The Proteasome Activators Blm10/PA200 Enhance the Proteasomal Degradation of N-Terminal Huntingtin

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Yeast Strains, Media, and Growth Conditions
2.3. Yeast Phenotype Analysis
2.4. Plasmids Constructs
2.5. Immunofluorescence and Image Analysis
2.6. Bacterial Expression and GST Pull-Down Assay
2.7. Filter Trap Assay from Human Neuroblastoma Cells
2.8. Filter Retardation Assay and Gradient Gel Analysis from Yeast Cells
2.9. RNA Extraction and cDNA Reverse Transcription
2.10. Quantitative Real-Time PCR
2.11. SDS-PAGE and Western Blot
2.12. Purification of Proteasome Complexes
2.13. Proteasome (20S CP) Activity Assay
2.14. In Vitro Protein Degradation Assay
2.15. Peptidomics Analysis
2.16. Statistical Analysis
3. Results
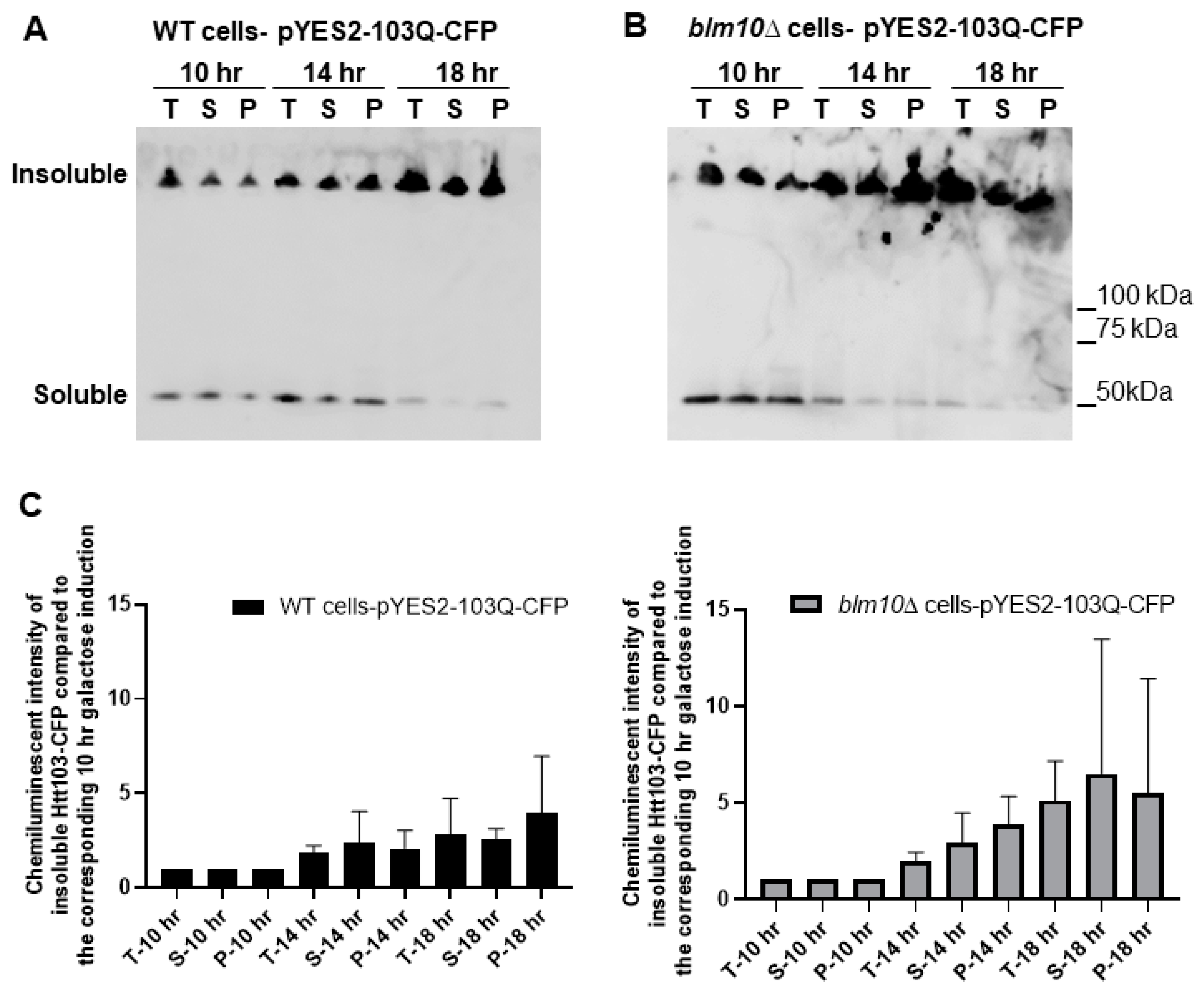
3.1. Blm10-Proteasomes Antagonize the Toxicity of Mutant Huntingtin Expression in Yeast
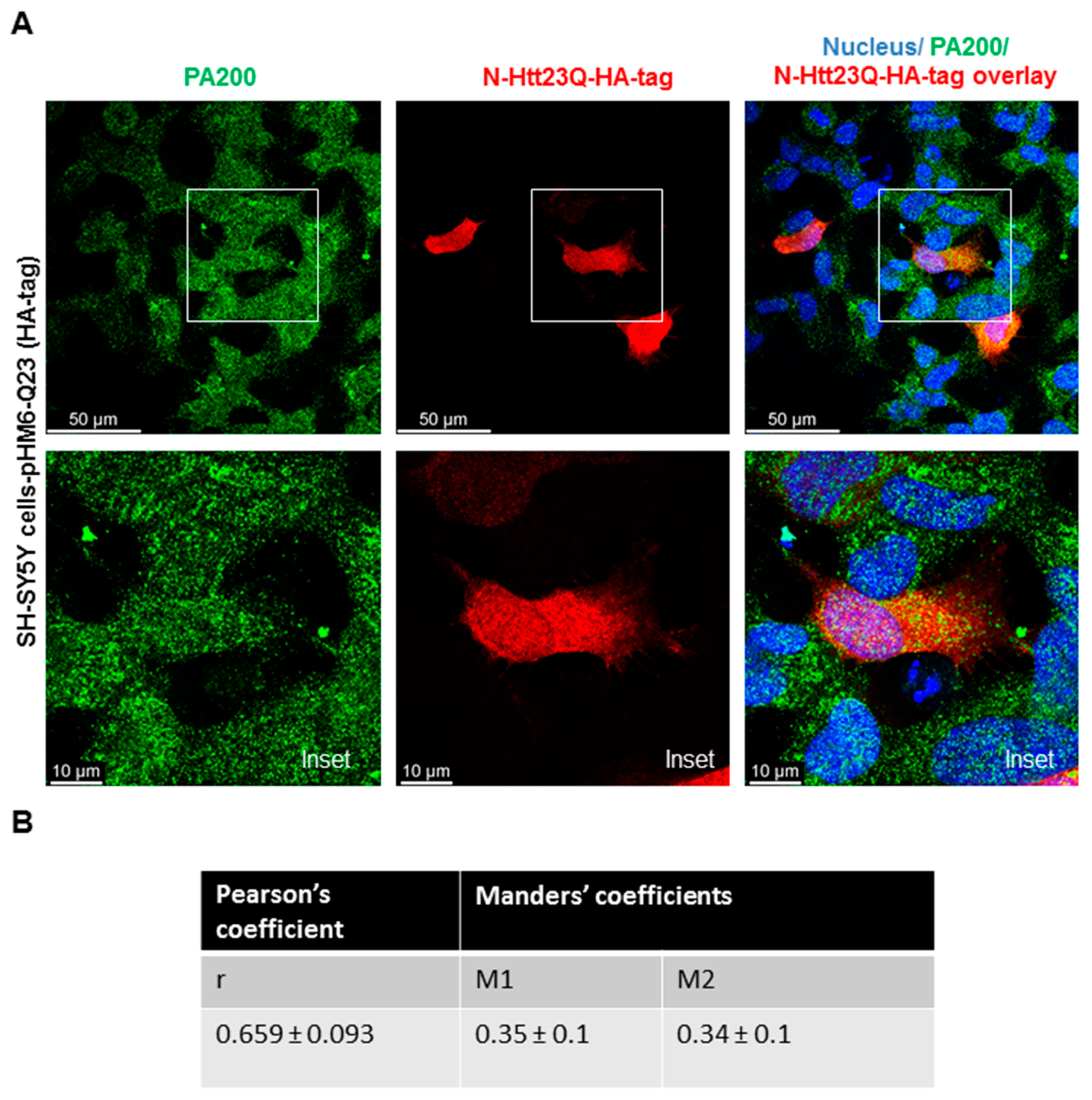
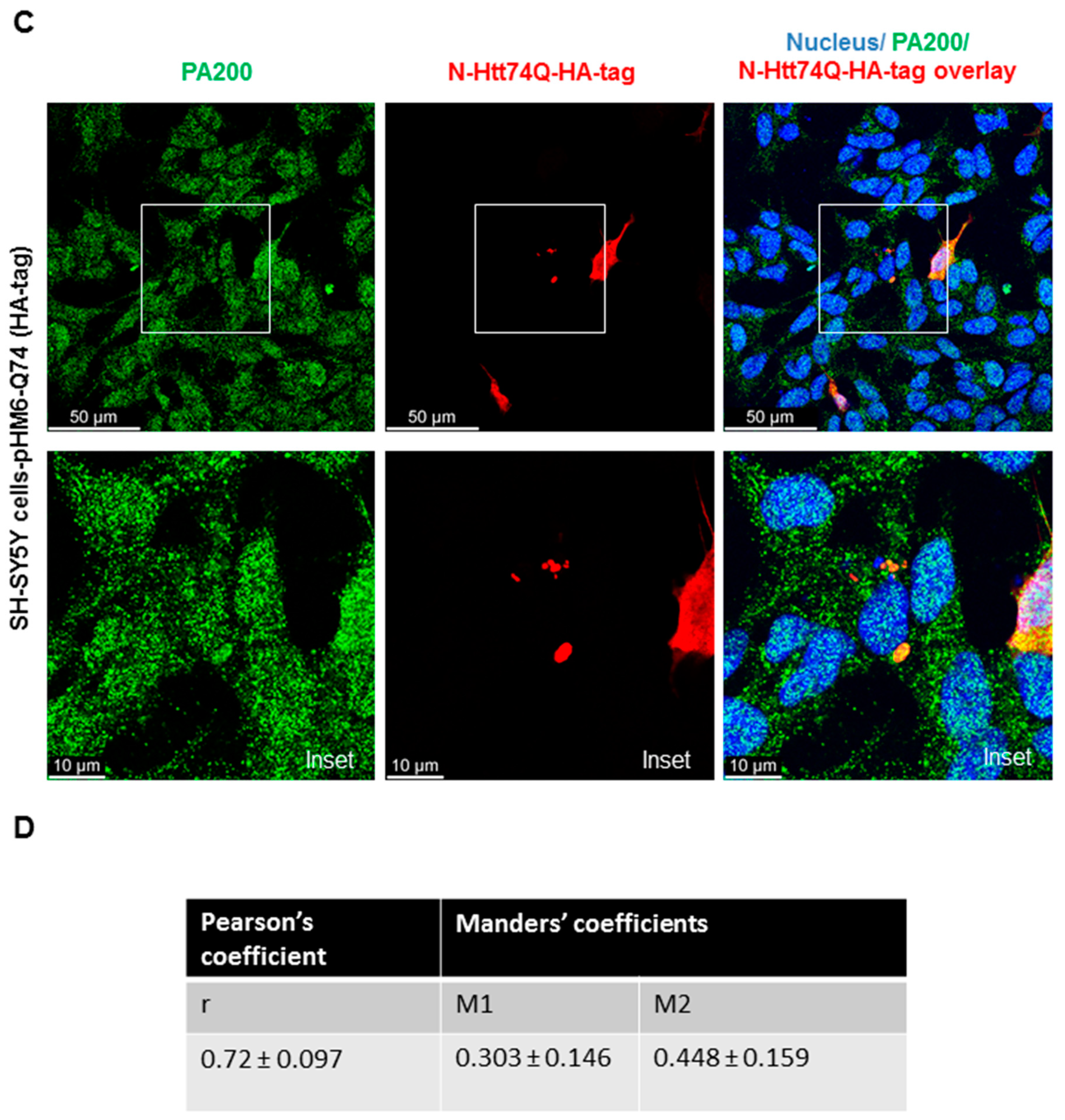
3.2. Endogenous PA200 Colocalizes with Wild Type (wt) and Mutant N-Htt in Human SH SY5Y Cells
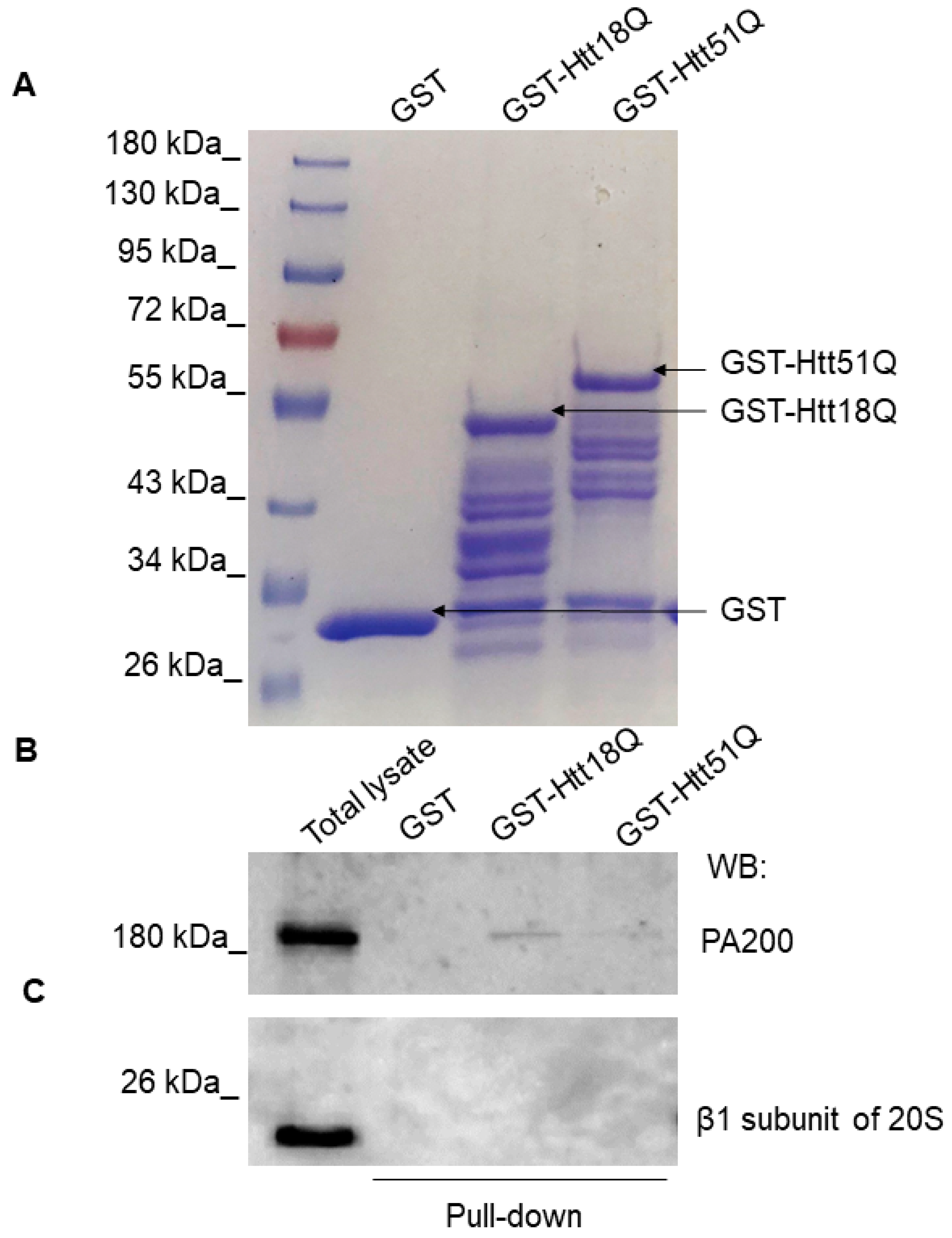
3.3. PA200 Interacts with wt and Mutant N-Htt In Vitro
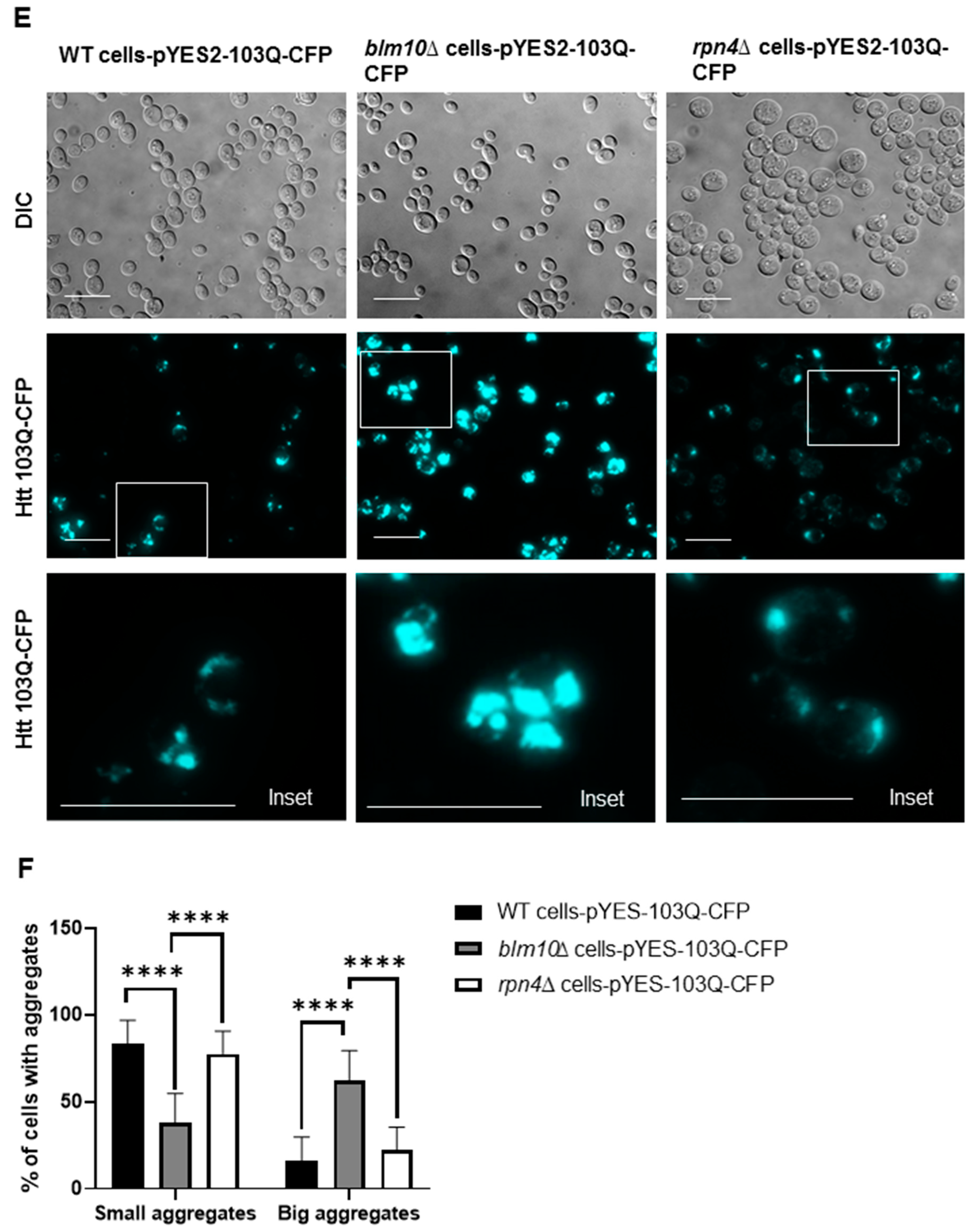
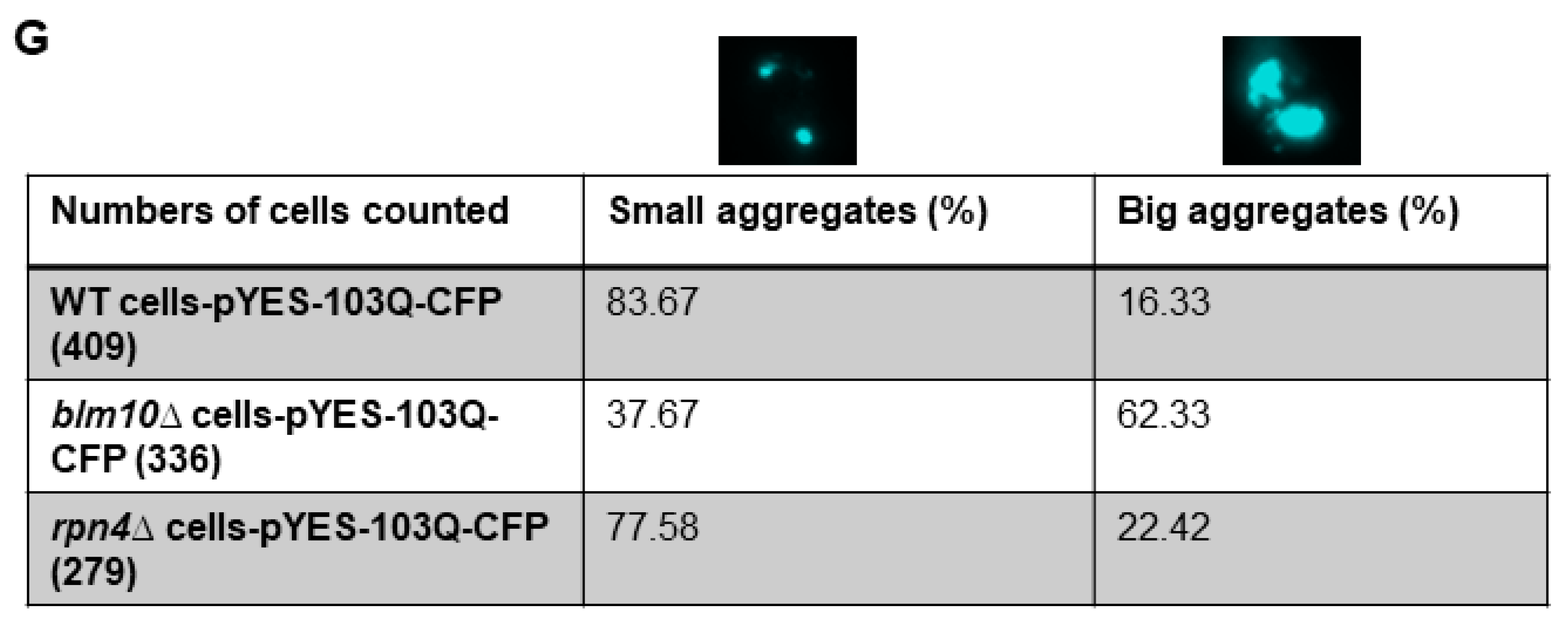
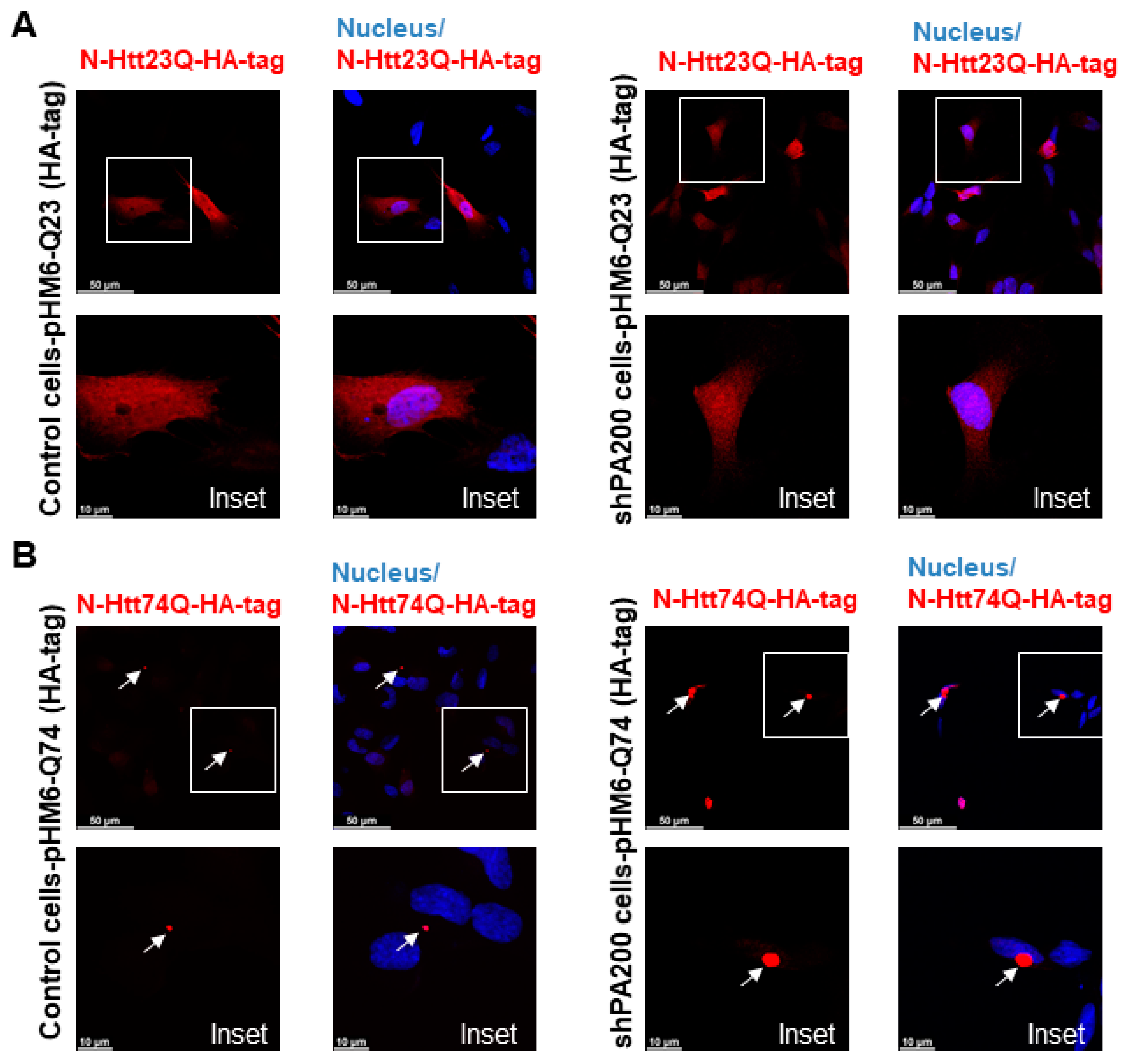
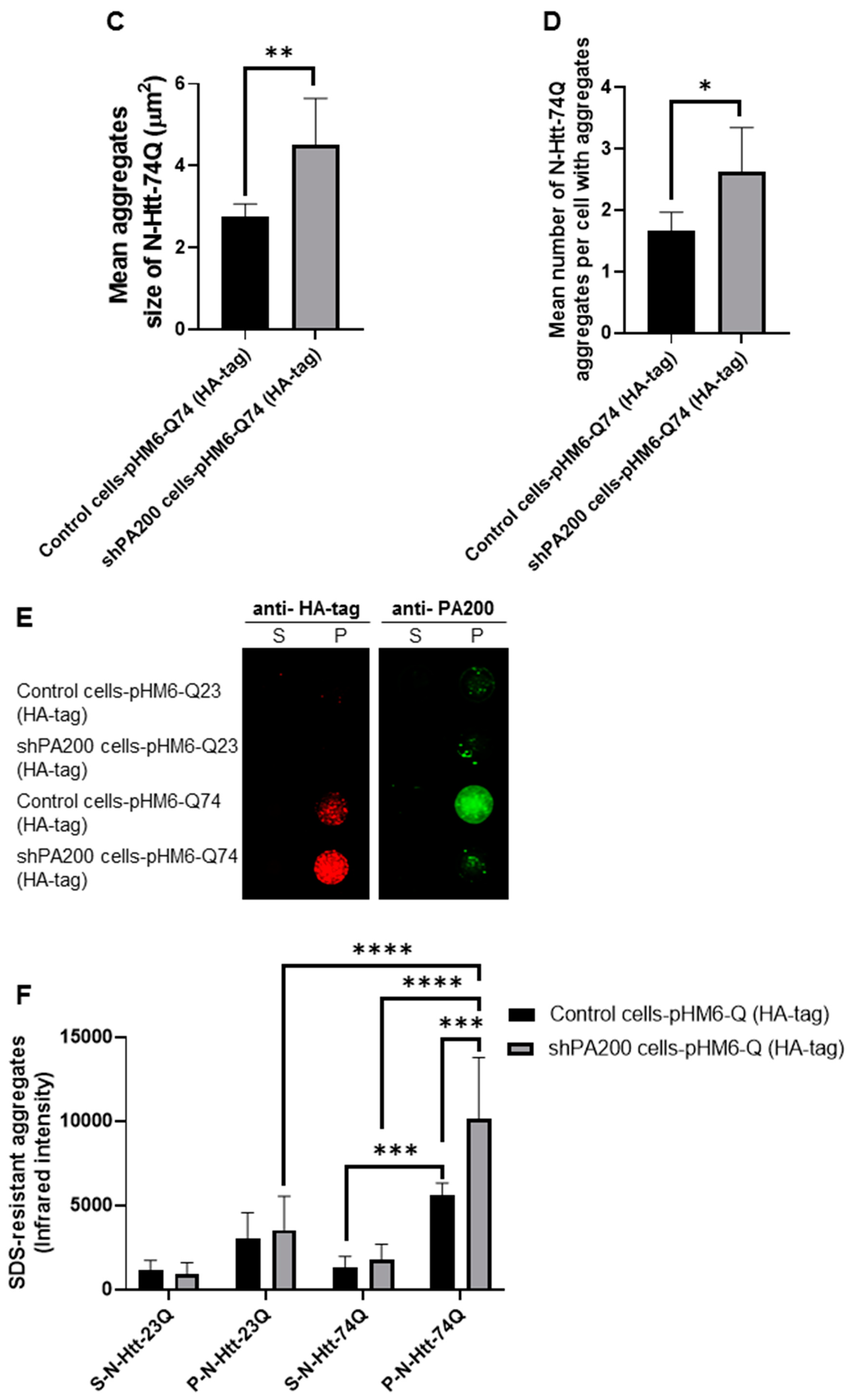
3.4. Loss of Mammalian PA200 Results in Increased Toxic N-Htt Aggregate Size and Number in Human Neuroblastoma Cells
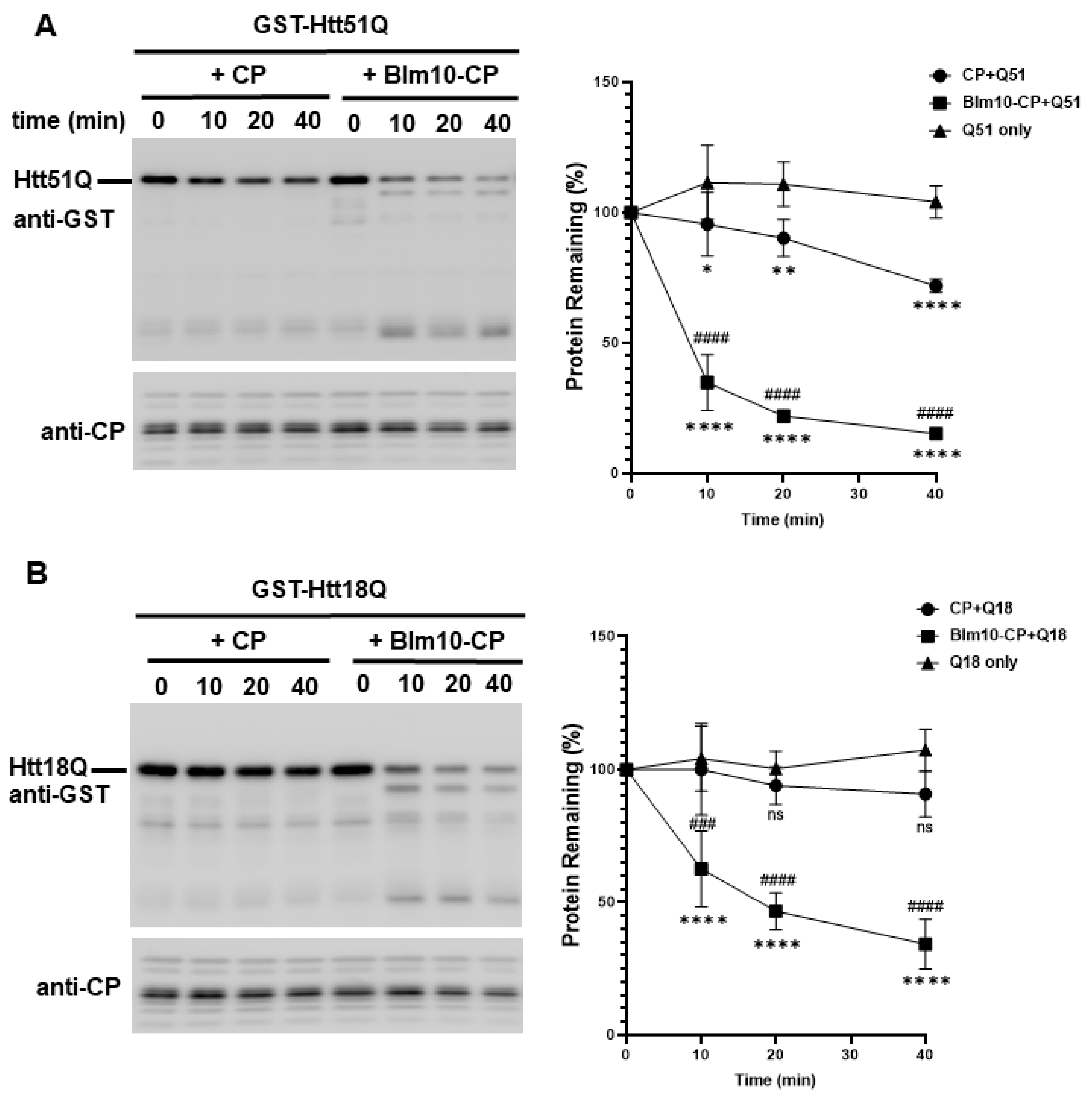
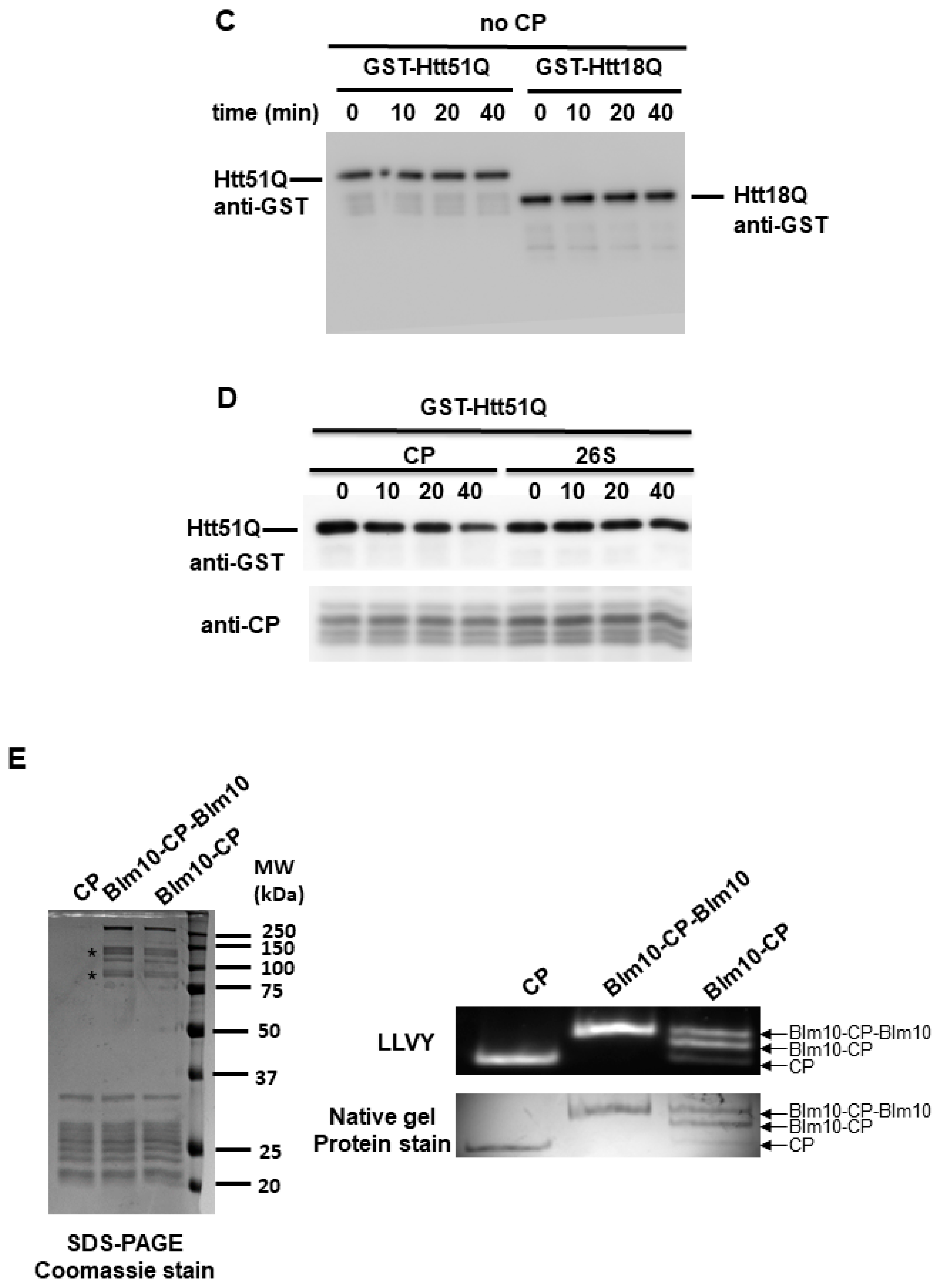
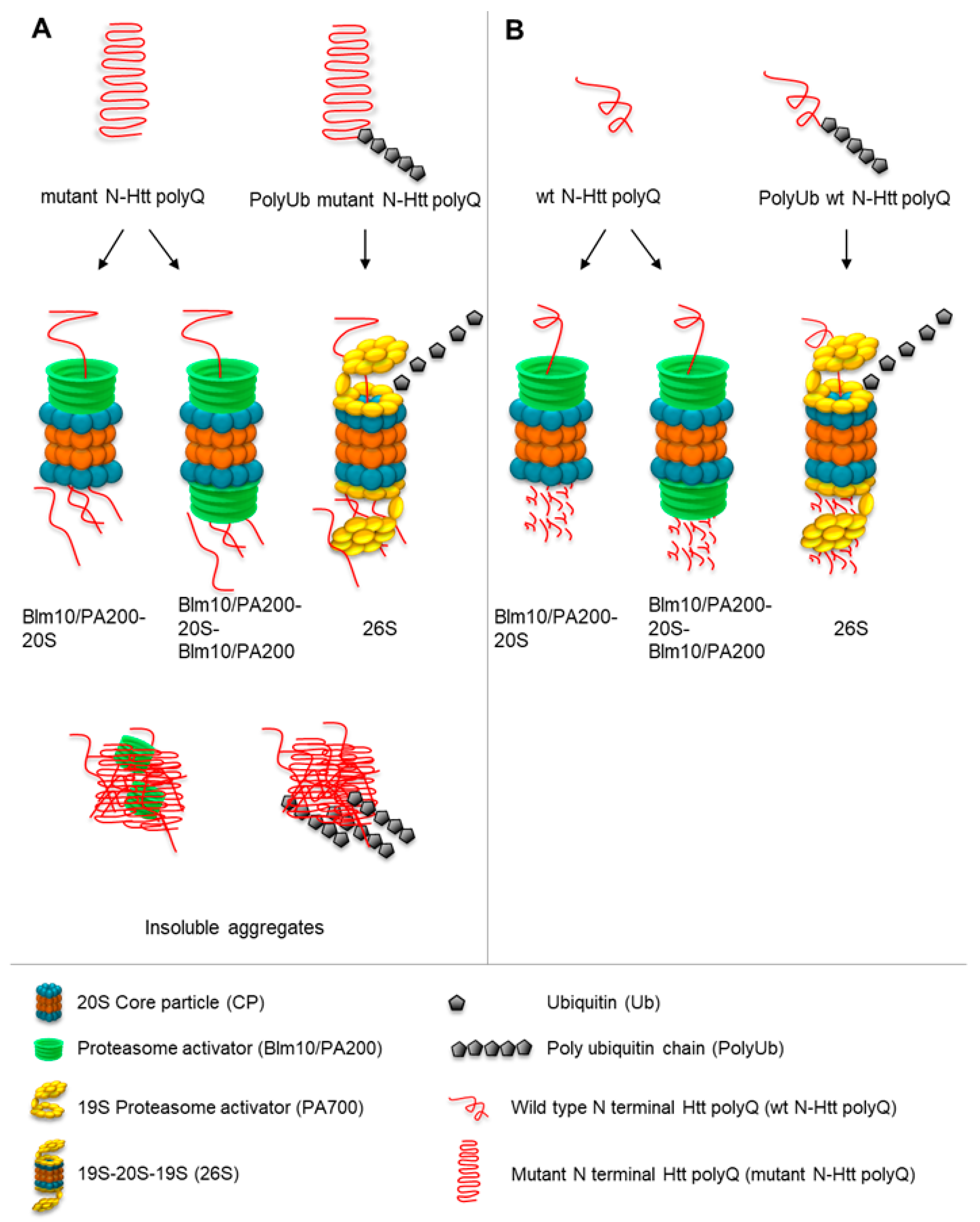
3.5. Blm10/PA200 Contribute to the Degradation of wt and Mutant N-Htt In Vitro
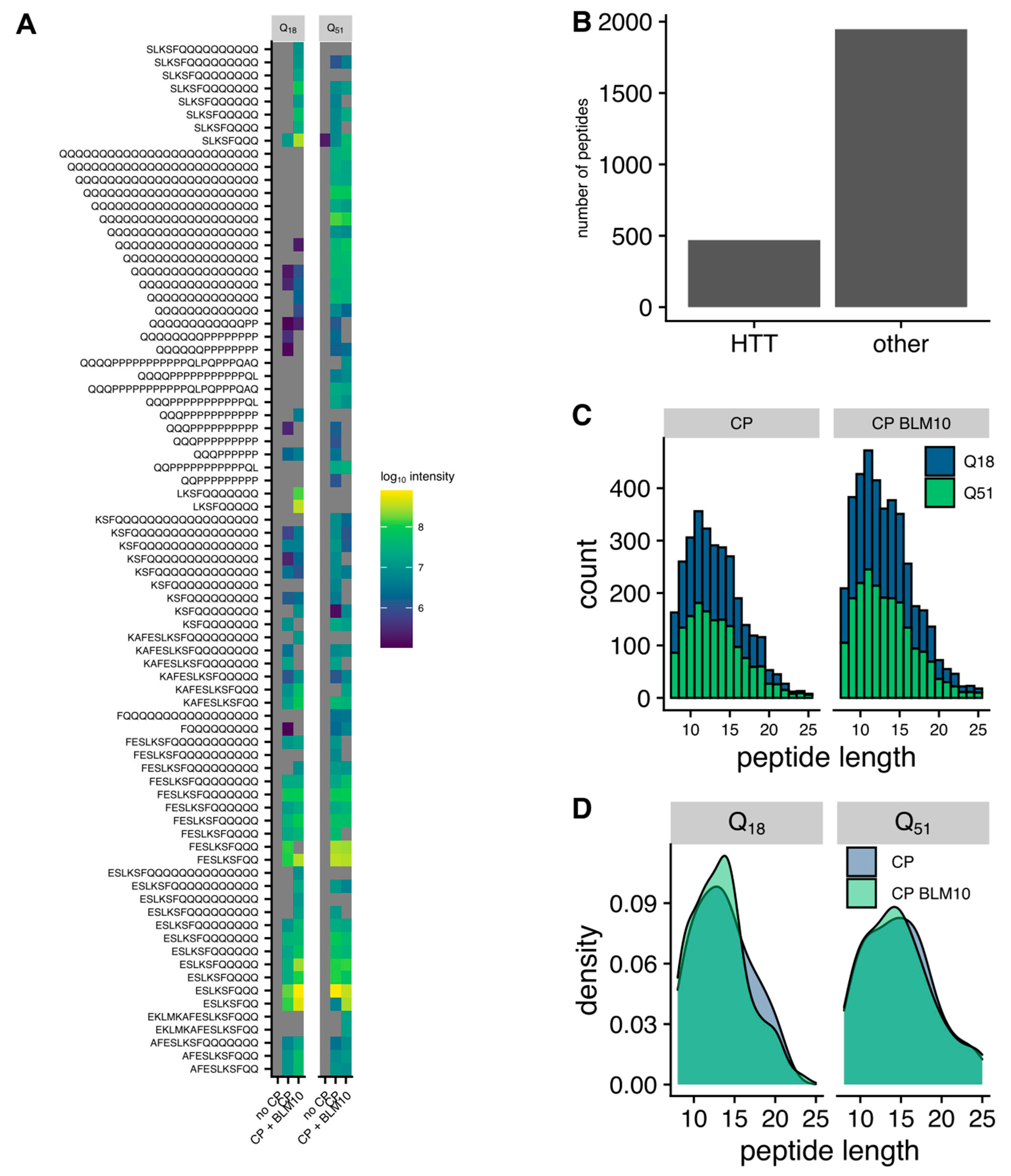
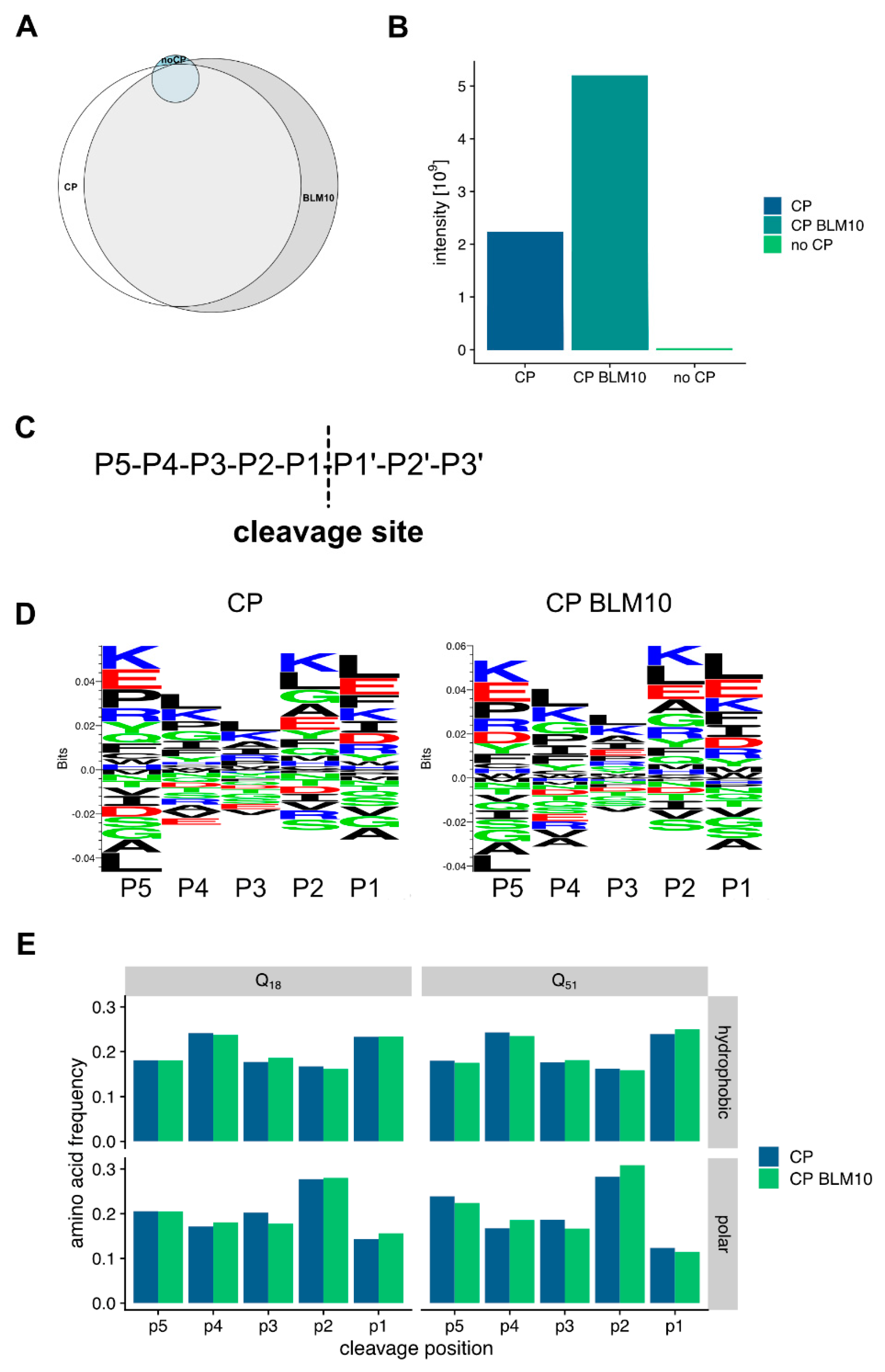
3.6. Blm10 Enhances the Ability of the Proteasome to Cut within Expanded polyQ Repeats
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HD | Huntington’s disease |
| CP | Core particle |
| N-Htt | N-terminal huntingtin fragments |
| Blm10 | Bleomycin resistance protein BLM10 |
| PA200 | Proteasome activator 200 |
| GST | Glutathione S-transferase |
| IB | Inclusion body |
| GFP | Green fluorescent protein |
References
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [PubMed]
