
VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases



Abstract
1. Mitochondria as Signaling Hubs
2. VDAC1 Structural Elements: The N-Terminal Domain and Its Oligomeric State
3. VDAC1 Extra-Mitochondrial Localization, Function, and Association with Pathological Conditions
4. VDAC1, a Multi-Functional Channel Controlling Cell Energy, Metabolism, and Oxidative Stress
5. VDAC1, at the Nexus of Mitochondria-Mediated Apoptosis, and mtDNA Release
5.1. Oligomerized VDAC1 Releases Cyto c, AIF, and SMAC/Diablo
5.2. Release of Mitochondrial DNA (mtDNA) through the VDAC1 Channel
5.3. VDAC1 Overexpression and Induction of Apoptosis
6. VDAC1, Metabolism, and a Link to Epigenetics
- Acetyl-CoA is a required co-factor for enzymes such as histone acetyltransferases (HATs) that catalyze the transfer of an acetyl group, to form ɛ-N-acetyl-lysine and acetylate histones in a process that is known to alter chromatin dynamics and to drive the epigenetic control of gene expression by activating transcriptional programs [193]. Practically, histone acetylation is dynamically regulated by the opposing actions of HATs and histone deacetylases (HDACs) that catalyze the addition and removal of the acetyl group, respectively. Intracellular concentrations of acetyl-CoA can vary roughly ~10-fold under normal physiological conditions, but they fall within the Km range of HATs. Histone acetylation activity is, thus, essentially regulated by the availability of acetyl CoA.
- NAD+ is essential for the deacetylation activity of sirtuins, a subgroup of HDAC, and changes in the NAD+/NADH ratio are thought to positively regulate sirtuin activity.
- Citrate in the cytosol can be converted to acetyl-CoA by the enzyme ATP-citrate lyase (ACLY), which catalyzes the ATP-dependent cleavage of mitochondrial-derived citrate into oxaloacetate and acetyl-CoA [194]. The acetyl-CoA can then serve as a donor for HAT-mediated histone acetylation. However, citrate can also be converted to acetyl-CoA in the nucleus by ACLY [194].
- α-ketoglutarate (α-KG) is an essential co-factor for 2-oxoglutarate-dependent dioxygenases (2-OGDD), including the histone demethylases with a Jumonji domain (JMJDs) and ten-eleven translocation (TET) DNA demethylases. α-KG has a direct impact on gene expression and, thus, can influence cellular fate by regulating histones and DNA demethylases. Succinate is the product of 2-oxoglutarate dependent dioxygenase (2-OGDD) enzyme reactions and, thus, when it accumulates, it works as an antagonist to the reaction.
- The 2-Hydroxyglutarate (2-HG) is not part of the TCA cycle, but it can be derived from α-KG by enzymes in the mitochondrial matrix and cytosol. The 2-HG competitively inhibits 2-OGDDs and together with fumarate, can rewire the epigenetic landscape of the cells through inhibition of histone and DNA demethylases.
- 6.
- Succinate and fumarate both behave as α-KG competitive antagonists and inhibit TETs and JMJDs. Both of these metabolites inhibit TET-catalyzed hydroxylation of 5 mC and the activity of histone demethylases KDM2A and KDM4A [195].
- 7.
- S-adenosylmethionine (SAM) serves as the methyl donor in reactions catalyzed by methionine adenosyl-transferase (MAT). DNA methylation at CpG sites represses gene expression by impeding access to transcription factors and inhibition of RNA polymerase II.
7. Proteins Interacting with VDAC1 Modulate VDAC1 Activity and They Are Regulated by VDAC1
7.1. VDAC1 Interacting Metabolism-Related Proteins
7.2. VDAC1 Interacting Apoptosis-Related Proteins
7.3. Interacting Cytoskeletal Proteins
7.4. VDAC1 Interacting Signaling Proteins
7.5. Viral Proteins Interact with VDAC1 to Modulate Expression Level and Function
- Influenza A Virus: the virus attacks the respiratory system including the nose, throat, and lungs. One of the virus proteins interacting with VDAC1 [268] is influenza A virus protein (PB1-F2). The protein is localized to both mitochondrial membranes, OMM and IMM, and is a 90-amino-acid protein expressed from the +1 open reading frame in the PB1 gene of influenza A viruses. PB1-F2 contributes to the pathogenesis of the virus by binding to the mitochondria, altering their morphology, and inducing apoptosis via direct interaction with VDAC1 [268].
- Hepatitis B Virus (HBV): one of five known human hepatitis viruses: hepatitis A, B, C, D, and E. This virus causes hepatitis B in which the virus attacks the liver and can cause acute and chronic liver disease such as cirrhosis and hepatocellular carcinoma. Hepatitis B viral protein (HBx) is a multifunctional protein encoded by the HBV genome that stimulates HBV replication. HBx binds to the mitochondria and co-localizes with VDAC-1, where it alters the mitochondrial transmembrane potential by creating together a hexamer that affects mitochondrial physiology [269].
- Hepatitis E Virus (HEV): HEV infections are often asymptomatic, although they can be severe and cause fulminant hepatitis and extra-hepatic manifestations including neurological and kidney injuries. Chronic HEV infections may also occur in immunocompromised patients causing inflammation of the liver. The open reading frame 3 (ORF3) protein is a small phosphoprotein of 113 or 114 aa proposed to act as an adaptor to link the intracellular transduction pathways, reduce the host inflammatory response, and protect virus-infected cells [270]. ORF3 protein interacts with several of the host’s cellular proteins, including VDAC1, where in a cross-linking study, ORF3-expressing cells were shown to produce higher levels of oligomeric VDAC1 [174,271].
- Human Immunodeficiency Virus type 1 (HIV-1): the virus causes acquired immunodeficiency syndrome, with progressive breakdown of the immune system, and promotes life-threatening opportunistic infections. A viral protein R (Vpr) is a 96-amino-acid (14-kDa) protein. This protein stimulates virus transcription and interacts with the host’s proteins, playing an active role in viral pathogenic factors. Vpr protein induces cell cycle arrest in proliferating cells, as well as regulating activation and apoptosis of infected T-lymphocytes via interaction with VDAC1 [239].
- Dengue Virus (DENV): the virus causes dengue fever characterized by headache, severe muscle and joint pain, and upper respiratory symptoms. The protein, DENV E, is an envelope protein (E protein) which is the key component of the dengue virion. The ENV E protein plays a vital role in the viral lifecycle by mediating interaction with host cells and facilitating invasion. It has been reported that this protein interacts with various proteins in host cells, including with the cellular chaperone GRP78 along with mitochondrial VDAC1. Furthermore, down-regulation of VDAC by siRNA reduced the expression of several DENV proteins: NS1, NS3, NS5, and DENVE [272]. Thus, VDAC plays a significant role in DENV infection.
- The Infectious Bursal Disease Virus (IBDV): the virus causes immunosuppressive disease in young chickens. The VP5 protein, a non-structural protein of IBDV, is associated within the plasma membrane of the host-infected cells and plays an essential part in pathogenesis of IBDV. VP5 induces apoptosis in host cells via interaction with VDAC [273], with the VDAC inhibitor DIDS [53] decreasing VP5 expression and apoptosis [274]. On the other hand, silencing VDAC1 expression decreases the expression of VP1, VP2, and VP5 [275]. VP1 and VP2 are essential structural proteins that participate in IBDV capsid assembly and penetration into the host cell, as well as replication of virus. The VP2 carries neutralizing epitopes which control antibody-mediated neutralization of IBDV infection. VP2 acts as an apoptotic inducer in infected cells.
- Japanese Encephalitis Virus infection (JEV): the virus causes Japanese encephalitis. The JEV invades the CNS, resulting in neuroinflammation, which negates the neuroprotective role of microglia, as characterized by increased microglial activation and neuronal death. The JEV envelope protein (JEV-E) is the major structural protein that facilitates the viral infection by recognizing host cellular receptors and mediating membrane fusion. The primary target of this protein is to neutralize antibodies; therefore, it is a key virulence factor involved in pathogenesis. The JEV-E protein interacts with VDAC, and in response to JEV infection, VDAC1 is overexpressed and co-localized with the ER protein GRP78, a multifunctional chaperone protein (HSP70) increasing MAM [276].
- Ostreid herpes virus-1 (OsHV-1): OsHV-1 is a variant of herpes virus, and it has been a major threat to Pacific cupped oysters. It contributes to the pathogenesis of mass mortality disease in the early life-stage of oysters. Infected oysters exhibit an increase in glycolysis and increased VDAC1 expression, as revealed in both mRNA and protein levels, while this increase in VDAC1 levels correlates with susceptibility of oysters to OsHV-1 [277].
