Cellular Dynamics of Transition Metal Exchange on Proteins: A Challenge but a Bonanza for Coordination Chemistry


Abstract
Foreword
1. Introduction
2. Biological Availability of Transition Metals: Not So Easy, even in an Ocean of Plenty
2.1. Evolution Could not Get Rid of Transition Metals
2.2. Transition Metals vs. Oxygen: Marriage or Divorce?
3. Getting Metals In and Out of Cells: Metal Exchange in Action
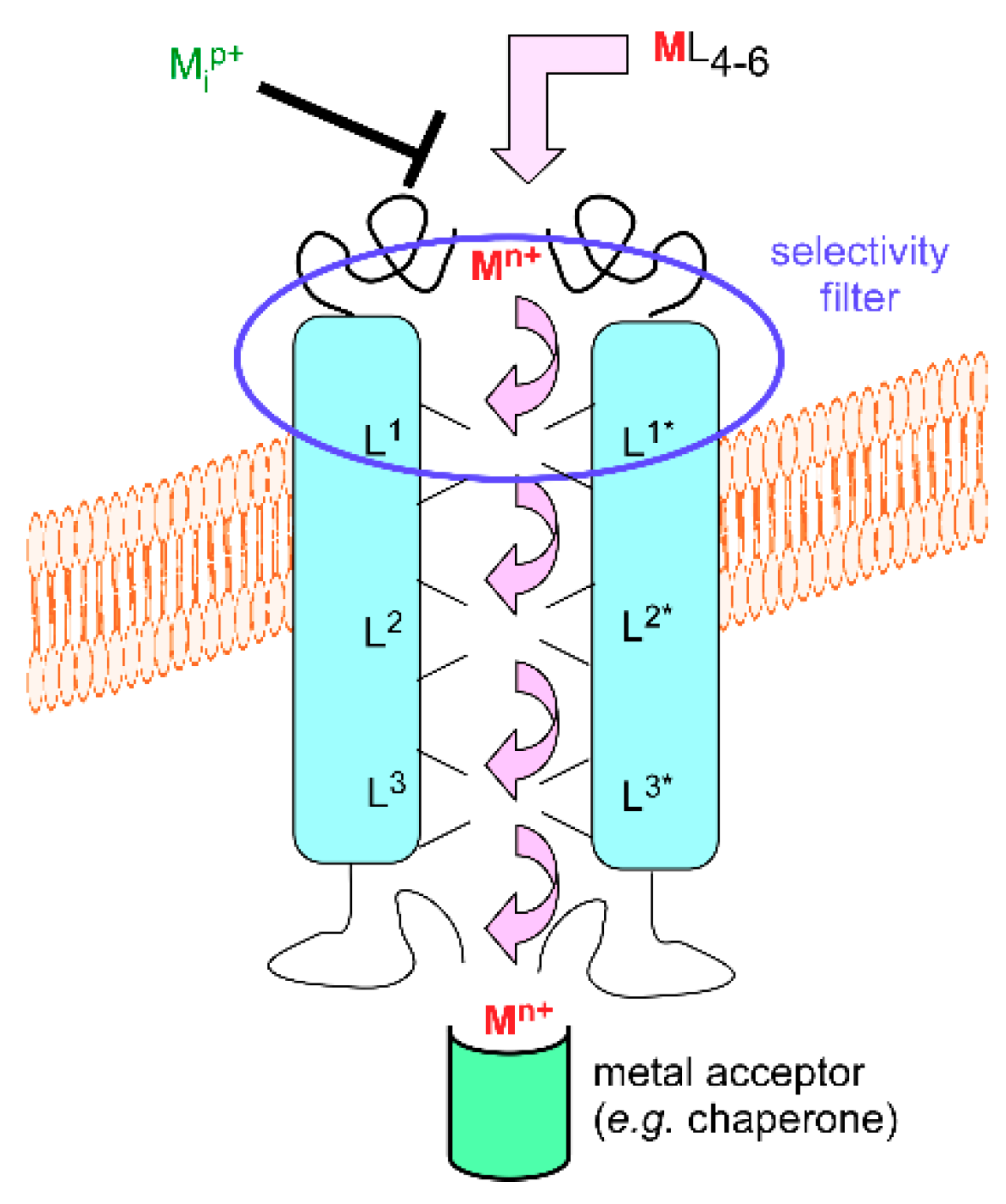
3.1. Metal Exchange through Biological Membranes
3.2. Crossing Membranes Alone or as a Party?
4. Methodological Hurdles in the Study of the Dynamics of Intracellular Transition Metal Trafficking
4.1. Transition Metals Biophysics: Diverse and Sophisticated
4.2. Transition Metals Biophysics: Necessarily Limited
5. Labeling Transition Metals in Cells: Pros and Cons
5.1. The Resolution Problem
5.2. The Specificity Problem
5.3. Monitoring Labeled Metals and Proteins: Work in Progress
6. Divide and (Try to) Conquer in Monitoring Transition Metals
6.1. Metal Exchange as a Probe
6.2. The Metal Trap of Model Organisms
6.3. Serendipitous and Understated Metal Binding
6.4. When Metals Spoil the Show
6.5. Questioning Metal ‘Reconstitution’
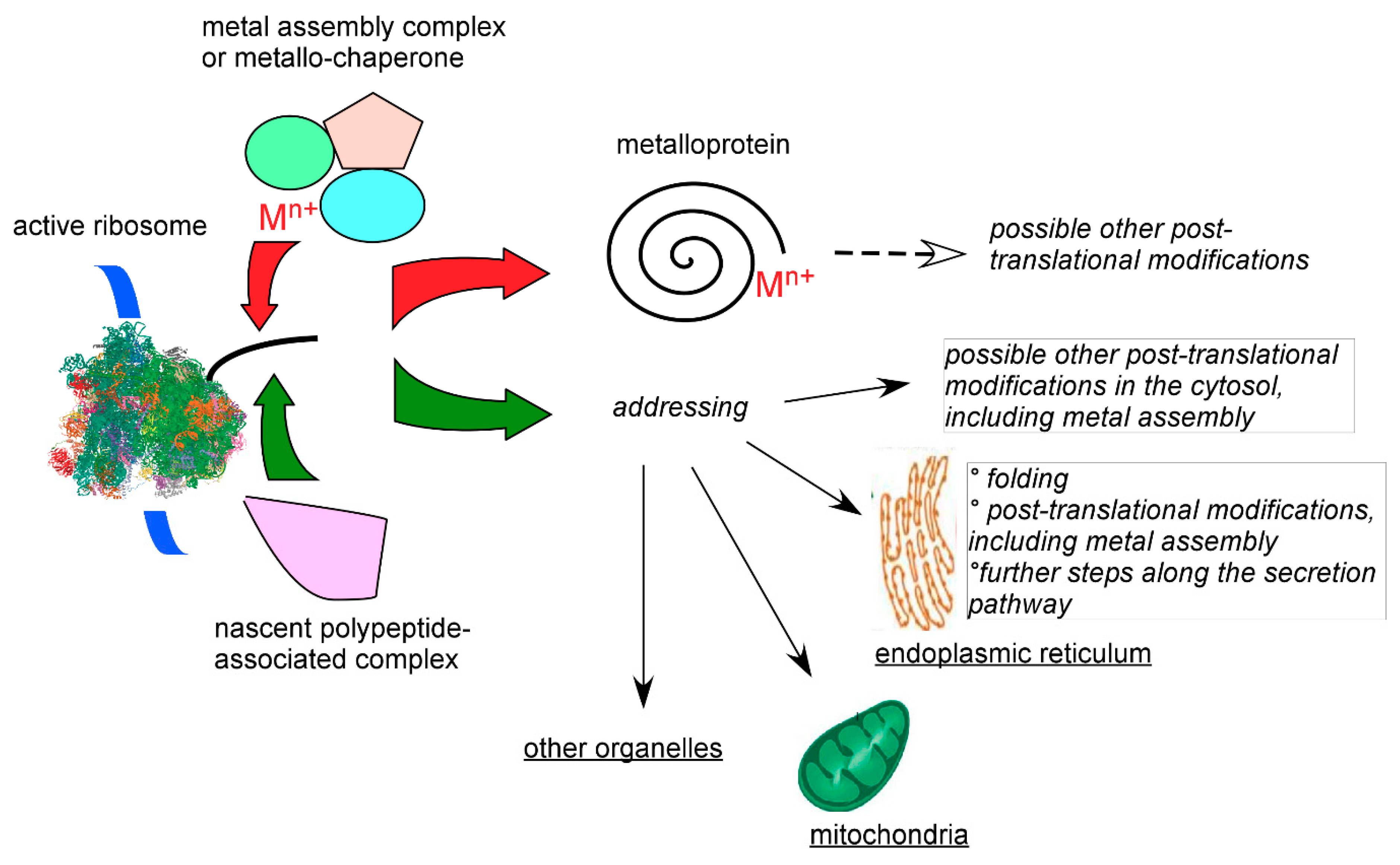
7. Cellular Targeting of Transition Metals
7.1. Different Metal Solutions to the Same Problem
7.2. Several Metal Problems with the Same Protein or Enzyme: Is Toxicity by Metal Replacement So Much Relevant?
7.3. Chaperone, Channeling, and Active Site Synthesis of Metalloproteins
7.4. Tuning Transition Metals’ Reactivity in Metalloproteins
8. Extreme Sensitivity of Transition Metal Exchange Reactions to Cellular Conditions
8.1. Coordinating Metal Assembly and Metalloprotein Folding: A Key Component of Stability
8.2. Transition Metals Turned Wild: A Major Cellular Threat
9. Puzzling Interactions of Transition Metals and Metal-Binding Molecules with Proteins
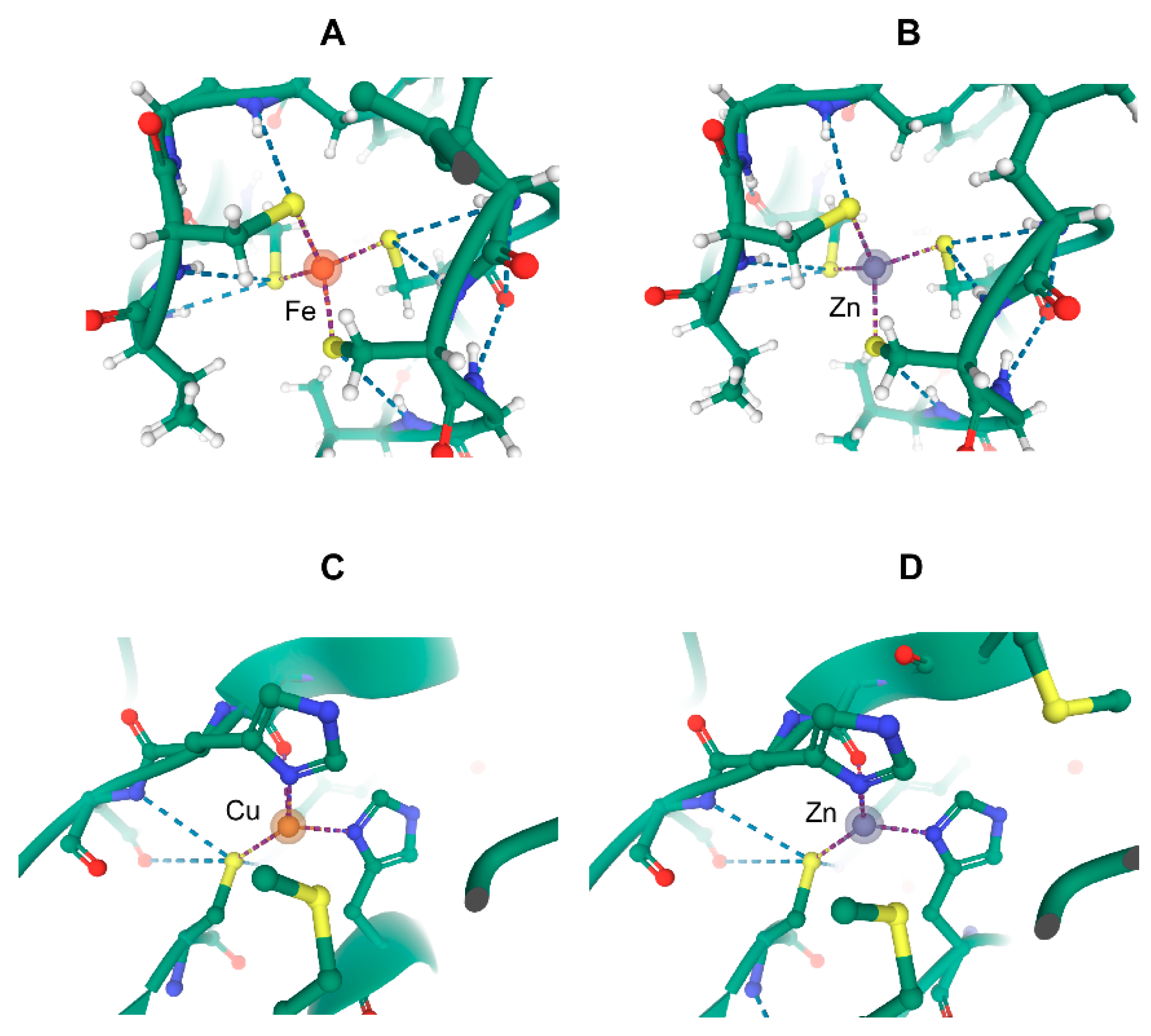
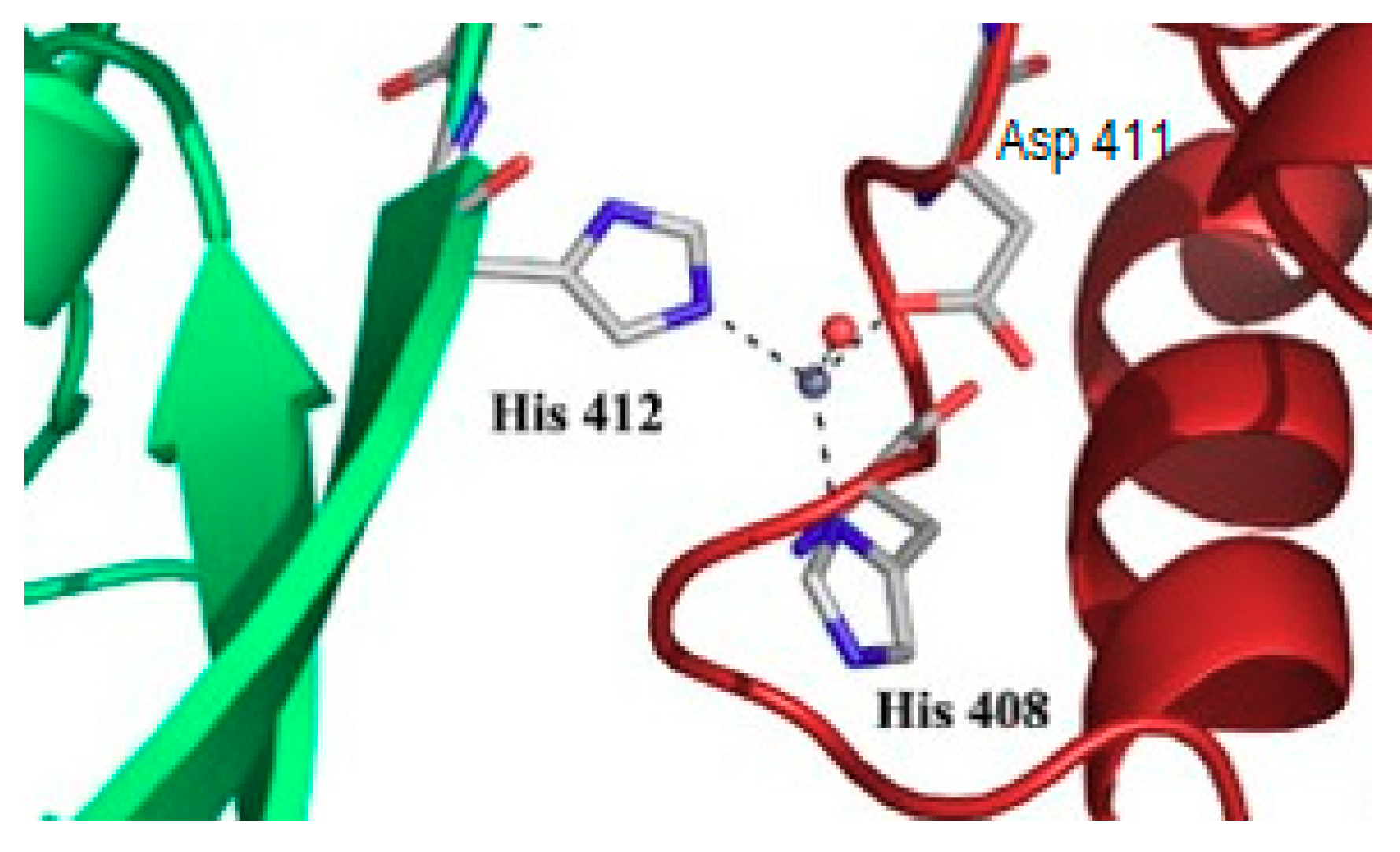
10. What May Be the Molecular Basis of Specificity for Cellular Transition Metals?
11. Conclusions and Perspectives
11.1. Metalloproteins: A Previous and Future Bonanza for Coordination Chemistry
11.2. Metal Exchange: A Large Array of Applications in Biology
11.3. Metals in Biology: A Plea for a Blended Flavor
Funding
Acknowledgments
Conflicts of Interest
References
- Andreini, C.; Bertini, I.; Rosato, A. Metalloproteomes: A Bioinformatic Approach. Acc. Chem. Res. 2009, 42, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Maret, W. Metalloproteomics, metalloproteomes, and the annotation of metalloproteins. Metallomics 2010, 2, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, J.; Darnault, C.; Brazzolotto, X.; Kühn, L.C.; Moulis, J.M.; Volbeda, A.; Fontecilla-Camps, J.C. Crystallization and preliminary X-ray diffraction data for the aconitase form of human iron-regulatory protein 1. Acta Cryst. Sect. F Struct. Biol. Cryst. Commun. 2005, 61, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.P.; Fedorov, A.A.; Sauder, J.M.; Burley, S.K.; Almo, S.C.; Ramagopal, U.A. The hidden treasure in your data: Phasing with unexpected weak anomalous scatterers from routine data sets. Acta Cryst. F Struct. Biol. Commun. 2017, 73, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Golyshina, O.V.; Beloqui, A.; Golyshin, P.N.; Timmis, K.N. The cellular machinery of Ferroplasma acidiphilum is iron-protein-dominated. Nature 2007, 445, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.E.; Musiani, F.; Huang, H.T.; Chivers, P.T.; Ciurli, S.; Maroney, M.J. Glutamate Ligation in the Ni(II)- and Co(II)-Responsive Escherichia coli Transcriptional Regulator, RcnR. Inorg. Chem. 2017, 56, 6459–6476. [Google Scholar] [CrossRef] [PubMed]
- Udagedara, S.R.; Wijekoon, C.J.K.; Xiao, Z.; Wedd, A.G.; Maher, M.J. The crystal structure of the CopC protein from Pseudomonas fluorescens reveals amended classifications for the CopC protein family. J. Inorg. Biochem. 2019, 195, 194–200. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G. Advanced Inorganic Chemistry a Comprehensive Text, 4th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1980; p. 1396. [Google Scholar]