- Gandhi, J.; Antonelli, A.C.; Afridi, A.; Vatsia, S.; Joshi, G.; Romanov, V.; Murray, I.V.J.; Khan, S.A. Protein misfolding and aggregation in neurodegenerative diseases: A review of pathogeneses, novel detection strategies, and potential therapeutics. Rev. Neurosci. 2019, 30, 339–358. [Google Scholar] [PubMed]
- Wang, Y.; Le, W.D. Autophagy and Ubiquitin-Proteasome System. Adv. Exp. Med. Biol. 2019, 1206, 527–550. [Google Scholar]
- Hanna, J.; Guerra-Moreno, A.; Ang, J.; Micoogullari, Y. Protein Degradation and the Pathologic Basis of Disease. Am. J. Pathol. 2019, 189, 94–103. [Google Scholar]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar]
- Golas, M.M. Human cellular models of medium spiny neuron development and Huntington disease. Life Sci. 2018, 209, 179–196. [Google Scholar]
- Creus-Muncunill, J.; Ehrlich, M.E. Cell-Autonomous and Non-cell-Autonomous Pathogenic Mechanisms in Huntington’s Disease: Insights from In Vitro and In Vivo Models. Neurotherapeutics 2019, 16, 957–978. [Google Scholar] [PubMed]
- Aladdin, A.; Kiraly, R.; Boto, P.; Regdon, Z.; Tar, K. Juvenile Huntington’s Disease Skin Fibroblasts Respond with Elevated Parkin Level and Increased Proteasome Activity as a Potential Mechanism to Counterbalance the Pathological Consequences of Mutant Huntingtin Protein. Int. J. Mol. Sci. 2019, 20, 5338. [Google Scholar]
- Demartino, G.N.; Gillette, T.G. Proteasomes: Machines for all reasons. Cell 2007, 129, 659–662. [Google Scholar] [PubMed]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [PubMed]
- Kunjappu, M.J.; Hochstrasser, M. Assembly of the 20S proteasome. Biochim. Biophys. Acta-Mol. Cell Res. 2014, 1843, 2–12. [Google Scholar]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar]
- Chow, W.N.; Luk, H.W.; Chan, H.Y.; Lau, K.F. Degradation of mutant huntingtin via the ubiquitin/proteasome system is modulated by FE65. Biochem. J. 2012, 443, 681–689. [Google Scholar]
- Jeong, H.; Then, F.; Melia, T.J.; Mazzulli, J.R.; Cui, L.; Savas, J.N.; Voisine, C.; Paganetti, P.; Tanese, N.; Hart, A.C.; et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009, 137, 60–72. [Google Scholar]
- Li, X.J.; Li, S. Proteasomal dysfunction in aging and Huntington disease. Neurobiol. Dis. 2011, 43, 4–8. [Google Scholar]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vincente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [PubMed]
- Holmberg, C.I.; Staniszewski, K.E.; Mensah, K.N.; Matouschek, A.; Morimoto, R.I. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 2004, 23, 4307–4318. [Google Scholar] [PubMed]