8. VDAC1 Overexpression in Disease States and Association with Apoptosis
8.1. Cancer and VDAC1
8.2. Over-Expression of VDAC1 in Neurodegenerative Diseases
8.3. Type 2 Diabetes (T2D) and VDAC1
8.4. Autoimmune and Inflammatory Diseases and Increased VDAC1 Expression Levels
8.5. Cardiovascular Diseases, Apoptosis, and VDAC
8.6. Non-Alcoholic Fatty Liver Disease (NAFLD)
8.7. Rheumatoid Arthritis, Acute Kidney and Spinal Cord Injury, and VDAC1 Expression
9. VDAC1-Based Strategies to Treat Diseases Such as Cancer, Neurodegenerative, Autoimmune, NASH, and T2D
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Zeth, K.; Zachariae, U. Ten Years of High Resolution Structural Research on the Voltage Dependent Anion Channel (VDAC)-Recent Developments and Future Directions. Front. Physiol. 2018, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Maldonado, E.N.; Krelin, Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress 2017, 1, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; De, S.; Meir, A. The Mitochondrial Voltage-Dependent Anion Channel 1, Ca2+ Transport, Apoptosis, and Their Regulation. Front. Oncol. 2017, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium. 2018, 69, 81–100. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Chen, Q. VDAC1 as a Player in Mitochondria-Mediated Apoptosis and Target for Modulating Apoptosis. Curr. Med. Chem. 2017, 24, 4435–4446. [Google Scholar] [CrossRef] [PubMed]
- Camara, A.K.S.; Zhou, Y.; Wen, P.C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Fang, D.; Maldonado, E.N. VDAC Regulation: A Mitochondrial Target to Stop Cell Proliferation. Adv. Cancer Res. 2018, 138, 41–69. [Google Scholar] [CrossRef] [PubMed]
- Karachitos, A.; Jordan, J.; Kmita, H. VDAC-Targeted Drugs Affecting Cytoprotection and Mitochondrial Physiology in Cerebrovascular and Cardiovascular Diseases. Curr. Med. Chem. 2017, 24, 4419–4434. [Google Scholar] [CrossRef]
- Mazure, N.M. VDAC in cancer. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 665–673. [Google Scholar] [CrossRef]
- Reina, S.; De Pinto, V. Anti-Cancer Compounds Targeted to VDAC: Potential and Perspectives. Curr. Med. Chem. 2017, 24, 4447–4469. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 as an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848 Pt B, 2547–2575. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- De Pinto, V.; Guarino, F.; Guarnera, A.A.; Messina, S.; Reina, F.M.; Palermo, T.V.; Mazzoni, C. Characterization of human VDAC isoforms: A peculiar function for VDAC3? Biochim. Biophys. Acta 2010, 1797, 1268–1275. [Google Scholar] [CrossRef]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. VDAC isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, A.; Sheiko, T.; Graham, B.H.; Craigen, W.J. Voltage-dependant anion channels: Novel insights into isoform function through genetic models. Biochim. Biophys. Acta 2012, 1818, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef] [PubMed]
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 2008, 321, 1206–1210. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Colletier, J.P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef] [PubMed]
- Hiller, S.; Wagner, G. The role of solution NMR in the structure determinations of VDAC-1 and other membrane proteins. Curr. Opin. Struct. Biol. 2009, 19, 396–401. [Google Scholar] [CrossRef][Green Version]
- Geula, S.; Ben-Hail, D.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1: N-terminus location, translocation, channel gating and association with anti-apoptotic proteins. Biochem. J. 2012, 444, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef] [PubMed]
- Neumann, D.; Buckers, J.; Kastrup, L.; Hell, S.W.; Jakobs, S. Two-color STED microscopy reveals different degrees of colocalization between hexokinase-I and the three human VDAC isoforms. PMC Biophys. 2010, 3, 4. [Google Scholar] [CrossRef]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004, 377 Pt 2, 347–355. [Google Scholar] [CrossRef]
- Arbel, N.; Ben-Hail, D.; Shoshan-Barmatz, V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J. Biol. Chem. 2012, 287, 23152–23161. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Arbel, N.; Arzoine, L. VDAC, the voltage-dependent anion channel: Function, regulation & mitochondrial signaling in cell life and death. Cell Sci. 2008, 4, 74–118. [Google Scholar]
- Zaid, H.; Abu-Hamad, S.; Israelson, A.; Nathan, I.; Shoshan-Barmatz, V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005, 12, 751–760. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Mizrachi, D. VDAC1: From structure to cancer therapy. Front. Oncol. 2012, 2, 164. [Google Scholar] [CrossRef]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The Voltage-dependent Anion Channel 1 Mediates Amyloid beta Toxicity and Represents a Potential Target for Alzheimer Disease Therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef]
- Thinnes, F.P. Apoptogenic interactions of plasmalemmal type-1 VDAC and Abeta peptides via GxxxG motifs induce Alzheimer’s disease—A basic model of apoptosis? Wien. Med. Wochenschr. 2011, 161, 274–276. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Arbel, N.; Calo, D.; Arzoine, L.; Israelson, A.; Keinan, N.; Ben-Romano, R.; Friedman, O.; Shoshan-Barmatz, V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J. Cell Sci. 2009, 122 Pt 11, 1906–1916. [Google Scholar] [CrossRef]
- Arbel, N.; Shoshan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J. Biol. Chem. 2010, 285, 6053–6062. [Google Scholar] [CrossRef]
- Malia, T.J.; Wagner, G. NMR structural investigation of the mitochondrial outer membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry 2007, 46, 514–525. [Google Scholar] [CrossRef]
- Shimizu, S.; Ide, T.; Yanagida, T.; Tsujimoto, Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 2000, 275, 12321–12325. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Narita, M.; Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 1999, 399, 483–487. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, J.; Weng, C.; Chen, R.; Zheng, Y.; Chen, Q.; Tang, H. Identification of the protein-protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins. Biochem. Biophys. Res. Commun. 2003, 305, 989–996. [Google Scholar] [CrossRef]
- Arzoine, L.; Zilberberg, N.; Ben-Romano, R.; Shoshan-Barmatz, V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J. Biol. Chem. 2009, 284, 3946–3955. [Google Scholar] [CrossRef] [PubMed]
- Keinan, N.; Tyomkin, D.; Shoshan-Barmatz, V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol. Cell Biol. 2010, 30, 5698–5709. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Israelson, A.; Brdiczka, D.; Sheu, S.S. The voltage-dependent anion channel (VDAC): Function in intracellular signalling, cell life and cell death. Curr. Pharm. Des. 2006, 12, 2249–2270. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Mizrachi, D.; Keinan, N. Oligomerization of the mitochondrial protein VDAC1: From structure to function and cancer therapy. Prog. Mol. Biol. Transl. Sci. 2013, 117, 303–334. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Golan, M. Mitochondrial VDAC1: Function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 2012, 19, 714–735. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Keinan, N.; Zaid, H. Uncovering the role of VDAC in the regulation of cell life and death. J. Bioenerg. Biomembr. 2008, 40, 183–191. [Google Scholar] [CrossRef]
- Zeth, K.; Meins, T.; Vonrhein, C. Approaching the structure of human VDAC1, a key molecule in mitochondrial cross-talk. J. Bioenerg. Biomembr. 2008, 40, 127–132. [Google Scholar] [CrossRef][Green Version]
- Schlattner, U.; Tokarska-Schlattner, M.; Wallimann, T. Mitochondrial creatine kinase in human health and disease. Biochim. Biophys. Acta 2006, 1762, 164–180. [Google Scholar] [CrossRef]
- Zalk, R.; Israelson, A.; Garty, E.S.; Azoulay-Zohar, H.; Shoshan-Barmatz, V. Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem. J. 2005, 386 Pt 1, 73–83. [Google Scholar] [CrossRef]
- Geula, S.; Naveed, H.; Liang, J.; Shoshan-Barmatz, V. Structure-based analysis of VDAC1 protein: Defining oligomer contact sites. J. Biol. Chem. 2012, 287, 2179–2190. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Chaptal, V.; Ping, P.; Abramson, J. Crystal packing analysis of murine VDAC1 crystals in a lipidic environment reveals novel insights on oligomerization and orientation. Channels (Austin) 2009, 3, 167–170. [Google Scholar] [CrossRef][Green Version]
- Keinan, N.; Pahima, H.; Ben-Hail, D.; Shoshan-Barmatz, V. The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2013, 1833, 1745–1754. [Google Scholar] [CrossRef]
- Weisthal, S.; Keinan, N.; Ben-Hail, D.; Arif, T.; Shoshan-Barmatz, V. Ca2+-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim. Biophys. Acta 2014, 1843, 2270–2281. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Han, J.; Ben-Hail, D.; He, L.; Li, B.; Chen, Z.; Wang, Y.; Yang, Y.; Liu, L.; Zhu, Y.; et al. A new fungal diterpene induces VDAC1-dependent apoptosis in Bax/Bak-deficient cells. J. Biol. Chem. 2015, 290, 23563–23578. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hail, D.; Shoshan-Barmatz, V. VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis. Biochim. Biophys. Acta 2016, 1863 Pt A, 1612–1623. [Google Scholar] [CrossRef]
- Akanda, N.; Tofighi, R.; Brask, J.; Tamm, C.; Elinder, F.; Ceccatelli, S. Voltage-dependent anion channels (VDAC) in the plasma membrane play a critical role in apoptosis in differentiated hippocampal neurons but not in neural stem cells. Cell Cycle 2008, 7, 3225–3234. [Google Scholar] [CrossRef]
- Bahamonde, M.I.; Valverde, M.A. Voltage-dependent anion channel localises to the plasma membrane and peripheral but not perinuclear mitochondria. Pflugers Arch. 2003, 446, 309–313. [Google Scholar] [CrossRef]
- Bathori, G.; Parolini, I.; Szabo, I.; Tombola, F.; Messina, A.; Oliva, M.; Sargiacomo, M.; De Pinto, V.; Zoratti, M. Extramitochondrial porin: Facts and hypotheses. J. Bioenerg. Biomembr. 2000, 32, 79–89. [Google Scholar] [CrossRef]
- Bathori, G.; Parolini, I.; Tombola, F.; Szabo, I.; Messina, A.; Oliva, M.; De Pinto, V.; Lisanti, M.; Sargiacomo, M.; Zoratti, M. Porin is present in the plasma membrane where it is concentrated in caveolae and caveolae-related domains. J. Biol. Chem. 1999, 274, 29607–29612. [Google Scholar] [CrossRef]
- Bouyer, G.; Cueff, A.; Egee, S.; Kmiecik, J.; Maksimova, Y.; Glogowska, E.; Gallagher, P.G.; Thomas, S.L. Erythrocyte peripheral type benzodiazepine receptor/voltage-dependent anion channels are upregulated by Plasmodium falciparum. Blood 2011, 118, 2305–2312. [Google Scholar] [CrossRef]
