Elevated Dkk1 Mediates Downregulation of the Canonical Wnt Pathway and Lysosomal Loss in an iPSC Model of Neuronopathic Gaucher Disease


Abstract
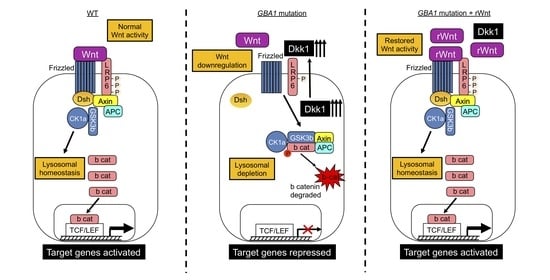
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Generation of iPSC-Derived Neuronal Progenitor Cells (NPCs)
2.2. Chemical Reagents and Treatments
2.3. Quantitative PCR (qPCR)
2.4. Immunocytochemistry/Immunofluorescence
2.5. Image Acquisition and Analysis
2.6. Western Blot Analysis
2.7. Statistical Analysis
3. Results
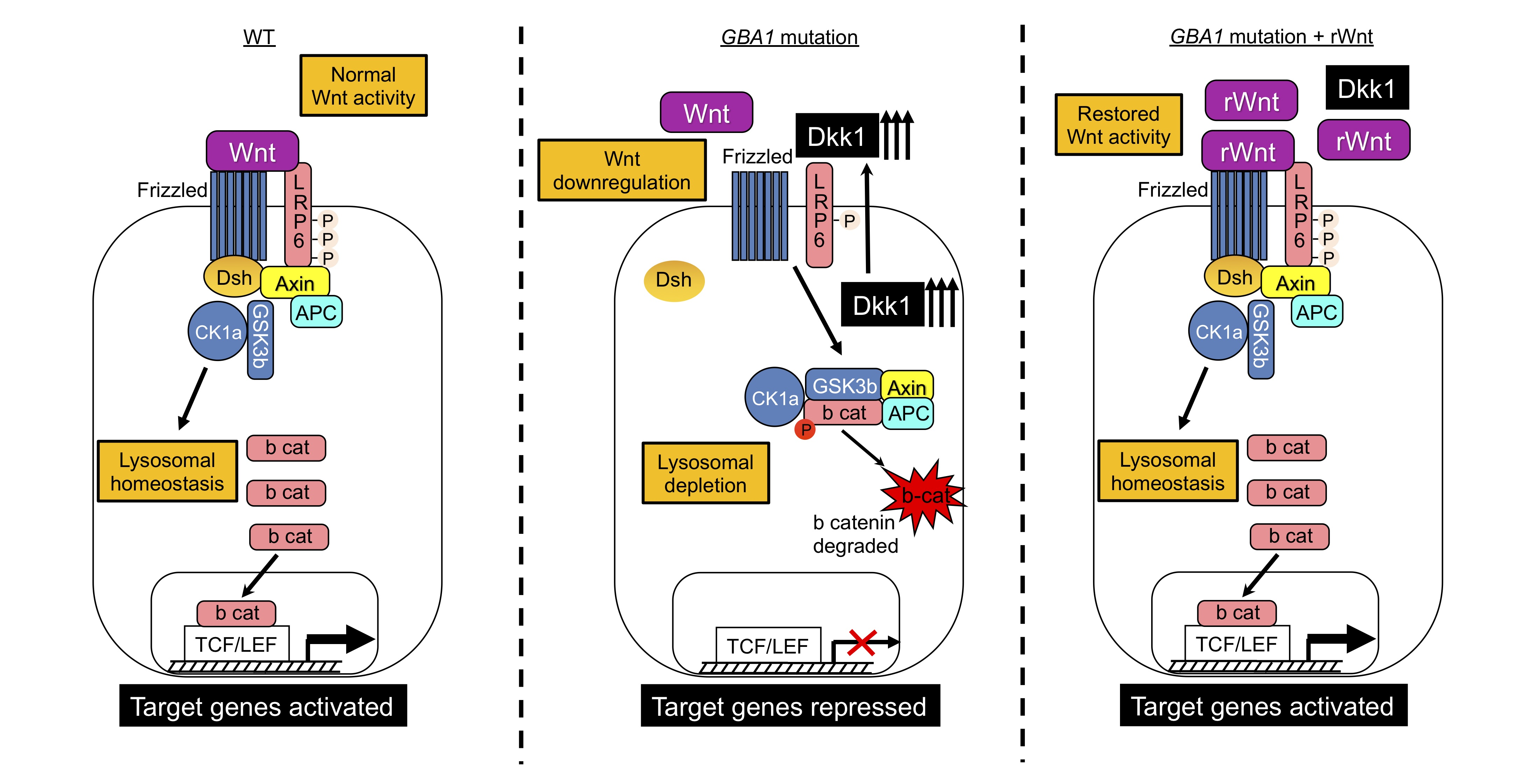
3.1. Upregulation of Dkk1 mRNA and Protein Levels in Neuronopathic GD NPCs
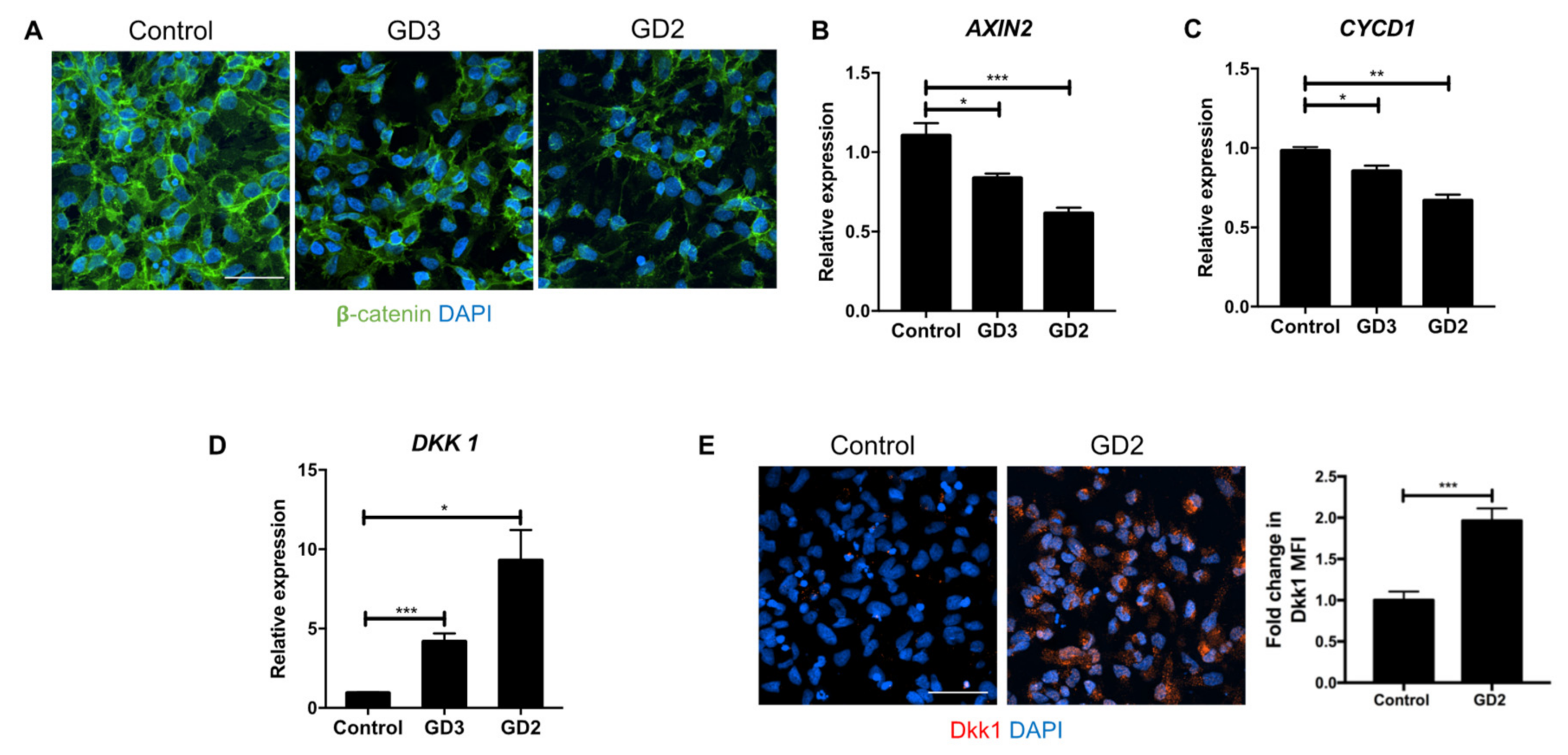
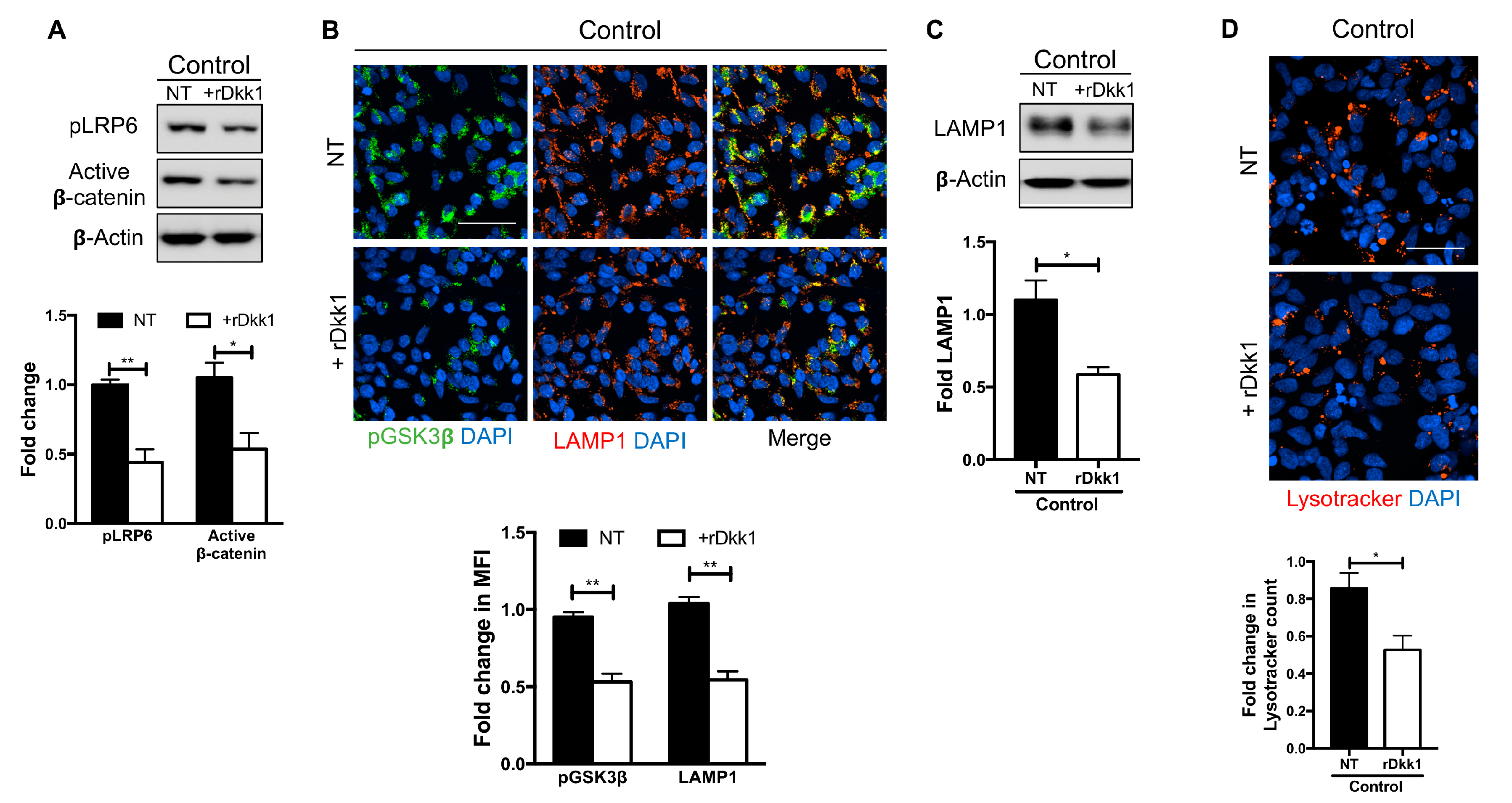
3.2. Recombinant Dkk1 Downregulates the Wnt/β-Catenin Pathway and Disrupts the Lysosomal Compartment in WT NPCs
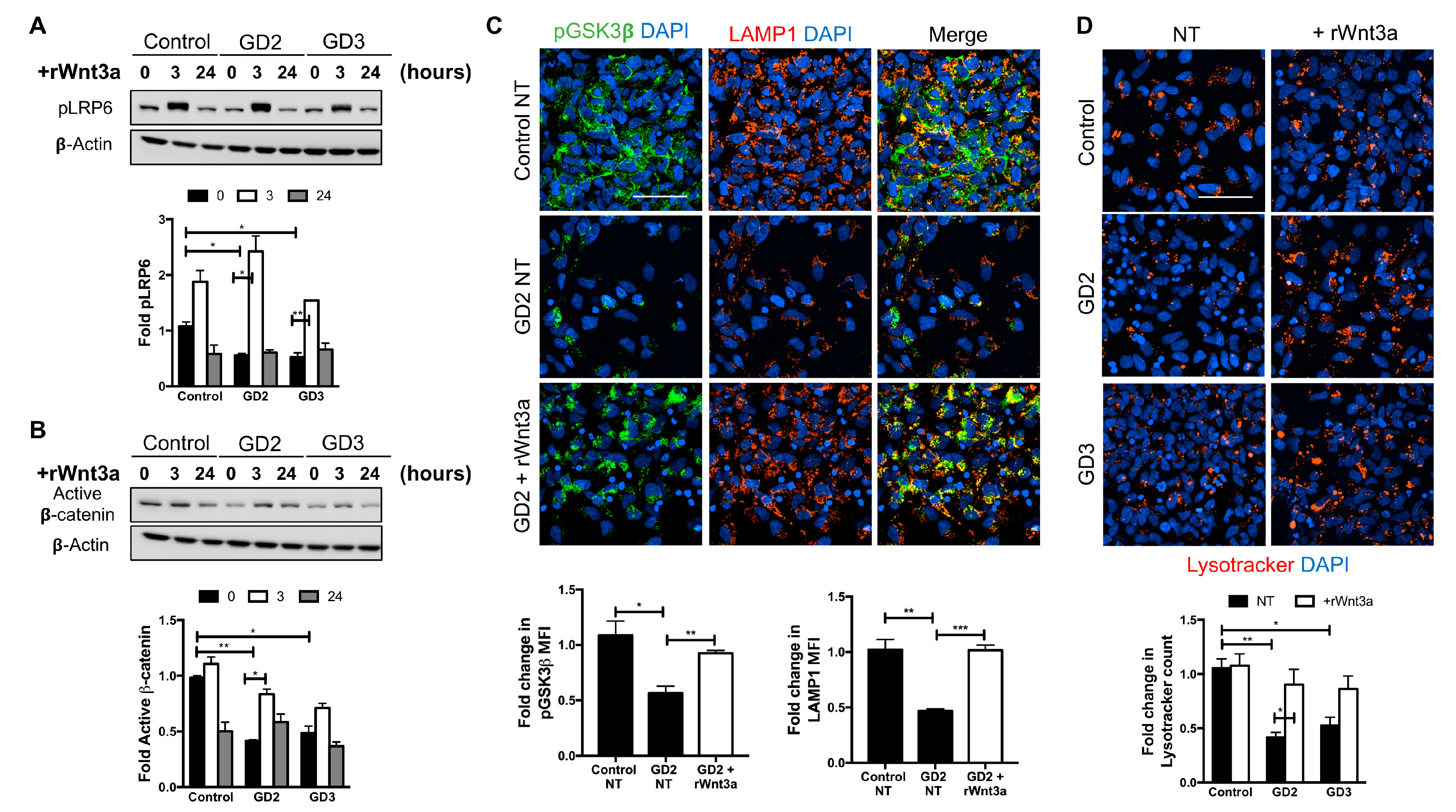
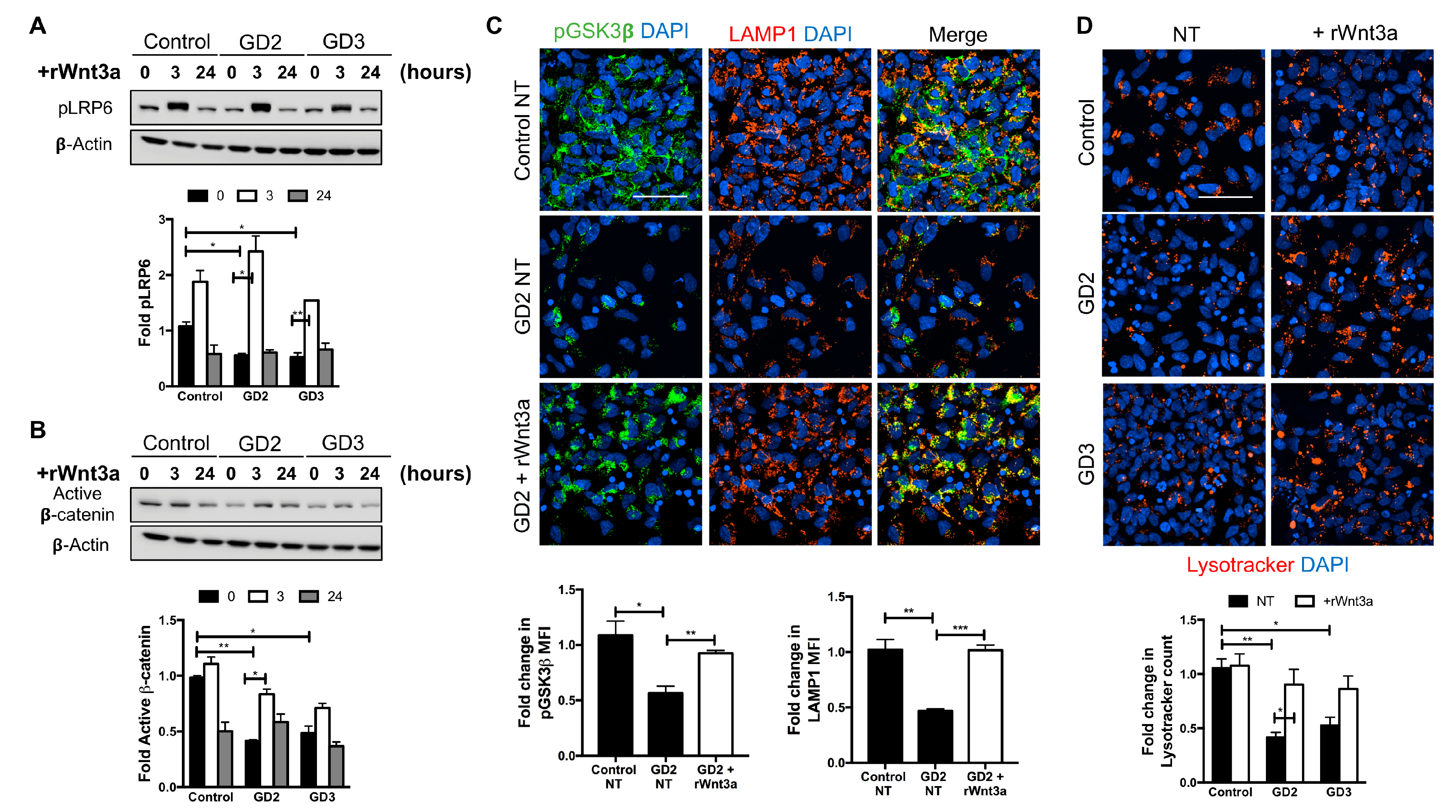
3.3. Recombinant Wnt3a Treatment of nGD NPCs Rescues Wnt/β-Catenin Signaling and Restores the Lysosomal Compartments
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Schmitz, M.; Alfalah, M.; Aerts, J.M.; Naim, H.Y.; Zimmer, K.P. Impaired trafficking of mutants of lysosomal glucocerebrosidase in gaucher’s disease. Int. J. Biochem. Cell. Biol. 2005, 37, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Bendikov-Bar, I.; Ron, I.; Filocamo, M.; Horowitz, M. Characterization of the erad process of the l444p mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 2011, 46, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, J. Gaucher disease: Review of the literature. Arch. Pathol. Lab. Med 2008, 132, 851–853. [Google Scholar] [PubMed]
- Cox, T.M. Gaucher disease: Clinical profile and therapeutic developments. Biologics 2010, 4, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, G.A.; Golembo, M.; Shaaltiel, Y. Taliglucerase alfa: An enzyme replacement therapy using plant cell expression technology. Mol. Genet. Metab. 2014, 112, 1–8. [Google Scholar] [CrossRef]
- Sidransky, E. Gaucher disease: Insights from a rare mendelian disorder. Discov. Med. 2012, 14, 273–281. [Google Scholar]
- Dandana, A.; Ben Khelifa, S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher disease: Clinical, biological and therapeutic aspects. Pathobiology 2016, 83, 13–23. [Google Scholar] [CrossRef]
- Alaei, M.R.; Tabrizi, A.; Jafari, N.; Mozafari, H. Gaucher disease: New expanded classification emphasizing neurological features. Iran. J. Child. Neurol. 2019, 13, 7–24. [Google Scholar]
- Eblan, M.J.; Goker-Alpan, O.; Sidransky, E. Perinatal lethal gaucher disease: A distinct phenotype along the neuronopathic continuum. Fetal Pediatr. Pathol. 2005, 24, 205–222. [Google Scholar] [CrossRef]