- Hipp, M.S.; Patel, C.N.; Bersuker, K.; Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Brandeis, M.; Kopito, R.R. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol. 2012, 196, 573–587. [Google Scholar] [PubMed]
- Strehl, B.; Seifert, U.; Kruger, E.; Heink, S.; Kuckelkorn, U.; Kloetzel, P.M. Interferon-gamma, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol. Rev. 2005, 207, 19–30. [Google Scholar]
- Cascio, P. PA28αβ: The enigmatic magic ring of the proteasome? Biomolecules 2014, 4, 566–584. [Google Scholar]
- Ustrell, V.; Hoffman, L.; Pratt, G.; Rechsteiner, M. PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 2002, 21, 3516–3525. [Google Scholar]
- Schmidt, M.; Haas, W.; Crosas, B.; Santamaria, P.G.; Gygi, S.P.; Walz, T.; Finley, D. The HEAT repeat protein Blm10 regulates the yeast proteasome by capping the core particle. Nat. Struct. Mol. Biol. 2005, 12, 294–303. [Google Scholar]
- Huang, L.; Haratake, K.; Miyahara, H.; Chiba, T. Proteasome activators, PA28γ and PA200, play indispensable roles in male fertility. Sci. Rep. 2016, 6, 23171. [Google Scholar]
- Dange, T.; Smith, D.; Noy, T.; Rommel, P.C.; Jurzitza, L.; Cordero, R.J.; Legendre, A.; Finley, D.; Goldberg, A.L.; Schmidt, M. Blm10 protein promotes proteasomal substrate turnover by an active gating mechanism. J. Biol. Chem. 2011, 286, 42830–42839. [Google Scholar]
- Qian, M.X.; Pang, Y.; Liu, C.H.; Haratake, K.; Du, B.Y.; Ji, D.Y.; Wang, G.F.; Zhu, Q.Q.; Song, W.; Yu, Y.; et al. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 2013, 153, 1012–1024. [Google Scholar]
- Tar, K.; Dange, T.; Yang, C.; Yao, Y.; Bulteau, A.L.; Salcedo, E.F.; Braigen, S.; Bouillaud, F.; Finley, D.; Schmidt, M. Proteasomes associated with the Blm10 activator protein antagonize mitochondrial fission through degradation of the fission protein Dnm1. J. Biol. Chem. 2014, 289, 12145–12156. [Google Scholar] [PubMed]
- Mandemaker, I.K.; Geijer, M.E.; Kik, I.; Bezstarosti, K.; Rijkers, E.; Raams, A.; Janssens, R.C.; Lans, H.; Hoeijmakers, J.H.; Demmers, J.A.; et al. DNA damage-induced replication stress results in PA200-proteasome-mediated degradation of acetylated histones. EMBO Rep. 2018, 19, e45566. [Google Scholar]
