From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data



Abstract
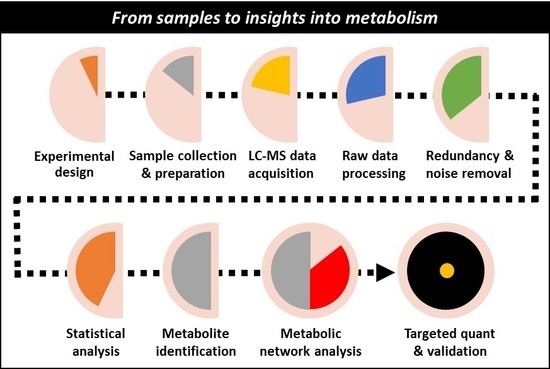
1. Introduction
2. Results
2.1. Considerations for Experimental Design
2.1.1. The Importance of Controls
- (a)
- Positive controls, where changes are expected. These can be used to check that experimental methods are working correctly, and could include a group of subjects (human or animal model) with a known disease, or a specific cell line.
- (b)
- Negative controls, where no change is expected. These can be used to check that unknown variables are not affecting the experiments, which could result in a false-positive conclusion.
- (c)
- Sham controls. These can be used to check effects induced by the procedure or treatment without actual use of the procedure (e.g., gastric bypass) or substance (e.g., drug).
- (d)
- Vehicle controls. These can be used to check effects induced by a solution of the experimental compound, e.g., when a drug is administered in dimethyl sulfoxide (DMSO), the effect of DMSO on its own should be studied.
- (e)
- Comparative controls. These act as a reference which is commonly accepted or an internal control/disease control. In cases where there is a drug treatment, it is important to test a sample of the drug to assess which (if any) signals observed in the metabolic profile arise from the drug, drug metabolites, or degradation products. Extraction blanks enable artefacts and contaminants to be assigned (e.g., from plastic tubes), and are particularly useful when extracting tissue samples.
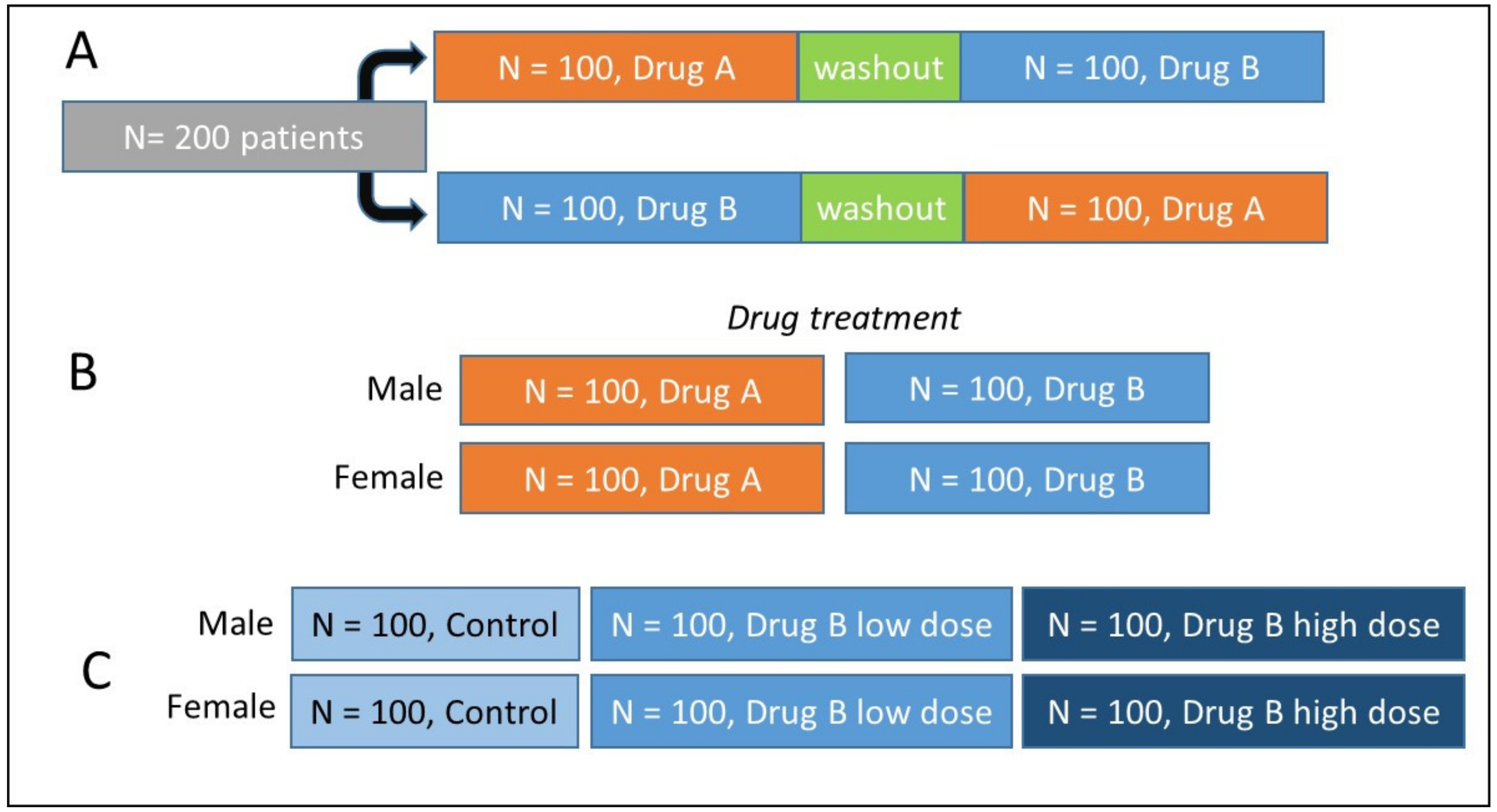
2.1.2. Confounding Factors and Variables
2.1.3. Which Experimental Design to Choose?
2.2. Sample Preparation Approaches
2.3. Data Acquisition Strategies to Facilitate Metabolite Quantification and Identification
2.3.1. LC Techniques