- Kaplan, P.; Andersson, H.C.; Kacena, K.A.; Yee, J.D. The clinical and demographic characteristics of nonneuronopathic gaucher disease in 887 children at diagnosis. Arch. Pediatr. Adolesc. Med. 2006, 160, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Pastores, G.M. Neuropathic gaucher disease. Wien. Med. Wochenschr. 2010, 160, 605–608. [Google Scholar] [CrossRef]
- Roshan Lal, T.; Sidransky, E. The spectrum of neurological manifestations associated with gaucher disease. Diseases 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Vellodi, A.; Tylki-Szymanska, A.; Davies, E.H.; Kolodny, E.; Bembi, B.; Collin-Histed, T.; Mengel, E.; Erikson, A.; Schiffmann, R. Management of neuronopathic gaucher disease: Revised recommendations. J. Inherit. Metab. Dis. 2009, 32, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Futerman, A.H. Neuronal forms of gaucher disease. Handb. Exp. Pharmacol. 2013, 216, 405–419. [Google Scholar]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef]
- Stone, D.L.; Tayebi, N.; Orvisky, E.; Stubblefield, B.; Madike, V.; Sidransky, E. Glucocerebrosidase gene mutations in patients with type 2 gaucher disease. Hum. Mutat. 2000, 15, 181–188. [Google Scholar] [CrossRef]
- Orvisky, E.; Sidransky, E.; McKinney, C.E.; Lamarca, M.E.; Samimi, R.; Krasnewich, D.; Martin, B.M.; Ginns, E.I. Glucosylsphingosine accumulation in mice and patients with type 2 gaucher disease begins early in gestation. Pediatr. Res. 2000, 48, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Weiss, K.; Gonzalez, A.; Lopez, G.; Pedoeim, L.; Groden, C.; Sidransky, E. The clinical management of type 2 gaucher disease. Mol. Genet. Metab. 2015, 114, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Lwin, A.; Orvisky, E.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 2004, 81, 70–73. [Google Scholar] [CrossRef] [Green Version]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutant gba to the development of parkinson disease in drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in parkinson’s patient midbrain neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef] [PubMed]
- Moors, T.; Paciotti, S.; Chiasserini, D.; Calabresi, P.; Parnetti, L.; Beccari, T.; van de Berg, W.D. Lysosomal dysfunction and alpha-synuclein aggregation in parkinson’s disease: Diagnostic links. Mov. Disord. 