- Guan, H.; Wang, Y.; Yu, T.; Huang, Y.; Li, M.; Saeed, A.F.U.H.; Perčulija, V.; Li, D.; Xiao, J.; Wang, D.; et al. Cryo-EM structures of the human PA200 and PA200-20S complex reveal regulation of proteasome gate opening and two PA200 apertures. PLoS Biol. 2020, 18, e3000654. [Google Scholar]
- Toste Rêgo, A.; da Fonseca, P.C.A. Characterization of Fully Recombinant Human 20S and 20S-PA200 Proteasome Complexes. Mol. Cell 2019, 76, 138–147.e135. [Google Scholar] [PubMed]
- Douida, A.; Batista, F.; Robaszkiewicz, A.; Boto, P.; Aladdin, A.; Szenykiv, M.; Czinege, R.; Virág, L.; Tar, K. The proteasome activator PA200 regulates expression of genes involved in cell survival upon selective mitochondrial inhibition in neuroblastoma cells. J. Cell Mol. Med. 2020, 24, 6716–6730. [Google Scholar] [PubMed]
- Brachmann, C.B.; Davies, A.; Cost, G.J.; Caputo, E.; Li, J.; Hieter, P.; Boeke, J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 1998, 14, 115–132. [Google Scholar]
- Longtine, M.S.; McKenzie, A.; Demarini, D.J.; Shah, N.G.; Wach, A.; Brachat, A.; Philippsen, P.; Pringle, J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998, 14, 953–961. [Google Scholar]
- Goldstein, A.L.; McCusker, J.H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 1999, 15, 1541–1553. [Google Scholar]
- Lopez, A.D.; Tar, K.; Krügel, U.; Dange, T.; Ros, I.G.; Schmidt, M. Proteasomal degradation of Sfp1 contributes to the repression of ribosome biogenesis during starvation and is mediated by the proteasome activator Blm10. Mol. Biol. Cell 2011, 22, 528–540. [Google Scholar]
- Kruegel, U.; Robison, B.; Dange, T.; Kahlert, G.; Delaney, J.R.; Kotireddy, S.; Tsuchiya, M.; Tsuchiyama, S.; Murakami, C.J.; Schleit, J.; et al. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 2011, 7, e1002253. [Google Scholar]
- Narain, Y.; Wyttenbach, A.; Rankin, J.; Furlong, R.A.; Rubinsztein, D.C. A molecular investigation of true dominance in Huntington’s disease. J. Med. Genet. 1999, 36, 739–746. [Google Scholar] [PubMed]
- Bennett, E.J.; Bence, N.F.; Jayakumar, R.; Kopito, R.R. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol. Cell 2005, 17, 351–365. [Google Scholar] [PubMed]
- Meriin, A.B.; Zhang, X.; He, X.; Newnam, G.P.; Chernoff, Y.O.; Sherman, M.Y. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J. Cell Biol. 2002, 157, 997–1004. [Google Scholar] [PubMed]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar]