- Haile, D.J.; Rouault, T.A.; Harford, J.B.; Kennedy, M.C.; Blondin, G.A.; Beinert, H.; Klausner, R.D. Cellular regulation of the iron-responsive element binding protein: Disassembly of the cubane iron-sulfur cluster results in high-affinity RNA binding. Proc. Natl. Acad. Sci. USA 1992, 89, 11735–11739. [Google Scholar] [CrossRef]
- Wang, H.; Shi, H.; Rajan, M.; Canarie, E.R.; Hong, S.; Simoneschi, D.; Pagano, M.; Bush, M.F.; Stoll, S.; Leibold, E.A.; et al. FBXL5 Regulates IRP2 Stability in Iron Homeostasis via an Oxygen-Responsive [2Fe2S] Cluster. Mol. Cell 2020, 78, 31–41.e5. [Google Scholar] [CrossRef]
- Martin, W.; Russell, M.J. On the origins of cells: A hypothesis for the evolutionary transitions from abiotic geochemistry to chemoautotrophic prokaryotes, and from prokaryotes to nucleated cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 59–83; discussion 83–85. [Google Scholar] [CrossRef]
- Sosa Torres, M.E.; Saucedo-Vazquez, J.P.; Kroneck, P.M. The magic of dioxygen. Met. Ions Life Sci. 2015, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C. Ceruloplasmin and other copper binding components of blood plasma and their functions: An update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.H.; Maryon, E.B. How Mammalian Cells Acquire Copper: An Essential but Potentially Toxic Metal. Biophys. J. 2016, 110, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Puig, S.; Lee, J.; Lau, M.; Thiele, D.J. Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake. J. Biol. Chem. 2002, 277, 26021–26030. [Google Scholar] [CrossRef] [PubMed]
- Elinder, F.; Århem, P. Metal ion effects on ion channel gating. Q. Rev. Biophys. 2003, 36, 373–427. [Google Scholar] [CrossRef] [PubMed]
- Hoch, E.; Lin, W.; Chai, J.; Hershfinkel, M.; Fu, D.; Sekler, I. Histidine pairing at the metal transport site of mammalian ZnT transporters controls Zn2+ over Cd2+ selectivity. Proc. Natl. Acad. Sci. USA 2012, 109, 7202–7207. [Google Scholar] [CrossRef] [PubMed]
- Ehrnstorfer, I.A.; Geertsma, E.R.; Pardon, E.; Steyaert, J.; Dutzler, R. Crystal structure of a SLC11 (NRAMP) transporter reveals the basis for transition-metal ion transport. Nat. Struct. Mol. Biol. 2014, 21, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Manatschal, C.; Pujol-Gimenez, J.; Poirier, M.; Reymond, J.L.; Hediger, M.A.; Dutzler, R. Mechanistic basis of the inhibition of SLC11/NRAMP-mediated metal ion transport by bis-isothiourea substituted compounds. eLife 2019, 8, e51913. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, J.; Fellner, M.; Zhang, C.; Sui, D.; Hu, J. Crystal structures of a ZIP zinc transporter reveal a binuclear metal center in the transport pathway. Sci. Adv. 2017, 3, e1700344. [Google Scholar] [CrossRef]
- Gibson, R.S.; Raboy, V.; King, J.C. Implications of phytate in plant-based foods for iron and zinc bioavailability, setting dietary requirements, and formulating programs and policies. Nutr. Rev. 2018, 76, 793–804. [Google Scholar] [CrossRef]
- Miethke, M. Molecular strategies of microbial iron assimilation: From high-affinity complexes to cofactor assembly systems. Metallomics 2013, 5, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Raymond, K.N.; Allred, B.E.; Sia, A.K. Coordination Chemistry of Microbial Iron Transport. Acc. Chem. Res. 2015, 48, 2496–2505. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Li, Q.; Wang, Y.; Cui, Z. MRP proteins as potential mediators of heavy metal resistance in zebrafish cells. Comp. Biochem. Physiol. C Toxicol. Pharm. 2011, 153, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Buratti, P.; Cairo, G.; Recalcati, S. The transferrin receptor: The cellular iron gate. Metallomics 2017, 9, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- van Renswoude, J.; Bridges, K.R.; Harford, J.B.; Klausner, R.D. Receptor-mediated endocytosis of transferrin and the uptake of Fe in K562 cells: Identification of a nonlysosomal acidic compartment. Proc. Natl. Acad. Sci. USA 1982, 79, 6186–6190. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Paw, B.H. Cellular and mitochondrial iron homeostasis in vertebrates. Biochim. Biophys. Acta 2012, 1823, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Cutsail, G.E., 3rd; Telser, J.; Hoffman, B.M. Advanced paramagnetic resonance spectroscopies of iron-sulfur proteins: Electron nuclear double resonance (ENDOR) and electron spin echo envelope modulation (ESEEM). Biochim. Biophys. Acta 2015, 1853, 1370–1394. [Google Scholar] [CrossRef]