2.3.2. Mass Spectrometry Acquisition Modes
2.4. Data (Pre)Processing: from Peak Detection to Profile Alignment
2.4.1. Software for Data Pre-processing
2.4.2. Important Steps in Data Pre-Processing
2.4.3. Dealing with Artefacts
2.4.4. The Importance of Quality Control
2.5. Univariate and Multivariate Statistical Data Analysis
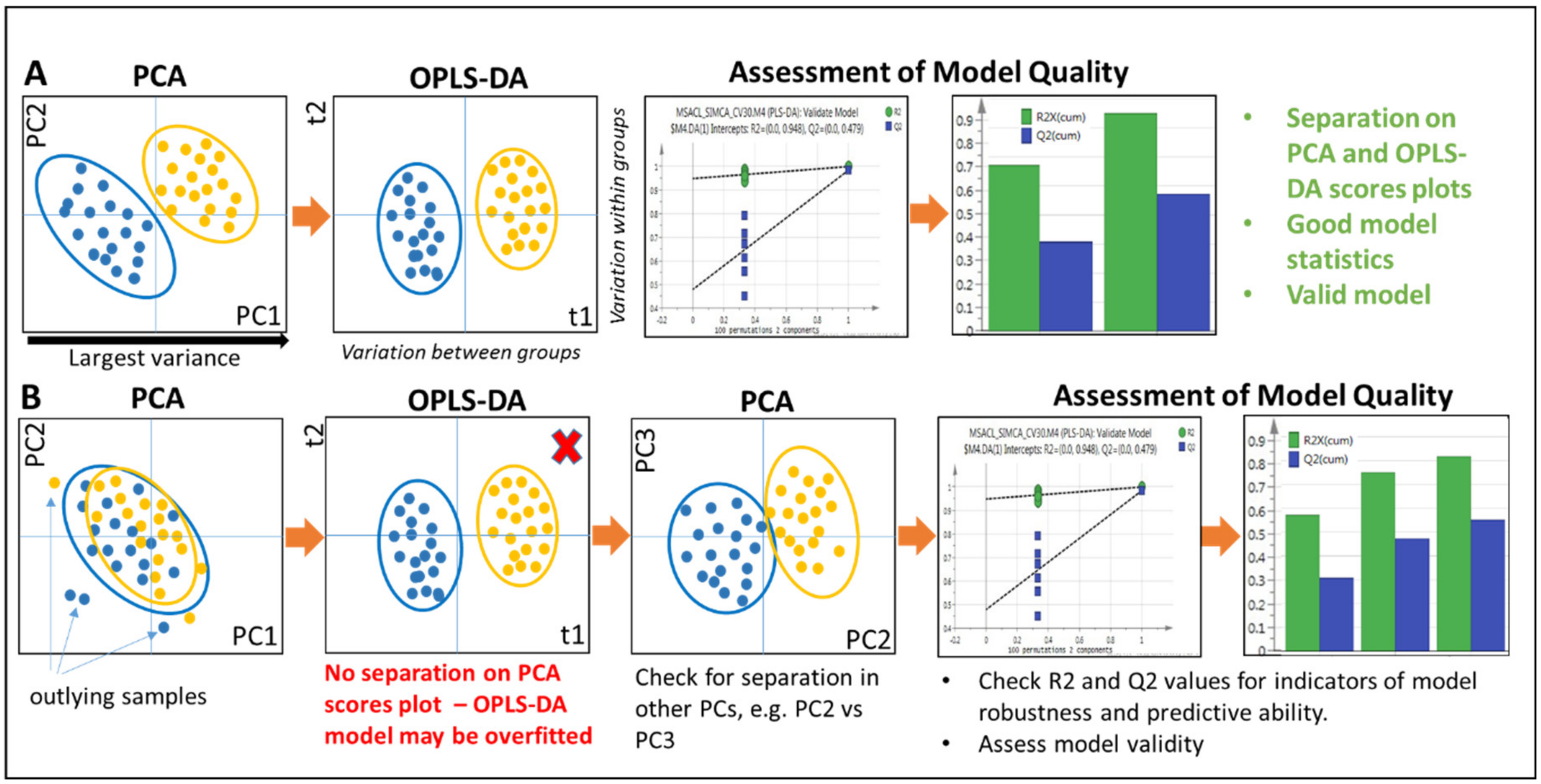
2.5.1. Multivariate Approaches
2.5.2. Principal Components Analysis
2.5.3. Supervised Approaches
2.5.4. Univariate Methods
2.5.5. Multiple Comparison Testing
2.6. Metabolite Identification: From Spectral Database Matching to Computational Approaches for Unknown Metabolite Annotation
2.7. Metabolite Features and/or Metabolites to Pathways and Metabolic Networks
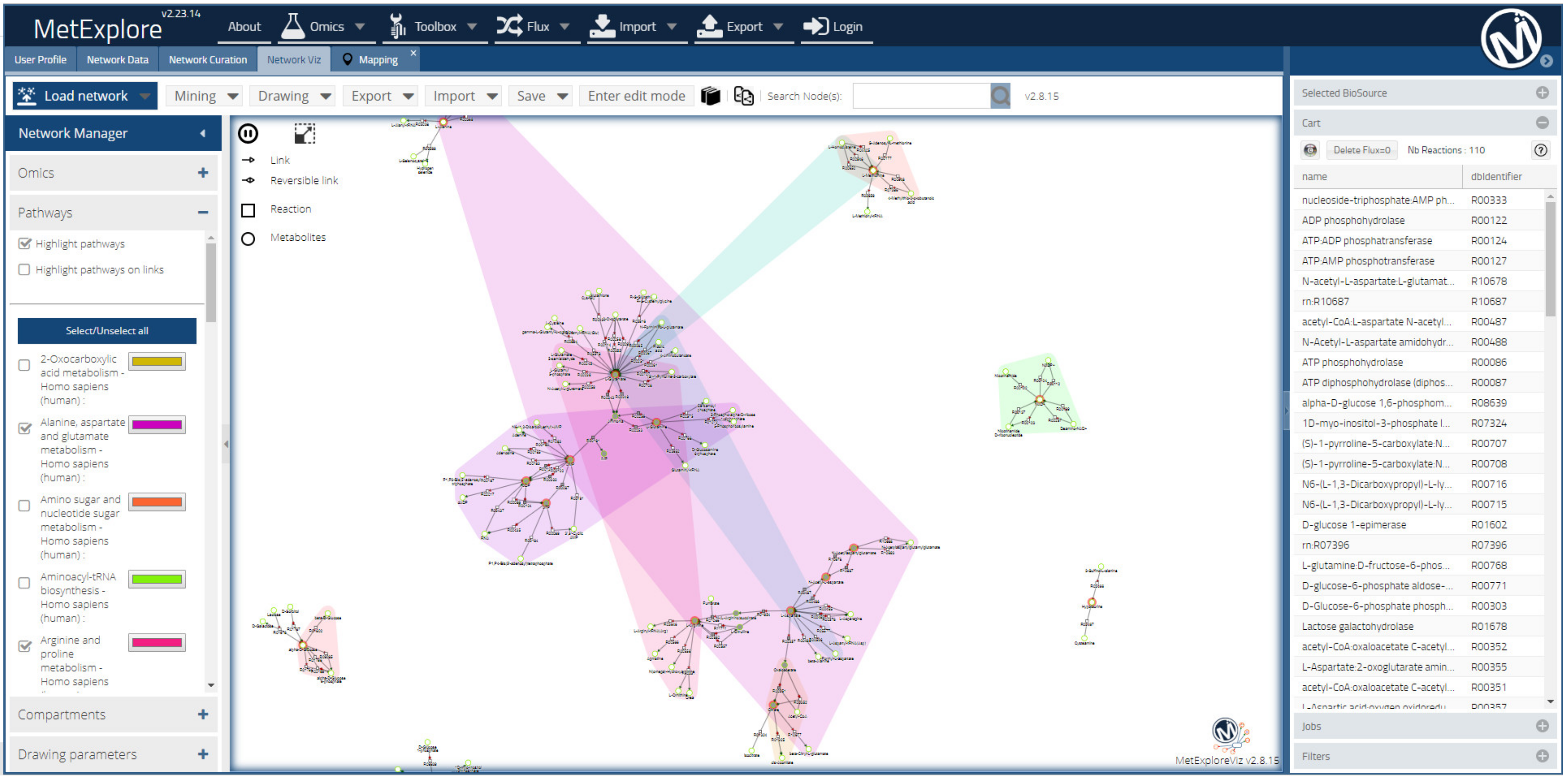
2.7.1. Metabolic Networking for Metabolite Identification
2.7.2. Metabolic Networking to Visualize and Interpret Metabolite Changes
2.8. From Untargeted to Targeted Assays
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patti, G.J.; Tautenhahn, R.; Siuzdak, G. Meta-analysis of untargeted metabolomic data from multiple profiling experiments. Nat. Protoc. 2012, 7, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Hayton, S.; Maker, G.L.; Mullaney, I.; Trengove, R.D. Experimental design and reporting standards for metabolomics studies of mammalian cell lines. Cell. Mol. Life Sci. 2017, 74, 4421–4441. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinformatics 2019, 68, e86. [Google Scholar]
- Blaise, B.J.; Correia, G.; Tin, A.; Young, J.H.; Vergnaud, A.-C.; Lewis, M.; Pearce, J.T.M.; Elliott, P.; Nicholson, J.K.; Holmes, E.; et al. Power Analysis and Sample Size Determination in Metabolic Phenotyping. Anal. Chem. 2016, 88, 5179–5188. [Google Scholar] [CrossRef] [PubMed]