- Sin, O.; Mata-Cabana, A.; Seinstra, R.I.; Nollen, E.A. Filter Retardation Assay for Detecting and Quantifying Polyglutamine Aggregates Using Caenorhabditis elegans Lysates. Bio-Protocol 2018, 8, e3042. [Google Scholar] [PubMed]
- Juenemann, K.; Wiemhoefer, A.; Reits, E.A. Detection of ubiquitinated huntingtin species in intracellular aggregates. Front. Mol. Neurosci. 2015, 8, 1. [Google Scholar]
- Ocampo, A.; Zambrano, A.; Barrientos, A. Suppression of polyglutamine-induced cytotoxicity in Saccharomyces cerevisiae by enhancement of mitochondrial biogenesis. FASEB J. 2010, 24, 1431–1441. [Google Scholar]
- Alberti, S.; Halfmann, R.; Lindquist, S. Biochemical, cell biological, and genetic assays to analyze amyloid and prion aggregation in yeast. Methods Enzymol. 2010, 470, 709–734. [Google Scholar]
- Elsasser, S.; Schmidt, M.; Finley, D. Characterization of the proteasome using native gel electrophoresis. Methods Enzymol. 2005, 398, 353–363. [Google Scholar]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; Wanker, E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar]
- Ishihama, Y.; Rappsilber, J.; Mann, M. Modular stop and go extraction tips with stacked disks for parallel and multidimensional Peptide fractionation in proteomics. J. Proteome Res. 2006, 5, 988–994. [Google Scholar] [PubMed]
- Hadley, W.; Mara, A.; Jennifer, B.; Winston, C.; Lucy, D.A.M.; Romain, F.; Garrett, G.; Alex, H.; Lionel, H.; Jim, H.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [PubMed]
- Mason, R.P.; Giorgini, F. Modeling Huntington disease in yeast: Perspectives and future directions. Prion 2011, 5, 269–276. [Google Scholar]
- Hofer, S.; Kainz, K.; Zimmermann, A.; Bauer, M.A.; Pendl, T.; Poglitsch, M.; Madeo, F.; Carmona-Gutierrez, D. Studying Huntington’s Disease in Yeast: From Mechanisms to Pharmacological Approaches. Front. Mol. Neurosci. 2018, 11, 318. [Google Scholar]
- Chen, L.B.; Ma, S.; Jiang, T.X.; Qiu, X.B. Transcriptional upregulation of proteasome activator Blm10 antagonizes cellular aging. Biochem. Biophys. Res. Commun. 2020, 532, 211–218. [Google Scholar]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol.-Cell Physiol. 2011, 300, C723–C742. [Google Scholar]
- Zou, K.H.; Tuncali, K.; Silverman, S.G. Correlation and simple linear regression. Radiology 2003, 227, 617–628. [Google Scholar]