- Sands, R.H.; Beinert, H. On the function of copper in cytochrome oxidase. Biochem. Biophys. Res. Commun. 1959, 1, 175–178. [Google Scholar] [CrossRef]
- Sands, R.H.; Beinert, H. Studies on mitochondria and submitochondrial particles by paramagnetic resonance (EPR) spectroscopy. Biochem. Biophys. Res. Commun. 1960, 3, 47–52. [Google Scholar] [CrossRef]
- Pandelia, M.E.; Lanz, N.D.; Booker, S.J.; Krebs, C. Mössbauer spectroscopy of Fe/S proteins. Biochim. Biophys. Acta 2015, 1853, 1395–1405. [Google Scholar] [CrossRef]
- Ghassaban, K.; Liu, S.; Jiang, C.; Haacke, E.M. Quantifying iron content in magnetic resonance imaging. NeuroImage 2019, 187, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Malanho Silva, J.; Cerofolini, L.; Giuntini, S.; Calderone, V.; Geraldes, C.; Macedo, A.L.; Parigi, G.; Fragai, M.; Ravera, E.; Luchinat, C. Metal centers in biomolecular solid-state NMR. J. Struct. Biol. 2019, 206, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, A.; Natile, G.; Rosato, A.; Tadini-Buoninsegni, F.; Arnesano, F. Monitoring Interactions inside Cells by Advanced Spectroscopies: Overview of Copper Transporters and Cisplatin. Curr. Med. Chem. 2018, 25, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.P.; Cole, N.; Zhang, T.; Doble, P.A.; Hare, D.J. A guide to integrating immunohistochemistry and chemical imaging. Chem. Soc. Rev. 2018, 47, 3770–3787. [Google Scholar] [CrossRef] [PubMed]
- Tsien, R.Y.; Pozzan, T.; Rink, T.J. Measuring and manipulating cytosolic Ca2+ with trapped indicators. Trends Biochem. Sci. 1984, 9, 263–266. [Google Scholar] [CrossRef]
- Carpenter, M.C.; Lo, M.N.; Palmer, A.E. Techniques for measuring cellular zinc. Arch. Biochem. Biophys. 2016, 611, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Fan, J.; Wang, B.; Peng, X. Fluorescent, MRI, and colorimetric chemical sensors for the first-row d-block metal ions. Chem. Soc. Rev. 2015, 44, 4337–4366. [Google Scholar] [CrossRef]
- Ando, T.; Bhamidimarri, S.P.; Brending, N.; Colin-York, H.; Collinson, L.; De Jonge, N.; de Pablo, P.J.; Debroye, E.; Eggeling, C.; Franck, C.; et al. The 2018 correlative microscopy techniques roadmap. J. Phys. D Appl. Phys. 2018, 51, 443001. [Google Scholar] [CrossRef]
- Zhang, A.S.; Sheftel, A.D.; Ponka, P. Intracellular kinetics of iron in reticulocytes: Evidence for endosome involvement in iron targeting to mitochondria. Blood 2005, 105, 368–375. [Google Scholar] [CrossRef]
- Sheftel, A.D.; Zhang, A.S.; Brown, C.; Shirihai, O.S.; Ponka, P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 2007, 110, 125–132. [Google Scholar] [CrossRef]
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta 2016, 1863, 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C.; Patel, S.J.; Protchenko, O. Management versus miscues in the cytosolic labile iron pool: The varied functions of iron chaperones. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118830. [Google Scholar] [CrossRef]
- Yien, Y.Y.; Shi, J.; Chen, C.; Cheung, J.T.M.; Grillo, A.S.; Shrestha, R.; Li, L.; Zhang, X.; Kafina, M.D.; Kingsley, P.D.; et al. FAM210B is an erythropoietin target and regulates erythroid heme synthesis by controlling mitochondrial iron import and ferrochelatase activity. J. Biol. Chem. 2018, 293, 19797–19811. [Google Scholar] [CrossRef] [PubMed]