2016, 31, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine promotes alpha-synuclein pathology in mutant gba-associated parkinson’s disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Grabowski, G.A. Impaired autophagosomes and lysosomes in neuronopathic gaucher disease. Autophagy 2010, 6, 648–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.H.; et al. Neuronopathic gaucher disease in the mouse: Viable combined selective saposin c deficiency and mutant glucocerebrosidase (v394l) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef] [Green Version]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Miller, D.; Zeng, X.; Sgambato, J.A.; Lipinski, M.M.; Feldman, R.A. Altered tfeb-mediated lysosomal biogenesis in gaucher disease ipsc-derived neuronal cells. Hum. Mol. Genet. 2015, 24, 5775–5788. [Google Scholar] [CrossRef] [Green Version]
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. Gba deficiency promotes snca/alpha-synuclein accumulation through autophagic inhibition by inactivated ppp2a. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef] [Green Version]
- Schondorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. Ipsc-derived neurons from gba1-associated parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.A.; Voit, A.; Srikanth, M.P.; Thayer, J.A.; Kingsbury, T.J.; Jacobson, M.A.; Lipinski, M.M.; Feldman, R.A.; Awad, O. Mtor hyperactivity mediates lysosomal dysfunction in gaucher’s disease ipsc-neuronal cells. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. Tfeb links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mtor and tfeb. Embo. J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolski, R.; Vick, P.; Ploper, D.; Gumper, I.; Snitkin, H.; Sabatini, D.D.; De Robertis, E.M. Presenilin deficiency or lysosomal inhibition enhances wnt signaling through relocalization of gsk3 to the late-endosomal compartment. Cell Rep. 2012, 2, 1316–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilić, J.; Huang, Y.-L.; Davidson, G.; Zimmermann, T.; Cruciat, C.-M.; Bienz, M.; Niehrs, C. Wnt induces lrp6 signalosomes and promotes dishevelled-dependent lrp6 phosphorylation. Science 2007, 316, 1619–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taelman, V.F.; Dobrowolski, R.; Plouhinec, J.-L.; Fuentealba, L.C.; Vorwald, P.P.; Gumper, I.; Sabatini, D.D.; De Robertis, E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 2010, 143, 1136–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin d1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusse, R.; Varmus, H. Three decades of wnts: A personal perspective on how a scientific field developed. Embo. J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inestrosa, N.C.; Arenas, E. Emerging roles of wnts in the adult nervous system. Nat. Rev. Neurosci. 2010, 11, 77–86. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Maguschak, K.A.; Ressler, K.J. Wnt signaling in amygdala-dependent learning and memory. J. Neurosci. 2011, 31, 13057–13067. [Google Scholar] [CrossRef]