- De Pinto, V.; Messina, A.; Lane, D.J.; Lawen, A. Voltage-dependent anion-selective channel (VDAC) in the plasma membrane. FEBS Lett. 2010, 584, 1793–1799. [Google Scholar] [CrossRef]
- Dermietzel, R.; Hwang, T.K.; Buettner, R.; Hofer, A.; Dotzler, E.; Kremer, M.; Deutzmann, R.; Thinnes, F.P.; Fishman, G.I.; Spray, D.C.; et al. Cloning and in situ localization of a brain-derived porin that constitutes a large-conductance anion channel in astrocytic plasma membranes. Proc. Natl. Acad. Sci. USA 1994, 91, 499–503. [Google Scholar] [CrossRef]
- Marin, R.; Ramirez, C.M.; Gonzalez, M.; Gonzalez-Munoz, E.; Zorzano, A.; Camps, M.; Alonso, R.; Diaz, M. Voltage-dependent anion channel (VDAC) participates in amyloid beta-induced toxicity and interacts with plasma membrane estrogen receptor alpha in septal and hippocampal neurons. Mol. Membr. Biol. 2007, 24, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.M.; Gonzalez, M.; D’iaz, M.; Alonso, R.; Marin, R. VDAC and ERalpha interaction in caveolae from human cortex is altered in Alzheimer’s disease. Mol. Cell Neurosci. 2009, 42, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Sabirov, R.Z.; Merzlyak, P. Plasmalemmal VDAC controversies and maxi-anion channel puzzle. Biochim. Biophys. Acta 2012, 1818, 1570–1580. [Google Scholar] [CrossRef]
- Thinnes, F.P. Neuroendocrine differentiation of LNCaP cells suggests: VDAC in the cell membrane is involved in the extrinsic apoptotic pathway. Mol. Genet. Metabol. 2009, 97, 241–243. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.N.; Hadad, F.I.; Shafir, I.; Orr, M.V.; Heilmeyer, L.M. VDAC/porin is present in sarcoplasmic reticulum from skeletal muscle. FEBS Lett. 1996, 386, 205–210. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.N.; Zalk, R.; Gincel, D.; Vardi, N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochim. Biophys. Acta 2004, 1657, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Thinnes, F.P. After all, plasmalemmal expression of type-1 VDAC can be understood. Phosphorylation, nitrosylation, and channel modulators work together in vertebrate cell volume regulation and either apoptotic pathway. Front. Physiol. 2015, 6, 126. [Google Scholar] [CrossRef] [PubMed]
- Stadtmuller, U.; Eben-Brunnen, J.; Schmid, A.; Hesse, D.; Klebert, S.; Kratzin, H.D.; Hesse, J.; Zimmermann, B.; Reymann, S.; Thinnes, F.P.; et al. Mitochondria-derived and extra-mitochondrial human type-1 porin are identical as revealed by amino acid sequencing and electrophysiological characterisation. Biol. Chem. 1999, 380, 1461–1466. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.F.; O’Neal, W.K.; Huang, P.; Nicholas, R.A.; Ostrowski, L.E.; Craigen, W.J.; Lazarowski, E.R.; Boucher, R.C. Voltage-dependent anion channel-1 (VDAC-1) contributes to ATP release and cell volume regulation in murine cells. J. Gen. Physiol. 2004, 124, 513–526. [Google Scholar] [CrossRef]
- Buettner, R.; Papoutsoglou, G.; Scemes, E.; Spray, D.C.; Dermietzel, R. Evidence for secretory pathway localization of a voltage-dependent anion channel isoform. Proc. Natl. Acad. Sci. USA 2000, 97, 3201–3206. [Google Scholar] [CrossRef]
- Thinnes, F.P.; Gotz, H.; Kayser, H.; Benz, R.; Schmidt, W.E.; Kratzin, H.D.; Hilschmann, N. Identification of human porins. I. Purification of a porin from human B-lymphocytes (Porin 31HL) and the topochemical proof of its expression on the plasmalemma of the progenitor cell. Biol. Chem. Hoppe. Seyler. 1989, 370, 1253–1264. [Google Scholar] [CrossRef]
- Gonzalez-Gronow, M.; Kalfa, T.; Johnson, C.E.; Gawdi, G.; Pizzo, S.V. The voltage-dependent anion channel is a receptor for plasminogen kringle 5 on human endothelial cells. J. Biol. Chem. 2003, 278, 27312–27318. [Google Scholar] [CrossRef]
- Li, L.; Yao, Y.C.; Gu, X.Q.; Che, D.; Ma, C.Q.; Dai, Z.Y.; Li, C.; Zhou, T.; Cai, W.B.; Yang, Z.H.; et al. Plasminogen kringle 5 induces endothelial cell apoptosis by triggering a voltage-dependent anion channel 1 (VDAC1) positive feedback loop. J. Biol. Chem. 2014, 289, 32628–32638. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Vilarino, M.; Cabodevilla, F.; Del Rio, J.; Frechilla, D.; Perez-Mediavilla, A. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer’s disease transgenic mice: An insight into the pathogenic effects of amyloid-beta. J. Alzheimers Dis. 2011, 23, 195–206. [Google Scholar] [CrossRef]
- Perez-Gracia, E.; Torrejon-Escribano, B.; Ferrer, I. Dystrophic neurites of senile plaques in Alzheimer’s disease are deficient in cytochrome c oxidase. Acta Neuropathol. 2008, 116, 261–268. [Google Scholar] [CrossRef]
- Hur, J.Y.; Teranishi, Y.; Kihara, T.; Yamamoto, N.G.; Inoue, M.; Hosia, W.; Hashimoto, M.; Winblad, B.; Frykman, S.; Tjernberg, L.O. Identification of novel gamma-secretase-associated proteins in detergent-resistant membranes from brain. J. Biol. Chem. 2012, 287, 11991–12005. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Hur, J.Y.; Kihara, T.; Teranishi, Y.; Yamamoto, N.G.; Ishikawa, T.; Wiehager, B.; Winblad, B.; Tjernberg, L.O.; Schedin-Weiss, S. Human brain proteins showing neuron-specific interactions with gamma-secretase. FEBS J. 2015, 282, 2587–2599. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Muhammed, S.; Kessler, B.; Salehi, A. Mitochondrial proteome analysis reveals altered expression of voltage dependent anion channels in pancreatic beta-cells exposed to high glucose. Islets 2010, 2, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Chen, X.; Middleditch, M.; Huang, L.; Vazhoor Amarsingh, G.; Reddy, S.; Lu, J.; Zhang, S.; Phillips, R.; Phillips, A.R.J.; et al. Quantitative proteomic profiling identifies new renal targets of copper(II)-selective chelation in the reversal of diabetic nephropathy in rats. Proteomics 2009, 9, 4309–4320. [Google Scholar] [CrossRef]
- Pittala, S.; Levy, I.; De, S.; Kumar Pandey, S.; Melnikov, N.; Shoshan-Barmatz, V. The VDAC1-based R-Tf-D-LP4 Peptide as a Potential Treatment for Diabetes Mellitus. Cells 2020, 9, 481. [Google Scholar] [CrossRef]
- Zhong, Z.; Lemasters, J.J. A Unifying Hypothesis Linking Hepatic Adaptations for Ethanol Metabolism to the Proinflammatory and Profibrotic Events of Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2018, 42, 2072–2089. [Google Scholar] [CrossRef]
- Zhang, E.; Al-Amily, I.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ben-Hail, D.; Ye, Y.; Soni, A.; Groop, L.; et al. Preserving Insulin Secretion in Diabetes by Inhibiting VDAC1 Overexpression and Surface Translocation in β Cells. Cell Metabol. 2019, 29, 64–77. [Google Scholar] [CrossRef]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef]
- Arif, T.; Vasilkovsky, L.; Refaely, K.A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef]
- Kim, J.R.; Gupta, L.P.; Blanco, S.; Yang, A.; Shteinfer-Kuzmine, W.; Kang, Z.; Zhu, X.; Park, S.-J.; Chung, J.H.; Shoshan-Barmatz, V.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- Pittala, S.; Krelin, Y.; Kuperman, Y.; Shoshan-Barmatz, V. A Mitochondrial VDAC1-Based Peptide Greatly Suppresses Steatosis and NASH-Associated Pathologies in a Mouse Model. Mol. Ther. 2019, 27, 1848–1862. [Google Scholar] [CrossRef] [PubMed]
- Branco, A.F.; Pereira, S.L.; Moreira, A.C.; Holy, J.; Sardao, V.A.; Oliveira, P.J. Isoproterenol cytotoxicity is dependent on the differentiation state of the cardiomyoblast H9c2 cell line. Cardiovasc. Toxicol. 2011, 11, 191–203. [Google Scholar] [CrossRef]
- Klapper-Goldstein, H.; Verma, A.; Elyagon, S.; Gillis, R.; Murninkas, M.; Pittala, S.; Paul, A.; Shoshan-Barmatz, V. VDAC1 overexpression in the diseased myocardium of humans and rats and the effect of VDAC1-interacting compound on atrial fibrosis induced by hyperaldosteronism. 2020. under revision. [Google Scholar]
- Shoshan-Barmatz, V.; Israelson, A. The voltage-dependent anion channel in endoplasmic/sarcoplasmic reticulum: Characterization, modulation and possible function. J. Membr. Biol. 2005, 204, 57–66. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. VDAC1, mitochondrial dysfunction, and Alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef]
- Leal Denis, M.F.; Alvarez, H.A.; Lauri, N.; Alvarez, C.L.; Chara, O.; Schwarzbaum, P.J. Dynamic Regulation of Cell Volume and Extracellular ATP of Human Erythrocytes. PLoS ONE 2016, 11, e0158305. [Google Scholar] [CrossRef]
- Sridharan, M.; Bowles, E.A.; Richards, J.P.; Krantic, M.; Davis, K.L.; Dietrich, K.A.; Stephenson, A.H.; Ellsworth, M.L.; Sprague, R.S. Prostacyclin receptor-mediated ATP release from erythrocytes requires the voltage-dependent anion channel. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H553–H559. [Google Scholar] [CrossRef] [PubMed]
- Marginedas-Freixa, I.; Alvarez, C.L.; Moras, M.; Leal Denis, M.F.; Hattab, C.; Halle, F.; Bihel, F.; Mouro-Chanteloup, I.; Lefevre, S.D.; Le Van Kim, C.; et al. Human erythrocytes release ATP by a novel pathway involving VDAC oligomerization independent of pannexin-1. Sci. Rep. 2018, 8, 11384. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 2012, 12, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F.; Pierri, C.L. Mitochondrial metabolite transport. Essays Biochem. 2010, 47, 37–52. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Chandel, N.S.; Li, X.X.; Schumacker, P.T.; Colombini, M.; Thompson, C.B. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. USA 2000, 97, 4666–4671. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef]
- Dolder, M.; Wendt, S.; Wallimann, T. Mitochondrial creatine kinase in contact sites: Interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage. Biol. Signals Recept. 2001, 10, 93–111. [Google Scholar] [CrossRef]
- Gurnev, P.A.; Rostovtseva, T.K.; Bezrukov, S.M. Tubulin-blocked state of VDAC studied by polymer and ATP partitioning. FEBS Lett. 2011, 585, 2363–2366. [Google Scholar] [CrossRef]
- Csordás, G.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Pinton, P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim. Biophys. Acta 2014, 1837, 461–469. [Google Scholar] [CrossRef]
- Rone, M.B.; Fan, J.; Papadopoulos, V. Cholesterol transport in steroid biosynthesis: Role of protein-protein interactions and implications in disease states. Biochim. Biophys. Acta 2009, 1791, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–62562. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef]