- Pourcelot, E.; Lénon, M.; Mobilia, N.; Cahn, J.-Y.; Arnaud, J.; Fanchon, E.; Moulis, J.-M.; Mossuz, P. Iron for proliferation of cell lines and hematopoietic progenitors: Nailing down the intracellular functional iron concentration. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 1596–1605. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vallee, B.L.; Altschule, M.D. Zinc in the mammalian organism, with particular reference to carbonic anhydrase. Physiol. Rev. 1949, 29, 370–388. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Anderson, I.; Dietrich, H.; Schneider-Bernlöhr, H.; Einarsson, R.; Zeppezauer, M. Site-Specific Substituted Cobalt(II) Horse Liver Alcohol Dehydrogenases. Eur. J. Biochem. 1979, 98, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Vallee, B.L.; Maret, W. Cobalt as probe and label of proteins. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1993; Volume 226, pp. 52–71. [Google Scholar]
- Meyer, J.; Moulis, J.-M. Rubredoxin. In Handbook of Metalloproteins; Messerschmidt, A., Huber, R.T., Wieghardt, K.P., Eds.; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar] [CrossRef]
- Mathieu, I.; Meyer, J.; Moulis, J.-M. Cloning, sequencing and expression in Escherichia coli of the rubredoxin gene from Clostridium pasteurianum. Biochem. J. 1992, 285, 2552–2562. [Google Scholar] [CrossRef][Green Version]
- Petillot, Y.; Forest, E.; Mathieu, I.; Meyer, J.; Moulis, J.-M. Analysis, by electrospray ionization mass spectrometry, of several forms of Clostridium pasteurianum rubredoxin. Biochem. J. 1993, 296, 657–661. [Google Scholar] [CrossRef]
- Dauter, Z.; Wilson, K.S.; Sieker, L.C.; Moulis, J.-M.; Meyer, J. Zinc- and iron-rubredoxins from Clostridium pasteurianum at atomic resolution: A high-precision model of a ZnS4 coordination unit in a protein. Proc. Natl. Acad. Sci. USA 1996, 93, 8836–8840. [Google Scholar] [CrossRef]
- Nar, H.; Huber, R.; Messerschmidt, A.; Filippou, A.C.; Barth, M.; Jaquinod, M.; van de Kamp, M.; Canters, G.W. Characterization and crystal structure of zinc azurin, a by-product of heterologous expression in Escherichia coli of Pseudomonas aeruginosa copper azurin. Eur. J. Biochem. 1992, 205, 1123–1129. [Google Scholar] [CrossRef]
- Leckner, J.; Bonander, N.; Wittung-Stafshede, P.; Malmström, B.G.; Karlsson, B.G. The effect of the metal ion on the folding energetics of azurin: A comparison of the native, zinc and apoprotein. Biochim. Biophys. Acta (Bba) Protein Struct. Mol. Enzym. 1997, 1342, 19–27. [Google Scholar] [CrossRef]
- Tabor, S. Expression Using the T7 RNA Polymerase/Promoter System. Curr. Protoc. Mol. Biol. 1990, 11, 16.12.1–16.12.11. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.C.W.; Garman, E.F.; Krojer, T.; von Delft, F.; Carpenter, E.P. An overview of heavy-atom derivatization of protein crystals. Acta Cryst. D Struct. Biol. 2016, 72, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Moulis, J.M. Zinc and cadmium specifically interfere with RNA-binding activity of human Iron Regulatory Protein 1. J. Inorg. Biochem. 2004, 98, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Daghino, S.; Di Vietro, L.; Petiti, L.; Martino, E.; Dallabona, C.; Lodi, T.; Perotto, S. Yeast expression of mammalian Onzin and fungal FCR1 suggests ancestral functions of PLAC8 proteins in mitochondrial metabolism and DNA repair. Sci. Rep. 2019, 9, 6629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Nguyen, N.; Breen, S.; Outram, M.A.; Dodds, P.N.; Kobe, B.; Solomon, P.S.; Williams, S.J. Production of small cysteine-rich effector proteins in Escherichia coli for structural and functional studies. Mol. Plant Pathol. 2017, 18, 141–151. [Google Scholar] [CrossRef]
- Horton, J.R.; Upadhyay, A.K.; Qi, H.H.; Zhang, X.; Shi, Y.; Cheng, X. Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat. Struct. Mol. Biol. 2010, 17, 38–43. [Google Scholar] [CrossRef]
- Nemeth, E.; Preza, G.C.; Jung, C.L.; Kaplan, J.; Waring, A.J.; Ganz, T. The N-terminus of hepcidin is essential for its interaction with ferroportin: Structure-function study. Blood 2006, 107, 328–333. [Google Scholar] [CrossRef]
- Farnaud, S.; Rapisarda, C.; Bui, T.; Drake, A.; Cammack, R.; Evans, R.W. Identification of an iron-hepcidin complex. Biochem. J. 2008, 413, 553–557. [Google Scholar] [CrossRef]
- Jordan, J.B.; Poppe, L.; Haniu, M.; Arvedson, T.; Syed, R.; Li, V.; Kohno, H.; Kim, H.; Schnier, P.D.; Harvey, T.S.; et al. Hepcidin revisited, disulfide connectivity, dynamics, and structure. J. Biol. Chem. 2009, 284, 24155–24167. [Google Scholar] [CrossRef]
