Graph Theory-Based Sequence Descriptors as Remote Homology Predictors

 ,
, 

 ,
, 
Abstract
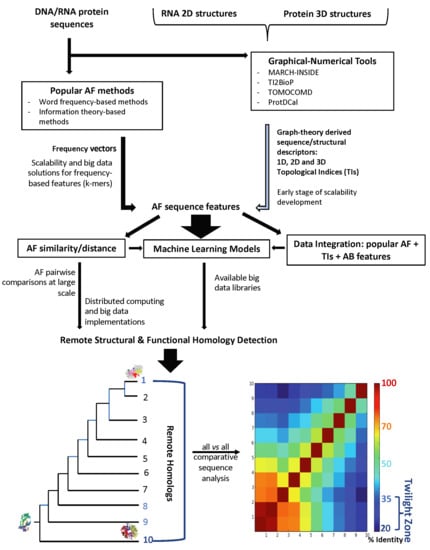
1. Introduction
2. The Twilight Zone for Protein and RNA Alignments
SeqDivA: Sequence Diversity Analysis for Detecting the Twilight Zone
3. Most Popular AF Approaches in the Twilight Zone
3.1. Word Frequency-Based Methods
3.2. Information Theory-Based Methods
4. Graphical–Numerical Approaches: Emerging AF Methodologies within the Twilight Zone
4.1. Brief Background of Graphical–Numerical Approaches
4.2. Graphical–Numerical-Based Methods in the Twilight Zone
4.2.1. MARCH-INSIDE Sequence Descriptors
The Pac1 Detection with 2D-Cartesian Maps and Stochastic Spectral Moments
2D-Cartesian Maps and Markovian Entropies to Detect Remote Homologs in Cellulase Complexes
2D-Cartesian Maps, Star Graphs, and Markovian TIs Characterizing Mycobacterial Promoters
4.2.2. S2SNet’s TIs: Star-Like Graphs Detecting Remote Enzymatic Signatures
4.2.3. Topological Indices to Biopolymers (TI2BioP)
Bacteriocin Remote Homologs Characterized with 2D-HP Maps and Simple TIs
RNase III Diversity Characterized by 1D and 2D Amino Acid Clustering Strategies
Internal Transcribed Spacer (ITS2) Region
Four Color-Maps and Simple TIs Characterizing NRPS’s A-Domains Diversity
4.2.4. TOpological MOlecular COMputer Design (TOMOCOMD) Descriptors
4.2.5. ProtDCal’s Descriptors
4.2.6. Amino Acid Sequence Autocorrelation Vectors (Descriptors)
5. Ensemble of AF, AB-Based Features and Machine Learning Classification Methods for the Detection of Remote Homology in the Twilight Zone
6. Scaling Up AB- and AF-Based Features/Measures for Homology Detection
7. Conclusions
Funding
Conflicts of Interest
References
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013, 42, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Krogh, A.B.; Brown, M.; Mian, I.S.; Sjeander, K.; Haussler, D. Hidden Markov models in computational biology. Applications to protein modeling. J. Mol. Biol. 1994, 235, 1501–1531. [Google Scholar] [CrossRef] [PubMed]
- Teodorescu, O.; Galor, T.; Pillardy, J.; Elber, R. Enriching the sequence substitution matrix by structural information. Proteins 2004, 54, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Mount, D.W. Using BLOSUM in Sequence Alignments. Csh. Protoc. 2008, 2008, pdb–top39. [Google Scholar] [CrossRef] [PubMed]
- Batzoglou, S. The many faces of sequence alignment. Brief. Bioinform. 2005, 6, 6–22. [Google Scholar] [CrossRef]
- Chatzou, M.; Magis, C.; Chang, J.-M.; Kemena, C.; Bussotti, G.; Erb, I.; Notredame, C. Multiple sequence alignment modeling: Methods and applications. Brief. Bioinform. 2015, bbv099. [Google Scholar] [CrossRef]
- Ivan, G.; Banky, D.; Grolmusz, V. Fast and exact sequence alignment with the Smith–Waterman algorithm: The SwissAlign webserver. Gene Rep. 2016, 4, 26–28. [Google Scholar] [CrossRef][Green Version]
- Torarinsson, E.; Lindgreen, S. WAR: Webserver for aligning structural RNAs. Nucleic Acids Res. 2008, 36, W79–W84. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.F.; Fletcher, W.; Förster, F.; Merget, B.; Wolf, M.; Schultz, J.; Markowetz, F. Evolutionary Distances in the Twilight Zone—A Rational Kernel Approach. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Pearson, W.R.; Sierk, M.L. The limits of protein sequence comparison? Curr. Opin. Strctural. Biol. 2005, 15, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Vinga, S.; Almeida, J. Alignment-free sequence comparison—a review. Bioinformatics 2003, 19, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef]
- Vinga, S. Editorial: Alignment-free methods in computational biology. Brief. Bioinform. 2014, 15, 341–342. [Google Scholar] [CrossRef]
- Davies, M.N.; Secker, A.; Freitas, A.A.; Timmis, J.; Clark, E.; Flower, D.R. Alignment-Independent Techniques for Protein Classification. Curr. Proteom. 2008, 5, 217–223. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; Sánchez-Rodríguez, A.; Hidalgo-Yanes, P.I.; Pérez-Castillo, Y.; Molina-Ruiz, R.; Marchal, K.; Vasconcelos, V.; Antunes, A. An alignment-free approach for eukaryotic ITS2 annotation and phylogenetic inference. PLoS ONE 2011, 6, e26638. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; Pérez-Machado, G.; Sánchez-Rodríguez, A.; Santos, M.M.; Antunes, A. Alignment-Free Methods for the Detection and Specificity Prediction of Adenylation Domains. In Nonribosomal Peptide and Polyketide Biosynthesis: Methods and Protocols; Evans, S.B., Ed.; Springer New York: New York, NY, USA, 2016. [Google Scholar]
- Agüero-Chapin, G.; Molina-Ruiz, R.; Pérez-Machado, G.; Vasconcelos, V.; Rodríguez-Negrin, Z.; Antunes, A. TI2BioP—Topological Indices to BioPolymers. A Graphical–Numerical Approach for Bioinformatics. In Recent Advances in Biopolymers; IntechOpen: Zagreb, Croatia, 2016. [Google Scholar]
- Gonzalez-Diaz, H.; Perez-Bello, A.; Uriarte, E.; Gonzalez-Diaz, Y. QSAR study for mycobacterial promoters with low sequence homology. Bioorg. Med. Chem. Lett. 2006, 16, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Diaz, H.; Gonzalez-Diaz, Y.; Santana, L.; Ubeira, F.M.; Uriarte, E. Proteomics, networks and connectivity indices. Proteomics 2008, 8, 750–778. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.R.; Gonzalez-Diaz, H.; Magalhaes, A.L. Enzymes/non-enzymes classification model complexity based on composition, sequence, 3D and topological indices. J. Biol. 2008, 254, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Ponce, Y.; Contreras-Torres, E.; García-Jacas, C.R.; Barigye, S.J.; Cubillán, N.; Alvarado, Y.J. Novel 3D bio-macromolecular bilinear descriptors for protein science: Predicting protein structural classes. J. Theor. Biol. 2015, 374, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Blanco, Y.B.; Paz, W.; Green, J.; Marrero-Ponce, Y. ProtDCal: A program to compute general-purpose-numerical descriptors for sequences and 3D-structures of proteins. BMC Bioinform. 2015, 16, 162. [Google Scholar] [CrossRef] [PubMed]
