Metabolic Effects of Selective Deletion of Group VIA Phospholipase A2 from Macrophages or Pancreatic Islet Beta-Cells



Abstract
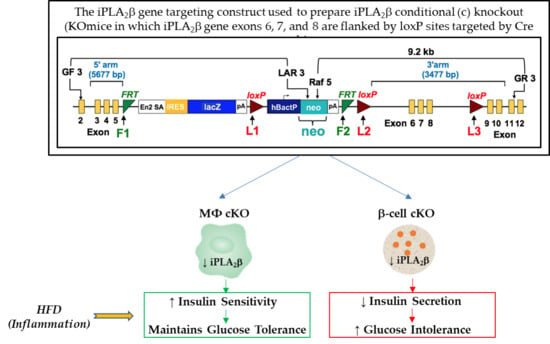
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
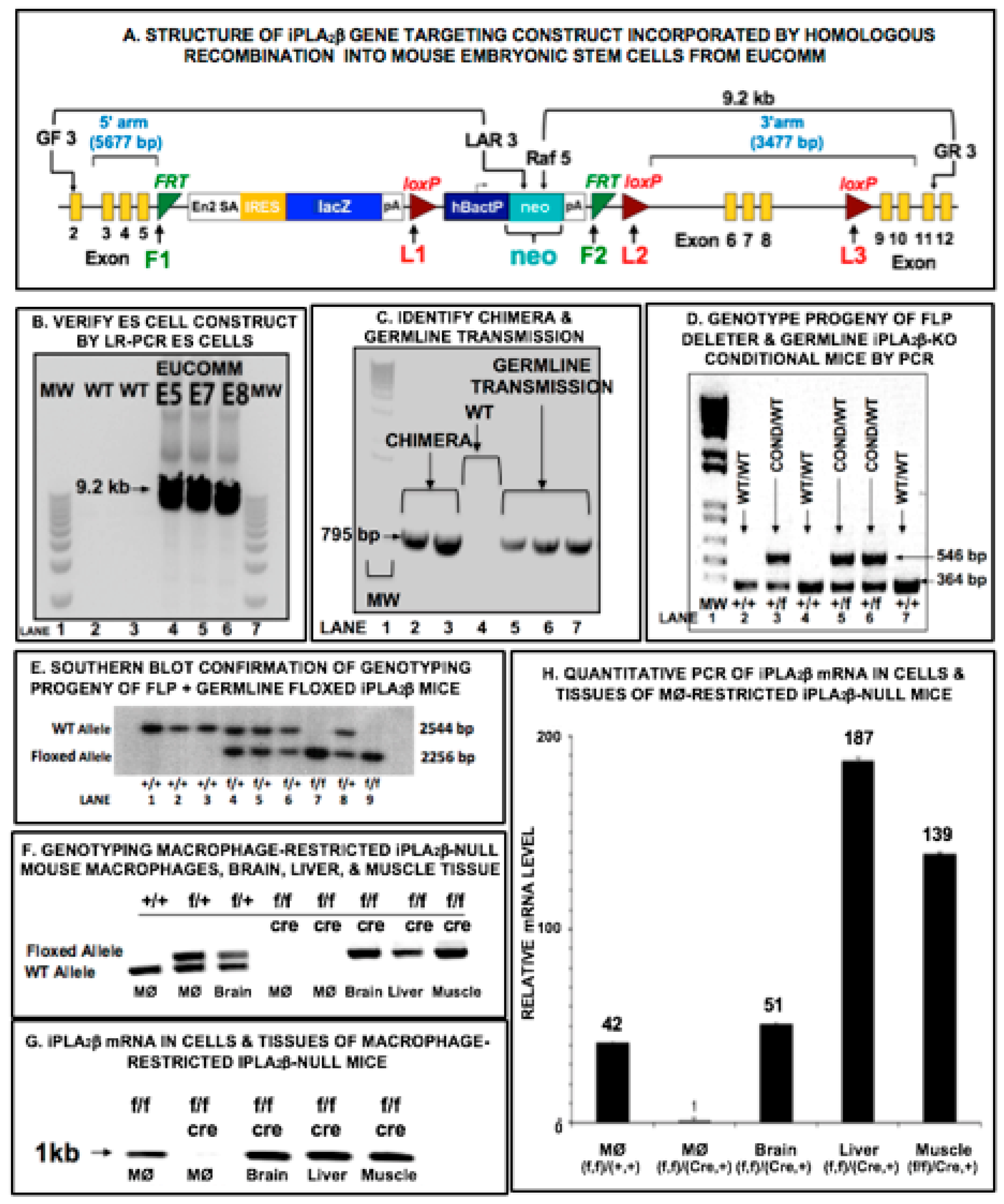
2.2. Preparation of Mice with Selective Deletion of iPLA2β from Restricted Cell Lineages
2.3. Analyses of iPLA2β mRNA in Mouse Tissues
2.4. High-Fat Dietary Intervention Studies
2.5. Blood Glucose and Insulin Concentrations
2.6. Glucose and Insulin Tolerance Tests
2.7. Area Under the Curve (AUC) Calculations for Glucose Tolerance Tests (GTTs)
2.8. Insulin Secretion In Vivo
2.9. Pancreatic Islet Isolation
2.10. Insulin Secretion Ex Vivo from Isolated Pancreatic Islets in Static Incubations
2.11. Other Analytical Procedures
2.12. Statistical Methods
3. Results
3.1. Mouse Genotype Characterization
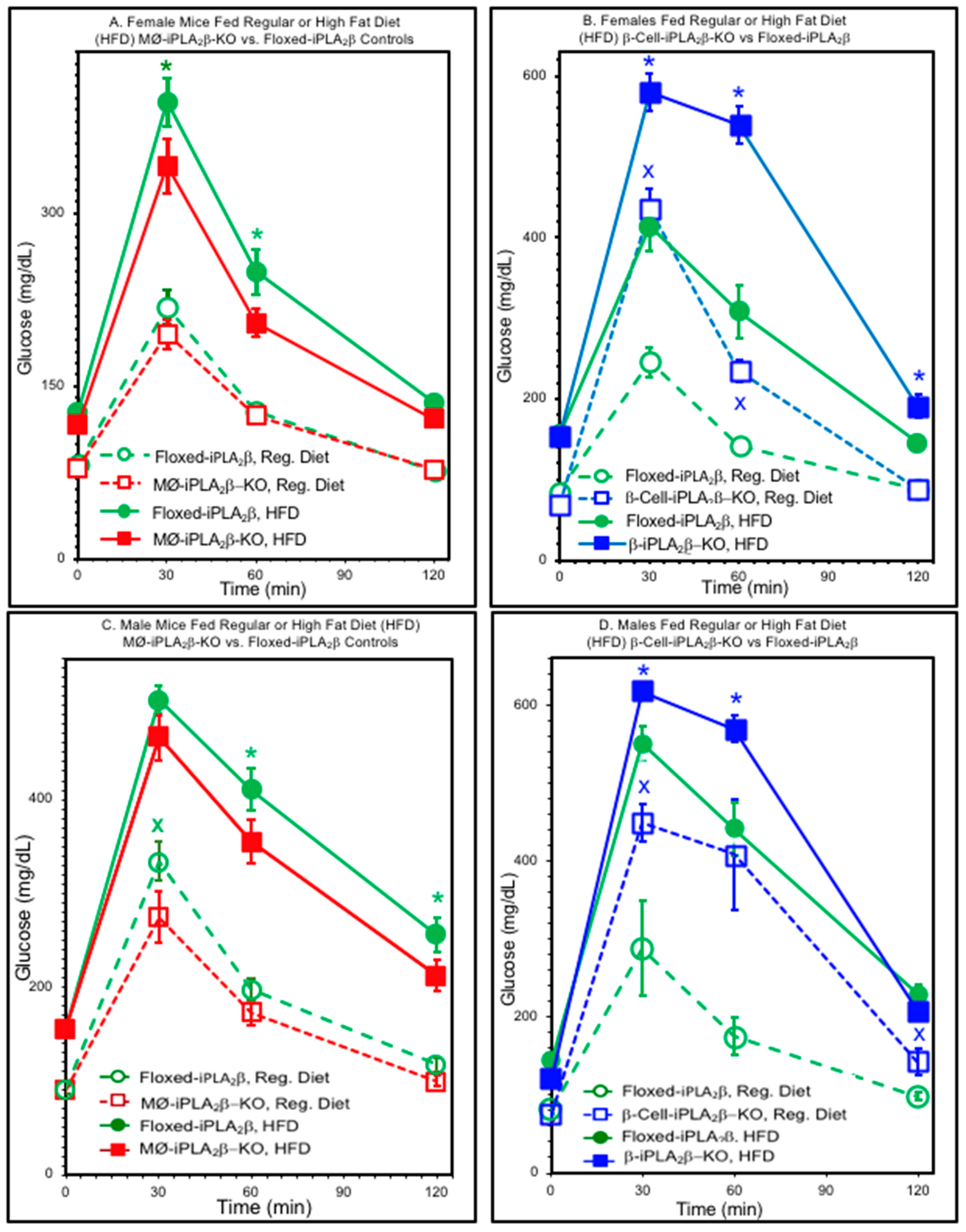
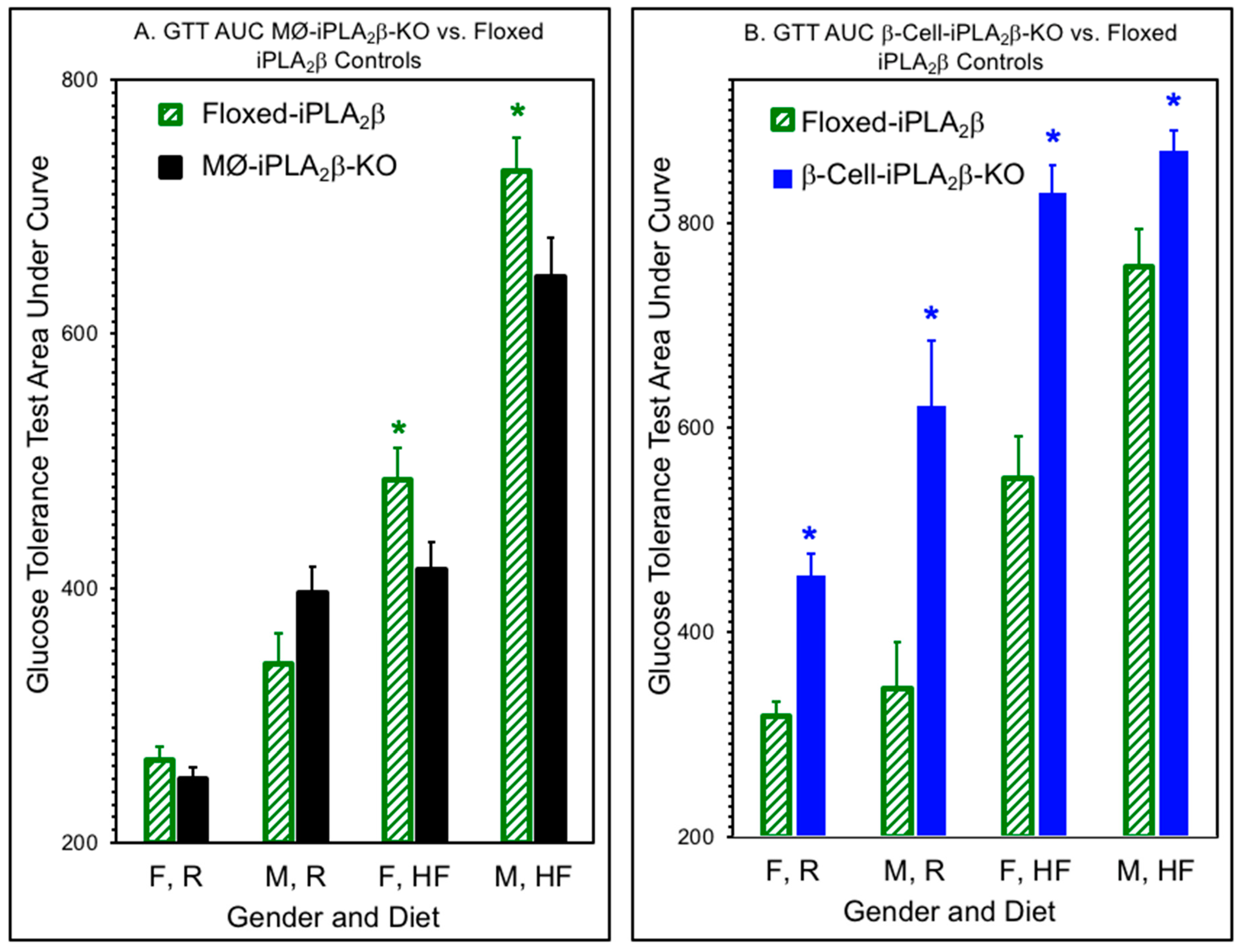
3.2. Glucose Tolerance Tests
3.3. Ex Vivo Insulin Secretion from Isolated Pancreatic Islets
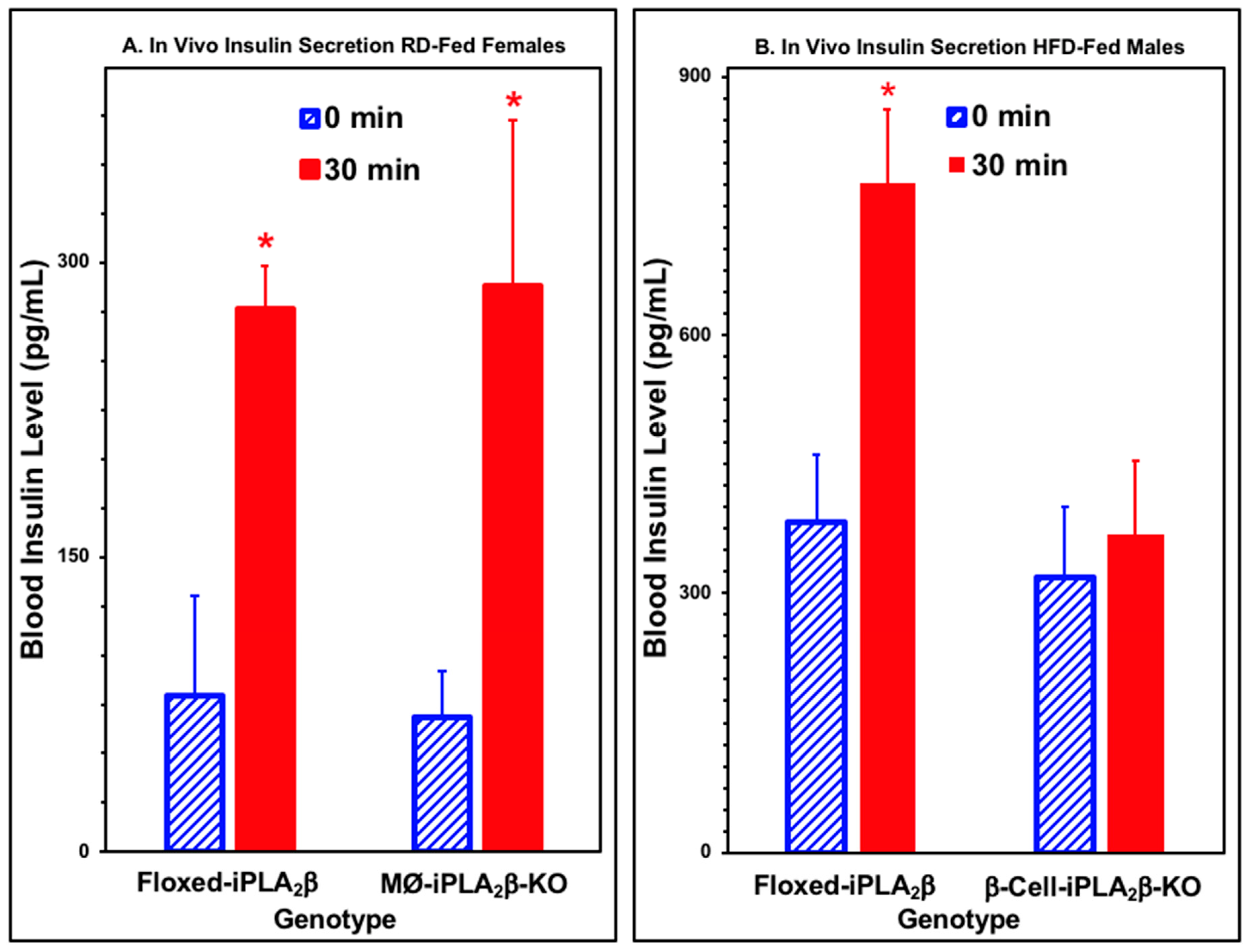
3.4. In Vivo Insulin Secretion in Mice After Intraperitoneal Glucose Administration
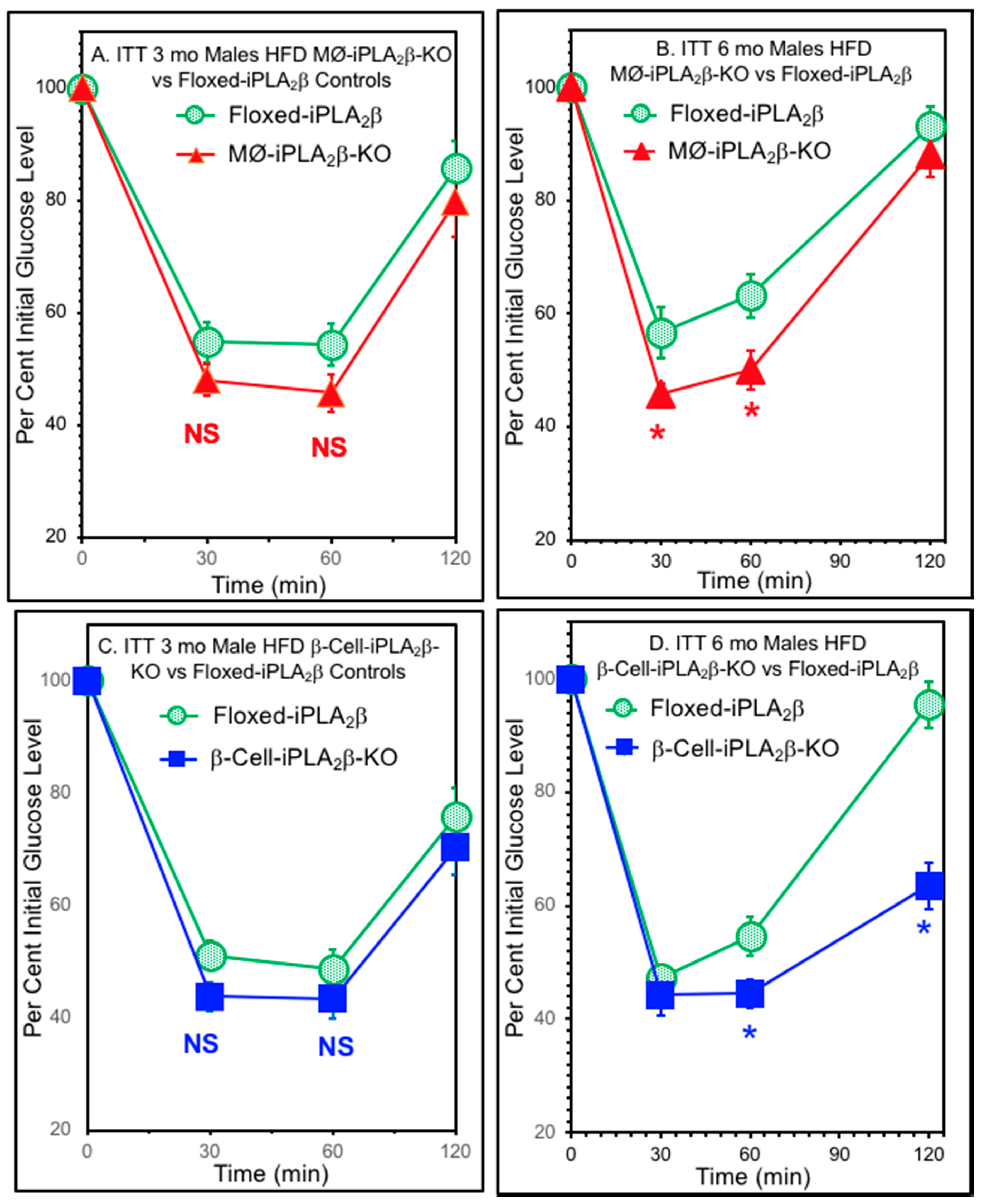
3.5. Insulin Tolerance Tests
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mouchlis, V.D.; Dennis, E.A. Phospholipase A2 catalysis and lipid mediator lipidomics. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Turk, J.; White, T.D.; Nelson, A.J.; Lei, X.; Ramanadham, S. iPLA2beta and its role in male fertility, neurological disorders, metabolic disorders, and inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Balboa, M.A.; Balsinde, J.; Jones, S.S.; Dennis, E.A. Identity between the Ca2+-independent phospholipase A2 enzymes from P388D1 macrophages and Chinese hamster ovary cells. J. Biol. Chem. 1997, 272, 8576–8580. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Ramanadham, S.; Kempe, K.; Chi, X.S.; Ladenson, J.; Turk, J. Pancreatic islets express a Ca2+-independent phospholipase A2 enzyme that contains a repeated structural motif homologous to the integral membrane protein binding domain of ankyrin. J. Biol. Chem. 1997, 272, 11118–11127. [Google Scholar] [CrossRef]
- Tang, J.; Kriz, R.W.; Wolfman, N.; Shaffer, M.; Seehra, J.; Jones, S.S. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J. Biol. Chem. 1997, 272, 8567–8575. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, D.J.; Jenkins, C.M.; Gross, R.W. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A2. J. Biol. Chem. 2000, 275, 9937–9945. [Google Scholar] [CrossRef] [PubMed]
- Rydel, T.J.; Williams, J.M.; Krieger, E.; Moshiri, F.; Stallings, W.C.; Brown, S.M.; Pershing, J.C.; Purcell, J.P.; Alibhai, M.F. The crystal structure, mutagenesis, and activity studies reveal that patatin is a lipid acyl hydrolase with a Ser-Asp catalytic dyad. Biochemistry 2003, 42, 6696–6708. [Google Scholar] [CrossRef]
- Wilson, P.A.; Gardner, S.D.; Lambie, N.M.; Commans, S.A.; Crowther, D.J. Characterization of the human patatin-like phospholipase family. J. Lipid Res. 2006, 47, 1940–1949. [Google Scholar] [CrossRef]
- Kienesberger, P.C.; Oberer, M.; Lass, A.; Zechner, R. Mammalian patatin domain containing proteins: A family with diverse lipolytic activities involved in multiple biological functions. J. Lipid Res. 2009, 50, S63–68. [Google Scholar] [CrossRef]