- Gagliardo, B.; Faye, A.; Jaouen, M.; Deschemin, J.C.; Canonne-Hergaux, F.; Vaulont, S.; Sari, M.A. Production of biologically active forms of recombinant hepcidin, the iron-regulatory hormone. FEBS J. 2008, 275, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Koliaraki, V.; Marinou, M.; Samiotaki, M.; Panayotou, G.; Pantopoulos, K.; Mamalaki, A. Iron regulatory and bactericidal properties of human recombinant hepcidin expressed in Pichia pastoris. Biochimie 2008, 90, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Gailer, J.; George, G.N.; Pickering, I.J.; Prince, R.C.; Kohlhepp, P.; Zhang, D.; Walker, F.A.; Winzerling, J.J. Human cytosolic Iron Regulatory Protein 1 contains a linear iron-sulfur cluster. J. Am. Chem. Soc. 2001, 123, 10121–10122. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.C.; Kent, T.A.; Emptage, M.; Merkle, H.; Beinert, H.; Munck, E. Evidence for the formation of a linear [3Fe-4S] cluster in partially unfolded aconitase. J. Biol. Chem. 1984, 259, 14463–14471. [Google Scholar] [PubMed]
- Joazeiro, C.A.P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 2019, 20, 368–383. [Google Scholar] [CrossRef]
- Phillips, B.P.; Gomez-Navarro, N.; Miller, E.A. Protein quality control in the endoplasmic reticulum. Curr. Opin. Cell Biol. 2020, 65, 96–102. [Google Scholar] [CrossRef]
- Cotruvo, J.A., Jr.; Stubbe, J. Metallation and mismetallation of iron and manganese proteins in vitro and in vivo: The class I ribonucleotide reductases as a case study. Metallomics 2012, 4, 1020–1036. [Google Scholar] [CrossRef]
- Mellor, D.P.; Maley, L. Order of Stability of Metal Complexes. Nature 1948, 161, 436–437. [Google Scholar] [CrossRef]
- Irving, H.; Williams, R.J.P. Order of Stability of Metal Complexes. Nature 1948, 162, 746–747. [Google Scholar] [CrossRef]
- Stubbe, J.; Cotruvo, J.A., Jr. Control of metallation and active cofactor assembly in the class Ia and Ib ribonucleotide reductases: Diiron or dimanganese? Curr. Opin. Chem. Biol. 2011, 15, 284–290. [Google Scholar] [CrossRef]
- Morel, F.M. The co-evolution of phytoplankton and trace element cycles in the oceans. Geobiology 2008, 6, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Moulis, J.M. Cellular mechanisms of cadmium toxicity related to the homeostasis of essential metals. Biometals 2010, 23, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, K.K.; Chan, M.C.; Thalhammer, A.; Demetriades, M.; Chowdhury, R.; Tian, Y.M.; Stolze, I.; McNeill, L.A.; Lee, M.K.; Woon, E.C.Y.; et al. Dual-action inhibitors of HIF prolyl hydroxylases that induce binding of a second iron ion. Org. Biomol. Chem. 2013, 11, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.M.; Yeoh, K.K.; Lee, M.K.; Eriksson, T.; Kessler, B.M.; Kramer, H.B.; Edelmann, M.J.; Willam, C.; Pugh, C.W.; Schofield, C.J.; et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J. Biol. Chem. 2011, 286, 13041–13051. [Google Scholar] [CrossRef] [PubMed]
- Miles, A.L.; Burr, S.P.; Grice, G.L.; Nathan, J.A. The vacuolar-ATPase complex and assembly factors, TMEM199 and CCDC115, control HIF1alpha prolyl hydroxylation by regulating cellular iron levels. eLife 2017, 6, e22693. [Google Scholar] [CrossRef]
- Weber, R.A.; Yen, F.S.; Nicholson, S.P.V.; Alwaseem, H.; Bayraktar, E.C.; Alam, M.; Timson, R.C.; La, K.; Abu-Remaileh, M.; Molina, H.; et al. Maintaining Iron Homeostasis Is the Key Role of Lysosomal Acidity for Cell Proliferation. Mol. Cell 2020, 77, 645–655.e764. [Google Scholar] [CrossRef]
- Davidson, T.L.; Chen, H.; Di Toro, D.M.; D’Angelo, G.; Costa, M. Soluble nickel inhibits HIF-prolyl-hydroxylases creating persistent hypoxic signaling in A549 cells. Mol. Carcinog. 2006, 45, 479–489. [Google Scholar] [CrossRef]
- Horton, J.R.; Upadhyay, A.K.; Hashimoto, H.; Zhang, X.; Cheng, X. Structural basis for human PHF2 Jumonji domain interaction with metal ions. J. Mol. Biol. 2011, 406, 1–8. [Google Scholar] [CrossRef]
- Muñoz-Sánchez, J.; Chánez-Cárdenas, M.E. The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 2019, 39, 556–570. [Google Scholar] [CrossRef]