- Borozan, I.; Watt, S.; Ferretti, V. Integrating alignment-based and alignment-free sequence similarity measures for biological sequence classification. Bioinformatics 2015, 31, 1396–1404. [Google Scholar] [CrossRef][Green Version]
- Galpert, D.; Fernandez, A.; Herrera, F.; Antunes, A.; Molina-Ruiz, R.; Aguero-Chapin, G. Surveying alignment-free features for Ortholog detection in related yeast proteomes by using supervised big data classifiers. BMC Bioinform. 2018, 19, 166. [Google Scholar] [CrossRef]
- Dai, Q.; Yang, Y.; Wang, T. Markov model plus k-word distributions: A synergy that produces novel statistical measures for sequence comparison. Bioinformatics 2008, 24, 2296–2302. [Google Scholar] [CrossRef]
- Sander, C.; Schneider, R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins 1991, 9, 56–68. [Google Scholar] [CrossRef]
- Capriotti, E.; Marti-Renom, M.A. Quantifying the relationship between sequence and three-dimensional structure conservation in RNA. BMC Bioinform. 2010, 11, 322. [Google Scholar] [CrossRef]
- Gardner, P.P.; Wilm, A.; Washietl, S. A benchmark of multiple sequence alignment programs upon structural RNAs. Nucleic Acids Res. 2005, 33, 2433–2439. [Google Scholar] [CrossRef] [PubMed]
- Bremges, A.; Schirmer, S.; Giegerich, R. Fine-tuning structural RNA alignments in the twilight zone. Bmc Bioinform. 2010, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J. Essential Bioinformatics; Cambridge University Press: Cambridge, UK, 2006. [Google Scholar]
- Stoye, J.; Evers, D.; Meyer, F. Rose: Generating sequence families. Bioinformatics 1998, 14, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, W.; Yang, Z. INDELible: A flexible simulator of biological sequence evolution. Mol. Biol. Evol. 2009, 26, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Agüero-Chapin, G.; Molina-Ruiz, R.; Maldonado, E.; de la Riva, G.; Sánchez-Rodríguez, A.; Vasconcelos, V.; Antunes, A. Exploring the adenylation domain repertoire of nonribosomal peptide synthetases using an ensemble of sequence-search methods. PLoS ONE 2013, 8, e65926. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Blanco, Y.B.; Aguero-Chapin, G.; Garcia-Hernandez, E.; Alvarez, O.; Antunes, A.; Green, J. Exploring general-purpose protein features for distinguishing enzymes and non-enzymes within the twilight zone. BMC Bioinform. 2017, 18, 349. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.-B.; Dong, C.; Hua, H.-L.; Liu, S.; Luo, H.; Zhang, H.-W.; Jin, Y.-T.; Zhang, K.-Y. Accurate prediction of human essential genes using only nucleotide composition and association information. Bioinformatics 2017, 33, 1758–1764. [Google Scholar] [CrossRef]
- Kumar, M.; Thakur, V.; Raghava, G.P. COPid: Composition based protein identification. In Silico Biol. 2008, 8, 121–128. [Google Scholar]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Gunasinghe, U.; Alahakoon, D.; Bedingfield, S. Extraction of high quality k-words for alignment-free sequence comparison. J. Theor. Biol. 2014, 358, 31–51. [Google Scholar] [CrossRef]
- Leimeister, C.-A.; Boden, M.; Horwege, S.; Lindner, S.; Morgenstern, B. Fast alignment-free sequence comparison using spaced-word frequencies. Bioinformatics 2014, 30, 1991–1999. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Lei, T.Y.; Jin, D.C.; Lin, H.; Chou, K.C. PseKNC: A flexible web server for generating pseudo K-tuple nucleotide composition. Anal. Biochem. 2014, 456, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Elrod, D.W.; Chou, K.C. A study on the correlation of G-protein-coupled receptor types with amino acid composition. Protein Eng. 2002, 15, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Suwa, M. Bioinformatics tools for predicting GPCR gene functions. Adv. Exp. Med. Biol. 2014, 796, 205–224. [Google Scholar] [CrossRef]
- Gu, Q.; Ding, Y.S.; Zhang, T.L. Prediction of G-protein-coupled receptor classes in low homology using Chou’s pseudo amino acid composition with approximate entropy and hydrophobicity patterns. Protein Pept. Lett. 2010, 17, 559–567. [Google Scholar] [CrossRef]
- Qiu, J.D.; Huang, J.H.; Liang, R.P.; Lu, X.Q. Prediction of G-protein-coupled receptor classes based on the concept of Chou’s pseudo amino acid composition: An approach from discrete wavelet transform. Anal. Biochem. 2009, 390, 68–73. [Google Scholar] [CrossRef]
- Chou, K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct. Funct. Bioinform. 2001, 43, 246–255. [Google Scholar] [CrossRef]
- Shen, H.B.; Chou, K.C. PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 2008, 373, 386–388. [Google Scholar] [CrossRef]
- Shen, H.B.; Chou, K.C. EzyPred: A top-down approach for predicting enzyme functional classes and subclasses. Biochem. Biophys. Res. Commun. 2007. [Google Scholar] [CrossRef]
- Ding, Y.S.; Zhang, T.L.; Chou, K.C. Prediction of protein structure classes with pseudo amino acid composition and fuzzy support vector machine network. Protein Pept. Lett. 2007, 14, 811–815. [Google Scholar] [CrossRef]
- Liu, B.; Wang, X.; Zou, Q.; Dong, Q.; Chen, Q. Protein Remote Homology Detection by Combining Chou’s Pseudo Amino Acid Composition and Profile-Based Protein Representation. Mol. Inf. 2013, 32, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Compeau, P.E.C.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987. [Google Scholar] [CrossRef] [PubMed]