- Nyamundanda, G.; Gormley, I.C.; Fan, Y.; Gallagher, W.M.; Brennan, L. MetSizeR: Selecting the optimal sample size for metabolomic studies using an analysis based approach. BMC Bioinform. 2013, 14, 338. [Google Scholar] [CrossRef]
- Leon, Z.; Garcia-Canaveras, J.C.; Donato, M.T.; Lahoz, A. Mammalian cell metabolomics: Experimental design and sample preparation. Electrophoresis 2013, 34, 2762–2775. [Google Scholar] [CrossRef]
- Jacyna, J.; Kordalewska, M.; Markuszewski, M.J. Design of Experiments in metabolomics-related studies: An overview. J. Pharm. Biomed. Anal. 2019, 164, 598–606. [Google Scholar] [CrossRef]
- Martins, M.C.M.; Caldana, C.; Wolf, L.D.; de Abreu, L.G.F. The Importance of Experimental Design, Quality Assurance, and Control in Plant Metabolomics Experiments. Methods Mol. Biol. 2018, 1778, 3–17. [Google Scholar] [CrossRef]
- Cruickshank-Quinn, C.; Zheng, L.K.; Quinn, K.; Bowler, R.; Reisdorph, R.; Reisdorph, N. Impact of Blood Collection Tubes and Sample Handling Time on Serum and Plasma Metabolome and Lipidome. Metabolites 2018, 8, 88. [Google Scholar] [CrossRef]
- Hernandes, V.V.; Barbas, C.; Dudzik, D. A review of blood sample handling and pre-processing for metabolomics studies. Electrophoresis 2017, 38, 2232–2241. [Google Scholar] [CrossRef] [PubMed]
- Khadka, M.; Todor, A.; Maner-Smith, K.M.; Colucci, J.K.; Tran, V.; Gaul, D.A.; Anderson, E.J.; Natrajan, M.S.; Rouphael, N.; Mulligan, M.J.; et al. The Effect of Anticoagulants, Temperature, and Time on the Human Plasma Metabolome and Lipidome from Healthy Donors as Determined by Liquid Chromatography-Mass Spectrometry. Biomolecules 2019, 9, 200. [Google Scholar] [CrossRef] [PubMed]
