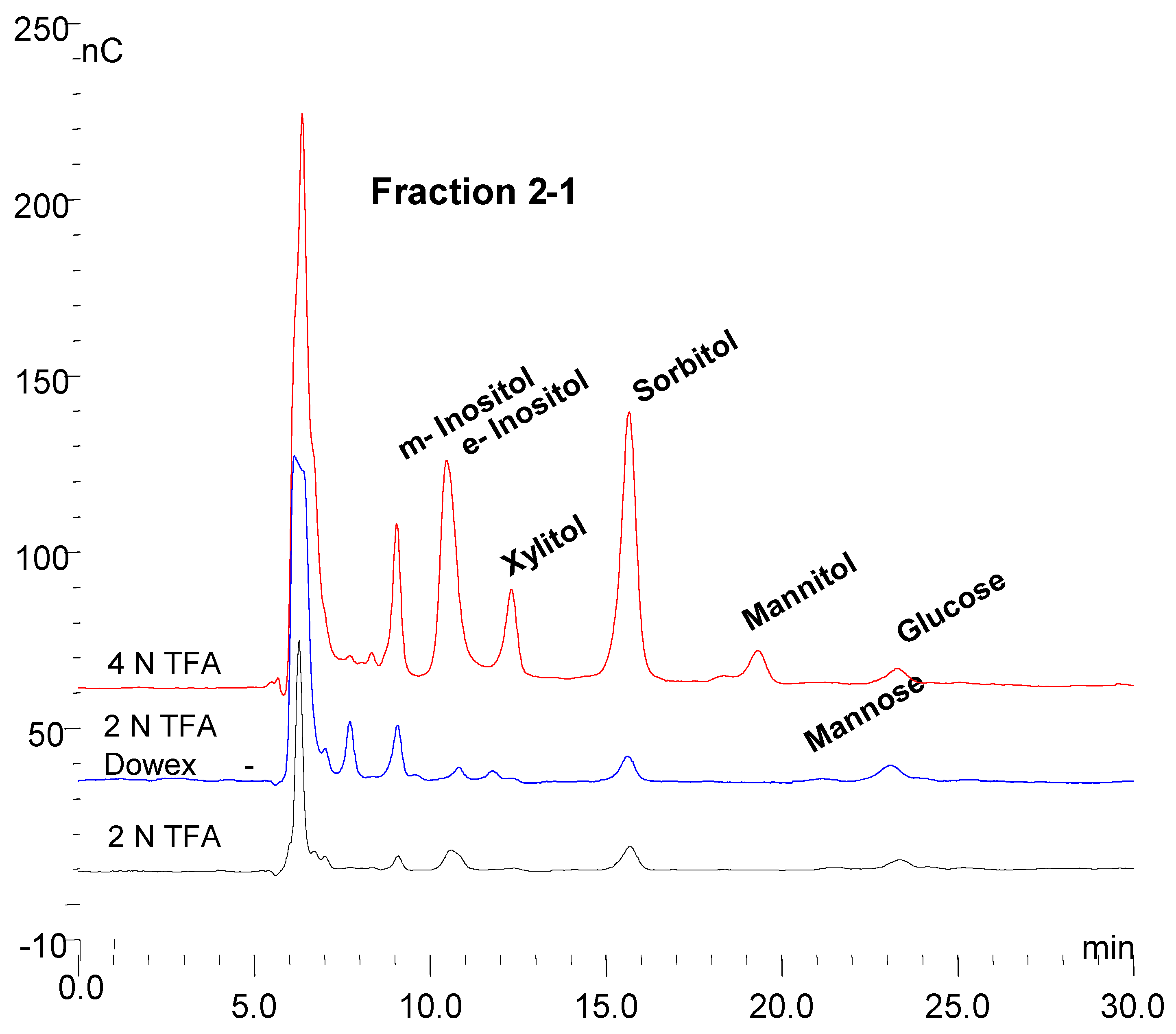
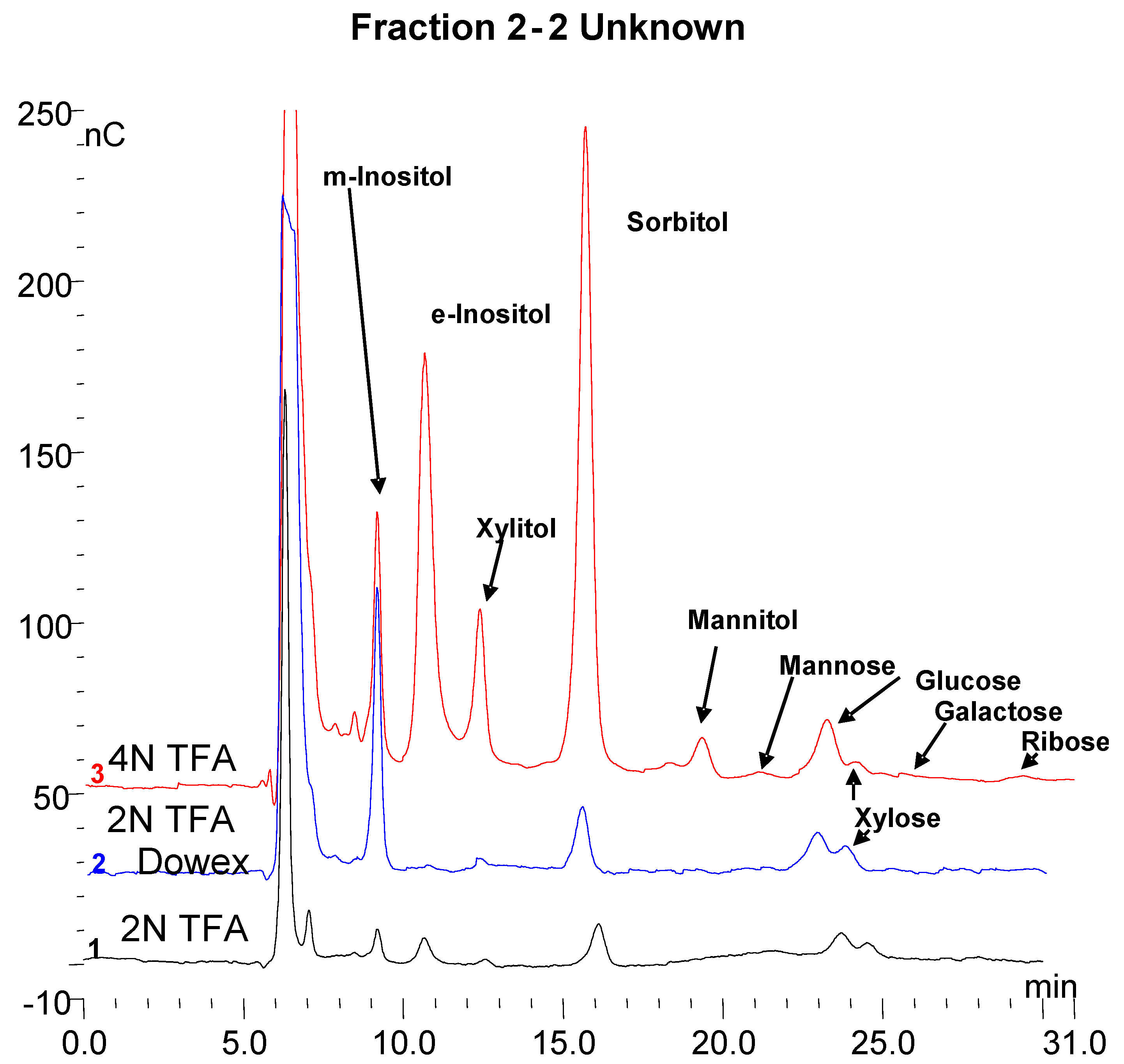
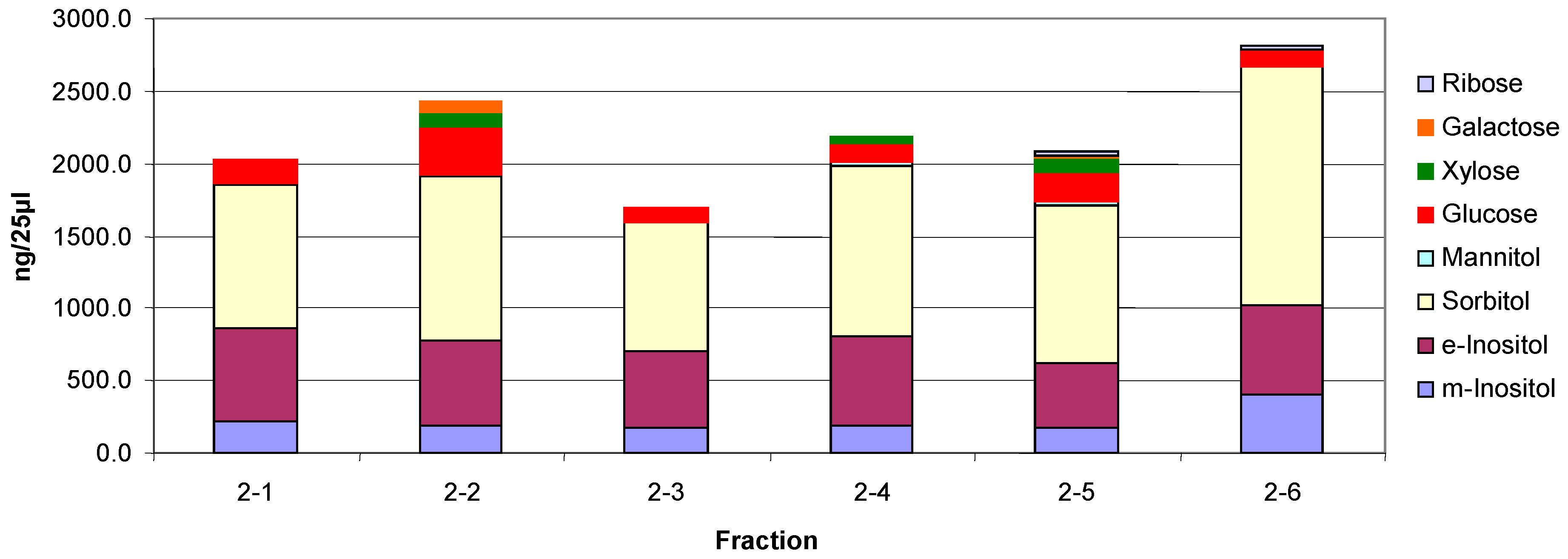
3.7. Monosaccharide Composition of Fractions
4. Discussion
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | bovine serum albumin |
| ERT | enzyme replacement therapy |
| GAA | Lysosomal α-glucosidase |
| rhGAA | recombinant human lysosomal α-glucosidase |
| HPAEC-PAD | high performance anion exchange chromatography-pulsed amperometric detection |
| HPLC | high performance liquid chromatography |
| GLC | gas liquid chromatography |
| TFA | trifluoroacetic acid |
| PAS | periodic acid Schiff stain. |
References
