4-Benzyloxylonchocarpin and Muracatanes A-C from Ranunculus muricatus L. and Their Biological Effects

 ,
,  , ,
, ,  , ,
, ,  ,
,  ,
,  , , ,
, , ,
Abstract
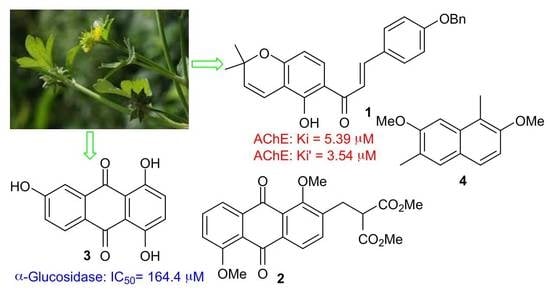
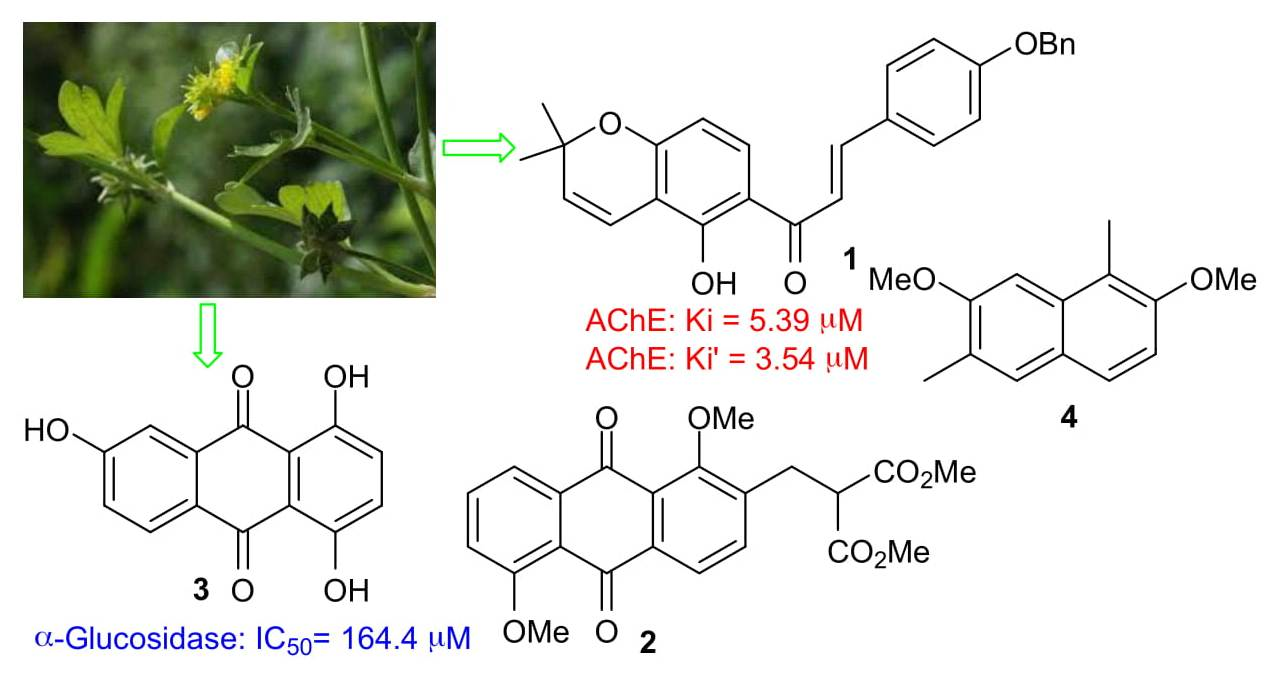
1. Introduction
2. Material and Methods
2.1. General Experimental Procedures and Chemicals
2.2. Plant Material
2.3. Extraction and Isolation
2.4. Solutions Preparation for AChE and BChE
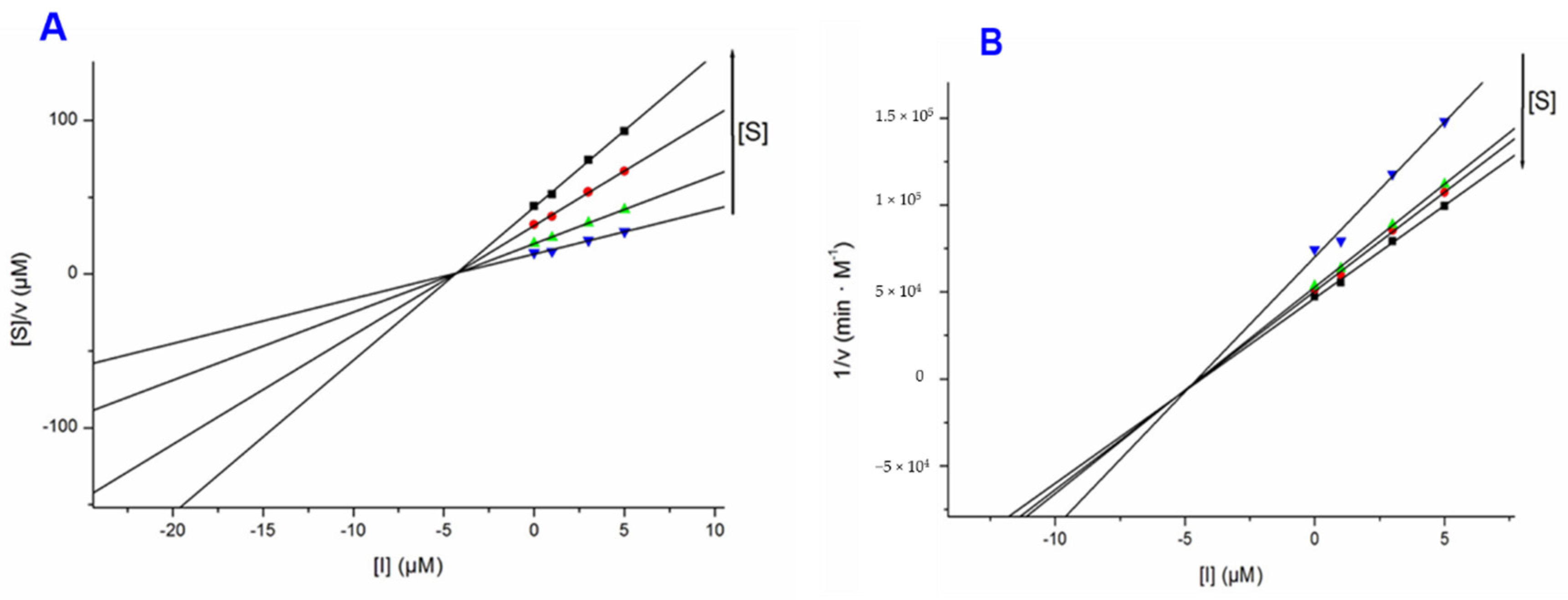
2.5. Cholinesterase Assay
2.6. α-Glucosidase Assay
2.7. Sulforhodamine B Assay
2.8. Molecular Docking
3. Results
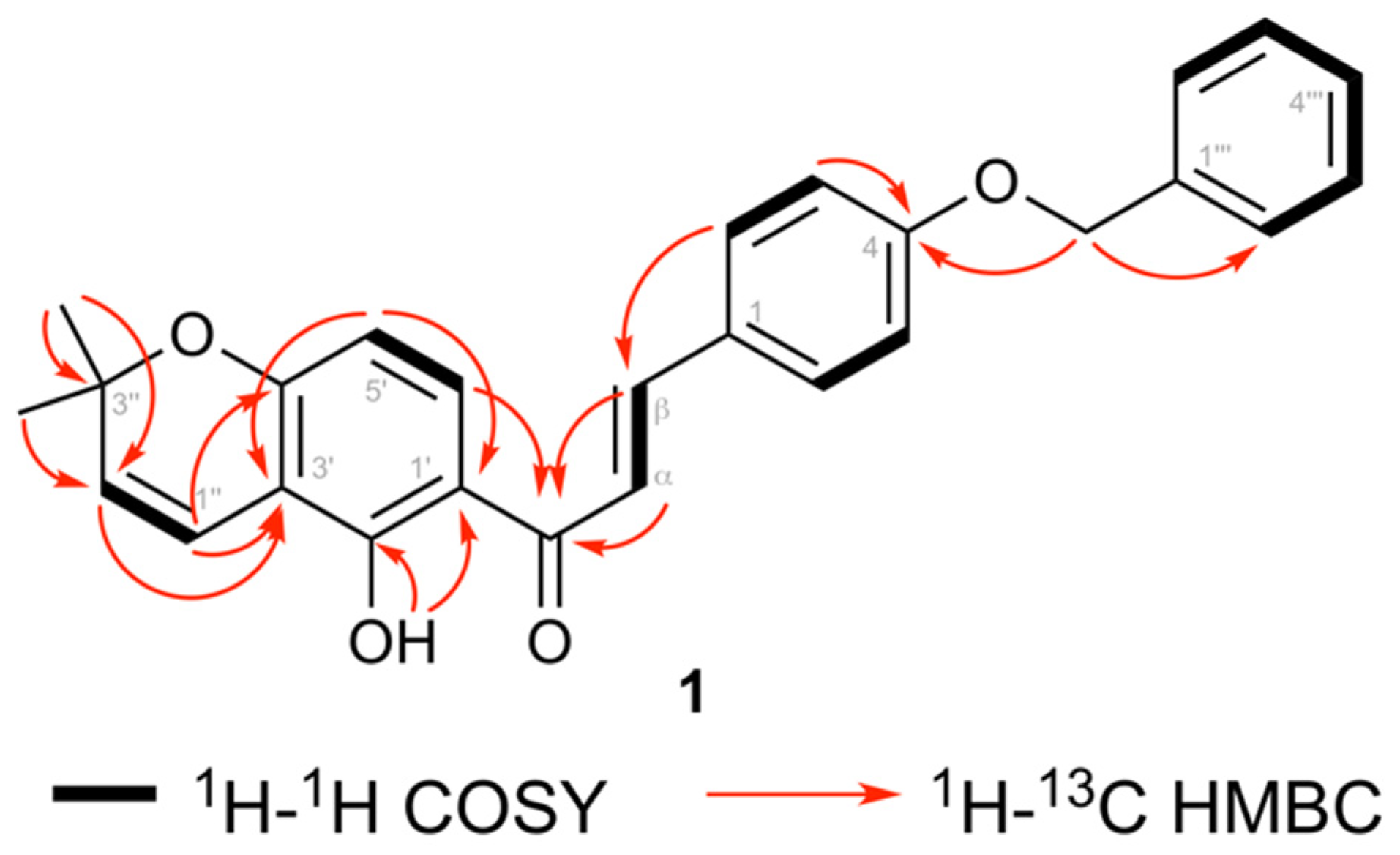
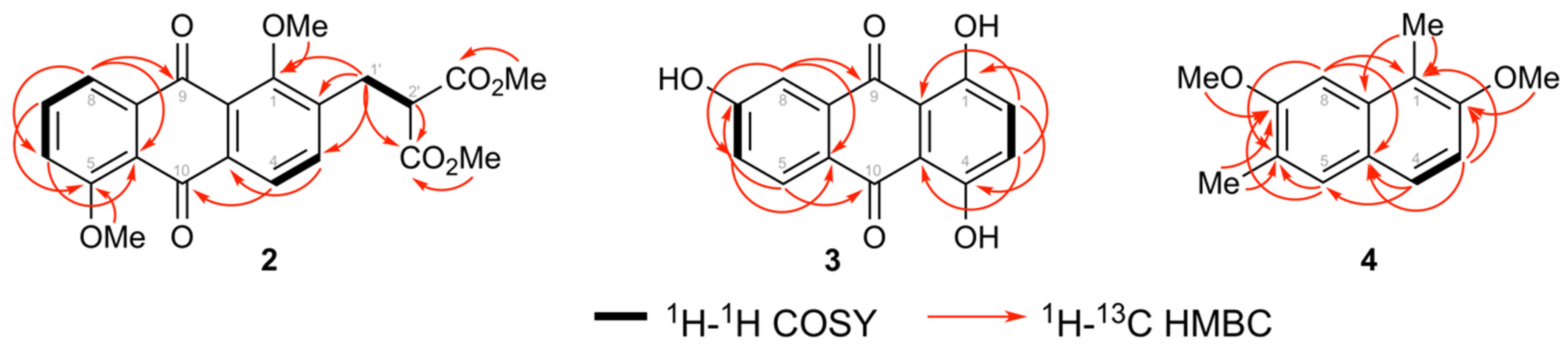
3.1. Structure Elucidation
3.2. Biological Evaluation
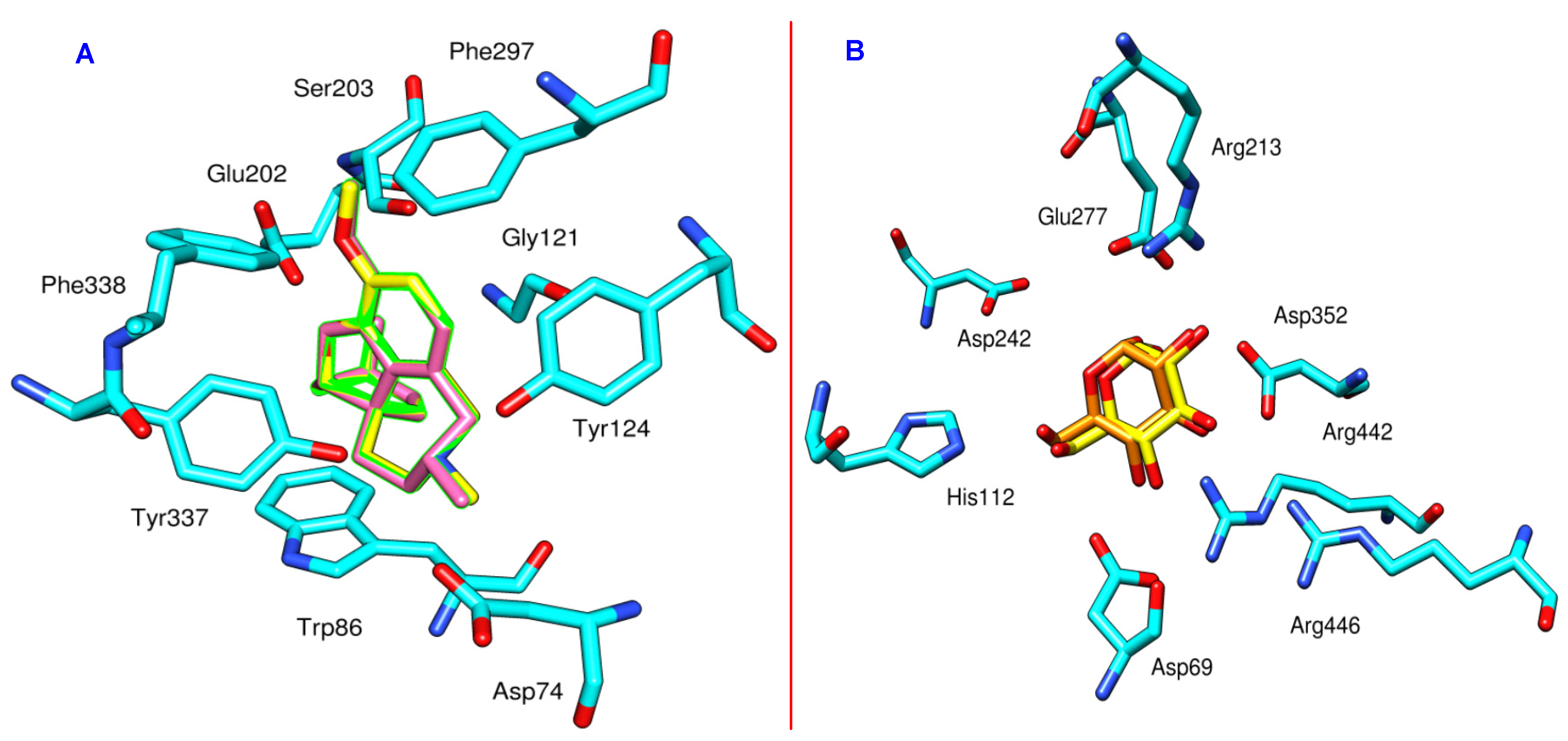
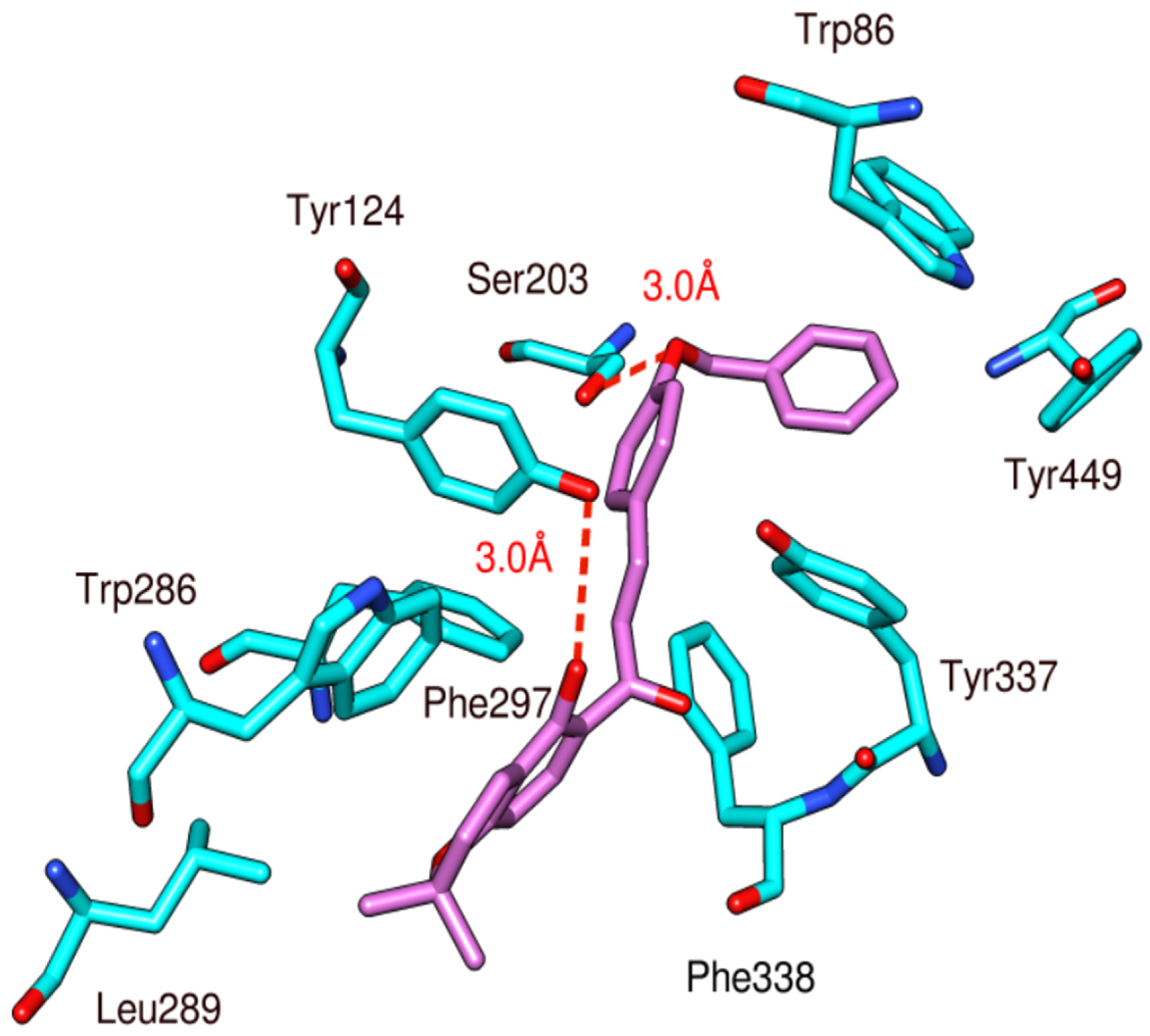
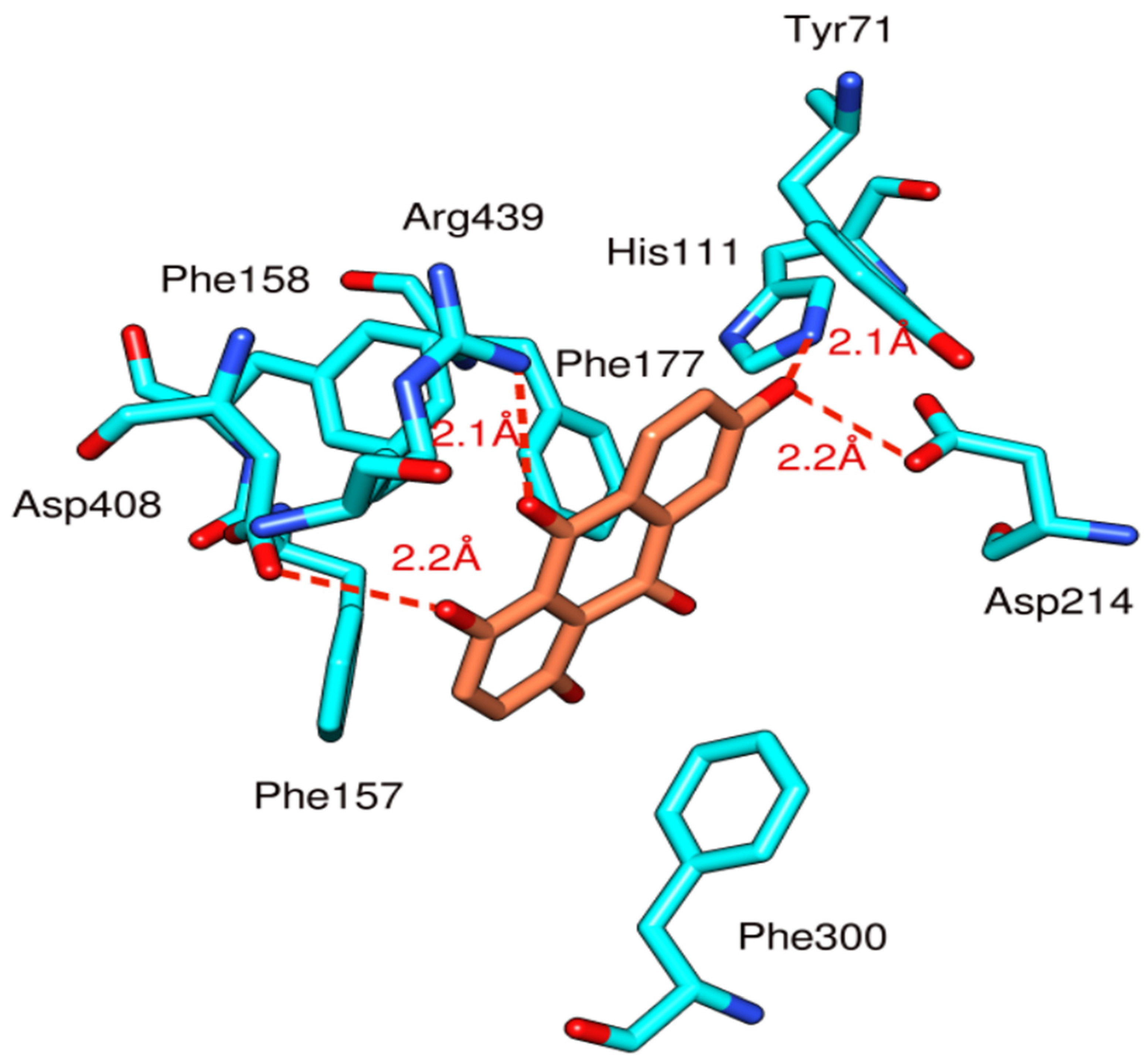
3.3. Docking Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on natural products for drug design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef]
- Clardy, J.; Fischbach, M.A.; Currie, C.R. The natural history of antibiotics. Curr. Biol. 2009, 19, 437–441. [Google Scholar] [CrossRef]
- Newman, D.J.; Giddings, L.-A. Natural products as leads to antitumor drugs. Phytochem. Rev. 2013, 13, 123–137. [Google Scholar] [CrossRef]
- Cragg, G.M.; Grothaus, P.G.; Newman, D.J. Impact of natural products on developing new anti-cancer agents. Chem Rev. 2009, 109, 3012–3043. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Wu, B.; Qin, F.; Zhou, G. Studies on chemical constituents of Ranunculus muricatus Linn. Nat. Prod. Res. Dev. 2013, 25, 736–741. [Google Scholar]
- Raziq, N.; Saeed, M.; Ali, M.S.; Lateef, M.; Shahid, M.; Akbar, S.; Zafar, S. Muriolide, a novel antioxidant lactone from Ranunculus muricatus. Nat. Prod. Res. 2020, 30. in print. [Google Scholar] [CrossRef]
- Khan, J.; Khan, R.; Qureshi, R. Ethnobotanical study of commonly used weeds of District Bannu, Khyber Pakhtunkhwa (Pakistan). J. Med. Plant Stud. 2013, 1, 1–6. [Google Scholar]
- Iqbal, H.; Sher, Z.; Khan, Z.U. Medicinal plants from salt range Pind Dadan Khan, district Jhelum, Punjab, Pakistan. J. Med. Plant Res. 2011, 5, 2157–2168. [Google Scholar]