- Ames, S.K.; Hysom, D.A.; Gardner, S.N.; Lloyd, G.S.; Gokhale, M.B.; Allen, J.E. Scalable metagenomic taxonomy classification using a reference genome database. Bioinformatics 2013, 29, 2253–2260. [Google Scholar] [CrossRef] [PubMed]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol 2004, 22, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Olson, R.; Disz, T.; Pusch, G.D.; Vonstein, V.; Stevens, R.; Overbeek, R. Real time metagenomics: Using k-mers to annotate metagenomes. Bioinformatics 2012, 28, 3316–3317. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Wang, T. Comparison study on k-word statistical measures for protein: From sequence to ‘sequence space’. Bmc Bioinform. 2008, 9, 394. [Google Scholar] [CrossRef]
- Lingner, T.; Meinicke, P. Remote homology detection based on oligomer distances. Bioinformatics 2006, 22, 2224–2231. [Google Scholar] [CrossRef]
- Qin, Y.F.; Wang, C.H.; Yu, X.Q.; Zhu, J.; Liu, T.G.; Zheng, X.Q. Predicting protein structural class by incorporating patterns of over-represented k-mers into the general form of Chou’s PseAAC. Protein Pept. Lett. 2012, 19, 388–397. [Google Scholar] [CrossRef]
- Domazet-Loso, M.; Haubold, B. Alignment-free detection of local similarity among viral and bacterial genomes. Bioinformatics 2011, 27, 1466–1472. [Google Scholar] [CrossRef]
- Hohl, M.; Ragan, M.A. Is multiple-sequence alignment required for accurate inference of phylogeny? Syst. Biol. 2007, 56, 206–221. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.X.; Ragan, M.A. Next-generation phylogenomics. Biol. Direct. 2013, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Luo, H.; Hao, B. CVTree: A phylogenetic tree reconstruction tool based on whole genomes. Nucleic Acids Res. 2004, 32, W45–W47. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Yang, X.; Lin, J.; Ye, K. PVTree: A Sequential Pattern Mining Method for Alignment Independent Phylogeny Reconstruction. Genes (Basel) 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Ren, J.; Zhai, Z.; Liu, X.; Deng, M.; Sun, F. Alignment-free sequence comparison based on next-generation sequencing reads. J. Comput. Biol. 2013, 20, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Ren, J.; Reinert, G.; Deng, M.; Waterman, M.S.; Sun, F. New developments of alignment-free sequence comparison: Measures, statistics and next-generation sequencing. Brief. Bioinform. 2014, 15, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Kantorovitz, M.R.; Robinson, G.E.; Sinha, S. A statistical method for alignment-free comparison of regulatory sequences. Bioinformatics 2007, 23, i249–i255. [Google Scholar] [CrossRef] [PubMed]
- Koohy, H.; Dyer, N.P.; Reid, J.E.; Koentges, G.; Ott, S. An alignment-free model for comparison of regulatory sequences. Bioinformatics 2010, 26, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Vitányi, P.M.B. An Introduction to Kolmogorov Complexity and its Applications, 3rd ed.; Springer: New York, NY, USA, 2008. [Google Scholar]
- Lempel, A.; Ziv, J. On the complexity of finite sequences. IEEE Trans. Inf. Theory 1976, 22, 75–81. [Google Scholar] [CrossRef]
- Otu, H.H.; Sayood, K. A new sequence distance measure for phylogenetic tree construction. Bioinformatics 2003, 19, 2122–2130. [Google Scholar] [CrossRef]
- Li, M.; Chen, X.; Li, X.; Ma, B.; Vitányi, P. The similarity metric. In Proceedings of the Fourteenth Anual ACM-SIAM Symposium on Discrete Algorithms, Baltimore, MD, USA, 12–14 January 2003; pp. 863–872. [Google Scholar]
- Kocsor, A.; Kertesz-Farkas, A.; Kajan, L.; Pongor, S. Application of compression-based distance measures to protein sequence classification: A methodological study. Bioinformatics 2006, 22, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Ferragina, P.; Giancarlo, R.; Greco, V.; Manzini, G.; Valiente, G. Compression-based classification of biological sequences and structures via the Universal Similarity Metric: Experimental assessment. BMC Bioinform. 2007, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Badger, J.H.; Chen, X.; Kwong, S.; Kearney, P.; Zhang, H. An information-based sequence distance and its application to whole mitochondrial genome phylogeny. Bioinformatics 2001, 17, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Krasnogor, N.; Pelta, D.A. Measuring the similarity of protein structures by means of the universal similarity metric. Bioinformatics 2004, 20, 1015–1021. [Google Scholar] [CrossRef]
- Strait, B.J.; Dewey, T.G. The Shannon information entropy of protein sequences. Biophys. J. 1996, 71, 148–155. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Nan, F.; Adjeroh, D. On complexity measures for biological sequences. In Proceedings of the 2004 IEEE Computational Systems Bioinformatics Conference, Stanford, CA, USA, 19 August 2004. [Google Scholar]
- Jani, M.; Azad, R.K. Information entropy based methods for genome comparison. ACM Sigbioinformatics Rec. 2013, 3, 2. [Google Scholar] [CrossRef]
- Wang, D.; Tapan, S. MISCORE: A new scoring function for characterizing DNA regulatory motifs in promoter sequences. BMC Syst. Biol. 2012, 6 Suppl 2, S4. [Google Scholar] [CrossRef]
- Comin, M.; Antonelli, M. Fast Alignment-free Comparison for Regulatory Sequences using Multiple Resolution Entropic Profiles. In Proceedings of BIOINFORMATICS, Methods and Algorithms (BIOSTEC 2015); SciTePress: Loire Valley, France, 2015; pp. 171–177. [Google Scholar]
- Erill, I.; O’Neill, M.C. A reexamination of information theory-based methods for DNA-binding site identification. BMC Bioinform. 2009, 10, 57. [Google Scholar] [CrossRef]
- Xu, M.; Su, Z. A novel alignment-free method for comparing transcription factor binding site motifs. PLoS ONE 2010, 5, e8797. [Google Scholar] [CrossRef]
- Vinga, S. Information theory applications for biological sequence analysis. Brief. Bioinform. 2014, 15, 376–389. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C. A key driving force in determination of protein structural classes. Biochem. Biophys. Res. Commun. 1999, 264, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Sierk, M.L.; Pearson, W.R. Sensitivity and selectivity in protein structure comparison. Protein Sci. 2004, 13, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Chew, L.P.; Kedem, K. Finding the consensus shape for a protein family. Algorithmica 2004, 38, 115–129. [Google Scholar] [CrossRef]