- Hers, H.G. α-Glucosidase deficiency in generalized glycogen-storage disease (Pompe’s disease). Biochem. J. 1963, 86, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Torres, H.N.; Olavarria, J.M. Liver α-Glucosidases. J. Biol. Chem. 1964, 239, 2427–2434. [Google Scholar] [PubMed]
- Brown, B.I.; Brown, D.H.; Jeffrey, P.L. Simultaneous Absence of α-1,4-Glucosidase and α-1,6-Glucosidase Activities (pH4) in Tissues of Children with Type II Glycogen Storage Disease. Biochemistry 1970, 9, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, P.L.; Brown, D.H.; Brown, B.I. Lysosomal α-glucosidase. I. Purification and properties of the rat liver enzyme. Biochemistry 1970, 9, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, P.L.; Brown, D.H.; Brown, B.I. Lysosomal α-glucosidase. II. Kinetics of action of the rat liver enzyme. Biochemistry 1970, 9, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.N. The substrate specificity of acid α-glucosidase from rabbit Mu muscle. Biochem. J. 1971, 124, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.N. The Maltase, Glucoamylase and Transglucosylase Activities of Acid α-Glucosidase from Rabbit Muscle. Biochem. J. 1971, 124, 713–724. [Google Scholar] [CrossRef]
- Hers, H.; Van Hoof, F. Enzymes of glycogen degradation in biopsy material. Methods Enzymol. 1966, 8, 525–532. [Google Scholar] [CrossRef]
- Murray, A.K. The action of recombinant lysosomal α-glucosidase [rhGAA] and amyloglucosidase on bovine and human liver glycogen. submitted.