- Tonazzi, A.; Giangregorio, N.; Console, L.; Indiveri, C. Mitochondrial carnitine/acylcarnitine translocase: Insights in structure/function relationships. Basis for drug therapy and side effects prediction. Mini Rev. Med. Chem. 2015, 15, 396–405. [Google Scholar] [CrossRef]
- Qiu, J.; Tan, Y.-W.; Hagenston, A.M.; Martel, M.-A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.A.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4, 2034. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal. Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Ahmad, A.; Ahmad, S.; Schneider, B.K.; Allen, C.B.; Chang, L.Y.; White, C.W. Elevated expression of hexokinase II protects human lung epithelial-like A549 cells against oxidative injury. Am. J. Physiol. Lung. Cell Mol. Physiol. 2002, 283, L573–L584. [Google Scholar] [CrossRef] [PubMed]
- Da-Silva, W.S.; Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shukair, S.; Naik, T.J.; Moazed, F.; Ardehali, H. Glucose phosphorylation and mitochondrial binding are required for the protective effects of hexokinases I and II. Mol. Cell Biol. 2008, 28, 1007–1017. [Google Scholar] [CrossRef]
- Bryson, J.M.; Coy, P.E.; Gottlob, K.; Hay, N.; Robey, R.B. Increased hexokinase activity, of either ectopic or endogenous origin, protects renal epithelial cells against acute oxidant-induced cell death. J. Biol. Chem. 2002, 277, 11392–11400. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Lacas-Gervais, S.; Adaixo, R.; Ilc, K.; Rouleau, M.; Notte, A.; Dieu, M.; Michiels, C.; Voeltzel, T.; Maguer-Satta, V.; et al. Local mitochondrial-endolysosomal microfusion cleaves voltage-dependent anion channel 1 to promote survival in hypoxia. Mol. Cell Biol. 2015, 35, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Mazure, N.M. News about VDAC1 in Hypoxia. Front. Oncol. 2016, 6, 193. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, M.C.; Ben-Hail, D.; Ilie, M.; Gounon, P.; Rouleau, M.; Hofman, V.; Doyen, J.; Mari, B.; Shoshan-Barmatz, V.; Hofman, P.; et al. Expression of a truncated active form of VDAC1 in lung cancer associates with hypoxic cell survival and correlates with progression to chemotherapy resistance. Cancer Res. 2012, 72, 2140–2150. [Google Scholar] [CrossRef]
- Aram, L.; Geula, S.; Arbel, N.; Shoshan-Barmatz, V. VDAC1 cysteine residues: Topology and function in channel activity and apoptosis. Biochem. J. 2010, 427, 445–454. [Google Scholar] [CrossRef][Green Version]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta 2012, 1818, 1457–1465. [Google Scholar] [CrossRef]
- Gincel, D.; Shoshan-Barmatz, V. Glutamate interacts with VDAC and modulates opening of the mitochondrial permeability transition pore. J. Bioenerg. Biomembr. 2004, 36, 179–186. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Komarov, A.; Bezrukov, S.M.; Colombini, M. VDAC channels differentiate between natural metabolites and synthetic molecules. J. Membr. Biol. 2002, 187, 147–156. [Google Scholar] [CrossRef]
- Yehezkel, G.; Hadad, N.; Zaid, H.; Sivan, S.; Shoshan-Barmatz, V. Nucleotide-binding sites in the voltage-dependent anion channel: Characterization and localization. J. Biol. Chem. 2006, 281, 5938–5946. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Gincel, D. The voltage-dependent anion channel: Characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem. Biophys. 2003, 39, 279–292. [Google Scholar] [CrossRef]
- Villinger, S.; Giller, K.; Bayrhuber, M.; Lange, A.; Griesinger, C.; Becker, S.; Zweckstetter, M. Nucleotide interactions of the human voltage-dependent anion channel. J. Biol. Chem. 2014, 289, 13397–13406. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, O.P.; Paz, A.; Adelman, J.L.; Colletier, J.P.; Abramson, J.; Grabe, M. Structure-guided simulations illuminate the mechanism of ATP transport through VDAC1. Nat. Struct. Mol. Biol. 2014, 21, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Bathori, G.; Csordas, G.; Garcia-Perez, C.; Davies, E.; Hajnoczky, G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 2006, 281, 17347–17358. [Google Scholar] [CrossRef] [PubMed]
- Gincel, D.; Zaid, H.; Shoshan-Barmatz, V. Calcium binding and translocation by the voltage-dependent anion channel: A possible regulatory mechanism in mitochondrial function. Biochem. J. 2001, 358 Pt 1, 147–155. [Google Scholar] [CrossRef]
- Israelson, A.; Abu-Hamad, S.; Zaid, H.; Nahon, E.; Shoshan-Barmatz, V. Localization of the voltage-dependent anion channel-1 Ca2+-binding sites. Cell Calcium. 2007, 41, 235–544. [Google Scholar] [CrossRef]
- Gincel, D.; Vardi, N.; Shoshan-Barmatz, V. Retinal voltage-dependent anion channel: Characterization and cellular localization. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2097–2104. [Google Scholar]
- Israelson, A.; Arzoine, L.; Abu-hamad, S.; Khodorkovsky, V.; Shoshan-Barmatz, V. A photoactivable probe for calcium binding proteins. Chem. Biol. 2005, 12, 1169–1178. [Google Scholar] [CrossRef][Green Version]
- Pittala, M.G.G.; Saletti, R.; Reina, S.; Cunsolo, V.; De Pinto, V.; Foti, S. A High Resolution Mass Spectrometry Study Reveals the Potential of Disulfide Formation in Human Mitochondrial Voltage-Dependent Anion Selective Channel Isoforms (hVDACs). Int. J. Mol. Sci. 2020, 21, 1468. [Google Scholar] [CrossRef]
- Baines, C.P.; Song, C.X.; Zheng, Y.T.; Wang, G.W.; Wang, Z.O.; Guo, Y.; Bolli, R.; Cardwell, E.M. Protein kinase Cepsilon interacts with and inhibits the permeability transition pore in cardiac mitochondria. Circ. Res. 2003, 92, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.K.; Ghosh, S.; Das, S. Mitochondrial VDAC can be phosphorylated by cyclic AMP-dependent protein kinase. Biochem. Biophys. Res. Commun. 1995, 209, 213–217. [Google Scholar] [CrossRef]
- Liberatori, S.; Canas, B.; Tani, C.; Bini, L.; Buonocore, G.; Godovac-Zimmermann, J.; Mishra, O.P.; Delivoria-Papadopoulos, M.; Bracci, R.; Pallini, V. Proteomic approach to the identification of voltage-dependent anion channel protein isoforms in guinea pig brain synaptosomes. Proteomics 2004, 4, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Kerner, J.; Lee, K.; Tandler, B.; Hoppel, C.L. VDAC proteomics: Post-translation modifications. Biochim. Biophys. Acta 2012, 1818, 1520–1525. [Google Scholar] [CrossRef]
- Aulak, K.S.; Koeck, T.; Crabb, J.W.; Stuehr, D.J. Dynamics of protein nitration in cells and mitochondria. Am. J. Physiol. Heart Circul. Physiol. 2004, 286, H30–H38. [Google Scholar] [CrossRef] [PubMed]
- Kanski, J.; Behring, A.; Pelling, J.; Schoneich, C. Proteomic identification of 3-nitrotyrosine-containing rat cardiac proteins. Effects Biol. Aging 2005, 288, H371–H381. [Google Scholar] [CrossRef]
- Turko, I.V.; Li, L.; Aulak, K.S.; Stuehr, D.J.; Chang, J.Y.; Murad, F. Protein tyrosine nitration in the mitochondria from diabetic mouse heart. Implications to dysfunctional mitochondria in diabetes. J. Biol. Chem. 2003, 278, 33972–33977. [Google Scholar] [CrossRef]
- Mello, C.F.; Sultana, R.; Piroddi, M.; Cai, J.; Pierce, W.M.; Klein, J.B.; Butterfield, D.A. Acrolein induces selective protein carbonylation in synaptosomes. Neuroscience 2007, 147, 674–967. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Antignani, A.; Youle, R.J. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol. 2006, 18, 685–689. [Google Scholar] [CrossRef]
- Lovell, J.F.; Billen, L.P.; Bindner, S.; Shamas-Din, A.; Fradin, C.; Leber, B.; Andrews, D.W. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 2008, 135, 1074–1084. [Google Scholar] [CrossRef]
- Banerjee, J.; Ghosh, S. Bax increases the pore size of rat brain mitochondrial voltage-dependent anion channel in the presence of tBid. Biochem. Biophys. Res. Commun. 2004, 323, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Tsujimoto, Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc. Natl. Acad. Sci. USA 2000, 97, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Madesh, M.; Hajnoczky, G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 2001, 155, 1003–1015. [Google Scholar] [CrossRef]
- Ghosh, T.; Pandey, N.; Maitra, A.; Brahmachari, S.K.; Pillai, B. A role for voltage-dependent anion channel Vdac1 in polyglutamine-mediated neuronal cell death. PLoS ONE 2007, 2, e1170. [Google Scholar] [CrossRef]
- Godbole, A.; Varghese, J.; Sarin, A.; Mathew, M.K. VDAC is a conserved element of death pathways in plant and animal systems. Biochim. Biophys. Acta 2003, 1642, 87–96. [Google Scholar] [CrossRef]
- Lu, A.J.; Dong, C.W.; Du, C.S.; Zhang, Q.Y. Characterization and expression analysis of Paralichthys olivaceus voltage-dependent anion channel (VDAC) gene in response to virus infection. Fish. Shellfish Immunol. 2007, 23, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Israelson, A.; Zaid, H.; Abu-Hamad, S.; Nahon, E.; Shoshan-Barmatz, V. Mapping the ruthenium red-binding site of the voltage-dependent anion channel-1. Cell Calcium. 2008, 43, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Tajeddine, N.; Galluzzi, L.; Kepp, O.; Hangen, E.; Morselli, E.; Senovilla, L.; Araujo, N.; Pinna, G.; Larochette, N.; Zamzami, N.; et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene 2008, 27, 4221–4232. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Fu, Y.; Wang, X.; Shi, H.; Huang, Y.; Song, X.; Li, L.; Song, N.; Luo, Y. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J. 2008, 22, 2809–2820. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, F.; Messina, A.; Lartigue, L.; Schembri, L.; Medina, C.; Reina, S.; Thoraval, D.; Crouzet, M.; Ichas, F.; De Pinto, V.; et al. Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res. 2009, 19, 1363–1376. [Google Scholar] [CrossRef] [PubMed]
- Betaneli, V.; Petrov, E.P.; Schwille, P. The role of lipids in VDAC oligomerization. Biophys. J. 2012, 102, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. P53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer’s disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef]
- Sureda, F.X.; Junyent, F.; Verdaguer, E.; Auladell, C.; Pelegri, C.; Vilaplana, J.; Folch, J.; Canudas, A.M.; Zarate, C.B.; Palles, M.; et al. Antiapoptotic drugs: A therapautic strategy for the prevention of neurodegenerative diseases. Curr. Pharm. Des. 2011, 17, 230–245. [Google Scholar] [CrossRef]
- Kostin, S.; Pool, L.; Elsasser, A.; Hein, S.; Drexler, H.C.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klovekorn, W.P.; et al. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef]
- Kroemer, G.; El-Deiry, W.S.; Golstein, P.; Peter, M.E.; Vaux, D.; Vandenabeele, P.; Zhivotovsky, B.; Blagosklonny, M.V.; Malorni, W.; Knight, R.A.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005, 12 (Suppl. S2), 1463–1467. [Google Scholar] [CrossRef]