- Ullah, M.; Khan, M.U.; Mahmood, A.; Malik, R.N.; Hussain, M.; Wazir, S.M.; Daud, M.; Shinwari, Z.K. An ethnobotanical survey of indigenous medicinal plants in Wana district South Waziristan agency. Pak. J. Ethnopharmacol. 2013, 150, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Chaudhry, B.A.; Ijaz, H.; Qadir, M.I. Caffeoyl-β-d-glucopyranoside and 1,3-dihydroxy-2-tetracosanoylamino-4-(E)-nonadecene isolated from Ranunculus muricatus exhibit antioxidant activity. Sci. Rep. 2019, 9, 15613. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.U.; Ijaz, F.; Iqbal, Z.; Afzal, A.; Ali, N.; Afzal, M.; Khan, M.A.; Muhammad, S.; Qadir, G.; Asif, M. A novel survey of the ethno medicinal knowledge of dental problems in Manoor valley (Northern Himalaya), Pakistan. J. Ethnopharmacol. 2016, 194, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Q.; Ahmad, T.; Mushtaq, M.N.; Malik, M.N.H.; Naz, H.; Ahsan, H.; Asif, H.; Noor, N.; Rahman, M.S.U.; Dar, U.; et al. Phytochemical analysis and cardiotonic activity of methanolic extract of Ranunculus muricatus Linn. In isolated rabbit heart. Acta Pol. Pharm. 2016, 73, 949–954. [Google Scholar]
- Khan, F.A.; Zahoor, M.; Khan, E. Chemical and biological evaluation of Ranunculus muricatus. Pak. J. Pharm. Sci. 2016, 29, 503–510. [Google Scholar]
- Nazir, S.; Tahir, K.; Naz, R.; Khan, Z.; Khan, A.; Islam, R.; Rehman, A.U. In vitro screening of Ranunculus muricatus for potential cytotoxic and antimicrobial activities. Glob. J. Pharmacol. 2014, 8, 427–431. [Google Scholar]
- Lal, S.D.; Yadav, B.K. Folk medicines of Kurukshetra district (Haryana). India. Econ. Bot. 1983, 37, 299–305. [Google Scholar] [CrossRef]
- Wang, A.W.; Wang, M.; Yuan, J.-R.; Tian, J.-K.; Wu, L.-M.; Geng, H. The study on antitumour effects in vitro of different extracts in Radix Ranunculus Ternati. J. Nat. Prod. Res. Dev. 2004, 16, 529–531. [Google Scholar]
- Xie, J.P.; He, Y.; Yue, J.; Hu, C.H.; Wang, H.-H. Identification of differential expression proteins of Mycobacterium tuberculosis strain isolated from clinical species treated with Radix Ranuncoli Ternati Extracts by comparative proteomics. Chin. Biochem. Mol. Biol. 2006, 22, 63–69. [Google Scholar]