- Bao, S.; Miller, D.J.; Ma, Z.; Wohltmann, M.; Eng, G.; Ramanadham, S.; Moley, K.; Turk, J. Male mice that do not express group VIA phospholipase A2 produce spermatozoa with impaired motility and have greatly reduced fertility. J. Biol. Chem. 2004, 279, 38194–38200. [Google Scholar] [CrossRef]
- Malik, I.; Turk, J.; Mancuso, D.J.; Montier, L.; Wohltmann, M.; Wozniak, D.F.; Schmidt, R.E.; Gross, R.W.; Kotzbauer, P.T. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am. J. Pathol. 2008, 172, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Khateeb, S.; Flusser, H.; Ofir, R.; Shelef, I.; Narkis, G.; Vardi, G.; Shorer, Z.; Levy, R.; Galil, A.; Elbedour, K.; et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am. J. Hum. Genet. 2006, 79, 942–948. [Google Scholar] [CrossRef]
- Morgan, N.V.; Westaway, S.K.; Morton, J.E.; Gregory, A.; Gissen, P.; Sonek, S.; Cangul, H.; Coryell, J.; Canham, N.; Nardocci, N.; et al. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 2006, 38, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Bohrer, A.; Ramanadham, S.; Jin, W.; Zhang, S.; Turk, J. Effects of stable suppression of Group VIA phospholipase A2 expression on phospholipid content and composition, insulin secretion, and proliferation of INS-1 insulinoma cells. J. Biol. Chem. 2006, 281, 187–198. [Google Scholar] [CrossRef]
- Gross, R.W.; Ramanadham, S.; Kruszka, K.K.; Han, X.; Turk, J. Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry 1993, 32, 327–336. [Google Scholar] [CrossRef]
- Ma, Z.; Ramanadham, S.; Wohltmann, M.; Bohrer, A.; Hsu, F.F.; Turk, J. Studies of insulin secretory responses and of arachidonic acid incorporation into phospholipids of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 (iPLA2b) indicate a signaling rather than a housekeeping role for iPLA2b. J. Biol. Chem. 2001, 276, 13198–13208. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, S.; Turk, J.; Ramanadham, S. Stimulation of insulin secretion and associated nuclear accumulation of iPLA2beta in INS-1 insulinoma cells. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E820–833. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Gross, R.W.; Han, X.; Turk, J. Inhibition of arachidonate release by secretagogue-stimulated pancreatic islets suppresses both insulin secretion and the rise in beta-cell cytosolic calcium ion concentration. Biochemistry 1993, 32, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Hsu, F.F.; Bohrer, A.; Ma, Z.; Turk, J. Studies of the role of group VI phospholipase A2 in fatty acid incorporation, phospholipid remodeling, lysophosphatidylcholine generation, and secretagogue-induced arachidonic acid release in pancreatic islets and insulinoma cells. J. Biol. Chem. 1999, 274, 13915–13927. [Google Scholar] [CrossRef]
- Ramanadham, S.; Song, H.; Hsu, F.F.; Zhang, S.; Crankshaw, M.; Grant, G.A.; Newgard, C.B.; Bao, S.; Ma, Z.; Turk, J. Pancreatic islets and insulinoma cells express a novel isoform of group VIA phospholipase A2 (iPLA2b) that participates in glucose-stimulated insulin secretion and is not produced by alternate splicing of the iPLA2b transcript. Biochemistry 2003, 42, 13929–13940. [Google Scholar] [CrossRef]