- Cammack, R. Iron-sulphur cluster interconversions and the activation of aconitase. Nature 1982, 298, 792–793. [Google Scholar] [CrossRef]
- Barth, C.; Weiss, M.C.; Roettger, M.; Martin, W.F.; Unden, G. Origin and phylogenetic relationships of [4Fe-4S]-containing O2 sensors of bacteria. Env. Microbiol. 2018, 20, 4567–4586. [Google Scholar] [CrossRef] [PubMed]
- Buren, S.; Jimenez-Vicente, E.; Echavarri-Erasun, C.; Rubio, L.M. Biosynthesis of Nitrogenase Cofactors. Chem. Rev. 2020, 120, 4921–4968. [Google Scholar] [CrossRef] [PubMed]
- Paul, V.D.; Lill, R. Biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability. Biochim. Biophys. Acta 2015, 1853, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.W.; Schut, G.J.; Boyd, E.S.; Mulder, D.W.; Shepard, E.M.; Broderick, J.B.; King, P.W.; Adams, M.W. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta 2015, 1853, 1350–1369. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.D.; Jones, C.E.; Solioz, M.; Dameron, C.T. Intracellular copper routing: The role of copper chaperones. Trends Biochem. Sci. 2000, 25, 29–32. [Google Scholar] [CrossRef]
- Öhrvik, H.; Aaseth, J.; Horn, N. Orchestration of dynamic copper navigation—New and missing pieces. Metallomics 2017, 9, 1204–1229. [Google Scholar] [CrossRef]
- Sachar, M.; Anderson, K.E.; Ma, X. Protoporphyrin IX: The Good, the Bad, and the Ugly. J. Pharm. Exp. 2016, 356, 267–275. [Google Scholar] [CrossRef]
- Hooper, P.L. COVID-19 and heme oxygenase: Novel insight into the disease and potential therapies. Cell Stress Chaperones 2020, 25, 707–710, Correction in Cell Stress Chaperones 2020, 25, 711, doi:10.1007/s12192-020-01130-z. [Google Scholar] [CrossRef]
- Peng, Y.Y.; Uprichard, J. Ferritin and iron studies in anaemia and chronic disease. Ann. Clin. Biochem. 2016, 54, 43–48. [Google Scholar] [CrossRef]
- Reiss, Y.; Gur, E.; Ravid, T. Releasing the Lockdown: An Emerging Role for the Ubiquitin-Proteasome System in the Breakdown of Transient Protein Inclusions. Biomolecules 2020, 10, 1168. [Google Scholar] [CrossRef]
- Haile, D.J.; Rouault, T.A.; Tang, C.K.; Chin, J.; Harford, J.B.; Klausner, R.D. Reciprocal control of RNA-binding and aconitase activity in the regulation of the iron-responsive element binding protein: Role of the iron-sulfur cluster. Proc. Natl. Acad. Sci. USA 1992, 89, 7536. [Google Scholar] [CrossRef]
- Pierro, A.; Etienne, E.; Gerbaud, G.; Guigliarelli, B.; Ciurli, S.; Belle, V.; Zambelli, B.; Mileo, E. Nickel and GTP Modulate Helicobacter pylori UreG Structural Flexibility. Biomolecules 2020, 10, 1062. [Google Scholar] [CrossRef] [PubMed]
- Lill, R. From the discovery to molecular understanding of cellular iron-sulfur protein biogenesis. Biol. Chem. 2020, 401, 855–876. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Rouault, T.A. Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis. Trends Biochem. Sci. 2020, 45, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Ciofi-Baffoni, S.; Nasta, V.; Banci, L. Protein networks in the maturation of human iron-sulfur proteins. Metallomics 2018, 10, 49–72. [Google Scholar] [CrossRef]
- Wachnowsky, C.; Fidai, I.; Cowan, J.A. Iron-sulfur cluster biosynthesis and trafficking—Impact on human disease conditions. Metallomics 2018, 10, 9–29. [Google Scholar] [CrossRef]
- Dai, S.; Schwendtmayer, C.; Schürmann, P.; Ramaswamy, S.; Eklund, H. Redox Signaling in Chloroplasts: Cleavage of Disulfides by an Iron-Sulfur Cluster. Science 2000, 287, 655. [Google Scholar] [CrossRef]
- Moulis, J.-M.; Meyer, J.; Lutz, M. Characterization of [4Fe-4S]2+, [4Fe-4Se]2+ and hybrid (S, Se) clusters in Clostridium pasteurianum ferredoxin. A resonance Raman study. Biochem. J. 1984, 219, 829–832. [Google Scholar] [CrossRef]
- Garcia, P.S.; Gribaldo, S.; Py, B.; Barras, F. The SUF system: An ABC ATPase-dependent protein complex with a role in Fe–S cluster biogenesis. Res. Microbiol. 2019, 170, 426–434. [Google Scholar] [CrossRef]