- Liao, L.; Noble, W.S. Combining pairwise sequence similarity and support vector machines for detecting remote protein evolutionary and structural relationships. J. Comput. Biol. 2003, 10, 857–868. [Google Scholar] [CrossRef]
- Leslie, C.S.; Eskin, E.; Cohen, A.; Weston, J.; Noble, W.S. Mismatch string kernels for discriminative protein classification. Bioinformatics 2004, 20, 467–476. [Google Scholar] [CrossRef]
- Randic, M.; Zupan, J.; Balaban, A.T.; Vikic-Topic, D.; Plavsic, D. Graphical representation of proteins. Chem. Rev. 2011, 111, 790–862. [Google Scholar] [CrossRef]
- Biggs, N.; Lloyd, E.; Wilson, R. Graph Theory; Oxford University Press: Oxford, UK, 1986; pp. 1736–1936. [Google Scholar]
- Estrada, E.; Uriarte, E. Recent advances on the role of topological indices in drug discovery research. Curr. Med. Chem. 2001, 8, 1573–1588. [Google Scholar] [CrossRef]
- Nandy, A.; Harle, M.; Basak, S.C. Mathematical descriptors of DNA sequences: Development and applications. Arkivoc 2006, 9, 211–238. [Google Scholar]
- Gonzalez-Diaz, H.; Perez-Montoto, L.G.; Duardo-Sanchez, A.; Paniagua, E.; Vazquez-Prieto, S.; Vilas, R.; Dea-Ayuela, M.A.; Bolas-Fernandez, F.; Munteanu, C.R.; Dorado, J.; et al. Generalized lattice graphs for 2D-visualization of biological information. J. Biol. 2009, 261, 136–147. [Google Scholar] [CrossRef]
- Randic, M.; Lers, N.; Plavšić, D.; Basak, S.; Balaban, A. Four-color map representation of DNA or RNA sequences and their numerical characterization. Chem. Phys. Lett. 2005, 407, 205–208. [Google Scholar] [CrossRef]
- Randic, M.; Zupan, J.; Vikic-Topic, D. On representation of proteins by star-like graphs. J. Mol. Graph. Model. 2007, 26, 290–305. [Google Scholar] [CrossRef] [PubMed]
- Aguero-Chapin, G.; Gonzalez-Diaz, H.; Molina, R.; Varona-Santos, J.; Uriarte, E.; Gonzalez-Diaz, Y. 2D maps and coupling numbers for protein sequences. The first QSAR study of polygalacturonases; isolation and prediction of a novel sequence from Psidium guajava L. Febs. Lett. 2006, 580, 723–730. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Aguero-Chapin, G.; Varona, J.; Molina, R.; Delogu, G.; Santana, L.; Uriarte, E.; Podda, G. 2D-RNA-coupling numbers: A new computational chemistry approach to link secondary structure topology with biological function. J. Comput. Chem. 2007, 28, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Aguero-Chapin, G.; Varona-Santos, J.; de la Riva, G.A.; Antunes, A.; Gonzalez-Vlla, T.; Uriarte, E.; Gonzalez-Diaz, H. Alignment-free prediction of polygalacturonases with pseudofolding topological indices: Experimental isolation from Coffea arabica and prediction of a new sequence. J. Proteome Res. 2009, 8, 2122–2128. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Diaz, H.; Uriarte, E.; Ramos de Armas, R. Predicting stability of Arc repressor mutants with protein stochastic moments. Bioorg. Med. Chem. 2005, 13, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Ponce, Y.; Marrero, R.; Castro, E.; Ramos de Armas, R.; Díaz, H.G.; Zaldivar, V.; Torrens, F. Protein quadratic indices of the “Macromolecular Pseudograph’s α-Carbon Atom Adjacency Matrix”. 1. Prediction of Arc repressor alanine-mutant’s stability. Molecules 2004, 9, 1124–1147. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Prado-Prado, F.; Ubeira, F.M. Predicting antimicrobial drugs and targets with the MARCH-INSIDE approach. Curr. Top Med. Chem. 2008, 8, 1676–1690. [Google Scholar] [CrossRef]
- Ponce, Y.; Nodarse, D.; Díaz, H.; De Armas, R.; Zaldivar, V.; Torrens, F.; Castro, E. Nucleic acid quadratic indices of the “macromolecular graph’s nucleotides adjacency matrix” modeling of footprints after the interaction of paromomycin with the HIV-1 Ψ-RNA Packaging Region. Int. J. Mol. Sci. 2004, 5, 276–293. [Google Scholar] [CrossRef]
- Aguiar-Pulido, V.; Munteanu, C.R.; Seoane, J.A.; Fernandez-Blanco, E.; Perez-Montoto, L.G.; Gonzalez-Diaz, H.; Dorado, J. Naive Bayes QSDR classification based on spiral-graph Shannon entropies for protein biomarkers in human colon cancer. Mol. Biosyst. 2012, 8, 1716–1722. [Google Scholar] [CrossRef]
- Randić, M.; Vračko, M.; Lerš, N.; Plavšić, D. Novel 2-D graphical representation of DNA sequences and their numerical characterization. Chem. Phys. Lett. 2003, 368, 1–6. [Google Scholar] [CrossRef]
- Nandy, A. Two-dimensional graphical representation of DNA sequences and intron-exon discrimination in intron-rich sequences. Comput. Appl. Biosci. 1996, 12, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Randic, M.; Mehulic, K.; Vukicevic, D.; Pisanski, T.; Vikic-Topic, D.; Plavsic, D. Graphical representation of proteins as four-color maps and their numerical characterization. J. Mol. Graph. Model. 2009, 27, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Aguero-Chapin, G.; Antunes, A.; Ubeira, F.M.; Chou, K.C.; Gonzalez-Diaz, H. Comparative study of topological indices of macro/supramolecular RNA complex networks. J. Chem. Inf. Model. 2008, 48, 2265–2277. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Monteagudo, M.; Gonzalez-Diaz, H.; Borges, F.; Dominguez, E.R.; Cordeiro, M.N. 3D-MEDNEs: An alternative “in silico” technique for chemical research in toxicology. 2. quantitative proteome-toxicity relationships (QPTR) based on mass spectrum spiral entropy. Chem. Res. Toxicol. 2008, 21, 619–632. [Google Scholar] [CrossRef]
- González-Díaz, H.; Molina-Ruiz, R.; Hernandez, I. MARCH-INSIDE v3.0 (MARkov CHains INvariants for SImulation & DEsign) 3.0 2007. p. Windows supported version under request to the main author contact email: gonzalezdiazh@yahoo.es.