- Ramanadham, S.; Wolf, M.J.; Jett, P.A.; Gross, R.W.; Turk, J. Characterization of an ATP-stimulatable Ca2+-independent phospholipase A2 from clonal insulin-secreting HIT cells and rat pancreatic islets: A possible molecular component of the beta-cell fuel sensor. Biochemistry 1994, 33, 7442–7452. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Jacobson, D.A.; Wohltmann, M.; Bohrer, A.; Jin, W.; Philipson, L.H.; Turk, J. Glucose homeostasis, insulin secretion, and islet phospholipids in mice that overexpress iPLA2b in pancreatic b-cells and in iPLA2b-null mice. Am. J. Physiol. Endocrinol. Metab. 2008, 294, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Song, H.; Wohltmann, M.; Ramanadham, S.; Jin, W.; Bohrer, A.; Turk, J. Insulin secretory responses and phospholipid composition of pancreatic islets from mice that do not express Group VIA phospholipase A2 and effects of metabolic stress on glucose homeostasis. J. Biol. Chem. 2006, 281, 20958–20973. [Google Scholar] [CrossRef]
- Song, H.; Wohltmann, M.; Tan, M.; Ladenson, J.H.; Turk, J. Group VIA phospholipase A2 mitigates palmitate-induced beta-cell mitochondrial injury and apoptosis. J. Biol. Chem. 2014, 289, 14194–14210. [Google Scholar] [CrossRef]
- Gil-de-Gomez, L.; Astudillo, A.M.; Guijas, C.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. Cytosolic group IVA and calcium-independent group VIA phospholipase A2s act on distinct phospholipid pools in zymosan-stimulated mouse peritoneal macrophages. J. Immunol. 2014, 192, 752–762. [Google Scholar] [CrossRef]
- Moran, J.M.; Buller, R.M.; McHowat, J.; Turk, J.; Wohltmann, M.; Gross, R.W.; Corbett, J.A. Genetic and pharmacologic evidence that calcium-independent phospholipase A2beta regulates virus-induced inducible nitric-oxide synthase expression by macrophages. J. Biol. Chem. 2005, 280, 28162–28168. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Balboa, M.A.; Balsinde, J. Involvement of group VIA calcium-independent phospholipase A2 in macrophage engulfment of hydrogen peroxide-treated U937 cells. J. Immunol. 2006, 176, 2555–2561. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Day, R.; Bao, S.; Turk, J.; Zhao, Q.D. Group VIA phospholipase A2 mediates enhanced macrophage migration in diabetes mellitus by increasing expression of nicotinamide adenine dinucleotide phosphate oxidase 4. Arter. Thromb. Vasc. Biol. 2014, 34, 768–778. [Google Scholar] [CrossRef]
- Ashley, J.W.; Hancock, W.D.; Nelson, A.J.; Bone, R.N.; Tse, H.M.; Wohltmann, M.; Turk, J.; Ramanadham, S. Polarization of Macrophages toward M2 Phenotype Is Favored by Reduction in iPLA2b (Group VIA Phospholipase A2). J. Biol. Chem. 2016, 291, 23268–23281. [Google Scholar] [CrossRef]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig. 2006, 116, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Carnevale, K.A.; Cathcart, M.K. Calcium-independent phospholipase A2 is required for human monocyte chemotaxis to monocyte chemoattractant protein 1. J. Immunol. 2001, 167, 3414–3421. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, M.K. Signal-activated phospholipase regulation of leukocyte chemotaxis. J. Lipid Res. 2009, 50, 231–236. [Google Scholar] [CrossRef]
- Mishra, R.S.; Carnevale, K.A.; Cathcart, M.K. iPLA2b: Front and center in human monocyte chemotaxis to MCP-1. J. Exp. Med. 2008, 205, 347–359. [Google Scholar] [CrossRef] [PubMed]
- DeChiara, T.M. Gene targeting in ES cells. Methods Mol. Biol. 2001, 158, 19–45. [Google Scholar] [CrossRef]
- Kuhn, R.; Torres, R.M. Cre/loxP recombination system and gene targeting. Methods Mol. Biol. 2002, 180, 175–204. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A. Cre recombinase: The universal reagent for genome tailoring. Genesis 2000, 26, 99–109. [Google Scholar] [CrossRef]