- Marunouchi, T.; Tanonaka, K. Cell Death in the Cardiac Myocyte. Biol. Pharm. Bull. 2015, 38, 1094–1097. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef]
- Nicholls, T.J.; Minczuk, M. In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Wiedmer, A.; Wang, P.; Zhou, J.; Rennekamp, A.J.; Tiranti, V.; Zeviani, M.; Lieberman, P.M. Epstein-Barr virus immediate-early protein Zta co-opts mitochondrial single-stranded DNA binding protein to promote viral and inhibit mitochondrial DNA replication. J. Virol. 2008, 82, 4647–4655. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Shao, L.; Bronstein, J.C.; Weber, P.C.; Weller, S.K. The product of a 1.9-kb mRNA which overlaps the HSV-1 alkaline nuclease gene (UL12) cannot relieve the growth defects of a null mutant. Virology 1996, 215, 152–164. [Google Scholar] [CrossRef]
- De Mendoza, C.; Martin-Carbonero, L.; Barreiro, P.; de Baar, M.; Zahonero, N.; Rodriguez-Novoa, S.; Benito, J.M.; Gonzalez-Lahoz, J.; Soriano, V. Mitochondrial DNA depletion in HIV-infected patients with chronic hepatitis C and effect of pegylated interferon plus ribavirin therapy. AIDS 2007, 21, 583–588. [Google Scholar] [CrossRef]
- Nawarak, J.; Huang-Liu, R.; Kao, S.H.; Liao, H.H.; Sinchaikul, S.; Chen, S.T.; Cheng, S.L. Proteomics analysis of A375 human malignant melanoma cells in response to arbutin treatment. Biochim. Biophys. Acta 2009, 1794, 159–167. [Google Scholar] [CrossRef]
- Jiang, N.; Kham, S.K.; Koh, G.S.; Suang Lim, J.Y.; Ariffin, H.; Chew, F.T.; Yeoh, A.E. Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (ALL). J. Proteomics 2011, 74, 843–857. [Google Scholar] [CrossRef]
- Castagna, A.; Antonioli, P.; Astner, H.; Hamdan, M.; Righetti, S.C.; Perego, P.; Zunino, F.; Righetti, P.G. A proteomic approach to cisplatin resistance in the cervix squamous cell carcinoma cell line A431. Proteomics 2004, 4, 3246–3267. [Google Scholar] [CrossRef]
- Zhang, Y.; Rosenberg, P.A. The essential nutrient pyrroloquinoline quinone may act as a neuroprotectant by suppressing peroxynitrite formation. Eur. J. Neurosci. 2002, 16, 1015–1024. [Google Scholar] [CrossRef]
- Ebeling, M.C.; Polanco, J.R.; Qu, J.; Tu, C.; Montezuma, S.R.; Ferrington, D.A. Improving retinal mitochondrial function as a treatment for age-related macular degeneration. Redox Biol. 2020, 34, 101552. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.L.; Liu, R.H.; Sheu, J.N.; Chen, S.T.; Sinchaikul, S.; Tsay, G.J. Toxicogenomics of A375 human malignant melanoma cells treated with arbutin. J. Biomed. Sci. 2007, 14, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Bengtsson, S.; Krogh, M.; Marquez, M.; Nilsson, S.; James, P.; Aliaya, A.; Holmberg, A.R. Somatostatin effects on the proteome of the LNCaP cell-line. Int. J. Oncol. 2007, 30, 1173–1179. [Google Scholar] [CrossRef][Green Version]
- Moin, S.M.; Panteva, M.; Jameel, S. The hepatitis E virus Orf3 protein protects cells from mitochondrial depolarization and death. J. Biol. Chem. 2007, 282, 21124–21133. [Google Scholar] [CrossRef]
- Voehringer, D.W.; Hirschberg, D.L.; Xiao, J.; Lu, Q.; Roederer, M.; Lock, C.B.; Herzenberg, L.A.; Steinman, L. Gene microarray identification of redox and mitochondrial elements that control resistance or sensitivity to apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 2680–2685. [Google Scholar] [CrossRef] [PubMed]
- Sharaf El Dein, O.; Gallerne, C.; Brenner, C.; Lemaire, C. Increased expression of VDAC1 sensitizes carcinoma cells to apoptosis induced by DNA cross-linking agents. Biochem. Pharmacol. 2012, 83, 1172–1182. [Google Scholar] [CrossRef]
- Lan, C.H.; Sheng, J.Q.; Fang, D.C.; Meng, Q.Z.; Fan, L.L.; Huang, Z.R. Involvement of VDAC1 and Bcl-2 family of proteins in VacA-induced cytochrome c release and apoptosis of gastric epithelial carcinoma cells. J. Dig. Dis. 2010, 11, 43–49. [Google Scholar] [CrossRef]
- Leone, A.; Roca, M.S.; Ciardiello, C.; Terranova-Barberio, M.; Vitagliano, C.; Ciliberto, G.; Mancini, R.; Di Gennaro, E.; Bruzzese, F.; Budillon, A. Vorinostat synergizes with EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation of the major mitochondrial porin VDAC1 and modulation of the c-Myc-NRF2-KEAP1 pathway. Free Radic. Biol. Med. 2015, 89, 287–299. [Google Scholar] [CrossRef]
- Yang, X.; Tang, S.; Dai, C.; Li, D.; Zhang, S.; Deng, S.; Zhou, Y.; Xiao, X. Quinocetone induces mitochondrial apoptosis in HepG2 cells through ROS-dependent promotion of VDAC1 oligomerization and suppression of Wnt1/beta-catenin signaling pathway. Food Chem. Toxicol. 2017, 105, 161–176. [Google Scholar] [CrossRef]
- Dehan, P.; Kustermans, G.; Guenin, S.; Horion, J.; Boniver, J.; Delvenne, P. DNA methylation and cancer diagnosis: New methods and applications. Expert. Rev. Mol. Diagn. 2009, 9, 651–657. [Google Scholar] [CrossRef]
- Hirst, M.; Marra, M.A. Epigenetics and human disease. Int. J. Biochem. Cell Biol. 2009, 41, 136–146. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef]
- Stefanska, B.; Karlic, H.; Varga, F.; Fabianowska-Majewska, K.; Haslberger, A. Epigenetic mechanisms in anti-cancer actions of bioactive food components--the implications in cancer prevention. Br. J. Pharmacol. 2012, 167, 279–297. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, L.; Di, L. Compartmentation of metabolites in regulating epigenome of cancer. Mol. Med. 2016, 22, 349–360. [Google Scholar] [CrossRef][Green Version]
- Gronbaek, K.; Treppendahl, M.; Asmar, F.; Guldberg, P. Epigenetic Changes in Cancer as Potential Targets for Prophylaxis and Maintenance Therapy. Basic Clin. Pharmacol. Toxicol. 2008, 103, 389–396. [Google Scholar] [CrossRef]
- Urvalek, A.; Laursen, K.B.; Gudas, L.J. The roles of retinoic acid and retinoic acid receptors in inducing epigenetic changes. Subcell. Biochem. 2014, 70, 129–149. [Google Scholar] [CrossRef]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef]
- Chandel, N.S. Evolution of Mitochondria as Signaling Organelles. Cell Metab. 2015, 22, 204–206. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Amsalem, Z.; Arif, T.; Shteinfer-Kuzmine, A.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers (Basel) 2020, 12, 1031. [Google Scholar] [CrossRef]
- Sivanand, S.; Viney, I.; Wellen, K.E. Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci. 2018, 43, 61–74. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Koren, I.; Raviv, Z.; Shoshan-Barmatz, V. Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo. Cancer Biol. Ther. 2010, 9, 1046–1052. [Google Scholar] [CrossRef]
- Ko, J.H.; Gu, W.; Lim, I.; Zhou, T.; Bang, H. Expression profiling of mitochondrial voltage-dependent anion channel-1 associated genes predicts recurrence-free survival in human carcinomas. PLoS ONE 2014, 9, e110094. [Google Scholar] [CrossRef]
- Caterino, M.; Ruoppolo, M.; Mandola, A.; Costanzo, M.; Orru, S.; Imperlini, E. Protein-protein interaction networks as a new perspective to evaluate distinct functional roles of voltage-dependent anion channel isoforms. Mol. Biosyst. 2017, 13, 2466–2476. [Google Scholar] [CrossRef]
- Prezma, T.; Shteinfer, A.; Admoni, L.; Raviv, Z.; Sela, I.; Levi, I.; Shoshan-Barmatz, V. VDAC1-based peptides: Novel pro-apoptotic agents and potential therapeutics for B-cell chronic lymphocytic leukemia. Cell Death Dis. 2013, 4, e809. [Google Scholar] [CrossRef][Green Version]
- Shteinfer-Kuzmine, A.; Arif, T.; Krelin, Y.; Tripathi, S.S.; Paul, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1-based peptides: Attacking oncogenic properties in glioblastoma. Oncotarget 2017, 8, 31329–31346. [Google Scholar] [CrossRef][Green Version]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L.; Mathupala, S.; Rempel, A.; Geschwind, J.F.; Ko, Y.H. Mitochondrial bound type II hexokinase: A key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim. Biophys. Acta 2002, 1555, 14–20. [Google Scholar] [CrossRef]
- Majewski, N.; Nogueira, V.; Robey, R.B.; Hay, N. Akt inhibits apoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases. Mol. Cell Biol. 2004, 24, 730–740. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005, 65, 10545–10554. [Google Scholar] [CrossRef]
- Allouche, M.; Pertuiset, C.; Robert, J.L.; Martel, C.; Veneziano, R.; Henry, C.; Dein, O.S.; Saint, N.; Brenner, C.; Chopineau, J. ANT-VDAC1 interaction is direct and depends on ANT isoform conformation in vitro. Biochem. Biophys. Res. Commun. 2012, 429, 12–17. [Google Scholar] [CrossRef]
- Bernardi, P.; Di Lisa, F.; Fogolari, F.; Lippe, G. From ATP to PTP and Back: A Dual Function for the Mitochondrial ATP Synthase. Circ. Res. 2015, 116, 1850–1862. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell. Biol. 2007, 9, 550–555. [Google Scholar] [CrossRef]
- Kokoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [CrossRef]
- Patterson, R.L.; van Rossum, D.B.; Kaplin, A.I.; Barrow, R.K.; Snyder, S.H. Inositol 1,4,5-trisphosphate receptor/GAPDH complex augments Ca2+ release via locally derived NADH. Proc. Natl. Acad. Sci. USA 2005, 102, 1357–1359. [Google Scholar] [CrossRef] [PubMed]
- Vyssokikh, M.; Zorova, L.; Zorov, D.; Heimlich, G.; Jurgensmeier, J.; Schreiner, D.; Brdiczka, D. The intra-mitochondrial cytochrome c distribution varies correlated to the formation of a complex between VDAC and the adenine nucleotide translocase: This affects Bax-dependent cytochrome c release. Biochim. Biophys. Acta 2004, 1644, 27–36. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maldonado, E.N. VDAC-Tubulin, an Anti-Warburg Pro-Oxidant Switch. Front. Oncol. 2017, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Azarashvili, T.; Krestinina, O.; Baburina, Y.; Odinokova, I.; Grachev, D.; Papadopoulos, V.; Akatov, V.; Lemasters, J.J.; Reiser, G. Combined effect of G3139 and TSPO ligands on Ca2+-induced permeability transition in rat brain mitochondria. Arch. Biochem. Biophys. 2015, 587, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Danial, N.N. BCL-2 family proteins: Critical checkpoints of apoptotic cell death. Clin. Cancer Res. 2007, 13, 7254–7263. [Google Scholar] [CrossRef]
- Huang, H.; Shah, K.; Bradbury, N.A.; Li, C.; White, C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 2014, 5, e1482. [Google Scholar] [CrossRef]
- Shimizu, S.; Konishi, A.; Kodama, T.; Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 3100–3105. [Google Scholar] [CrossRef]