- Mukaka, M.M. Statistics corner: A guide to appropriate use of correlation coefficient in medical research. Malawi Med. J. 2012, 24, 69–71. [Google Scholar]
- Zinchuk, V.; Zinchuk, O.; Okada, T. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: Pushing pixels to explore biological phenomena. Acta Histochem. Cytochem. 2007. [Google Scholar] [CrossRef]
- Berger, A.; Schechter, I. Mapping the active site of papain with the aid of peptide substrates and inhibitors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1970, 257, 249–264. [Google Scholar] [PubMed]
- Turk, B.E.; Huang, L.L.; Piro, E.T.; Cantley, L.C. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat. Biotechnol. 2001, 19, 661–667. [Google Scholar] [PubMed]
- Coradin, M.; Karch, K.R.; Garcia, B.A. Monitoring proteolytic processing events by quantitative mass spectrometry. Expert Rev. Proteomics 2017, 14, 409–418. [Google Scholar] [PubMed]
- Blickwedehl, J.; Agarwal, M.; Seong, C.; Pandita, R.K.; Melendy, T.; Sung, P.; Pandita, T.K.; Bangia, N. Role for proteasome activator PA200 and postglutamyl proteasome activity in genomic stability. Proc. Natl. Acad. Sci. USA 2008, 105, 16165–16170. [Google Scholar]
- Theodoraki, M.A.; Nillegoda, N.B.; Saini, J.; Caplan, A.J. A Network of Ubiquitin Ligases Is Important for the Dynamics of Misfolded Protein Aggregates in Yeast. J. Biol. Chem. 2012, 287, 23911–23922. [Google Scholar]
- Juenemann, K.; Schipper-Krom, S.; Wiemhoefer, A.; Kloss, A.; Sanz Sanz, A.; Reits, E.A. Expanded polyglutamine-containing N-terminal huntingtin fragments are entirely degraded by mammalian proteasomes. J. Biol. Chem. 2013, 288, 27068–27084. [Google Scholar]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar]
- Lontay, B.; Kiss, A.; Virág, L.; Tar, K. How Do Post-Translational Modifications Influence the Pathomechanistic Landscape of Huntington’s Disease? A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 4282. [Google Scholar]
- Harding, R.J.; Tong, Y.F. Proteostasis in Huntington’s disease: Disease mechanisms and therapeutic opportunities. Acta Pharmacol. Sin. 2018, 39, 754–769. [Google Scholar]
- Sadre-Bazzaz, K.; Whitby, F.G.; Robinson, H.; Formosa, T.; Hill, C.P. Structure of a Blm10 complex reveals common mechanisms for proteasome binding and gate opening. Mol. Cell 2010, 37, 728–735. [Google Scholar]