- Takeuchi, T.; Nomura, T.; Tsujita, M.; Suzuki, M.; Fuse, T.; Mori, H.; Mishina, M. Flp recombinase transgenic mice of C57BL/6 strain for conditional gene targeting. Biochem. Biophys Res. Commun. 2002, 293, 953–957. [Google Scholar] [CrossRef]
- Turan, S.; Galla, M.; Ernst, E.; Qiao, J.; Voelkel, C.; Schiedlmeier, B.; Zehe, C.; Bode, J. Recombinase-mediated cassette exchange (RMCE): Traditional concepts and current challenges. J. Mol. Biol. 2011, 407, 193–221. [Google Scholar] [CrossRef] [PubMed]
- Friedel, R.H.; Wurst, W.; Wefers, B.; Kuhn, R. Generating conditional knockout mice. Methods Mol. Biol. 2011, 693, 205–231. [Google Scholar] [CrossRef]
- Fex, M.; Wierup, N.; Nitert, M.D.; Ristow, M.; Mulder, H. Rat insulin promoter 2-Cre recombinase mice bred onto a pure C57BL/6J background exhibit unaltered glucose tolerance. J. Endocrinol. 2007, 194, 551–555. [Google Scholar] [CrossRef]
- Gannon, M.; Shiota, C.; Postic, C.; Wright, C.V.; Magnuson, M. Analysis of the Cre-mediated recombination driven by rat insulin promoter in embryonic and adult mouse pancreas. Genesis 2000, 26, 139–142. [Google Scholar] [CrossRef]
- Kalis, M.; Bolmeson, C.; Esguerra, J.L.; Gupta, S.; Edlund, A.; Tormo-Badia, N.; Speidel, D.; Holmberg, D.; Mayans, S.; Khoo, N.K.; et al. Beta-cell specific deletion of Dicer1 leads to defective insulin secretion and diabetes mellitus. PLoS ONE 2011, 6, e29166. [Google Scholar] [CrossRef]
- Pappan, K.L.; Pan, Z.; Kwon, G.; Marshall, C.A.; Coleman, T.; Goldberg, I.J.; McDaniel, M.L.; Semenkovich, C.F. Pancreatic beta-cell lipoprotein lipase independently regulates islet glucose metabolism and normal insulin secretion. J. Biol. Chem. 2005, 280, 9023–9029. [Google Scholar] [CrossRef]
- Sun, G.; Tarasov, A.I.; McGinty, J.A.; French, P.M.; McDonald, A.; Leclerc, I.; Rutter, G.A. LKB1 deletion with the RIP2.Cre transgene modifies pancreatic beta-cell morphology and enhances insulin secretion in vivo. Am. J. Physiol. Endocrinol. Metab. 2010, 298, 1261–1273. [Google Scholar] [CrossRef]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Forster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.G.; Yang, Z.; Chakravarthy, M.V.; Lodhi, I.J.; Wei, X.; Turk, J.; Semenkovich, C.F. Macrophage fatty-acid synthase deficiency decreases diet-induced atherosclerosis. J. Biol. Chem. 2010, 285, 23398–23409. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Ristow, M.; Lin, X.; White, M.F.; Magnuson, M.A.; Hennighausen, L. RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J. Biol. Chem. 2006, 281, 2649–2653. [Google Scholar] [CrossRef]
- Chakravarthy, M.V.; Pan, Z.; Zhu, Y.; Tordjman, K.; Schneider, J.G.; Coleman, T.; Turk, J.; Semenkovich, C.F. “New” hepatic fat activates PPARalpha to maintain glucose, lipid, and cholesterol homeostasis. Cell Metab. 2005, 1, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Cheverud, J.M.; Ehrich, T.H.; Hrbek, T.; Kenney, J.P.; Pletscher, L.S.; Semenkovich, C.F. Quantitative trait loci for obesity- and diabetes-related traits and their dietary responses to high-fat feeding in LGXSM recombinant inbred mouse strains. Diabetes 2004, 53, 3328–3336. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bernal-Mizrachi, E.; Fatrai, S.; Johnson, J.D.; Ohsugi, M.; Otani, K.; Han, Z.; Polonsky, K.S.; Permutt, M.A. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J. Clin. Investig. 2004, 114, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wohltmann, M.; Bao, S.; Ladenson, J.H.; Semenkovich, C.F.; Turk, J. Mice deficient in group VIB phospholipase A2 (iPLA2g) exhibit relative resistance to obesity and metabolic abnormalities induced by a Western diet. Am. J. Physiol. Endocrinol. Metab. 2010, 298, 1097–1114. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Takeda, K.; Maeda, M.; Ogawa, W.; Sato, T.; Okada, S.; Ohnishi, Y.; Nakajima, H.; Kashiwagi, A. Glucose area under the curve during oral glucose tolerance test as an index of glucose intolerance. Diabetol. Int. 2016, 7, 53–58. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, M.L.; Colca, J.R.; Kotagal, N.; Lacy, P.E. A subcellular fractionation approach for studying insulin release mechanisms and calcium metabolism in islets of Langerhans. Methods Enzymol. 1983, 98, 182–200. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Guyenet, S.J.; Cirulli, V. The hypothalamus and ss-cell connection in the gene-targeting era. Diabetes 2010, 59, 2991–2993. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Q.; Wang, X.; Zhu, J.; Xu, K.; Okada, H.; Wang, R.; Woo, M. Survivin is required for beta-cell mass expansion in the pancreatic duct-ligated mouse model. PLoS ONE 2012, 7, e41976. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Lim, Y.M.; Lee, M.S. Role of autophagy in diabetes and endoplasmic reticulum stress of pancreatic beta-cells. Exp. Mol. Med. 2012, 44, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Cai, E.P.; Schroer, S.A.; Wang, L.; Woo, M. Vhl is required for normal pancreatic beta cell function and the maintenance of beta cell mass with age in mice. Lab. Investig. 2011, 91, 527–538. [Google Scholar] [CrossRef]
- Cui, J.; Wang, Z.; Cheng, Q.; Lin, R.; Zhang, X.M.; Leung, P.S.; Copeland, N.G.; Jenkins, N.A.; Yao, K.M.; Huang, J.D. Targeted inactivation of kinesin-1 in pancreatic beta-cells in vivo leads to insulin secretory deficiency. Diabetes 2011, 60, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Lu, S.Y.; Nguyen, K.T.; Schroer, S.A.; Suzuki, A.; Mak, T.W.; Gaisano, H.; Woo, M. Deletion of Pten in pancreatic ss-cells protects against deficient ss-cell mass and function in mouse models of type 2 diabetes. Diabetes 2010, 59, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Choi, C.S.; Ghoshal, P.; Pliss, L.; Pandya, J.D.; Hill, D.; Cline, G.; Patel, M.S. β-Cell-specific pyruvate dehydrogenase deficiency impairs glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 2010, 299, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Tokumoto, S.; Yabe, D.; Tatsuoka, H.; Usui, R.; Fauzi, M.; Botagarova, A.; Goto, H.; Herrera, P.L.; Ogura, M.; Inagaki, N. Generation and characterization of a novel mouse model that allows spatiotemporal quantification of pancreatic beta-cell proliferation. Diabetes 2020. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Tang, L.H.; Davidson, C.; Vosburgh, E.; Chen, W.; Foran, D.J.; Notterman, D.A.; Levine, A.J.; Xu, E.Y. Two well-differentiated pancreatic neuroendocrine tumor mouse models. Cell Death Differ. 2020, 27, 269–283. [Google Scholar] [CrossRef]