- Sugiyama, T.; Shimizu, S.; Matsuoka, Y.; Yoneda, Y.; Tsujimoto, Y. Activation of mitochondrial voltage-dependent anion channel by apro-apoptotic BH3-only protein Bim. Oncogene 2002, 21, 4944–4956. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. The voltage-dependent anion channel: An essential player in apoptosis. Biochimie 2002, 84, 187–193. [Google Scholar] [CrossRef]
- Yamagata, H.; Shimizu, S.; Nishida, Y.; Watanabe, Y.; Craigen, W.J.; Tsujimoto, Y. Requirement of voltage-dependent anion channel 2 for pro-apoptotic activity of Bax. Oncogene 2009, 28, 3563–3572. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Vervliet, T.; Akl, H.; Bultynck, G. The selective BH4-domain biology of Bcl-2-family members: IP3Rs and beyond. Cell Mol. Life Sci. 2013, 70, 1171–1183. [Google Scholar] [CrossRef]
- Eckenrode, E.F.; Yang, J.; Velmurugan, G.V.; Foskett, J.K.; White, C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J. Biol. Chem. 2010, 285, 13678–13684. [Google Scholar] [CrossRef] [PubMed]
- Distelhorst, C.W.; Bootman, M.D. Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: Role in Ca2+ signaling and disease. Cell Calcium. 2011, 50, 234–241. [Google Scholar] [CrossRef]
- Monaco, G.; Decrock, E.; Arbel, N.; van Vliet, A.R.; La Rovere, R.M.; De Smedt, H.; Parys, J.B.; Agostinis, P.; Leybaert, L.; Shoshan-Barmatz, V.; et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J. Biol. Chem. 2015, 290, 9150–9161. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Higuchi, H.; Miura, S.; Azuma, T.; Inokuchi, S.; Saito, H.; Kato, S.; Ishii, H. Bax interacts with the voltage-dependent anion channel and mediates ethanol-induced apoptosis in rat hepatocytes. Am. J. Physiol. Gastrointest Liver. Physiol. 2004, 287, G695–G705. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Shi, Y.; Tian, C.; Jiang, C.; Jin, H.; Chen, J.; Almasan, A.; Tang, H.; Chen, Q. Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 2004, 23, 1239–1247. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Pittala, S.; Mizrachi, D. VDAC1 and the TSPO: Expression, Interactions, and Associated Functions in Health and Disease States. Int. J. Mol. Sci. 2019, 20, 3348. [Google Scholar] [CrossRef]
- Levin, E.; Premkumar, A.; Veenman, L.; Kugler, W.; Leschiner, S.; Spanier, I.; Weisinger, G.; Lakomek, M.; Weizman, A.; Snyder, S.H.; et al. The peripheral-type benzodiazepine receptor and tumorigenicity: Isoquinoline binding protein (IBP) antisense knockdown in the C6 glioma cell line. Biochemistry 2005, 44, 9924–9935. [Google Scholar] [CrossRef]
- Veenman, L.; Levin, E.; Weisinger, G.; Leschiner, S.; Spanier, I.; Snyder, S.H.; Weizman, A.; Gavish, M. Peripheral-type benzodiazepine receptor density and in vitro tumorigenicity of glioma cell lines. Biochem. Pharmacol. 2004, 68, 689–698. [Google Scholar] [CrossRef]
- Veenman, L.; Papadopoulos, V.; Gavish, M. Channel-like functions of the 18-kDa translocator protein (TSPO): Regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 2007, 13, 2385–2405. [Google Scholar] [CrossRef] [PubMed]
- Veenman, L.; Shandalov, Y.; Gavish, M. VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J. Bioenerg. Biomembr. 2008, 40, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Gatliff, J.; Campanella, M. TSPO is a REDOX regulator of cell mitophagy. Biochem. Society Transact. 2015, 43, 543–552. [Google Scholar] [CrossRef]
- Gatliff, J.; East, D.; Crosby, J.; Abeti, R.; Harvey, R.; Craigen, W.; Parker, P.; Campanella, M. TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 2014, 10, 2279–2296. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Craigen, W.J.; Riley, D.J. Nek1 regulates cell death and mitochondrial membrane permeability through phosphorylation of VDAC1. Cell Cycle 2009, 8, 257–267. [Google Scholar] [CrossRef]
- Chen, Y.; Gaczynska, M.; Osmulski, P.; Polci, R.; Riley, D.J. Phosphorylation by Nek1 regulates opening and closing of voltage dependent anion channel 1. Biochem. Biophys. Res. Commun. 2010, 394, 798–803. [Google Scholar] [CrossRef]
- Zhang, X.; Bian, X.; Kong, J. The proapoptotic protein BNIP3 interacts with VDAC to induce mitochondrial release of endonuclease G. PLoS ONE 2014, 9, e113642. [Google Scholar] [CrossRef]
- Kusano, H.; Shimizu, S.; Koya, R.C.; Fujita, H.; Kamada, S.; Kuzumaki, N.; Tsujimoto, Y. Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene 2000, 19, 4807–4814. [Google Scholar] [CrossRef]
- Qiao, H.; McMillan, J.R. Gelsolin segment 5 inhibits HIV-induced T-cell apoptosis via Vpr-binding to VDAC. FEBS Lett. 2007, 581, 535–540. [Google Scholar] [CrossRef]
- Li, G.H.; Arora, P.D.; Chen, Y.; McCulloch, C.A.; Liu, P. Multifunctional roles of gelsolin in health and diseases. Med. Res. Rev. 2012, 32, 999–1025. [Google Scholar] [CrossRef]
- Miura, N.; Takemori, N.; Kikugawa, T.; Tanji, N.; Higashiyama, S.; Yokoyama, M. Adseverin: A novel cisplatin-resistant marker in the human bladder cancer cell line HT1376 identified by quantitative proteomic analysis. Mol. Oncol. 2012, 6, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef] [PubMed]
- Carre, M.; Andre, N.; Carles, G.; Borghi, H.; Brichese, L.; Briand, C.; Braguer, D. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J. Biol. Chem. 2002, 277, 33664–33669. [Google Scholar] [CrossRef] [PubMed]
- Puurand, M.; Tepp, K.; Timohhina, N.; Aid, J.; Shevchuk, I.; Chekulayev, V.; Kaambre, T. Tubulin betaII and betaIII Isoforms as the Regulators of VDAC Channel Permeability in Health and Disease. Cells 2019, 8, 239. [Google Scholar] [CrossRef]
- Rostovtseva, T.K.; Bezrukov, S.M. VDAC inhibition by tubulin and its physiological implications. Biochim. Biophys. Acta 2012, 1818, 1526–1535. [Google Scholar] [CrossRef]
- Saks, V.; Guzun, R.; Timohhina, N.; Tepp, K.; Varikmaa, M.; Monge, C.; Beraud, N.; Kaambre, T.; Kuznetsov, A.; Kadaja, L.; et al. Structure-function relationships in feedback regulation of energy fluxes in vivo in health and disease: Mitochondrial interactosome. Biochim. Biophys. Acta 2010, 1797, 678–697. [Google Scholar] [CrossRef]
- Linden, M.; Karlsson, G. Identification of porin as a binding site for MAP2. Biochem. Biophys. Res. Commun. 1996, 218, 833–836. [Google Scholar] [CrossRef]
- Schwarzer, C.; Barnikol-Watanabe, S.; Thinnes, F.P.; Hilschmann, N. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 2002, 34, 1059–1070. [Google Scholar] [CrossRef]
- Sasaki, S.; Yui, N.; Noda, Y. Actin directly interacts with different membrane channel proteins and influences channel activities: AQP2 as a model. Biochim. Biophys. Acta 2014, 1838, 514–520. [Google Scholar] [CrossRef]
- Sun, J.; Liao, J.K. Functional interaction of endothelial nitric oxide synthase with a voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2002, 99, 13108–13113. [Google Scholar] [CrossRef]
- Bergemalm, D.; Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brannstrom, T.; Rehnmark, A.; Marklund, S.L. Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J. Neurosci. 2006, 26, 4147–4154. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Shi, Y.; Furukawa, Y.; Zhai, H.; Fu, R.; Liu, E.; Gorrie, G.H.; Khan, M.S.; Hung, W.Y.; Bigio, E.H.; et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7142–7147. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Shteinfer-Kuzmine, A.; Argueti, S.; Gupta, R.; Shvil, N.; Abu-Hamad, S.; Gropper, Y.; Hoeber, J.; Magri, A.; Messina, A.; Kozlova, E.N.; et al. A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS. Front. Cell Neurosci. 2019, 13, 346. [Google Scholar] [CrossRef]
- Tan, W.; Naniche, N.; Bogush, A.; Pedrini, S.; Trotti, D.; Pasinelli, P. Small peptides against the mutant SOD1/Bcl-2 toxic mitochondrial complex restore mitochondrial function and cell viability in mutant SOD1-mediated ALS. J. Neurosci. 2013, 33, 11588–11598. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell. 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Guan, K.; Zheng, Z.; Song, T.; He, X.; Xu, C.; Zhang, Y.; Ma, S.; Wang, Y.; Xu, Q.; Cao, Y.; et al. MAVS regulates apoptotic cell death by decreasing K48-linked ubiquitination of voltage-dependent anion channel 1. Mol. Cell Biol. 2013, 33, 3137–3149. [Google Scholar] [CrossRef]
- Mitra, A.; Basak, T.; Datta, K.; Naskar, S.; Sengupta, S.; Sarkar, S. Role of alpha-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 2013, 4, e582. [Google Scholar] [CrossRef]
- Shen, J.; Du, T.; Wang, X.; Duan, C.; Gao, G.; Zhang, J.; Lu, L.; Yang, H. Alpha-Synuclein amino terminus regulates mitochondrial membrane permeability. Brain. Res. 2014, 1591, 14–26. [Google Scholar] [CrossRef]
- Jacobs, D.; Hoogerheide, D.P.; Rovini, A.; Jiang, Z.; Lee, J.C.; Rostovtseva, T.K.; Bezrukov, S.M. Probing Membrane Association of alpha-Synuclein Domains with VDAC Nanopore Reveals Unexpected Binding Pattern. Sci. Rep. 2019, 9, 4580. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, C.; Cai, Q.; Lu, Q.; Duan, C.; Zhu, Y.; Yang, H. Voltage-dependent anion channel involved in the alpha-synuclein-induced dopaminergic neuron toxicity in rats. Acta Biochim. Biophys. Sin (Shanghai) 2013, 45, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Chernyshov, V.; Kryukova, E.; Tsetlin, V.; Komisarenko, S.; Skok, M. Mitochondria express alpha7 nicotinic acetylcholine receptors to regulate Ca2+ accumulation and cytochrome c release: Study on isolated mitochondria. PLoS ONE 2012, 7, e31361. [Google Scholar] [CrossRef]
- Skok, M.; Gergalova, G.; Lykhmus, O.; Kalashnyk, O.; Koval, L.; Uspenska, K. Nicotinic acetylcholine receptors in mitochondria: Subunit composition, function and signaling. Neurotransmitter 2016, 3, e1290. [Google Scholar] [CrossRef]
- Breydo, L.; Wu, J.W.; Uversky, V.N. Alpha-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 261–285. [Google Scholar] [CrossRef]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. Alpha-Synuclein Shows High Affinity Interaction with Voltage-dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 2015, 290, 18467–18477. [Google Scholar] [CrossRef]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005, 1, e4. [Google Scholar] [CrossRef]
- Rahmani, Z.; Huh, K.W.; Lasher, R.; Siddiqui, A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef]
- Holla, R.P.; Ahmad, I.; Ahmad, Z.; Jameel, S. Molecular virology of hepatitis E virus. Semin. Liver. Dis. 2013, 33, 3–14. [Google Scholar] [CrossRef]
- Deniaud, A.; Brenner, C.; Kroemer, G. Mitochondrial membrane permeabilization by HIV-1 Vpr. Mitochondrion 2004, 4, 223–233. [Google Scholar] [CrossRef]