- Raben, N.; NFukuda, O.; Gilbert, A.L.; de Jong, D.; Thurberg, B.L.; Mattaliano, R.J.; Meikle, P.; Hopwood, J.J.; Nagashima, K.; Nagaraju, K.; et al. Replacing Acid α-Glucosidase in Pompe Disease: Recombinant and Transgenic Enzymes are Equipotent, but Neither Completely Clears Glycogen from Type II Muscle Fibers. Mol. Ther. 2005, 11, 48–56. [Google Scholar] [CrossRef]
- Del Rizzo, M.; Fanin, M.; Cerutti, A.; Cazzorla, C.; Milanesi, O.; Nascimbeni, A.C.; Angelini, C.; Giordano, L.; Bordugo, A.; Burlina, A. Long-term follow-up results in enzyme replacement therapy for Pompe disease: A case report. J. Inherit. Metab. Dis. 2010, 33, 389–393. [Google Scholar] [CrossRef]
- Van Der Ploeg, A.; Carlier, P.G.; Carlier, R.-Y.; Kissel, J.; Schoser, B.; Wenninger, S.; Pestronk, A.; Barohn, R.J.; Dimachkie, M.M.; Goker-Alpan, O.; et al. Prospective exploratory muscle biopsy, imaging, and functional assessment in patients with late-onset Pompe disease treated with alglucosidase alfa: The EMBASSY Study. Mol. Genet. Metab. 2016, 119, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Dapson, R.W. Histochemistry of mucus in the skin of the frog, Rana pipiens. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 1970, 166, 615–625. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Leger, R.S.; Cooper, R.; Charnley, A. Cuticle-degrading enzymes of entomopathogenic fungi: Cuticle degradation in vitro by enzymes from entomopathogens. J. Invertebr. Pathol. 1986, 47, 167–177. [Google Scholar] [CrossRef]
- Montgomery, R. Determination of glycogen. Arch. Biochem. Biophys. 1957, 67, 378–386. [Google Scholar] [CrossRef]
- Bell, D.J.; Young, F.G. Observations on the Chemistry of Liver Glycogen. Biochem. J. 1934, 28, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Mordoh, J.; Krisman, C.R.; Leloir, L.F. Further studies on high molecular weight liver glycogen. Arch. Biochem. Biophys. 1966, 113, 265–272. [Google Scholar] [CrossRef]
- De Wulf, H.; Hers, H.G. Paracrystalline Glycogen. In Biochemistry of the Glycosidic Linkage; PAABS Symposium Vol. 2; Piras, R., Pontis, H.G., Eds.; Academic Press: New York, NY, USA, 1972; pp. 399–402. [Google Scholar]
- Metzenberg, A.B. Structural Feathers of Stored Glycogen in a Case of Pompe’s Disease [Glycogenosis Type II]. Master’s Thesis, University of California, Irvine, CA, USA, 1980. [Google Scholar]
- Murray, A.K.; Metzenberg, A.B.; Nichols, R.L. Polysaccharide similarities: Glycosylated protein cores of glycogen, starch and cellulose. submitted.