- Qasem, J.R. Fungicidal activity of Ranunculus asiaticus and other weeds against Fusarium oxysporum f. sp. lycopersici. Ann. Appl. Biol. 1996, 128, 533–540. [Google Scholar] [CrossRef]
- Ibrar, M.; Samreen, U. Phytochemical screening and evaluation of cytotoxic and phytotoxic effects of Ranunculus muricatus L. Pak. J. Plant Sci. 2012, 18, 35–45. [Google Scholar]
- Aslam, M.S.; Choudhary, B.A.; Uzair, M.; Ijaz, A.S. Phytochemical study of Ariel parts of Ranunculus muricatus for the pharmacological active compounds. J. Appl. Pharm. 2013, 5, 827–832. [Google Scholar]
- Cavdar, H.; Senturk, M.; Guney, M.; Durdagi, S.; Kayik, G.; Supuran, C.T.; Ekinci, D. Inhibition of acetylcholinesterase and butyrylcholinesterase with uracil derivatives: Kinetic and computational studies. J. Enzyme Inhib. Med. Chem. 2019, 34, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, G.; Wu, D.; Yu, Y.; Hu, N.; Wang, H.; Li, X.; Wu, Y. Emerging strategies for the activity assay and inhibitor screening of alpha-glucosidase. Food Funct. 2020, 11, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.S.; Lauridsen, M.B.; Dragsted, L.O.; Nielsen, J.; Staerk, D. Development of a bioassay-coupled HPLC-SPE-ttNMR platform for identification of α-glucosidase inhibitors in apple peel (Malus domestica Borkh). Food Chem. 2012, 135, 1692–1699. [Google Scholar] [CrossRef]
- Shah, S.; Javaid, K.; Zafar, H.; Khan, K.M.; Khalil, R.; Ul-Haq, Z.; Choudhary, M.I. Synthesis, and In vitro and in silico α-glucosidase inhibitory studies of 5-chloro-2-aryl benzo [d] thiazoles. Bioorg. Chem. 2018, 78, 269–279. [Google Scholar] [CrossRef]
- Chemical Computing Group (CCG) Inc. Molecular Operating Environment (MOE); Chemical Computing Group: Montreal, QC, Canada, 2019. [Google Scholar]
- Kuete, V.; Noumedem, J.A.K.; Nana, F. Chemistry and Pharmacology of 4-Hydroxylonchocarpin: A Review. Chin. J. Integr. Med. 2013, 19, 475–480. [Google Scholar] [CrossRef]