- Tauscher, S.; Nakagawa, H.; Volker, K.; Werner, F.; Krebes, L.; Potapenko, T.; Doose, S.; Birkenfeld, A.L.; Baba, H.A.; Kuhn, M. beta Cell-specific deletion of guanylyl cyclase A, the receptor for atrial natriuretic peptide, accelerates obesity-induced glucose intolerance in mice. Cardiovasc. Diabetol. 2018, 17, 103. [Google Scholar] [CrossRef]
- Otten, J.J.; de Jager, S.C.; Kavelaars, A.; Seijkens, T.; Bot, I.; Wijnands, E.; Beckers, L.; Westra, M.M.; Bot, M.; Busch, M.; et al. Hematopoietic G-protein-coupled receptor kinase 2 deficiency decreases atherosclerotic lesion formation in LDL receptor-knockout mice. FASEB J. 2013, 27, 265–276. [Google Scholar] [CrossRef]
- Klose, A.; Zigrino, P.; Mauch, C. Monocyte/macrophage MMP-14 modulates cell infiltration and T-cell attraction in contact dermatitis but not in murine wound healing. Am. J. Pathol. 2013, 182, 755–764. [Google Scholar] [CrossRef]
- Pello, O.M.; Chevre, R.; Laoui, D.; De Juan, A.; Lolo, F.; Andres-Manzano, M.J.; Serrano, M.; Van Ginderachter, J.A.; Andres, V. In vivo inhibition of c-MYC in myeloid cells impairs tumor-associated macrophage maturation and pro-tumoral activities. PLoS ONE 2012, 7, e45399. [Google Scholar] [CrossRef]
- Luiz, J.P.M.; Toller-Kawahisa, J.E.; Viacava, P.R.; Nascimento, D.C.; Pereira, P.T.; Saraiva, A.L.; Prado, D.S.; LeBert, M.; Giurisato, E.; Tournier, C.; et al. MEK5/ERK5 signaling mediates IL-4-induced M2 macrophage differentiation through regulation of c-Myc expression. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef]
- Otto, N.A.; de Vos, A.F.; van Heijst, J.W.J.; Roelofs, J.; van der Poll, T. Myeloid Liver Kinase B1 depletion is associated with a reduction in alveolar macrophage numbers and an impaired host defense during gram-negative pneumonia. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D. What if Minkowski had been ageusic? An alternative angle on diabetes. Science 1992, 258, 766–770. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Otero, Y.F.; Stafford, J.M.; McGuinness, O.P. Pathway-selective insulin resistance and metabolic disease: The importance of nutrient flux. J. Biol. Chem. 2014, 289, 20462–20469. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. Lipid oversupply, selective insulin resistance, and lipotoxicity: Molecular mechanisms. Biochim. Biophys. Acta 2010, 1801, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Siddique, M.M.; Wang, S.T.; Ching, J.; Shayman, J.A.; Summers, S.A. Ceramides and glucosylceramides are independent antagonists of insulin signaling. J. Biol. Chem. 2014, 289, 723–734. [Google Scholar] [CrossRef]
- Unger, R.H. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 1995, 44, 863–870. [Google Scholar] [CrossRef]
- Miki, T.; Nagashima, K.; Tashiro, F.; Kotake, K.; Yoshitomi, H.; Tamamoto, A.; Gonoi, T.; Iwanaga, T.; Miyazaki, J.; Seino, S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 10402–10406. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Minami, K.; Zhang, L.; Morita, M.; Gonoi, T.; Shiuchi, T.; Minokoshi, Y.; Renaud, J.M.; Seino, S. ATP-sensitive potassium channels participate in glucose uptake in skeletal muscle and adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2002, 283, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wang, J.; Jiao, L.; Utaipan, T.; Tuma-Kellner, S.; Schmitz, G.; Liebisch, G.; Stremmel, W.; Chamulitrat, W. iPLA2beta deficiency attenuates obesity and hepatic steatosis in ob/ob mice through hepatic fatty-acyl phospholipid remodeling. Biochim. Biophys. Acta 2016, 1861, 449–461. [Google Scholar] [CrossRef]