- Williams, J.C.; Murray, A.K. Enzyme replacement in Pompe disease with an alpha-glucosidase-low density lipoprotein complex. Birth defects Orig. Artic. Ser. 1980, 16, 415–423. [Google Scholar]
- Alandy-Dy, J.; Wencel, M.; Hall, K.; Simon, J.; Chen, Y.; Valenti, E.; Yang, J.; Bali, D.; Lakatos, A.; Goyal, N.; et al. Variable clinical features and genotype-phenotype correlations in 18 patients with late-onset Pompe disease. Ann. Transl. Med. 2019, 7, 276. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional Activation of Lysosomal Exocytosis Promotes Cellular Clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs Involved in Synaptotagmin VII-regulated Lysosomal Exocytosis. J. Biol. Chem. 2004, 279, 20471–20479. [Google Scholar] [CrossRef] [PubMed]
- Spampanato, C.; Feeney, E.; Li, L.; Cardone, M.; Lim, J.-A.; Zare, F.A.H.; Polishchuk, R.; Puertollano, R.; Parenti, G.; Ballabio, A.; et al. Transcription factor EB [TFEB] is a new.therapeutic target for Pompe disease. EMBO Mol. Med. 2013, 5, 691–706. [Google Scholar] [CrossRef] [PubMed]
- Li, H.M.; Feeney, E.; Li, L.; Zare, H.; Puertollano, R.; Raben, N. WITHDRAWN: Clearance of lysosomal glycogen accumulation by Transcription factor EB (TFEB) in muscle cells from lysosomal alpha-glucosidase deficient mice. Biochem. Biophys. Res. Commun. 2013. [Google Scholar] [CrossRef]
- Paigen, K.; Peterson, J. Coordinacy of lysosomal enzyme excretion in human urine. J. Clin. Investig. 1978, 61, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.H.; Waindle, L.M.; Brown, B.I. The apparent activity in vivo of the lysosomal pathway of glycogen catabolism in cultured human skin fibroblasts from patients with type III glycogen storage disease. J. Biol. Chem. 1978, 253, 5005–5011. [Google Scholar] [PubMed]
- Brown, B.I.; Brown, D.H. The subcellular distribution of enzymes in type II glycogenosis and the occurrence of an oligo-α-1,4-glucan glucohydrolase in human tissues. Biochim. Biophys. Acta 1965, 110, 124–133. [Google Scholar] [CrossRef]
- Roach, P.J.; DePaoli-Roach, A.A.; Hurley, T.D.; Tagliabracci, V.S. Glycogen and its metabolism: Some new developments and old themes. Biochem. J. 2012, 441, 763–787. [Google Scholar] [CrossRef]
- Rodén, L.; Ananth, S.; Campbell, P.; Manzella, S.; Meezan, E. Xylosyl transfer to an endogenous renal acceptor. Purification of the transferase and the acceptor and their identification as glycogenin. J. Biol. Chem. 1994, 269, 11509–11513. [Google Scholar]
- Bukovac, M.J.; Olien, W.C. Ethephon-Induced Gummosis in Sour Cherry (Prunus cerasus L.): 1. Effect on xylem function and shoot water status. Plant Physiol. 1982, 70, 547–555. [Google Scholar]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murray, A.K. The Release of a Soluble Glycosylated Protein from Glycogen by Recombinant Lysosomal α-Glucosidase (rhGAA) In Vitro and Its Presence in Serum In Vivo. Biomolecules 2020, 10, 1613. https://doi.org/10.3390/biom10121613
Murray AK. The Release of a Soluble Glycosylated Protein from Glycogen by Recombinant Lysosomal α-Glucosidase (rhGAA) In Vitro and Its Presence in Serum In Vivo. Biomolecules. 2020; 10(12):1613. https://doi.org/10.3390/biom10121613
Chicago/Turabian StyleMurray, Allen K. 2020. "The Release of a Soluble Glycosylated Protein from Glycogen by Recombinant Lysosomal α-Glucosidase (rhGAA) In Vitro and Its Presence in Serum In Vivo" Biomolecules 10, no. 12: 1613. https://doi.org/10.3390/biom10121613
APA StyleMurray, A. K. (2020). The Release of a Soluble Glycosylated Protein from Glycogen by Recombinant Lysosomal α-Glucosidase (rhGAA) In Vitro and Its Presence in Serum In Vivo. Biomolecules, 10(12), 1613. https://doi.org/10.3390/biom10121613