- Krohn, K.; Farooq, U.; Hussain, H.; Ahmed, I.; Rheinheimer, J.; Draeger, S.; Schulz, B.; van Ree, T. Phomosines H–J, Novel Highly Substituted Biaryl Ethers, Isolated from the Endophytic Fungus Phomopsis sp. from Ligustrum vulgare. Nat. Prod. Commun. 2011, 6, 1907–1912. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, S.; Lin, S.; Zhu, C.; Yue, Z.; Yu, Y.; Liu, B.; Wu, X.; Yang, Y.; Li, Y.; et al. Natural and unnatural anthraquinones isolated from the ethanol extract of the roots of Knoxia valerianoides. Acta Pharm. Sin. B 2012, 2, 260–266. [Google Scholar] [CrossRef]
- Farina, F.; Vega, J.C.; Prados, P. 1,4,6-Trihydroxyantbraquinone. J. Am Chem. Soc. 1918, 40, 404–406. [Google Scholar]
- Farina, F.; Vega, J.C.; Prados, P. Polycyclic hydroxyquinones. X. Synthesis of substituted tetrahydroquinizarins by Diels-Alder reaction with naphthazarin and its diacetate. Quim. Org. Y Bioquim. 1982, 78, 344–453. [Google Scholar]
- Echavarren, A.; Prados, P.; Farina, F. Polycyclic hydroxyquinones. Part 17. Regiospecific Diels-Alder cycloadditions with chloronaphthoquinones as model reactions for regiospecific construction of the A-ring of anthracyclinones. J. Chem. Res. Synop. 1986, 364–365. [Google Scholar] [CrossRef]
- Ng’ang’a, M.M.; Hussain, H.; Chhabra, S.; Langat-Thoruwa, C.; Krohn, K.; Hussain, J.; Al-Harrasi, A.; Green, I. Eucleanal: A newnaphthalene derivative from Euclea divinorum. Nat. Prod. Commun. 2012, 7, 193–194. [Google Scholar]
- Ng’ang’a, M.M.; Hussain, H.; Chhabra, S.; Langat-Thoruwa, C.; Krohn, K.; Hussain, J.; Al-Harrasi, A.; Green, I. Eucleanal A and B: Two newnapthalene derivatives from Euclea divinorum. Chin. Chem. Lett. 2012, 23, 576–578. [Google Scholar] [CrossRef]
- Mahabusarakam, W.; Hemtasin, C.; Chakthong, S.; Voravuthikunchai, S.P.; Olawumi, I.B. Naphthoquinones, Anthraquinones and Naphthalene Derivatives from the Bulbs of Eleutherine americana. Planta Med. 2010, 76, 345–349. [Google Scholar] [CrossRef]
- Lin, C.N.; Lu, C.M.; Lin, H.C.; Ko, F.N.; Teng, C.M. Novel antiplatelet naphthalene from Rhamnus nakaharai. J. Nat. Prod. 1995, 58, 1934–1940. [Google Scholar] [CrossRef]
- Ganapaty, S.; Thomas, P.S.; Karagianis, G.; Waterman, P.G.; Brun, R. Antiprotozoal and cytotoxic naphthalene derivatives from Diospyros assimilis. Phytochemistry 2006, 67, 1950–1956. [Google Scholar] [CrossRef]
- Ngadjui, B.T.; Kapche, G.W.; Tamboue, H.; Abegaz, B.M.; Connolly, J.D. Prenylated flavonoids and a dihydro-4-phenylcoumarinfrom Dorstenia poinsettifolia. Phytochemistry 1999, 51, 119–123. [Google Scholar] [CrossRef]