- Sivanandam, V.N.; Jayaraman, M.; Hoop, C.L.; Kodali, R.; Wetzel, R.; van der Wel, P.C. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 2011, 133, 4558–4566. [Google Scholar] [PubMed]
- Kim, M.W.; Chelliah, Y.; Kim, S.W.; Otwinowski, Z.; Bezprozvanny, I. Secondary structure of Huntingtin amino-terminal region. Structure 2009, 17, 1205–1212. [Google Scholar] [PubMed]
- Kish-Trier, E.; Hill, C.P. Structural biology of the proteasome. Annu. Rev. Biophys. 2013, 42, 29–49. [Google Scholar] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Genotypes | Ref. |
|---|---|---|
| BY4742 | MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 | [36] |
| yMS122 | MATa his3 Δ1 leu2Δ0 met15Δ0 ura3Δ0 PRE1tevProA (HIS3) (KanMX)GAL1pHA3BLM10 | [27] |
| yMS131 | MATα his3∆1 leu2∆0 met15∆0 ura3∆0 blm10∆::NatMX | [39] |
| yMS268 | MATα his3∆1 leu2∆0 met15∆0 lys2∆0 ura3∆0 | [39] |
| yMS285 | MATα his3∆1 leu2∆0 ura3∆0 rpn4∆::KanMX | [31] |
| yMS476 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 PRE1TEVProA(HIS3) | [29] |
| yMS1371 | MATa his3∆1 leu2∆0 met15∆0 ura3∆0+ pYES2-Htt25Q-CFP | [40] |
| yMS1372 | MATa his3 ∆1 leu2Δ0 lys2Δ0 ura3Δ0 rpn4Δ::HphMX+ pYES2-Htt25Q-CFP | This study |
| yMS1377 | MATa his3∆1 leu2∆0 met15∆0 ura3∆0+ pYES2-Htt103Q-CFP | [40] |
| yMS1378 | MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 rpn4Δ::HphMX+ pYES2-Htt103Q-CFP | This study |
| yMS1392 | MATa MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 blm10∆::NatMX+ pYES2-Htt25Q-CFP | This study |
| yMS1393 | MATa MATα his3∆1 leu2∆0 lys2∆0 ura3∆0 blm10∆::NatMX+ pYES2-Htt103Q-CFP | This study |
| Primary Antibody (Source) | Catalog No. | Host | Dilution | Method |
| Anti-HA-Tag (Cell Signaling Technology, Danvers, MA, USA) | 3724S | Rabbit | 1:1000 | Immunoblot |
| Immunofluorescence | ||||
| Anti-HA-Tag (Cell Signaling Technology, Danvers, MA, USA) | 2367 | Mouse | 1:1000 | Immunoblot |
| 1:500 | Immunofluorescence | |||
| PSME4/PA200 (Novus Biologicals, Littleton, CO, USA) | NBP2-22236 | Rabbit | 1:2000 | Immunoblot |
| 1:1000 | Immunofluorescence | |||
| Anti-GFP (Clontech, Mountain View, CA, USA) | 632592 | Rabbit | 1:1000 | Immunoblot |
| Anti-GST Tag (Upstate Biotechnology, Lake Placid, NY, USA) | 05-311 | Mouse | 1:1000 | Immunoblot |
| β1 subunit of Proteasome 20S (human) (Enzo Biochem, Farmingdale, NY, USA). | BML-PW8140-0100 | Mouse | 1:1000 | Immunoblot |
| Proteasome 20S (Yeast) core subunits (Enzo Biochem, Farmingdale, NY, USA) | BML-PW9355-0100 | Rabbite | 1:1000 | Immunoblot |