- Marzo, A.; Galli, S.; Lopes, D.; McLeod, F.; Podpolny, M.; Segovia-Roldan, M.; Ciani, L.; Purro, S.; Cacucci, F.; Gibb, A.; et al. Reversal of synapse degeneration by restoring wnt signaling in the adult hippocampus. Curr. Biol. 2016, 26, 2551–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, F.; Salinas, P.C. Wnt proteins as modulators of synaptic plasticity. Curr. Opin. Neurobiol. 2018, 53, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Toledo, E.M. The role of wnt signaling in neuronal dysfunction in alzheimer’s disease. Mol. Neurodegener. 2008, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephano, F.; Nolte, S.; Hoffmann, J.; El-Kholy, S.; von Frieling, J.; Bruchhaus, I.; Fink, C.; Roeder, T. Impaired wnt signaling in dopamine containing neurons is associated with pathogenesis in a rotenone triggered drosophila parkinson’s disease model. Sci. Rep. 2018, 8, 2372. [Google Scholar] [CrossRef] [Green Version]
- Dun, Y.; Li, G.; Yang, Y.; Xiong, Z.; Feng, M.; Wang, M.; Zhang, Y.; Xiang, J.; Ma, R. Inhibition of the canonical wnt pathway by dickkopf-1 contributes to the neurodegeneration in 6-ohda-lesioned rats. Neurosci. Lett. 2012, 525, 83–88. [Google Scholar] [CrossRef]
- Caricasole, A.; Copani, A.; Caraci, F.; Aronica, E.; Rozemuller, A.J.; Caruso, A.; Storto, M.; Gaviraghi, G.; Terstappen, G.C.; Nicoletti, F. Induction of dickkopf-1, a negative modulator of the wnt pathway, is associated with neuronal degeneration in alzheimer’s brain. J. Neurosci. 2004, 24, 6021–6027. [Google Scholar] [CrossRef]
- L’Episcopo, F.; Tirolo, C.; Testa, N.; Caniglia, S.; Morale, M.C.; Cossetti, C.; D’Adamo, P.; Zardini, E.; Andreoni, L.; Ihekwaba, A.E.; et al. Reactive astrocytes and wnt/β-catenin signaling link nigrostriatal injury to repair in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of parkinson’s disease. Neurobiol. Dis. 2011, 41, 508–527. [Google Scholar] [CrossRef] [Green Version]
- Mikels, A.J.; Nusse, R. Wnts as ligands: Processing, secretion and reception. Oncogene 2006, 25, 7461–7468. [Google Scholar] [CrossRef] [Green Version]
- Verheyen, E.M.; Gottardi, C.J. Regulation of wnt/beta-catenin signaling by protein kinases. Dev. Dyn. 2010, 239, 34–44. [Google Scholar]
- Willert, K.; Nusse, R. Beta-catenin: A key mediator of wnt signaling. Curr. Opin. Genet. Dev. 1998, 8, 95–102. [Google Scholar] [CrossRef]