- Gonzalez Diaz, H.; Olazabal, E.; Castanedo, N.; Sanchez, I.H.; Morales, A.; Serrano, H.S.; Gonzalez, J.; de Armas, R.R. Markovian chemicals “in silico” design (MARCH-INSIDE), a promising approach for computer aided molecular design II: Experimental and theoretical assessment of a novel method for virtual screening of fasciolicides. J. Mol. Model. 2002, 8, 237–245. [Google Scholar] [CrossRef]
- Estrada, E. Spectral Moments of the Edge Adjacency Matrix in Molecular Graphs. 1. Definition and Applications to the Prediction of Physical Properties of Alkanes. J. Chem. Inf. Comput. Sci. 1996, 36, 844–849. [Google Scholar] [CrossRef]
- Ramos de Armas, R.; Gonzalez Diaz, H.; Molina, R.; Uriarte, E. Markovian Backbone Negentropies: Molecular descriptors for protein research. I. Predicting protein stability in Arc repressor mutants. Proteins 2004, 56, 715–723. [Google Scholar] [CrossRef]
- Ramos de Armas, R.; Gonzalez Diaz, H.; Molina, R.; Perez Gonzalez, M.; Uriarte, E. Stochastic-based descriptors studying peptides biological properties: Modeling the bitter tasting threshold of dipeptides. Bioorg. Med. Chem. 2004, 12, 4815–4822. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; Molina, R.; Uriarte, E. Markov entropy backbone electrostatic descriptors for predicting proteins biological activity. Bioorg. Med. Chem. Lett. 2004, 14, 4691–4695. [Google Scholar] [CrossRef]
- Gonzalez-Diaz, H.; de Armas, R.R.; Molina, R. Markovian negentropies in bioinformatics. 1. A picture of footprints after the interaction of the HIV-1 Psi-RNA packaging region with drugs. Bioinformatics 2003, 19, 2079–2087. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, F.; Sun, X.; Shi, X.; Zhai, H.; Tian, C.; Kong, F.; Liu, B.; Yuan, X. A Global Analysis of the Polygalacturonase Gene Family in Soybean (Glycine max). PLoS ONE 2016, 11, e0163012. [Google Scholar] [CrossRef] [PubMed]
- Aguero-Chapin, G.; Gonzalez-Diaz, H.; de la Riva, G.; Rodriguez, E.; Sanchez-Rodriguez, A.; Podda, G.; Vazquez-Padron, R.I. MMM-QSAR recognition of ribonucleases without alignment: Comparison with an HMM model and isolation from Schizosaccharomyces pombe, prediction, and experimental assay of a new sequence. J. Chem. Inf. Model. 2008, 48, 434–448. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lamontagne, B.; Elela, S.A. Evaluation of the RNA determinants for bacterial and yeast RNase III binding and cleavage. J. Biol. Chem. 2004, 279, 2231–2241. [Google Scholar] [CrossRef]
- Rotondo, G.; Frendewey, D. Purification and characterization of the Pac1 ribonuclease of Schizosaccharomyces pombe. Nucleic Acids Res. 1996, 24, 2377–2386. [Google Scholar] [CrossRef]
- Percival Zhang, Y.H.; Himmel, M.E.; Mielenz, J.R. Outlook for cellulase improvement: Screening and selection strategies. Biotechnol. Adv. 2006, 24, 452–481. [Google Scholar] [CrossRef]
- Henrissat, B.; Claeyssens, M.; Tomme, P.; Lemesle, L.; Mornon, J.P. Cellulase families revealed by hydrophobic cluster analysis. Gene 1989, 81, 83–95. [Google Scholar] [CrossRef]
- Agüero-Chapin, G.; Sánchez-Rodríguez, A.; Antunes, A.; de la Riva, G.A.; González-Díaz, H. Network entropies classification of fungi and bacteria cellulases of interest for biotechnology. In Topological Indices for Medicinal Chemistry, Biology, Parasitology, Neurological and Social Networks; Munteanu, C.R., González-Díaz, H., Eds.; Transworld Research Network: Kerala, India, 2010. [Google Scholar]
- R Munteanu, C.; L Magalhaes, A.; Duardo-Sánchez, A.; Pazos, A.; González-Díaz, H. S2Snet: A tool for transforming characters and numeric sequences into star network topological indices in chemoinformatics, bioinformatics, biomedical, and social-legal sciences. Curr. Bioinform. 2013, 8, 429–437. [Google Scholar] [CrossRef]
- Perez-Bello, A.; Munteanu, C.R.; Ubeira, F.M.; De Magalhaes, A.L.; Uriarte, E.; Gonzalez-Diaz, H. Alignment-free prediction of mycobacterial DNA promoters based on pseudo-folding lattice network or star-graph topological indices. J. Biol. 2009, 256, 458–466. [Google Scholar] [CrossRef]
- Concu, R.; Podda, G.; Uriarte, E.; Gonzalez-Diaz, H. Computational chemistry study of 3D-structure-function relationships for enzymes based on Markov models for protein electrostatic, HINT, and van der Waals potentials. J. Comput. Chem. 2009, 30, 1510–1520. [Google Scholar] [CrossRef]
- Munteanu, C.R.; Gonzalez-Diaz, H.; Borges, F.; de Magalhaes, A.L. Natural/random protein classification models based on star network topological indices. J. Biol. 2008, 254, 775–783. [Google Scholar] [CrossRef]
- Dobson, P.D.; Doig, A.J. Distinguishing Enzyme Structures from Non-enzymes Without Alignments. J. Mol. Biol. 2003, 330, 771–783. [Google Scholar] [CrossRef]
- Naik, P.K.; Mishra, V.S.; Gupta, M.; Jaiswal, K. Prediction of enzymes and non-enzymes from protein sequences based on sequence derived features and PSSM matrix using artificial neural network. Bioinformation 2007, 2, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Aguero-Chapin, G.; Perez-Machado, G.; Molina-Ruiz, R.; Perez-Castillo, Y.; Morales-Helguera, A.; Vasconcelos, V.; Antunes, A. TI2BioP: Topological Indices to BioPolymers. Its practical use to unravel cryptic bacteriocin-like domains. Amino Acids 2011, 40, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Aguero-Chapin, G.; de la Riva, G.A.; Molina-Ruiz, R.; Sanchez-Rodriguez, A.; Perez-Machado, G.; Vasconcelos, V.; Antunes, A. Non-linear models based on simple topological indices to identify RNase III protein members. J. Biol. 2011, 273, 167–178. [Google Scholar] [CrossRef]
- Cotter, P.; Hill, C.; Ross, R. What’s in a name? Class distinction for bacteriocins. Nat. Rev. Microbiol. 2006, 4. [Google Scholar] [CrossRef]