| Actin (Santa Cruz Biotechnology, Dallas, TX, USA) | Sc-1616 | Goat | 1:10,000 | Immunoblot |
| Secondary Antibody (Source) | Catalog No. | Host | Dilution | Method |
| Alexa fluor 594 (Thermo Fisher Scientific, Waltham, MA, USA) | A11005 | Mouse | 1:1000 | Immunofluorescence |
| A31631 | Rabbit | |||
| Alexa fluor 488 (Thermo Fisher Scientific, Waltham, MA, USA) | A11001 | Mouse | 1:1000 | Immunofluorescence |
| A11034 | Rabbit | |||
| IRDye 800CW (LI-COR, Lincoln, NE, USA) | 926-32211 | Rabbit | 1:10,000 | Immunoblot |
| IRDye 680RD (LI-COR, Lincoln, NE, USA) | 926-68072 | Mouse | 1:5000 | Immunoblot |
| Anti-Mouse IgG Antibody, HRP conjugate | A9044 | Rabbit | 1:3000 | Immunoblot |
| Anti-Goat IgG Antibody, HRP conjugate | A8919 | Rabbit | 1:3000 | Immunoblot |
| Anti-Rabbit IgG Antibody, HRP conjugate | A0545 | Goat | 1:3000 | Immunoblot |
| Gene | Forward Primer | Reverse Primer | |
|---|---|---|---|
| PMSE4(PA200) | Human | 5′-ATGGAGAGTGCCTGAACTATTG-3′ | 5′-GTAGGTCAGCACACTTCCTATTC-3′ |
| GAPDH | Human | 5′-GAGTCAACGGATTTGGTCGT-3′ | 5′-GATCTCGCTCCTGGAAGATG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aladdin, A.; Yao, Y.; Yang, C.; Kahlert, G.; Ghani, M.; Király, N.; Boratkó, A.; Uray, K.; Dittmar, G.; Tar, K. The Proteasome Activators Blm10/PA200 Enhance the Proteasomal Degradation of N-Terminal Huntingtin. Biomolecules 2020, 10, 1581. https://doi.org/10.3390/biom10111581
Aladdin A, Yao Y, Yang C, Kahlert G, Ghani M, Király N, Boratkó A, Uray K, Dittmar G, Tar K. The Proteasome Activators Blm10/PA200 Enhance the Proteasomal Degradation of N-Terminal Huntingtin. Biomolecules. 2020; 10(11):1581. https://doi.org/10.3390/biom10111581
Chicago/Turabian StyleAladdin, Azzam, Yanhua Yao, Ciyu Yang, Günther Kahlert, Marvi Ghani, Nikolett Király, Anita Boratkó, Karen Uray, Gunnar Dittmar, and Krisztina Tar. 2020. "The Proteasome Activators Blm10/PA200 Enhance the Proteasomal Degradation of N-Terminal Huntingtin" Biomolecules 10, no. 11: 1581. https://doi.org/10.3390/biom10111581
APA StyleAladdin, A., Yao, Y., Yang, C., Kahlert, G., Ghani, M., Király, N., Boratkó, A., Uray, K., Dittmar, G., & Tar, K. (2020). The Proteasome Activators Blm10/PA200 Enhance the Proteasomal Degradation of N-Terminal Huntingtin. Biomolecules, 10(11), 1581. https://doi.org/10.3390/biom10111581