- Inupakutika, M.A.; Sengupta, S.; Nechushtai, R.; Jennings, P.A.; Onuchic, J.N.; Azad, R.K.; Padilla, P.; Mittler, R. Phylogenetic analysis of eukaryotic NEET proteins uncovers a link between a key gene duplication event and the evolution of vertebrates. Sci. Rep. 2017, 7, 42571. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Bouton, C. Nitrosative and oxidative modulation of iron regulatory proteins. Cell. Mol. Life Sci. CMLS 1999, 55, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Recalcati, S.; Pietrangelo, A.; Minotti, G. The iron regulatory proteins: Targets and modulators of free radical reactions and oxidative damage. Free Radic. Biol. Med. 2002, 32, 1237–1243. [Google Scholar] [CrossRef]
- Fillebeen, C.; Pantopoulos, K. Redox control of iron regulatory proteins. Redox Rep. 2002, 7, 15–22. [Google Scholar] [CrossRef]
- Hanson, E.S.; Leibold, E.A. Regulation of the iron regulatory proteins by reactive nitrogen and oxygen species. Gene Expr. 1999, 7, 367–376. [Google Scholar]
- Soum, E.; Brazzolotto, X.; Goussias, C.; Bouton, C.; Moulis, J.-M.; Mattioli, T.A.; Drapier, J.-C. Peroxynitrite and Nitric Oxide Differently Target the Iron−Sulfur Cluster and Amino Acid Residues of Human Iron Regulatory Protein 1. Biochemistry 2003, 42, 7648–7654. [Google Scholar] [CrossRef]
- Oliveira, L.; Bouton, C.; Drapier, J.-C. Thioredoxin activation of iron regulatory proteins. Redox regulation of RNA binding after exposure to nitric oxide. J. Biol. Chem. 1999, 274, 516–521. [Google Scholar] [CrossRef]
- Berndt, C.; Hanschmann, E.-M.; Urbainsky, C.; Jordt, L.M.; Müller, C.S.; Bodnar, Y.; Schipper, S.; Handorf, O.; Nowack, R.; Moulis, J.-M.; et al. FeS-cluster coordination of vertebrate thioredoxins regulates suppression of hypoxia-induced factor 2α through Iron Regulatory Protein 1. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mittler, R.; Darash-Yahana, M.; Sohn, Y.S.; Bai, F.; Song, L.; Cabantchik, I.Z.; Jennings, P.A.; Onuchic, J.N.; Nechushtai, R. NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer. Antioxid. Redox Signal. 2019, 30, 1083–1095. [Google Scholar] [CrossRef]
- Hood, M.I.; Skaar, E.P. Nutritional immunity: Transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 2012, 10, 525–537. [Google Scholar] [CrossRef]
- Zygiel, E.M.; Nolan, E.M. Transition Metal Sequestration by the Host-Defense Protein Calprotectin. Annu. Rev. Biochem. 2018, 87, 621–643. [Google Scholar] [CrossRef] [PubMed]
- Damo, S.M.; Kehl-Fie, T.E.; Sugitani, N.; Holt, M.E.; Rathi, S.; Murphy, W.J.; Zhang, Y.; Betz, C.; Hench, L.; Fritz, G.; et al. Molecular basis for manganese sequestration by calprotectin and roles in the innate immune response to invading bacterial pathogens. Proc. Natl. Acad. Sci. USA 2013, 110, 3841–3846. [Google Scholar] [CrossRef] [PubMed]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.G.; Dubuisson, A.; Derosa, L.; Almire, C.; Henon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418. [Google Scholar] [CrossRef] [PubMed]
- Boal, A.K.; Rosenzweig, A.C. Crystal Structures of Cisplatin Bound to a Human Copper Chaperone. J. Am. Chem. Soc. 2009, 131, 14196–14197. [Google Scholar] [CrossRef]
- Belviso, B.D.; Galliani, A.; Lasorsa, A.; Mirabelli, V.; Caliandro, R.; Arnesano, F.; Natile, G. Oxaliplatin Binding to Human Copper Chaperone Atox1 and Protein Dimerization. Inorg. Chem. 2016, 55, 6563–6573. [Google Scholar] [CrossRef]
- Chen, J.-J.; Zhang, S. Heme-regulated eIF2α kinase in erythropoiesis and hemoglobinopathies. Blood 2019, 134, 1697–1707. [Google Scholar] [CrossRef]
- Pradhan, P.; Vijayan, V.; Gueler, F.; Immenschuh, S. Interplay of Heme with Macrophages in Homeostasis and Inflammation. Int. J. Mol. Sci. 2020, 21, 740. [Google Scholar] [CrossRef]
- Taketani, S.; Adachi, Y.; Kohno, H.; Ikehara, S.; Tokunaga, R.; Ishii, T. Molecular characterization of a newly identified heme-binding protein induced during differentiation of murine erythroleukemia cells. J. Biol. Chem. 1998, 273, 31388–31394. [Google Scholar] [CrossRef]
- Blackmon, B.J.; Dailey, T.A.; Lianchun, X.; Dailey, H.A. Characterization of a human and mouse tetrapyrrole-binding protein. Arch. Biochem. Biophys. 2002, 407, 196–201. [Google Scholar] [CrossRef]