- Habib, M.R.; Nikkon, F.; Rahman, M.; Haque, M.E.; Karim, M.R. Isolation of stigmasterol and beta-sitosterol from methanolic extract of root bark of Calotropis gigantea (Linn). Pak. J. Biol. Sci. 2007, 10, 4174–4176. [Google Scholar]
- Seo, S.; Tomita, Y.; Tori, K.; Yoshimura, Y. Determination of the absolute configuration of a secondary hydroxy group in a chiral secondary alcohol using glycosidation shifts in carbon-13 nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1978, 100, 3331–3339. [Google Scholar] [CrossRef]
- Burmaoglu, S.; Yilmaz, A.O.; Polat, M.F.; Kaya, R.; Gulcin, İ.; Algul, O. Synthesis and biological evaluation of novel tris-chalcones as potent carbonic anhydrase, acetylcholinesterase, butyrylcholinesterase and α-glycosidase inhibitors. Bioorg. Chem. 2019, 85, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Aslan, H.E.; Demir, Y.; Özaslan, M.S.; Türkan, F.; Beydemir, Ş.; Küfrevioğlu, Ö.I. The behavior of some chalconeson acetylcholinesterase and carbonic anhydrase activity. Drug Chem. Toxicol. 2019, 42, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Liua, H.R.; Liua, X.J.; Fana, H.Q.; Tanga, J.J.; Gaob, X.H.; Liu, W.K. Design, synthesis and pharmacological evaluation of chalconederivatives as acetylcholinesterase inhibitors. Bioorg. Med. Chem. 2014, 22, 6124–6133. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Rangarajan, T.M.; Chaudhary, R.; Singh 2, R.P.; Singh, M.; Singh 3, R.P.; Tondo 6, A.R.; Gambacorta, N.; Nicolotti, O.; Mathew, B.; et al. Novel Class of Chalcone Oxime Ethers as Potent Monoamine Oxidase-B and Acetylcholinesterase Inhibitors. Molecules 2020, 25, 2356. [Google Scholar] [CrossRef] [PubMed]
- Sribuhoma, T.; Saraphona, C.; Decharchoocharta, P.; Boonyaratb, C.; Yenjai, C. Acetylcholinesterase inhibition and cytotoxicity of flavonoids and chalcones from Derris indica. ScienceAsia 2016, 42, 247–251. [Google Scholar] [CrossRef][Green Version]
- Fosso, M.Y.; LeVine, H., 3rd; Green, K.D.; Tsodikov, O.V.; Garneau-Tsodikov, S. Effects of structural modifications on the metal binding, anti-amyloid activity, and cholinesterase inhibitory activity of chalcones. Org. Biomol. Chem. 2015, 13, 9418–9426. [Google Scholar] [CrossRef]