- Jitobaom, K.; Tongluan, N.; Smith, D.R. Involvement of voltage-dependent anion channel (VDAC) in dengue infection. Sci. Rep. 2016, 6, 35753. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, Z.; Xu, Z.; Wang, B.; Li, X.; Cao, H.; Wang, Y.; Zheng, S.J. The association of receptor of activated protein kinase C 1(RACK1) with infectious bursal disease virus viral protein VP5 and voltage-dependent anion channel 2 (VDAC2) inhibits apoptosis and enhances viral replication. J. Biol. Chem. 2015, 290, 8500–8510. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Xue, Y.; Li, X.; Cao, H.; Zheng, S.J. Critical role for voltage-dependent anion channel 2 in infectious bursal disease virus-induced apoptosis in host cells via interaction with VP5. J. Virol. 2012, 86, 1328–1338. [Google Scholar] [CrossRef]
- Han, C.; Zeng, X.; Yao, S.; Gao, L.; Zhang, L.; Qi, X.; Duan, Y.; Yang, B.; Gao, Y.; Liu, C.; et al. Voltage-Dependent Anion Channel 1 Interacts with Ribonucleoprotein Complexes To Enhance Infectious Bursal Disease Virus Polymerase Activity. J. Virol. 2017, 91, e00584-17. [Google Scholar] [CrossRef]
- Fongsaran, C.; Phaonakrop, N.; Roytrakul, S.; Thepparit, C.; Kuadkitkan, A.; Smith, D.R. Voltage dependent anion channel is redistributed during Japanese encephalitis virus infection of insect cells. Sci. World J. 2014, 2014, 976015. [Google Scholar] [CrossRef]
- Delisle, L.; Fuhrmann, M.; Quere, C.; Pauletto, M.; Pichereau, V.; Pernet, F.; Corporeau, C. The Voltage-Dependent Anion Channel (VDAC) of Pacific Oysters Crassostrea gigas Is Upaccumulated During Infection by the Ostreid Herpesvirus-1 (OsHV-1): An Indicator of the Warburg Effect. Mar. Biotechnol. (NY) 2018, 20, 87–97. [Google Scholar] [CrossRef]
- Borgne-Sanchez, A.; Dupont, S.; Langonne, A.; Baux, L.; Lecoeur, H.; Chauvier, D.; Lassalle, M.; Deas, O.; Briere, J.J.; Brabant, M.; et al. Targeted Vpr-derived peptides reach mitochondria to induce apoptosis of alphaVbeta3-expressing endothelial cells. Cell Death Differ. 2007, 14, 422–435. [Google Scholar] [CrossRef]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef]
- Shirakata, Y.; Koike, K. Hepatitis B virus X protein induces cell death by causing loss of mitochondrial membrane potential. J. Biol. Chem. 2003, 278, 22071–22078. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Shelton, L.M. Cancer as a metabolic disease. Nutr. Metab. (Lond.) 2010, 7, 7. [Google Scholar] [CrossRef]
- Seyfried, T.N. Cancer as a mitochondrial metabolic disease. Front. Cell Dev. Biol. 2015, 3, 43. [Google Scholar] [CrossRef]
- Pittala, S.; Krelin, Y.; Shoshan-Barmatz, V. Targeting Liver Cancer and Associated Pathologies in Mice with a Mitochondrial VDAC1-Based Peptide. Neoplasia 2018, 20, 594–609. [Google Scholar] [CrossRef]
- Pedersen, P.L. Voltage dependent anion channels (VDACs): A brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J. Bioenerg. Biomembr. 2008, 40, 123–126. [Google Scholar] [CrossRef]
- Shi, Y.; Jiang, C.; Chen, Q.; Tang, H. One-step on-column affinity refolding purification and functional analysis of recombinant human VDAC1. Biochem. Biophys. Res. Commun. 2003, 303, 475–482. [Google Scholar] [CrossRef]
- Li, Q.Q.; Zhang, L.; Wan, H.Y.; Liu, M.; Li, X.; Tang, H. CREB1-driven expression of miR-320a promotes mitophagy by down-regulating VDAC1 expression during serum starvation in cervical cancer cells. Oncotarget 2015, 6, 34924–34940. [Google Scholar] [CrossRef]
- Zhang, G.; Jiang, G.; Wang, C.; Zhong, K.; Zhang, J.; Xue, Q.; Li, X.; Jin, H.; Li, B. Decreased expression of microRNA-320a promotes proliferation and invasion of non-small cell lung cancer cells by increasing VDAC1 expression. Oncotarget 2016, 7, 49470–49480. [Google Scholar] [CrossRef]
- Wang, F.; Qiang, Y.; Zhu, L.; Jiang, Y.; Wang, Y.; Shao, X.; Yin, L.; Chen, J.; Chen, Z. MicroRNA-7 downregulates the oncogene VDAC1 to influence hepatocellular carcinoma proliferation and metastasis. Tumour. Biol. 2016, 37, 10235–10246. [Google Scholar] [CrossRef]
- Chaudhuri, A.D.; Choi, D.C.; Kabaria, S.; Tran, A.; Junn, E. MicroRNA-7 Regulates the Function of Mitochondrial Permeability Transition Pore by Targeting VDAC1 Expression. J. Biol. Chem. 2016, 291, 6483–6493. [Google Scholar] [CrossRef]
- Yuan, J.; Yankner, B.A. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar] [CrossRef]
- Friedlander, R.M. Apoptosis and caspases in neurodegenerative diseases. N. Eng. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef]
- Gervais, F.G.; Xu, D.; Robertson, G.S.; Vaillancourt, J.P.; Zhu, Y.; Huang, J.; LeBlanc, A.; Rigby, S.M.; Shearman, M.S.; Clarke, E.E.; et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999, 97, 395–406. [Google Scholar] [CrossRef]
- Li, M.; Ona, V.O.; Guegan, C.; Chen, M.; Jackson-Lewis, V.; Andrews, L.J.; Olszewski, A.J.; Stieg, P.E.; Lee, J.P.; Przedborski, S.; et al. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 2000, 288, 335–339. [Google Scholar] [CrossRef]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef]
- Petrozzi, L.; Ricci, G.; Giglioli, N.J.; Siciliano, G.; Mancuso, M. Mitochondria and neurodegeneration. Biosci. Rep. 2007, 27, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42 (Suppl. S3), S125–S152. [Google Scholar] [CrossRef]
- Ferrer, I. Altered mitochondria, energy metabolism, voltage-dependent anion channel, and lipid rafts converge to exhaust neurons in Alzheimer’s disease. J. Bioenerg. Biomembr. 2009, 41, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer’s disease? Biochim. Biophys. Acta 2013, 1832, 67–75. [Google Scholar] [CrossRef]
- Yoo, B.C.; Fountoulakis, M.; Cairns, N.; Lubec, G. Changes of voltage-dependent anion-selective channel proteins VDAC1 and VDAC2 brain levels in patients with Alzheimer’s disease and Down syndrome. Electrophoresis 2001, 22, 172–179. [Google Scholar] [CrossRef]
- Fukada, K.; Zhang, F.; Vien, A.; Cashman, N.R.; Zhu, H. Mitochondrial proteomic analysis of a cell line model of familial amyotrophic lateral sclerosis. Mol. Cell Proteom. 2004, 3, 1211–1223. [Google Scholar] [CrossRef]
- Kielar, C.; Wishart, T.M.; Palmer, A.; Dihanich, S.; Wong, A.M.; Macauley, S.L.; Chan, C.H.; Sands, M.S.; Pearce, D.A.; Cooper, J.D.; et al. Molecular correlates of axonal and synaptic pathology in mouse models of Batten disease. Hum. Mol. Genet. 2009, 18, 4066–4080. [Google Scholar] [CrossRef] [PubMed]
- Bueno, K.O.; de Souza Resende, L.; Ribeiro, A.F.; Dos Santos, D.M.; Goncalves, E.C.; Vigil, F.A.; de Oliveira Silva, I.F.; Ferreira, L.F.; de Castro Pimenta, A.M.; Ribeiro, A.M. Spatial cognitive deficits in an animal model of Wernicke-Korsakoff syndrome are related to changes in thalamic VDAC protein concentrations. Neuroscience 2015, 294, 29–37. [Google Scholar] [CrossRef]
- Lezi, E.; Swerdlow, R.H. Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; Cardoso, S.M.; Swerdlow, R.H. Mitochondrial abnormalities in Alzheimer’s disease: Possible targets for therapeutic intervention. Adv. Pharmacol. 2012, 64, 83–126. [Google Scholar] [CrossRef]
- Colurso, G.J.; Nilson, J.E.; Vervoort, L.G. Quantitative assessment of DNA fragmentation and beta-amyloid deposition in insular cortex and midfrontal gyrus from patients with Alzheimer’s disease. Life Sci. 2003, 73, 1795–1803. [Google Scholar] [CrossRef]
- Magri, A.; Belfiore, S.; Reina, M.F.; Tomasello, M.C.; Di Rosa, F.; Guarino, L.; Leggio, V.; De Pinto, V.; Messina, A.; Hexokinase, I. N-terminal based peptide prevents the VDAC1-SOD1 G93A interaction and re-establishes ALS cell viability. Sci. Rep. 2016, 6, 34802. [Google Scholar] [CrossRef]
- Sasaki, K.; Donthamsetty, R.; Heldak, M.; Cho, Y.E.; Scott, B.T.; Makino, A. VDAC: Old protein with new roles in diabetes. Am. J. Physiol. Cell Physiol. 2012, 303, C1055–C1060. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, H.; Chen, G.; Feng, Y.; Zou, J.; Liu, M.; Liu, K.; Wang, N.; Zhang, H.; Wang, K.; et al. WDR26/MIP2 interacts with VDAC1 and regulates VDAC1 expression levels in H9c2 cells. Free Radic. Biol. Med. 2018, 117, 58–65. [Google Scholar] [CrossRef]
- Liao, Z.; Liu, D.; Tang, L.; Yin, D.; Yin, S.; Lai, S.; Yao, J.; He, M. Long-term oral resveratrol intake provides nutritional preconditioning against myocardial ischemia/reperfusion injury: Involvement of VDAC1 downregulation. Mol. Nutr. Food Res. 2015, 59, 454–464. [Google Scholar] [CrossRef]
- Lim, D.S.; Roberts, R.; Marian, A.J. Expression profiling of cardiac genes in human hypertrophic cardiomyopathy: Insight into the pathogenesis of phenotypes. J. Am. Coll. Cardiol. 2001, 38, 1175–1180. [Google Scholar] [CrossRef]
- Schwertz, H.; Carter, J.M.; Abdudureheman, M.; Russ, M.; Buerke, U.; Schlitt, A.; Muller-Werdan, U.; Prondzinsky, R.; Werdan, K.; Buerke, M. Myocardial ischemia/reperfusion causes VDAC phosphorylation which is reduced by cardioprotection with a p38 MAP kinase inhibitor. Proteomics 2007, 7, 4579–4588. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Xie, Y.; Meng, Y.; Ma, W.; Tong, Z.; Yang, X.; Lai, S.; Zhou, Y.; He, M.; Liao, Z. Resveratrol protects cardiomyocytes against anoxia/reoxygenation via dephosphorylation of VDAC1 by Akt-GSK3 beta pathway. Eur. J. Pharmacol. 2019, 843, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Xie, Y.; He, M.; Ma, W.; Zhou, Y.; Lai, S.; Meng, Y.; Liao, Z. VDAC1 deacetylation is involved in the protective effects of resveratrol against mitochondria-mediated apoptosis in cardiomyocytes subjected to anoxia/reoxygenation injury. Biomed Pharmacother. 2017, 95, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Xu, Y.; Heisner, J.S.; Sun, J.; Stowe, D.F.; Kwok, W.M.; Camara, A.K.S. Peroxynitrite nitrates adenine nucleotide translocase and voltage-dependent anion channel 1 and alters their interactions and association with hexokinase II in mitochondria. Mitochondrion 2019, 46, 380–392. [Google Scholar] [CrossRef]
- Zeng, F.; Wen, W.; Cui, W.; Zheng, W.; Liu, Y.; Sun, X.; Hou, N.; Ma, D.; Yuan, Y.; Shi, H.; et al. Central role of RIPK1-VDAC1 pathway on cardiac impairment in a non-human primate model of rheumatoid arthritis. J. Mol. Cell Cardiol. 2018, 125, 50–60. [Google Scholar] [CrossRef]
- Paschon, V.; Morena, B.C.; Correia, F.F.; Beltrame, G.R.; Dos Santos, G.B.; Cristante, A.F.; Kihara, A.H. VDAC1 is essential for neurite maintenance and the inhibition of its oligomerization protects spinal cord from demyelination and facilitates locomotor function recovery after spinal cord injury. Sci. Rep. 2019, 9, 14063. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol. Aging. 2001, 22, 187–194. [Google Scholar] [CrossRef]
- Weeber, E.J.; Levy, M.; Sampson, M.J.; Anflous, K.; Armstrong, D.L.; Brown, S.E.; Sweatt, J.D.; Craigen, W.J. The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J. Biol. Chem. 2002, 277, 18891–18897. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Rothstein, J.D. From Charcot to Lou Gehrig: Deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef]