- Otto, A.C.; Gan-Schreier, H.; Zhu, X.; Tuma-Kellner, S.; Staffer, S.; Ganzha, A.; Liebisch, G.; Chamulitrat, W. Group VIA phospholipase A2 deficiency in mice chronically fed with high-fat-diet attenuates hepatic steatosis by correcting a defect of phospholipid remodeling. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Zhang, S.; Bohrer, A.; Bao, S.; Song, H.; Ramanadham, S. The group VIA calcium-independent phospholipase A2 participates in ER stress-induced INS-1 insulinoma cell apoptosis by promoting ceramide generation via hydrolysis of sphingomyelins by neutral sphingomyelinase. Biochemistry 2007, 46, 10170–10185. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, S.; Bohrer, A.; Ramanadham, S. Calcium-independent phospholipase A2 (iPLA2b)-mediated ceramide generation plays a key role in the cross-talk between the endoplasmic reticulum (ER) and mitochondria during ER stress-induced insulin-secreting cell apoptosis. J. Biol. Chem. 2008, 283, 34819–34832. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, S.; Barbour, S.E.; Bohrer, A.; Ford, E.L.; Koizumi, A.; Papa, F.R.; Ramanadham, S. Spontaneous development of endoplasmic reticulum stress that can lead to diabetes mellitus is associated with higher calcium-independent phospholipase A2 expression: A role for regulation by SREBP-1. J. Biol. Chem. 2010, 285, 6693–6705. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, S.; Emani, B.; Barbour, S.E.; Ramanadham, S. A link between endoplasmic reticulum stress-induced beta-cell apoptosis and the group VIA Ca2+-independent phospholipase A2 (iPLA2b). Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 93–98. [Google Scholar] [CrossRef]
- Li, H.; Zhao, Z.; Wei, G.; Yan, L.; Wang, D.; Zhang, H.; Sandusky, G.E.; Turk, J.; Xu, Y. Group VIA phospholipase A2 in both host and tumor cells is involved in ovarian cancer development. FASEB J. 2010, 24, 4103–4116. [Google Scholar] [CrossRef]
- Moon, S.H.; Jenkins, C.M.; Mancuso, D.J.; Turk, J.; Gross, R.W. Smooth muscle cell arachidonic acid release, migration, and proliferation are markedly attenuated in mice null for calcium-independent phospholipase A2beta. J. Biol. Chem. 2008, 283, 33975–33987. [Google Scholar] [CrossRef]
- Nelson, A.J.; Stephenson, D.J.; Cardona, C.L.; Lei, X.; Almutairi, A.; White, T.D.; Tusing, Y.G.; Park, M.A.; Barbour, S.E.; Chalfant, C.E.; et al. Macrophage polarization is linked to Ca2+-independent phospholipase A2beta-derived lipids and cross-cell signaling in mice. J. Lipid Res. 2020, 61, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Bone, R.N.; Gai, Y.; Magrioti, V.; Kokotou, M.G.; Ali, T.; Lei, X.; Tse, H.M.; Kokotos, G.; Ramanadham, S. Inhibition of Ca2+-independent phospholipase A2beta (iPLA2b) ameliorates islet infiltration and incidence of diabetes in NOD mice. Diabetes 2015, 64, 541–554. [Google Scholar] [CrossRef]
- Nelson, A.J.; Stephenson, D.J.; Bone, R.N.; Cardona, C.L.; Park, M.A.; Tusing, Y.G.; Lei, X.; Kokotos, G.; Graves, C.L.; Mathews, C.E.; et al. Lipid mediators and biomarkers associated with type 1 diabetes development. JCI Insight 2020, 5, 138034. [Google Scholar] [CrossRef]
- Balsinde, J.; Balboa, M.A.; Dennis, E.A. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J. Biol. Chem. 1997, 272, 29317–29321. [Google Scholar] [CrossRef]