- Cummings, J.L. Alzheimer’s disease. N. Engl. J. Med. 2004, 351, 56–67. [Google Scholar] [CrossRef]
- Mohd, S.; Rizvi, D.; Shaikh, S.; Naaz, D.; Shakil, S.; Ahmad, A.; Haneef, M.; Abuzenada, A.M. Kinetics and Molecular Docking Study of an Anti-diabetic Drug Glimepiride as Acetylcholinesterase Inhibitor: Implication for Alzheimer’s Disease-Diabetes Dual Therapy. Neurochem. Res. 2016, 41, 1475–1482. [Google Scholar]
- Forstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef]
- Hitzeman, N. Cholinesterase inhibitors for Alzheimer’s disease. Am. Fam. Physician. 2006, 74, 747–759. [Google Scholar] [PubMed]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N. Drug development for Alzheimer’s disease: Where are we now and where are we headed. Am. J. Geriatr. Pharmacother. 2009, 7, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Dowarah, J.; Singh, V.P. Anti-diabetic drugs recent approaches and advancements. Bioorg. Med. Chem. 2020, 28, 115263. [Google Scholar] [CrossRef]
- Jörgens, V.; Grüsser, M. Happy Birthday, Claude Bernard. Diabetes 2013, 62, 2181–2182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hussain, H.; Abbas, G.; Green, I.R.; Ali, I. Dipeptidyl peptidase IV inhibitors as a potential target for diabetes: Patent review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 535–553. [Google Scholar] [CrossRef]
- Copeland, R.A. Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; Wiley: Hobokin, NJ, USA, 2013. [Google Scholar]
- Chiasson, J.L. Acarbose for the prevention of diabetes, hypertension, and cardiovascular disease in subjects with impaired glucose tolerance: The Study to Prevent Non-Insulin-Dependent Diabetes Mellitus (STOP-NIDDM) Trial. Endocr. Pract. 2006, 12, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Van de Laar, F.A.; Lucassen, P.L.; Akkermans, R.P.; van de Lisdonk, E.H.; Rutten, G.E.; van Weel, C. α-Glucosidase Inhibitors for Patients With Type 2 Diabetes. Diabetes Care 2005, 28, 154–163. [Google Scholar]