- Carri, M.T.; Cozzolino, M. SOD1 and mitochondria in ALS: A dangerous liaison. J. Bioenerg. Biomembr. 2011, 43, 593–599. [Google Scholar] [CrossRef]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [CrossRef] [PubMed]
- Magrane, J.; Sahawneh, M.A.; Przedborski, S.; Estevez, A.G.; Manfredi, G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J. Neurosci. 2012, 32, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Wei, Y.M.; Zhang, J.Y.; Gal, J.; Zhu, H.N. Mitochondrial Dysfunction is a Converging Point of Multiple Pathological Pathways in Amyotrophic Lateral Sclerosis. J. Alzheimers Dis. 2010, 20, S311–S324. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Kahn, J.; Leyton-Jaimes, M.F.; Rosenblatt, J.; Israelson, A. Misfolded SOD1 Accumulation and Mitochondrial Association Contribute to the Selective Vulnerability of Motor Neurons in Familial ALS: Correlation to Human Disease. ACS Chem. Neurosci. 2017, 8, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.G.; Shaw, J. Global and societal implications of the diabetes epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef]
- Li, X.; Wang, H.; Yao, B.; Xu, W.; Chen, J.; Zhou, X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci. Rep. 2016, 6, 36340. [Google Scholar] [CrossRef]
- Liu, Y.; Yin, H.; Zhao, M.; Lu, Q. TLR2 and TLR4 in autoimmune diseases: A comprehensive review. Clin. Rev. Allergy Immunol. 2014, 47, 136–147. [Google Scholar] [CrossRef]
- Libby, P. Inflammatory mechanisms: The molecular basis of inflammation and disease. Nutr. Rev. 2007, 65 Pt 2, S140–S146. [Google Scholar] [CrossRef]
- Yang, M.; Li, K.; Sun, J.; Stowe, D.; Tajkhorshid, E.; Kwok, W.; Camara, A.K.S. Knockout of VDAC1 in H9c2 Cells Promotes tBHP-induced Cell Apoptosis Through Decreased Mitochondrial HK II Binding and Enhanced Glycolytic Stress. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Kar, D.; Bandyopadhyay, A. Targeting Peroxisome Proliferator Activated Receptor alpha (PPAR alpha) for the Prevention of Mitochondrial Impairment and Hypertrophy in Cardiomyocytes. Cell Physiol. Biochem. 2018, 49, 245–259. [Google Scholar] [CrossRef]
- Law, K.; Brunt, E.M. Nonalcoholic fatty liver disease. Clin. Liver. Dis. 2010, 14, 591–604. [Google Scholar] [CrossRef]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. S1), S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Holmuhamedov, E.L.; Czerny, C.; Beeson, C.C.; Lemasters, J.J. Ethanol suppresses ureagenesis in rat hepatocytes: Role of acetaldehyde. J. Biol. Chem. 2012, 287, 7692–7700. [Google Scholar] [CrossRef] [PubMed]
- Turkaly, P.; Kerner, J.; Hoppel, C. A 22 kDa polyanion inhibits carnitine-dependent fatty acid oxidation in rat liver mitochondria. FEBS Lett. 1999, 460, 241–245. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Weinberg, J.M. Recent advances in the pathophysiology of ischemic acute renal failure. J. Am. Soc. Nephrol. 2003, 14, 2199–2210. [Google Scholar] [CrossRef]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Renal. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef]
- Ralto, K.M.; Parikh, S.M. Mitochondria in Acute Kidney Injury. Semin Nephrol. 2016, 36, 8–16. [Google Scholar] [CrossRef]
- Nowak, G.; Megyesi, J.; Craigen, W.J. Deletion of VDAC1 Hinders Recovery of Mitochondrial and Renal Functions After Acute Kidney Injury. Biomolecules 2020, 10, 585. [Google Scholar] [CrossRef]
- Jeong, J.J.; Park, N.; Kwon, Y.J.; Ye, D.J.; Moon, A.; Chun, Y.J. Role of annexin A5 in cisplatin-induced toxicity in renal cells: Molecular mechanism of apoptosis. J. Biol. Chem. 2014, 289, 2469–2481. [Google Scholar] [CrossRef]
- Sekhon, L.H.; Fehlings, M.G. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine (Phila Pa 1976) 2001, 26 (Suppl. S24), S2–S12. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Paul, A.; Krelin, Y.; Shteinfer-Kuzmine, A.; Shoshan-Barmatz, V. Mitochondrial VDAC1 Silencing Leads to Metabolic Rewiring and the Reprogramming of Tumour Cells into Advanced Differentiated States. Cancers (Basel) 2018, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Stern, O.; Pittala, S.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept. Cells 2019, 8, 1330. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Takehara, T.; Liu, X.; Fujimoto, J.; Friedman, S.L.; Takahashi, H. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 2001, 34, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Yang, X.; Pater, A.; Tang, S.C. Resistance to apoptosis is correlated with the reduced caspase-3 activation and enhanced expression of antiapoptotic proteins in human cervical multidrug-resistant cells. Biochem. Biophys. Res. Commun. 2000, 270, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Grobholz, R.; Zentgraf, H.; Kohrmann, K.U.; Bleyl, U. Bax, Bcl-2, fas and Fas-L antigen expression in human seminoma: Correlation with the apoptotic index. Apmis 2002, 110, 724–732. [Google Scholar] [CrossRef]
- Krajewska, M.; Moss, S.F.; Krajewski, S.; Song, K.; Holt, P.R.; Reed, J.C. Elevated expression of Bcl-X and reduced Bak in primary colorectal adenocarcinomas. Cancer Res. 1996, 56, 2422–2427. [Google Scholar] [PubMed]
- Pedersen, P.L. Warburg, me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J. Bioenerg. Biomembr. 2007, 39, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase-2 bound to mitochondria: Cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin. Cancer Biol. 2009, 19, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef]
- Shteinfer-Kuzmine, A.; Amsalem, Z.; Arif, T.; Zooravlov, A.; Shoshan-Barmatz, V. Selective induction of cancer cell death by VDAC1-based peptides and their potential use in cancer therapy. Mol. Oncol. 2018, 12, 1077–1103. [Google Scholar] [CrossRef]






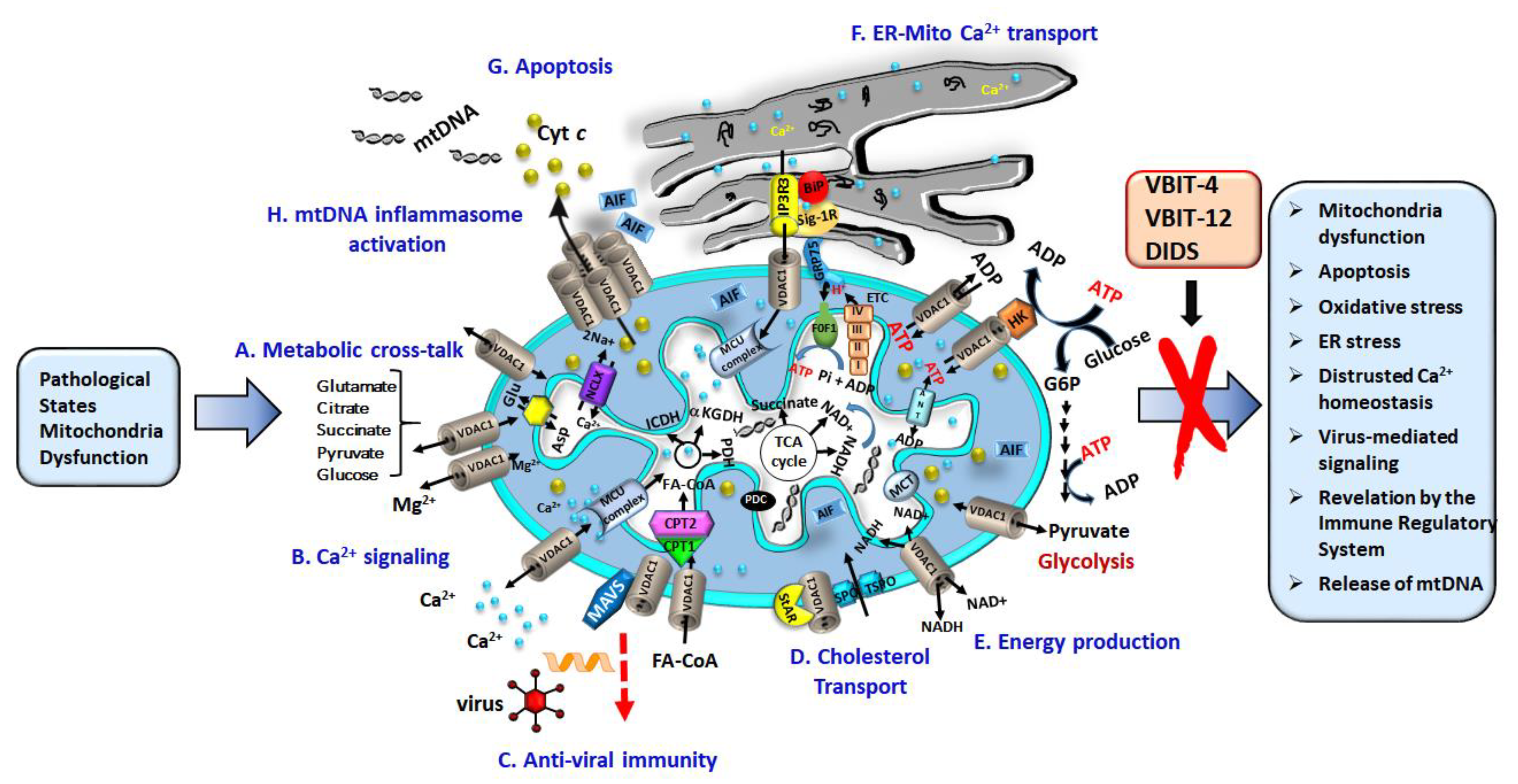{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound | Ref. | No. | Compound | Ref. |
|---|---|---|---|---|---|
| 1 | Prednisolone1 | [168] | 11 | Etoposide | [51] |
| 2 | Cisplatin | [51,169] | 12 | Selenite | [51] |
| 3 | Arsenic trioxide (As2O3) | [51] | 13 | A23187 | [51] |
| 4 | Arbutin (hydroquinone -O-β-D glucopyranoside) | [167,172] | 14 | Somatostatin | [65,173] |
| 5 | Hepatitis E virus ORF3 protein | [174] | 15 | H2O2 | [51] |
| 6 | UV irradiation | [175] | 16 | Thapsigargin | [51] |
| 7 | Mechlorethamine and its derivative, melphalan | [176] | 17 | Ionomycin | [51] |
| 8 | Vacuolating cytotoxin | [177] | 18 | Vorinostat | [51,178] |
| 9 | Quinocetone | [179] | 19 | DSS | (unpublished data) |
| 10 | Endostatin | [151] | 20 | Pyrroloquinoline quinone (PQQ) | [171] |
| 21 | Plasminogen kringle 5 (PK5) | [74] |
| Virus | Virus Type | Viral Protein | Action on VDAC1 | Reference |
|---|---|---|---|---|
| HIV-1 Human Immunodeficiency Virus type 1 | Single-stranded positive RNA | Vpr | Interacts with VDAC1 and induces apoptosis | [239,278] |
| DENV Dengue Virus | Single-stranded positive RNA | DENV-E | Interacts with GRP78, a chaperone interacting with VDAC1. Down-regulation of VDAC reduced DENV protein expression | [272] |
| Influenza A Virus | Single-stranded negative-sense RNA | PB1-F2 | Induces apoptosis via interaction with VDAC1 | [268] |
| IBDV Infectious Bursal Disease Virus | Double-stranded RNA | VP5 | Induces apoptosis in DF-1 cells via interaction with VDAC1 | [53,273,274,279] |
| HBV Hepatitis B Virus | DNA | HBx | HBx interaction with VDAC1 to form a hexamer, causing mitochondria dysfunction | [269,280] |
| HEV Hepatitis E Virus | Single-stranded positive sense RNA | ORF3 | Expression of ORF3 increases VDAC1 levels, siRNA against ORF3 reduces VDAC1 levels | [174,271] |
| Japanese Encephalitis Virus | Single-stranded, positive-sense RNA | JEV E | JEV E protein interacts with VDAC. Upon JEV infection, VDAC1 was co-localized with the ER protein GRP78 (HSP-70) | [276] |
| OsHV-1 Ostreid Herpes Virus-1 | DNA | Increases VDAC1 expression at the mRNA and protein levels | [277] |
| Disease | VDAC1 State | Function | Reference |
|---|---|---|---|
| Cancer | Overexpressed | Increased cancer cell metabolic activity | [3,15,85,86,99,196,283] |
| Alzheimer’s disease | Overexpressed | Neuronal cell death | [75,76,77] |
| ALS—Amyotrophic lateral sclerosis | Interacted with mutated SOD | Enhanced cell survival | [254,255,306] |
| Type 2 diabetes | Overexpressed | Impaired generation of cellular ATP | [80,81,84,307] |
| Systemic lupus erythematosus | Overexpressed | Mediated release of mtDNA and triggered type-Ι interferon responses | [87] |
| Cardiovascular diseases | Overexpressed | Cardiomyocyte cell death | [90,308,309,310,311,312,313,314] |
| Non-alcoholic fatty liver disease (NAFLD) | Overexpressed | Mediated transport of fatty acids across the OMM | [88,105,107] |
| Rheumatoid arthritis | Overexpressed | Cardiac cell death and functional impairment | [315], Figure 5 |
| Acute kidney injury | Deletion of VDAC1 hinders recovery of mitochondrial and renal functions | Required for the recovery of renal mitochondrial function | [315] |
| Spinal cord injury | Overexpressed | Oligomerization and apoptosis | [316] |
| Peptide | Sequence | No. of AA | Molecular Mass, Da |
|---|---|---|---|
| Tf-D-LP4 | HAIYPRHSWTWE-199-KKLETAVNLAWTA GNSN-216-KWTWK | 34 | 4111 |
| R-Tf-D-LP4 | KWTWK-216-NSNGATWALNVATELKK-199-EWTWSHRPYIAH | 34 | 4111 |
| N-Ter | 1-MAVPPTYADLGKSARDVFTKGYGFGL-26- | 26 | 2762 |
| N-Ter-Antp | 1-MAVPPTYADLGKSARDVFTKGYGFGL-26-RQIKIWFQNRRMKWKK | 42 | 4990 |
| D-Δ(1-14)N-Ter-Antp | 15-RDVFTKGYGFGL-26-RQIKIWFQNRRMKWKK | 28 | 3588 |
| ΔN-Ter Δ(21-26)-Antp | 1-MAVPPTYADLGKSARDVFTK-20-RQIKIWFQNRRMKWKK | 36 | 4396 |
| Δ(1-4)N-Ter Δ(21-26)-Antp | 5-PTYADLGKSARDVFTK-20-RQIKIWFQNRRMKWKK | 32 | 3997 |
| Δ(1-9)N-Ter Δ(21-26)-Antp | 10-LGKSARDVFTK-20-RQIKIWFQNRRMKWKK | 27 | 3450 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. https://doi.org/10.3390/biom10111485
Shoshan-Barmatz V, Shteinfer-Kuzmine A, Verma A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules. 2020; 10(11):1485. https://doi.org/10.3390/biom10111485
Chicago/Turabian StyleShoshan-Barmatz, Varda, Anna Shteinfer-Kuzmine, and Ankit Verma. 2020. "VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases" Biomolecules 10, no. 11: 1485. https://doi.org/10.3390/biom10111485
APA StyleShoshan-Barmatz, V., Shteinfer-Kuzmine, A., & Verma, A. (2020). VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules, 10(11), 1485. https://doi.org/10.3390/biom10111485