- Dirix, G.; Monsieurs, P.; Dombrecht, B.; Daniels, R.; Marchal, K.; Vanderleyden, J.; Michiels, J. Peptide signal molecules and bacteriocins in Gram-negative bacteria: A genome-wide in silico screening for peptides containing a double-glycine leader sequence and their cognate transporters. Peptides 2004, 25, 1425–1440. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef]
- Vazquez-Padron, R.I.; de la Riva, G.; Aguero, G.; Silva, Y.; Pham, S.M.; Soberon, M.; Bravo, A.; Aitouche, A. Cryptic endotoxic nature of Bacillus thuringiensis Cry1Ab insecticidal crystal protein. Febs. Lett. 2004, 570, 30–36. [Google Scholar] [CrossRef][Green Version]
- Schultz, J.; Maisel, S.; Gerlach, D.; Müller, T.; Wolf, M. A common core of secondary structure of the internal transcribed spacer 2 (ITS2) throughout the Eukaryota. RNA 2005, 11, 361–364. [Google Scholar] [CrossRef]
- Mathews, D.H. Predicting a set of minimal free energy RNA secondary structures common to two sequences. Bioinformatics 2005, 21, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Kirk, P.M.; Cannon, P.F.; Stalpers, J.A. The Dictionary of the Fungi, 10th ed.; CABI: Wallingford, UK, 2008; p. 784. [Google Scholar]
- Jenke-Kodama, H.; Dittmann, E. Bioinformatic perspectives on NRPS/PKS megasynthases: Advances and challenges. Nat. Prod. Rep. 2009, 26, 874–883. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Ponce, Y.; Castillo-Garit, J.A.; Olazabal, E.; Serrano, H.S.; Morales, A.; Castanedo, N.; Ibarra-Velarde, F.; Huesca-Guillen, A.; Jorge, E.; del Valle, A.; et al. TOMOCOMD-CARDD, a novel approach for computer-aided ‘rational’ drug design: I. Theoretical and experimental assessment of a promising method for computational screening and in silico design of new anthelmintic compounds. J. Comput. Aided Mol. Des. 2004, 18, 615–634. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Ponce, Y.; Marrero, R.M.; Torrens, F.; Martinez, Y.; Bernal, M.G.; Zaldivar, V.R.; Castro, E.A.; Abalo, R.G. Non-stochastic and stochastic linear indices of the molecular pseudograph’s atom-adjacency matrix: A novel approach for computational in silico screening and “rational” selection of new lead antibacterial agents. J. Mol. Model 2005, 1–17. [Google Scholar] [CrossRef]
- Marrero-Ponce, Y.; Castillo-Garit, J.A.; Nodarse, D. Linear indices of the “macromolecular graph’s nucleotides adjacency matrix” as a promising approach for bioinformatics studies. Part 1: Prediction of paromomycin’s affinity constant with HIV-1 W-RNA packaging region. Bioorg. Med. Chem. 2005, 13, 3397–3404. [Google Scholar] [CrossRef]
- Marrero-Ponce, Y.; Medina-Marrero, R.; Castillo-Garit, J.A.; Romero-Zaldivar, V.; Torrens, F.; Castro, E.A. Protein linear indices of the ‘macromolecular pseudograph alpha-carbon atom adjacency matrix’ in bioinformatics. Part 1: Prediction of protein stability effects of a complete set of alanine substitutions in Arc repressor. Bioorg. Med. Chem. 2005, 13, 3003–3015. [Google Scholar] [CrossRef]
- Ortega-Broche, S.E.; Marrero-Ponce, Y.; Diaz, Y.E.; Torrens, F.; Perez-Gimenez, F. TOMOCOMD-CAMPS and protein bilinear indices--novel bio-macromolecular descriptors for protein research: I. Predicting protein stability effects of a complete set of alanine substitutions in the Arc repressor. Febs. J. 2010, 277, 3118–3146. [Google Scholar] [CrossRef]
- Marrero-Ponce, Y.; R Garcia-Jacas, C.; J Barigye, S.; R Valdés-Martiní, J.; Miguel Rivera-Borroto, O.; W Pino-Urias, R.; Cubillán, N.; J Alvarado, Y.; Le-Thi-Thu, H. Optimum search strategies or novel 3D molecular descriptors: Is there a stalemate? Curr. Bioinform. 2015, 10, 533–564. [Google Scholar] [CrossRef]
- Garcia-Jacas, C.R.; Contreras-Torres, E.; Marrero-Ponce, Y.; Pupo-Merino, M.; Barigye, S.J.; Cabrera-Leyva, L. Examining the predictive accuracy of the novel 3D N-linear algebraic molecular codifications on benchmark datasets. J. Cheminform. 2016, 8, 10. [Google Scholar] [CrossRef]
- Terán, J.E.; Marrero-Ponce, Y.; Contreras-Torres, E.; García-Jacas, C.R.; Vivas-Reyes, R.; Terán, E.; Torres, F.J. Tensor Algebra-based Geometrical (3D) Biomacro-Molecular Descriptors for Protein Research: Theory, Applications and Comparison with other Methods. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Moreau, G.; Broto, P. The Autocorrelation of a topological structure. A new molecular descriptor. Nouv. J. Chim. 1980, 4, 359–360. [Google Scholar]
- Kier, L.B.; Hall, L.H. An electrotopological-state index for atoms in molecules. Pharm. Res. 1990, 7, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Ivanciuc, O. Building–Block Computation of the Ivanciuc–Balaban Indices for the Virtual Screening of Combinatorial Libraries. Internet Electron. J. Mol. Des. 2002, 1, 1–9. [Google Scholar]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors, 1st ed.; Wiley-VCH: Mannheim, Germany, 2000. [Google Scholar]
- Heidelberg, J.F.; Paulsen, I.T.; Nelson, K.E.; Gaidos, E.J.; Nelson, W.C.; Read, T.D.; Eisen, J.A.; Seshadri, R.; Ward, N.; Methe, B. Genome sequence of the dissimilatory metal ion–reducing bacterium Shewanella oneidensis. Nat. Biotechnol. 2002, 20, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Romero-Molina, S.; Ruiz-Blanco, Y.B.; Green, J.R.; Sanchez-Garcia, E. ProtDCal-Suite: A web server for the numerical codification and functional analysis of proteins. Protein Sci. 2019, 28, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Biggar, K.K.; Ruiz-Blanco, Y.B.; Charih, F.; Fang, Q.; Connolly, J.; Frensemier, K.; Adhikary, H.; Li, S.S.; Green, J.R. MethylSight: Taking a wider view of lysine methylation through computer-aided discovery to provide insight into the human methyl-lysine proteome. bioRxiv 2018, 274688. [Google Scholar] [CrossRef]