- Abbas, G.; Hassan, Z.; Al-Harrasi, A.; Muhammad, S.A.; Al-Quraini, A.J.; Al-Maani, Z.K.; Al-Adawai, A.M. Synthesis, molecular docking, and pharmacological evaluation of halobenzodithiophene derivatives against alpha-glucosidase, urease, and free radical production. Turk. J. Chem. 2018, 42, 1113–1123. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | ||||
|---|---|---|---|---|---|
| No | δH (nH, Multiplicity, J in Hz) | δC | No | δH (nH, Multiplicity, J in Hz) | δC |
| 1′ | - | 109.4 | 1 | - | 158.7 |
| 2′ | - | 160.9 | 2 | - | 138.4 |
| 3′ | - | 114.1 | 3 | 7.60 (1H, d, J = 8.0 Hz) | 136.4 |
| 4′ | - | 159.6 | 4 | 8.00 (1H, d, J = 8.0 Hz) | 123.2 |
| 5′ | 6.37 (1H, d, J = 9.0 Hz) | 108.1 | 4a | - | 137.0 |
| 6′ | 7.72 (1H, d, J = 9.0 Hz) | 130.5 | 5 | - | 159.8 |
| α | 7.45 (1H, d, J = 16 Hz, 1H) | 127.8 | 6 | 7.30 (1H, dd, J = 8.0, 2.0 Hz) | 117.1 |
| β | 7.86 (1H, d, J = 16.0 Hz, 1H) | 144.0 | 7 | 7.70 (1H, t, J = 8.0 Hz) | 135.0 |
| keto | - | 191.9 | 8 | 7.90 (1H, dd, J = 8.0, 2.0 Hz) | 119.6 |
| OH-2′ | 13.77 | 8a | - | 137.0 | |
| 1 | - | 128.0 | 9 | - | 182.7 |
| 2,6 | 7.60 (2H, BB′) | 130.3 | 9a | - | 124.8 |
| 3,5 | 7.00 (2H, AA′) | 115.3 | 10 | - | 182.1 |
| 4 | - | 160.9 | 10a | - | 120.9 |
| 1′′ | 6.76 (1H, d, J = 10 Hz) | 115.9 | 1′ | 3.34 (1H, d, J = 8.0 Hz) | 30.1 |
| 2′′ | 5.59 (1H, d, J = 10 Hz) | 127.4 | 2′ | 3.90 (1H, t, J = 8.0 Hz) | 51.3 |
| 3′′ | - | 77.7 | CO2Me | 3.70 (6H, s) | 52.6 |
| 4′′,5′′ | 1.47 (6H, s) | 28.3 | CO2Me | - | 169.0 |
| 1′′′ | - | 136.3 | 1-OMe | 3.97 (3H, s) | 62.0 |
| 2′′′–6′′′ | 7.46 (2H, m) | 127.4 | 5-OMe | 4.04 (3H, s) | 56.3 |
| 3′′′,5′′′ | 7.40 (2H, m) | 128.6 | |||
| 4′′′ | 7.32 (1H, m) | 127.8 | |||
| CH2Bn | 5.12 (2H, s) | 70.1 |
| 3 | 4 | ||||
|---|---|---|---|---|---|
| No | δH (nH, Multiplicity, J in Hz) | δC | No | δH (nH, Multiplicity, J in Hz) | δC |
| 1 | - | 156.4 | 1 | - | 117.9 |
| 2 | 7.28 (1H, m) | 128.3 | 2 | - | 154.2 |
| 3 | 7.28 (1H, m) | 129.2 | 3 | 7.09 (1H, d, J = 8.0 Hz) | 111.0 |
| 4 | 156.5 | 4 | 7.56 (1H, d, J = 8.0 Hz) | 125.9 | |
| 4a | 112.0 | 4a | 124.3 | ||
| 5 | 7.98 (1H, d, J = 8.0 Hz) | 129.5 | 5 | 7.49 (1H, s) | 129.1 |
| 6 | 7.20 (1H, dd, J = 8.0, 2.0 Hz) | 121.8 | 6 | - | 125.6 |
| 7 | 163.6 | 7 | - | 157.2 | |