- Balsinde, J.; Bianco, I.D.; Ackermann, E.J.; Conde-Frieboes, K.; Dennis, E.A. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc. Natl. Acad. Sci. USA 1995, 92, 8527–8531. [Google Scholar] [CrossRef]
- Monge, P.; Garrido, A.; Rubio, J.M.; Magrioti, V.; Kokotos, G.; Balboa, M.A.; Balsinde, J. The Contribution of Cytosolic Group IVA and Calcium-Independent Group VIA Phospholipase A2s to Adrenic Acid Mobilization in Murine Macrophages. Biomolecules 2020, 10, 542. [Google Scholar] [CrossRef]
- Balboa, M.A.; Perez, R.; Balsinde, J. Calcium-independent phospholipase A2 mediates proliferation of human promonocytic U937 cells. FEBS J. 2008, 275, 1915–1924. [Google Scholar] [CrossRef]
- Bao, S.; Li, Y.; Lei, X.; Wohltmann, M.; Jin, W.; Bohrer, A.; Semenkovich, C.F.; Ramanadham, S.; Tabas, I.; Turk, J. Attenuated free cholesterol loading-induced apoptosis but preserved phospholipid composition of peritoneal macrophages from mice that do not express group VIA phospholipase A2. J. Biol. Chem. 2007, 282, 27100–27114. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, D.M.; Gong, M.C.; Turk, J.; Post, S.R. Class A scavenger receptor-mediated macrophage adhesion requires coupling of calcium-independent phospholipase A2 and 12/15-lipoxygenase to Rac and Cdc42 activation. J. Biol. Chem. 2007, 282, 33405–33411. [Google Scholar] [CrossRef]
- Carper, M.J.; Zhang, S.; Turk, J.; Ramanadham, S. Skeletal muscle group VIA phospholipase A2 (iPLA2b): Expression and role in fatty acid oxidation. Biochemistry 2008, 47, 12241–12249. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Su, X.; Mancuso, D.J.; Bickel, P.E.; Jenkins, C.M.; Gross, R.W. Small interfering RNA knockdown of calcium-independent phospholipases A2 beta or gamma inhibits the hormone-induced differentiation of 3T3-L1 preadipocytes. J. Biol. Chem. 2004, 279, 21740–21748. [Google Scholar] [CrossRef] [PubMed]
- Zisman, A.; Peroni, O.D.; Abel, E.D.; Michael, M.D.; Mauvais-Jarvis, F.; Lowell, B.B.; Wojtaszewski, J.F.; Hirshman, M.F.; Virkamaki, A.; Goodyear, L.J.; et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 2000, 6, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M.; Michael, M.D.; Peroni, O.D.; Ueki, K.; Carter, N.; Kahn, B.B.; Kahn, C.R. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev. Cell 2002, 3, 25–38. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turk, J.; Song, H.; Wohltmann, M.; Frankfater, C.; Lei, X.; Ramanadham, S. Metabolic Effects of Selective Deletion of Group VIA Phospholipase A2 from Macrophages or Pancreatic Islet Beta-Cells. Biomolecules 2020, 10, 1455. https://doi.org/10.3390/biom10101455
Turk J, Song H, Wohltmann M, Frankfater C, Lei X, Ramanadham S. Metabolic Effects of Selective Deletion of Group VIA Phospholipase A2 from Macrophages or Pancreatic Islet Beta-Cells. Biomolecules. 2020; 10(10):1455. https://doi.org/10.3390/biom10101455
Chicago/Turabian StyleTurk, John, Haowei Song, Mary Wohltmann, Cheryl Frankfater, Xiaoyong Lei, and Sasanka Ramanadham. 2020. "Metabolic Effects of Selective Deletion of Group VIA Phospholipase A2 from Macrophages or Pancreatic Islet Beta-Cells" Biomolecules 10, no. 10: 1455. https://doi.org/10.3390/biom10101455
APA StyleTurk, J., Song, H., Wohltmann, M., Frankfater, C., Lei, X., & Ramanadham, S. (2020). Metabolic Effects of Selective Deletion of Group VIA Phospholipase A2 from Macrophages or Pancreatic Islet Beta-Cells. Biomolecules, 10(10), 1455. https://doi.org/10.3390/biom10101455