- Dias, J.S.; Macedo, A.L.; Ferreira, G.C.; Peterson, F.C.; Volkman, B.F.; Goodfellow, B.J. The first structure from the SOUL/HBP family of heme-binding proteins, murine P22HBP. J. Biol. Chem. 2006, 281, 31553–31561. [Google Scholar] [CrossRef]
- Gell, D.A.; Westman, B.J.; Gorman, D.; Liew, C.; Welch, J.J.; Weiss, M.J.; Mackay, J.P. A novel haem-binding interface in the 22 kDa haem-binding protein p22HBP. J. Mol. Biol. 2006, 362, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Fortunato, A.E.; Sordino, P.; Andreakis, N. Evolution of the SOUL Heme-Binding Protein Superfamily across Eukarya. J. Mol. Evol. 2016, 82, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Yagensky, O.; Kohansal-Nodehi, M.; Gunaseelan, S.; Rabe, T.; Zafar, S.; Zerr, I.; Härtig, W.; Urlaub, H.; Chua, J.J.E. Increased expression of heme-binding protein 1 early in Alzheimer’s disease is linked to neurotoxicity. eLife 2019, 8, e47498. [Google Scholar] [CrossRef] [PubMed]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9, 579. [Google Scholar] [CrossRef]
- Osman, D.; Martini, M.A.; Foster, A.W.; Chen, J.; Scott, A.J.P.; Morton, R.J.; Steed, J.W.; Lurie-Luke, E.; Huggins, T.G.; Lawrence, A.D.; et al. Bacterial sensors define intracellular free energies for correct enzyme metalation. Nat. Chem. Biol. 2019, 15, 241–249. [Google Scholar] [CrossRef]
- Robinson, N.J.; Glasfeld, A. Metalation: Nature’s challenge in bioinorganic chemistry. J. Biol. Inorg. Chem. 2020, 25, 543–545. [Google Scholar] [CrossRef]
- Colvin, R.A.; Holmes, W.R.; Fontaine, C.P.; Maret, W. Cytosolic zinc buffering and muffling: Their role in intracellular zinc homeostasis. Metallomics 2010, 2, 306–317. [Google Scholar] [CrossRef]
- Thomas, R.C.; Coles, J.A.; Deitmer, J.W. Homeostatic muffling. Nature 1991, 350, 564. [Google Scholar] [CrossRef]
- Lindahl, P.A.; Moore, M.J. Labile Low-Molecular-Mass Metal Complexes in Mitochondria: Trials and Tribulations of a Burgeoning Field. Biochemistry 2016, 55, 4140–4153. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Dziuba, N.; Lindahl, P.A. Isolated Saccharomyces cerevisiae vacuoles contain low-molecular-mass transition-metal polyphosphate complexes. Metallomics 2019, 11, 1298–1309. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Kim, J.E.; Brawley, H.N.; Lindahl, P.A. Chromatographic detection of low-molecular-mass metal complexes in the cytosol of Saccharomyces cerevisiae. Metallomics 2020, 12, 1094–1105. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.A. A comprehensive mechanistic model of iron metabolism in Saccharomyces cerevisiae. Metallomics 2019, 11, 1779–1799. [Google Scholar] [CrossRef] [PubMed]
- Wofford, J.D.; Lindahl, P.A. A mathematical model of iron import and trafficking in wild-type and Mrs3/4ΔΔ yeast cells. BMC Syst. Biol. 2019, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Rousselet, E.; Dycke, C.; Bouron, A.; Moulis, J.M. Cadmium toxicity in animal cells by interference with essential metals. Biochimie 2006, 88, 1807–1814. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium | % Replacement of [4Fe-4S] by [4Fe-4Se] | % Replacement of [4Fe-4Se] by [4Fe-4S] |
|---|---|---|
| No urea | 0 | 22 |
| 8M urea | 28 | 87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moulis, J.-M. Cellular Dynamics of Transition Metal Exchange on Proteins: A Challenge but a Bonanza for Coordination Chemistry. Biomolecules 2020, 10, 1584. https://doi.org/10.3390/biom10111584
Moulis J-M. Cellular Dynamics of Transition Metal Exchange on Proteins: A Challenge but a Bonanza for Coordination Chemistry. Biomolecules. 2020; 10(11):1584. https://doi.org/10.3390/biom10111584
Chicago/Turabian StyleMoulis, Jean-Marc. 2020. "Cellular Dynamics of Transition Metal Exchange on Proteins: A Challenge but a Bonanza for Coordination Chemistry" Biomolecules 10, no. 11: 1584. https://doi.org/10.3390/biom10111584
APA StyleMoulis, J.-M. (2020). Cellular Dynamics of Transition Metal Exchange on Proteins: A Challenge but a Bonanza for Coordination Chemistry. Biomolecules, 10(11), 1584. https://doi.org/10.3390/biom10111584