- Nishiumi, S.; Suzuki, M.; Kobayashi, T.; Yoshida, M. Differences in metabolite profiles caused by pre-analytical blood processing procedures. J. Biosci. Bioeng. 2018, 125, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Teahan, O.; Gamble, S.; Holmes, E.; Waxman, J.; Nicholson, J.K.; Bevan, C.; Keun, H.C. Impact of analytical bias in metabonomic studies of human blood serum and plasma. Anal. Chem. 2006, 78, 4307–4318. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Maher, A.D.; Want, E.J.; Elliott, P.; Stamler, J.; Hawkes, G.E.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Large-scale human metabolic phenotyping and molecular epidemiological studies via 1H NMR spectroscopy of urine: Investigation of borate preservation. Anal. Chem. 2009, 81, 4847–4856. [Google Scholar] [CrossRef] [PubMed]
- La Frano, M.R.; Carmichael, S.L.; Ma, C.; Hardley, M.; Shen, T.; Wong, R.; Rosales, L.; Borkowski, K.; Pedersen, T.L.; Shaw, G.M.; et al. Impact of post-collection freezing delay on the reliability of serum metabolomics in samples reflecting the California mid-term pregnancy biobank. Metabolomics 2018, 14, 151. [Google Scholar] [CrossRef]
- Jonasdottir, H.S.; Brouwers, H.; Toes, R.E.M.; Ioan-Facsinay, A.; Giera, M. Effects of anticoagulants and storage conditions on clinical oxylipid levels in human plasma. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2018, 1863, 1511–1522. [Google Scholar] [CrossRef]
- Gibon, Y.; Rolin, D. Aspects of experimental design for plant metabolomics experiments and guidelines for growth of plant material. Methods Mol. Biol. 2012, 860, 13–30. [Google Scholar] [CrossRef]
- DeBoer, M.D.; Platts-Mills, J.A.; Scharf, R.J.; McDermid, J.M.; Wanjuhi, A.W.; Gratz, J.; Svensen, E.; Swann, J.R.; Donowitz, J.R.; Jatosh, S.; et al. Early Life Interventions for Childhood Growth and Development in Tanzania (ELICIT): A protocol for a randomised factorial, double-blind, placebo-controlled trial of azithromycin, nitazoxanide and nicotinamide. BMJ Open 2018, 8, e021817. [Google Scholar] [CrossRef]