| 8 | 7.40 (1H, d, J = 2.0 Hz) | 112.1 | 8 | 7.08 (1H, s) | 100.3 |
| 8a | - | 134.8 | 8a | - | 133.5 |
| 9 | - | 186.3 | 1-Me | 2.50 (3H, s) | 10.7 |
| 9a | - | 112.5 | 6-Me | 2.33 (3H, s) | 16.5 |
| 10 | - | 185.3 | 7-OMe | 3.95 (3H, s) | 55.1 |
| 10a | - | 124.3 | 2-OMe | 3.91 (3H, s) | 56.6 |
| OH-1 | 12.53 (1H, s) | - | |||
| OH-4 | 12.84 (1H, s) | - | |||
| OH-7 | 11.18 (1H, s) | - |
| AChE | BChE | α-Glucosidase | |||
|---|---|---|---|---|---|
| Compound | % Inhibition a | Ki (µM) (Ki’ (µM)) | % Inhibition a | % Inhibition a | IC50 (µM) |
| 1 | 97.4 | 5.39 ± 0.51 (3.54 ± 0.24) | 27.3 | n.d. | n.d. |
| 2 | n.d. | n.d. | n.d. | 13.4 | n.d. |
| 3 | n.d. | n.d. | n.d. | 80.4 | 164.46 ± 83.04 |
| 4 | n.d. | n.d. | n.d. | n.d. | n.d. |
| 5 | 49.9 | n.d. | 8.7 | 36.1 | n.d. |
| GH | 90.5 | 0.54 | 54.8 | Acarbose | 1072.5 ± 453.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, H.; Ali, I.; Wang, D.; Mamadalieva, N.Z.; Hussain, W.; Csuk, R.; Loesche, A.; Fischer, L.; Staerk, D.; Anam, S.; et al. 4-Benzyloxylonchocarpin and Muracatanes A-C from Ranunculus muricatus L. and Their Biological Effects. Biomolecules 2020, 10, 1562. https://doi.org/10.3390/biom10111562
Hussain H, Ali I, Wang D, Mamadalieva NZ, Hussain W, Csuk R, Loesche A, Fischer L, Staerk D, Anam S, et al. 4-Benzyloxylonchocarpin and Muracatanes A-C from Ranunculus muricatus L. and Their Biological Effects. Biomolecules. 2020; 10(11):1562. https://doi.org/10.3390/biom10111562
Chicago/Turabian StyleHussain, Hidayat, Iftikhar Ali, Daijie Wang, Nilufar Z. Mamadalieva, Wahid Hussain, René Csuk, Anne Loesche, Lucie Fischer, Dan Staerk, Syariful Anam, and et al. 2020. "4-Benzyloxylonchocarpin and Muracatanes A-C from Ranunculus muricatus L. and Their Biological Effects" Biomolecules 10, no. 11: 1562. https://doi.org/10.3390/biom10111562
APA StyleHussain, H., Ali, I., Wang, D., Mamadalieva, N. Z., Hussain, W., Csuk, R., Loesche, A., Fischer, L., Staerk, D., Anam, S., AlZain, M. N., Mushtaq, M., Ul-Haq, Z., Ullah, R., Noman, O. M., Abbas, G., & Green, I. R. (2020). 4-Benzyloxylonchocarpin and Muracatanes A-C from Ranunculus muricatus L. and Their Biological Effects. Biomolecules, 10(11), 1562. https://doi.org/10.3390/biom10111562