- Mao, B.; Wu, W.; Li, Y.; Hoppe, D.; Stannek, P.; Glinka, A.; Niehrs, C. Ldl-receptor-related protein 6 is a receptor for dickkopf proteins. Nature 2001, 411, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. Function and biological roles of the dickkopf family of wnt modulators. Oncogene 2006, 25, 7469–7481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Liu, L.; Liu, A. Dickkopf-1: Current knowledge and related diseases. Life Sci. 2018, 209, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, L.G. Alzheimer disease. Continuum 2016, 22, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Rosi, M.C.; Luccarini, I.; Grossi, C.; Fiorentini, A.; Spillantini, M.G.; Prisco, A.; Scali, C.; Gianfriddo, M.; Caricasole, A.; Terstappen, G.C.; et al. Increased dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J. Neurochem. 2010, 112, 1539–1551. [Google Scholar] [CrossRef]
- Lecourt, S.; Mouly, E.; Freida, D.; Cras, A.; Ceccaldi, R.; Heraoui, D.; Chomienne, C.; Marolleau, J.-P.; Arnulf, B.; Porcher, R.; et al. A prospective study of bone marrow hematopoietic and mesenchymal stem cells in type 1 gaucher disease patients. PLoS ONE 2013, 8, e69293. [Google Scholar] [CrossRef]
- Zancan, I.; Bellesso, S.; Costa, R.; Salvalaio, M.; Stroppiano, M.; Hammond, C.; Argenton, F.; Filocamo, M.; Moro, E. Glucocerebrosidase deficiency in zebrafish affects primary bone ossification through increased oxidative stress and reduced wnt/beta-catenin signaling. Hum. Mol. Genet. 2015, 24, 1280–1294. [Google Scholar] [CrossRef] [Green Version]
- Panicker, L.M.; Miller, D.; Park, T.S.; Patel, B.; Azevedo, J.L.; Awad, O.; Masood, M.A.; Veenstra, T.D.; Goldin, E.; Stubblefield, B.K.; et al. Induced pluripotent stem cell model recapitulates pathologic hallmarks of gaucher disease. Proc. Natl. Acad. Sci. USA 2012, 109, 18054–18059. [Google Scholar] [CrossRef] [Green Version]
- Panicker, L.M.; Miller, D.; Awad, O.; Bose, V.; Lun, Y.; Park, T.S.; Zambidis, E.T.; Sgambato, J.A.; Feldman, R.A. Gaucher ipsc-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells 2014, 32, 2338–2349. [Google Scholar] [CrossRef] [Green Version]
- Sgambato, J.A.; Park, T.S.; Miller, D.; Panicker, L.M.; Sidransky, E.; Lun, Y.; Awad, O.; Bentzen, S.M.; Zambidis, E.T.; Feldman, R.A. Gaucher disease-induced pluripotent stem cells display decreased erythroid potential and aberrant myelopoiesis. Stem Cells Transl. Med. 2015, 4, 878–886. [Google Scholar] [CrossRef]
- Awad, O.; Panicker, L.M.; Deranieh, R.M.; Srikanth, M.P.; Brown, R.A.; Voit, A.; Peesay, T.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Altered differentiation potential of gaucher’s disease ipsc neuronal progenitors due to wnt/beta-catenin downregulation. Stem Cell Rep. 2017, 9, 1853–1867. [Google Scholar] [CrossRef] [Green Version]