- Khan, S.R.; Whiteman, D.C.; Kimlin, M.G.; Janda, M.; Clarke, M.W.; Lucas, R.M.; Neale, R.E. Effect of solar ultraviolet radiation exposure on serum 25(OH)D concentration: A pilot randomised controlled trial. Photochem. Photobiol. Sci. 2018, 17, 570–577. [Google Scholar] [CrossRef]
- Roager, H.M.; Vogt, J.K.; Kristensen, M.; Hansen, L.B.S.; Ibrugger, S.; Maerkedahl, R.B.; Bahl, M.I.; Lind, M.V.; Nielsen, R.L.; Frokiaer, H.; et al. Whole grain-rich diet reduces body weight and systemic low-grade inflammation without inducing major changes of the gut microbiome: A randomised cross-over trial. Gut 2019, 68, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.G.; Hu, J.; Wu, X.; Xu, Y.J. The Recent Developments in Sample Preparation for Mass Spectrometry-Based Metabolomics. Crit. Rev. Anal. Chem. 2017, 47, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Patejko, M.; Jacyna, J.; Markuszewski, M.J. Sample preparation procedures utilized in microbial metabolomics: An overview. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1043, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Deda, O.; Chatziioannou, A.C.; Fasoula, S.; Palachanis, D.; Raikos, N.; Theodoridis, G.A.; Gika, H.G. Sample preparation optimization in fecal metabolic profiling. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1047, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Drouin, N.; Rudaz, S.; Schappler, J. Sample preparation for polar metabolites in bioanalysis. Analyst 2017, 143, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite Measurement: Pitfalls to Avoid and Practices to Follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef]
- Li, N.; Song, Y.; Tang, H.; Wang, Y. Recent developments in sample preparation and data pre-treatment in metabonomics research. Arch. Biochem. Biophys. 2016, 589, 4–9. [Google Scholar] [CrossRef]