- Caballero, J.; Fernandez, L.; Abreu, J.I.; Fernandez, M. Amino Acid Sequence Autocorrelation vectors and ensembles of Bayesian-Regularized Genetic Neural Networks for prediction of conformational stability of human lysozyme mutants. J. Chem. Inf. Model. 2006, 46, 1255–1268. [Google Scholar] [CrossRef]
- Fernandez, L.; Caballero, J.; Abreu, J.I.; Fernandez, M. Amino acid sequence autocorrelation vectors and Bayesian-regularized genetic neural networks for modeling protein conformational stability: Gene V protein mutants. Proteins 2007, 67, 834–852. [Google Scholar] [CrossRef]
- Fernandez, M.; Kumagai, Y.; Standley, D.M.; Sarai, A.; Mizuguchi, K.; Ahmad, S. Prediction of dinucleotide-specific RNA-binding sites in proteins. BMC Bioinform. 2011, 12, S5. [Google Scholar] [CrossRef]
- Fernandez, M.; Caballero, J.; Fernandez, L.; Sarai, A. Graphical Representations of Protein Sequences for Alignment-Free Comparative and Predictive Studies. Recognition of Protease Inhibition Pattern from H-Depleted Molecular Graph Representation of Protease Sequences. Curr. Bioinform. 2010, 5, 241–252. [Google Scholar] [CrossRef]
- Nandini, C.; Aroquiaraj, I.L. A Survey on Protein Sequence Classification with Data Mining Techniques. Int. J. Sci. Eng. Res. 2016, 7, 1442–1449. [Google Scholar]
- Saigo, H.; Vert, J.-P.; Ueda, N.; Akutsu, T. Protein homology detection using string alignment kernels. Bioinformatics 2004, 20, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Salichos, L.; Rokas, A. Evaluating ortholog prediction algorithms in a yeast model clade. PLoS ONE 2011, 6, e18755. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, K.; Webb, G.I.; Song, J.; Whisstock, J.C.; Konagurthu, A.S. Efficient large-scale protein sequence comparison and gene matching to identify orthologs and co-orthologs. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Byma, S.; Dhasade, A.; Altenhoff, A.; Dessimoz, C.; Larus, J.R. Parallel and Scalable Precise Clustering for Homologous Protein Discovery. bioRxiv 2019. [Google Scholar] [CrossRef]
- Glover, N.; Dessimoz, C.; Ebersberger, I.; Forslund, S.K.; Gabaldón, T.; Huerta-Cepas, J.; Maria-Jesus, M.; Muffato, M.; Patricio, M.; Pereira, C.; et al. Advances and Applications in the Quest for Orthologs. Mol. Biol. Evol. 2019, 10. [Google Scholar] [CrossRef]
- Chen, J.; Liu, B.; Huang, D. Protein Remote Homology Detection Based on an Ensemble Learning Approach. Biomed Res. Int. Hindawi Publ. Corp. 2016, 11. [Google Scholar] [CrossRef]
- Tyson, G.W.; Chapman, J.; Hugenholtz, P.; Allen, E.E.; Ram, R.J.; Richardson, P.M.; Solovyev, V.V.; Rubin, E.M.; Rokhsar, D.S.; Banfield, J.F. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428, 37. [Google Scholar] [CrossRef]
- Meng, X.; Bradley, J.; Yavuz, B.; Sparks, E.; Venkataraman, S.; Liu, D.; Freeman, J.; Tsai, D.; Amde, M.; Owen, S. Mllib: Machine learning in apache spark. J. Mach. Learn. Res. 2016, 17, 1235–1241. [Google Scholar]
- Kashyap, H.; Ahmed, H.A.; Hoque, N.; Roy, S.; Bhattacharyya, D.K. Big data analytics in bioinformatics: A machine learning perspective. arXiv 2015, arXiv:1506.05101v1. [Google Scholar]
- Galpert, D.; García, S.d.R.; Herrera, F.; Ancede-Gallardo, E.; Antunes, A.; Agüero-Chapin, G. Big Data Supervised Pairwise Ortholog Detection in Yeasts. In Yeast-Industrial Applications; IntechOpen: Zagreb, Croatia, 2017; pp. 41–43. [Google Scholar]
- Elloumi, M.; Zomaya, A.Y. Algorithms in Computational Molecular Biology: Techniques, Approaches and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Cattaneo, G.; Petrillo, U.F.; Giancarlo, R.; Roscigno, G. Alignment-free sequence comparison over Hadoop for computational biology. In Proceedings of the 44th International Conference on Parallel Processing Workshops, Washington, DC, USA, 1–4 September 2015. [Google Scholar]
- Matsunaga, A.; Tsugawa, M.; Fortes, J. Cloudblast: Combining mapreduce and virtualization on distributed resources for bioinformatics applications. In Proceedings of the 2008 IEEE Fourth International Conference on eScience, Indianapolis, IN, USA, 7–12 December 2008. [Google Scholar]
- Steinegger, M.; Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Galpert, D.; Del Rio, S.; Herrera, F.; Ancede-Gallardo, E.; Antunes, A.; Aguero-Chapin, G. An Effective Big Data Supervised Imbalanced Classification Approach for Ortholog Detection in Related Yeast Species. Biomed Res. Int. 2015, 2015, 748681. [Google Scholar] [CrossRef] [PubMed]
- Zielezinski, A.; Girgis, H.Z.; Bernard, G.; Leimeister, C.A.; Tang, K.; Dencker, T.; Lau, A.K.; Rohling, S.; Choi, J.J.; Waterman, M.S.; et al. Benchmarking of alignment-free sequence comparison methods. Genome Biol. 2019, 20, 144. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Word-Frequency Methods | |||
|---|---|---|---|
| AF Feature | Low-Similarity Dataset | Web-Implementation | Ref. |
| Amino Acid Composition (ACC) | G-protein coupled receptor superfamily | COPid https://webs.iiitd.edu.in/raghava/COPid/ | [46] |
| Pseudo Amino Acid (PseACC) | G-protein coupled receptor superfamily | http://www.csbio.sjtu.edu.cn/bioinf/PseAA/ | [48,49] |
| PseACC | Designed dataset identity from ENZYME SwissPro database in [52] | http://chou.med.harvard.edu/bioinf/EzyPred/ | [52] |
| PseACC | Chou’s designed dataset [89] from SCOP structural classes | http://www.csbio.sjtu.edu.cn/bioinf/PseAA/ | [53] |
| k-mers | Benchmark Structural data designed based on [90,91] | No publicly available for proteins | [60] |