- Doble, B.W.; Woodgett, J.R. Gsk-3: Tricks of the trade for a multi-tasking kinase. J. Cell. Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niehrs, C.; Acebron, S.P. Wnt signaling: Multivesicular bodies hold gsk3 captive. Cell 2010, 143, 1044–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, N.P.; Kamireddy, A.; Van Nostrand, J.L.; Eichner, L.J.; Shokhirev, M.N.; Dayn, Y.; Shaw, R.J. Ampk governs lineage specification through tfeb-dependent regulation of lysosomes. Genes. Dev. 2016, 30, 535–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glinka, A.; Wu, W.; Delius, H.; Monaghan, A.P.; Blumenstock, C.; Niehrs, C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature 1998, 391, 357–362. [Google Scholar] [CrossRef]
- Haaber, J.; Abildgaard, N.; Knudsen, L.M.; Dahl, I.M.; Lodahl, M.; Thomassen, M.; Kerndrup, G.B.; Rasmussen, T. Myeloma cell expression of 10 candidate genes for osteolytic bone disease. Only overexpression of dkk1 correlates with clinical bone involvement at diagnosis. Br. J. Haematol. 2008, 140, 25–35. [Google Scholar] [CrossRef]
- Tian, E.; Zhan, F.; Walker, R.; Rasmussen, E.; Ma, Y.; Barlogie, B.; Shaughnessy, J.D., Jr. The role of the wnt-signaling antagonist dkk1 in the development of osteolytic lesions in multiple myeloma. N. Engl. J. Med. 2003, 349, 2483–2494. [Google Scholar] [CrossRef]
- Ko, J.-Y.; Wang, F.-S.; Wang, C.-J.; Wong, T.; Chou, W.-Y.; Tseng, S.-L. Increased dickkopf-1 expression accelerates bone cell apoptosis in femoral head osteonecrosis. Bone 2010, 46, 584–591. [Google Scholar] [CrossRef]
- Tsentidis, C.; Gourgiotis, D.; Kossiva, L.; Marmarinos, A.; Doulgeraki, A.; Karavanaki, K. Increased levels of dickkopf-1 are indicative of wnt/β-catenin downregulation and lower osteoblast signaling in children and adolescents with type 1 diabetes mellitus, contributing to lower bone mineral density. Osteoporos. Int. 2017, 28, 945–953. [Google Scholar] [CrossRef]
- Cappuccio, I.; Calderone, A.; Busceti, C.L.; Biagioni, F.; Pontarelli, F.; Bruno, V.; Storto, M.; Terstappen, G.T.; Gaviraghi, G.; Fornai, F.; et al. Induction of dickkopf-1, a negative modulator of the wnt pathway, is required for the development of ischemic neuronal death. J. Neurosci. 2005, 25, 2647–2657. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.D.; Ferreira, N.S.; Ben-Dor, S.; Duan, J.; Hardy, J.; Cox, T.M.; Merrill, A.H., Jr.; Futerman, A.H. Identification of modifier genes in a mouse model of gaucher disease. Cell Rep. 2016, 16, 2546–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, L.V.; Ploper, D.; Tejeda-Muñoz, N.; De Robertis, E.M. Arginine methylation is required for canonical wnt signaling and endolysosomal trafficking. Proc. Natl. Acad. Sci. USA 2018, 115, E5317–E5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redelman-Sidi, G.; Binyamin, A.; Gaeta, I.; Palm, W.; Thompson, C.B.; Romesser, P.B.; Lowe, S.W.; Bagul, M.; Doench, J.G.; Root, D.E.; et al. The canonical wnt pathway drives macropinocytosis in cancer. Cancer Res. 2018, 78, 4658–4670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito-Diaz, K.; Benchabane, H.; Tiwari, A.; Tian, A.; Li, B.; Thompson, J.J.; Hyde, A.S.; Sawyer, L.M.; Jodoin, J.N.; Santos, E.; et al. Apc inhibits ligand-independent wnt signaling by the clathrin endocytic pathway. Dev. Cell 2018, 44, 566–581.e568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, L.V.; Tejeda-Muñoz, N.; Bui, M.H.; Cicchetto, A.C.; Di Biagio, D.; Colozza, G.; Schmid, E.; Piccolo, S.; Christofk, H.R.; De Robertis, E.M. Gsk3 inhibits macropinocytosis and lysosomal activity through the wnt destruction complex machinery. Cell Rep. 2020, 32, 107973. [Google Scholar] [CrossRef]
- Park, J.H.; Min, J.; Baek, S.R.; Kim, S.W.; Kwon, I.K.; Jeon, S.R. Enhanced neuroregenerative effects by scaffold for the treatment of a rat spinal cord injury with wnt3a-secreting fibroblasts. Acta. Neurochir. 2013, 155, 809–816. [Google Scholar] [CrossRef]
- Gao, K.; Wang, Y.S.; Yuan, Y.J.; Wan, Z.H.; Yao, T.C.; Li, H.H.; Tang, P.F.; Mei, X.F. Neuroprotective effect of rapamycin on spinal cord injury via activation of the wnt/β-catenin signaling pathway. Neural. Regen. Res. 2015, 10, 951–957. [Google Scholar]
- González-Fernández, C.; Fernández-Martos, C.M.; Shields, S.D.; Arenas, E.; Javier Rodríguez, F. Wnts are expressed in the spinal cord of adult mice and are differentially induced after injury. J. Neurotrauma 2014, 31, 565–581. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Y.; Lee, J.H.; Gu, X.; Wei, Z.Z.; Harris, M.J.; Yu, S.P.; Wei, L. Intranasally delivered wnt3a improves functional recovery after traumatic brain injury by modulating autophagic, apoptotic, and regenerative pathways in the mouse brain. J. Neurotrauma 2018, 35, 802–813. [Google Scholar] [CrossRef]
- Matei, N.; Camara, J.; McBride, D.; Camara, R.; Xu, N.; Tang, J.; Zhang, J.H. Intranasal wnt3a attenuates neuronal apoptosis through frz1/piwil1a/foxm1 pathway in mcao rats. J. Neurosci. 2018, 38, 6787–6801. [Google Scholar] [CrossRef] [Green Version]