| k-mers | Benchmark Structural data designed in [92] and also used by [93] | No publicly available for proteins | [61] |
| Information theory-based methods | |||
| Lempel-Ziv complexity | Subset of SCOP designed by [92] | No publicly available | [76] |
| Kolmogorov complexity | Subset of SCOP designed by [92] | No publicly available | [76] |
| Kolmogorov complexity (Universal Similarity Metric) | Benchmark Structural data < 25% designed based on [90,91] | No publicly available | [77] |
| Kolmogorov complexity (Universal Similarity Metric) | Clustering protein structures using at low sequence similarity Benchmark Structural data [91] | http://www.cs.nott.ac.uk/~nxk/USM/protocol.html | [79] |
| Graph-Theory-Based Sequence Descriptors | ||||
|---|---|---|---|---|
| AF Feature | Low-Similarity Dataset | Graphical Representation | New Member Detected | Ref. |
| Stochastic spectral moments (MARCH-INSIDE) | RNase III family | 2D Cartesian protein maps | Pac1 brk Accession DQ647826 | [122] |
| Markovian entropies (MARCH-INSIDE) | Cellulase complex | 2D Cartesian protein maps | - | [127] |
| Markovian entropies, spectral moments and electrostatic potentials (MARCH-INSIDE) | Mycobacterial promoters | 2D Cartesian DNA maps | - | [129] |
| 3D-Markovian descriptors (MARCH-INSIDE) | D&D benchmark dataset [132] | 3D protein representation from PDB files considering distances between Cα of aa | - | [130] |
| Set of TIs for Star Networks (S2SNet) | Natural and unnatural proteins | 2D star protein graphs | [131] | |
| Set of TIs for Star Networks (S2SNet) | D&D benchmark dataset [132] | 2D star protein graphs | [25] | |
| Spectral moments (TI2BioP) | Bacteriocin proteins | 2D Cartesian protein maps | Bacteriocin-like protein in the Cry 1Ab C-terminal domain | [134] |
| Spectral moments (TI2BioP) | RNase III family | 2D Cartesian protein maps | RNase III GU190214 | [135] |
| Spectral moments (TI2BioP) | ITS2 family | 2D Cartesian DNA maps | ITS2 from Petrakia sp. FJ892749 | [20] |
| Spectral moments (TI2BioP) | A-domains from NRPSs | Four-colour maps | Remote homologous in the proteome of Microcystis aeruginosa | [38] |
| 3D protein bilinear indices TOMOCOMD (QuBiLS-MIDAS) | Chou’s designed dataset [89] from SCOP structural classes | 3D PDB graphical information considering Cα and non-covalent interactions | - | [26] |
| 3D protein three-linear indices TOMOCOMD (QuBiLS-MIDAS) | Chou’s designed dataset [89] from SCOP structural classes | 3D PDB graphical information considering Cα, Cβ and average of the coordinates of all atoms in the amino acid | - | [151] |
| 3D and 1D descriptors (ProtDCal) | D&D benchmark dataset [132] | 1D Sequence information 3D PDB information | [27] | |
| AB and AF Features/Measures Integrated under the Same Model/Algorithm | |||
|---|---|---|---|
| AB/AF Features-Methods | Low-Similarity Dataset | Integrative Algorithm | Ref. |
| BLAST-bitscores (AB) Smith-Waterman scores (AB) k-mers (AF) Kolmogorov complexity (AF) | - Complete viral genomes - Short reads from metagenomic data [170] - Subset of SCOP designed by [92] | k-NN algorithm provides a combined score resulted from the combination/weighting of the individual scores resulting from AB and AF-based classifications | [28] |
| Profile-based sequence representation based on PSI-BLAST alignments Pseudo Amino Acid (PseACC) | Benchmark dataset - SCOP structural classes [61,164] | Original sequences are replaced by their profile-based representation containing evolutionary information of the family, then the PseACC concept is applied to generate AF predictors | [48,49] |
| Smith-Waterman (AB) Needleman–Wunsch (AB) Physicochemical profile of aligned regions (AB) ACC (AF) PseACC (AF) Composition, Transition and Distribution (AF) | Benchmark dataset reported in [165] (Saccharomycete yeast proteome pairs). Ortholog detection in the twilight zone | Decision Tree Models (DTM) implemented in the Big Data Spark platform | [29] |
| Integration of Models/Algorithms Using AB and AF Features as Predictors | |||
| Multi-template BLASTp (AB) HMM (AB) DTM using four-colour maps (AF) | Real dataset made up of NRPS’s A-domains (10–40% of identity) and CATH domains | Assembling the predictions from AB and AF sequence similarity searches. The consensus prediction is more sensitive and reliable for detecting A-domain remote homologous. | [21,38] |
| Support Vector Machines (SVM) SVM-kmers (AF) SVM-Auto-cross Covariance (AF) SVM-PseACC (AF) | Subset of SCOP structural classes designed by [92] | SVM-Ensemble weighted voting strategy | [169] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agüero-Chapin, G.; Galpert, D.; Molina-Ruiz, R.; Ancede-Gallardo, E.; Pérez-Machado, G.; De la Riva, G.A.; Antunes, A. Graph Theory-Based Sequence Descriptors as Remote Homology Predictors. Biomolecules 2020, 10, 26. https://doi.org/10.3390/biom10010026
Agüero-Chapin G, Galpert D, Molina-Ruiz R, Ancede-Gallardo E, Pérez-Machado G, De la Riva GA, Antunes A. Graph Theory-Based Sequence Descriptors as Remote Homology Predictors. Biomolecules. 2020; 10(1):26. https://doi.org/10.3390/biom10010026
Chicago/Turabian StyleAgüero-Chapin, Guillermin, Deborah Galpert, Reinaldo Molina-Ruiz, Evys Ancede-Gallardo, Gisselle Pérez-Machado, Gustavo A. De la Riva, and Agostinho Antunes. 2020. "Graph Theory-Based Sequence Descriptors as Remote Homology Predictors" Biomolecules 10, no. 1: 26. https://doi.org/10.3390/biom10010026
APA StyleAgüero-Chapin, G., Galpert, D., Molina-Ruiz, R., Ancede-Gallardo, E., Pérez-Machado, G., De la Riva, G. A., & Antunes, A. (2020). Graph Theory-Based Sequence Descriptors as Remote Homology Predictors. Biomolecules, 10(1), 26. https://doi.org/10.3390/biom10010026