- Ke, H.Z.; Richards, W.G.; Li, X.; Ominsky, M.S. Sclerostin and dickkopf-1 as therapeutic targets in bone diseases. Endocr. Rev. 2012, 33, 747–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulciniti, M.; Tassone, P.; Hideshima, T.; Vallet, S.; Nanjappa, P.; Ettenberg, S.A.; Shen, Z.; Patel, N.; Tai, Y.T.; Chauhan, D.; et al. Anti-dkk1 mab (bhq880) as a potential therapeutic agent for multiple myeloma. Blood 2009, 114, 371–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.P.; Beck, J.T.; Stewart, A.K.; Shah, J.; Kelly, K.R.; Isaacs, R.; Bilic, S.; Sen, S.; Munshi, N.C. A phase ib multicentre dose-determination study of bhq880 in combination with anti-myeloma therapy and zoledronic acid in patients with relapsed or refractory multiple myeloma and prior skeletal-related events. Br. J. Haematol. 2014, 167, 366–375. [Google Scholar] [CrossRef]
- Iozzi, S.; Remelli, R.; Lelli, B.; Diamanti, D.; Pileri, S.; Bracci, L.; Roncarati, R.; Caricasole, A.; Bernocco, S. Functional characterization of a small-molecule inhibitor of the dkk1-lrp6 interaction. ISRN Mol. Biol. 2012, 2012, 823875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thysiadis, S.; Katsamakas, S.; Mpousis, S.; Avramidis, N.; Efthimiopoulos, S.; Sarli, V. Design and synthesis of gallocyanine inhibitors of dkk1/lrp6 interactions for treatment of alzheimer’s disease. Bioorg. Chem. 2018, 80, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Panicker, L.M.; Srikanth, M.P.; Castro-Gomes, T.; Miller, D.; Andrews, N.W.; Feldman, R.A. Gaucher disease ipsc-derived osteoblasts have developmental and lysosomal defects that impair bone matrix deposition. Hum. Mol. Genet. 2018, 27, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srikanth, M.P.; Feldman, R.A. Elevated Dkk1 Mediates Downregulation of the Canonical Wnt Pathway and Lysosomal Loss in an iPSC Model of Neuronopathic Gaucher Disease. Biomolecules 2020, 10, 1630. https://doi.org/10.3390/biom10121630
Srikanth MP, Feldman RA. Elevated Dkk1 Mediates Downregulation of the Canonical Wnt Pathway and Lysosomal Loss in an iPSC Model of Neuronopathic Gaucher Disease. Biomolecules. 2020; 10(12):1630. https://doi.org/10.3390/biom10121630
Chicago/Turabian StyleSrikanth, Manasa P., and Ricardo A. Feldman. 2020. "Elevated Dkk1 Mediates Downregulation of the Canonical Wnt Pathway and Lysosomal Loss in an iPSC Model of Neuronopathic Gaucher Disease" Biomolecules 10, no. 12: 1630. https://doi.org/10.3390/biom10121630
APA StyleSrikanth, M. P., & Feldman, R. A. (2020). Elevated Dkk1 Mediates Downregulation of the Canonical Wnt Pathway and Lysosomal Loss in an iPSC Model of Neuronopathic Gaucher Disease. Biomolecules, 10(12), 1630. https://doi.org/10.3390/biom10121630