- Chetwynd, A.J.; Dunn, W.B.; Rodriguez-Blanco, G. Collection and Preparation of Clinical Samples for Metabolomics. Adv. Exp. Med. Biol. 2017, 965, 19–44. [Google Scholar] [CrossRef]
- Masson, P.; Alves, A.C.; Ebbels, T.M.; Nicholson, J.K.; Want, E.J. Optimization and evaluation of metabolite extraction protocols for untargeted metabolic profiling of liver samples by UPLC-MS. Anal. Chem. 2010, 82, 7779–7786. [Google Scholar] [CrossRef]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using UPLC-MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, L.; Stahlman, M.; Forsberg, G.B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Löfgren, L.; Forsberg, G.-B.; Ståhlman, M. The BUME method: A new rapid and simple chloroform-free method for total lipid extraction of animal tissue. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Zhang, W.; Wolters, J.C.; Permentier, H.; Boer, T.; Horvatovich, P.; Heiner-Fokkema, M.R.; Reijngoud, D.-J.; Bischoff, R. One- vs two-phase extraction: Re-evaluation of sample preparation procedures for untargeted lipidomics in plasma samples. Anal. Bioanal. Chem. 2018, 410, 5859–5870. [Google Scholar] [CrossRef] [PubMed]
- Sarafian, M.H.; Gaudin, M.; Lewis, M.R.; Martin, F.-P.; Holmes, E.; Nicholson, J.K.; Dumas, M.-E. Objective Set of Criteria for Optimization of Sample Preparation Procedures for Ultra-High Throughput Untargeted Blood Plasma Lipid Profiling by Ultra Performance Liquid Chromatography–Mass Spectrometry. Anal. Chem. 2014, 86, 5766–5774. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. Trends Anal. Chem. 2014, 61, 192–206. [Google Scholar] [CrossRef]
- Vorkas, P.A.; Isaac, G.; Anwar, M.A.; Davies, A.H.; Want, E.J.; Nicholson, J.K.; Holmes, E. Untargeted UPLC-MS Profiling Pipeline to Expand Tissue Metabolome Coverage: Application to Cardiovascular Disease. Anal. Chem. 2015, 87, 4184–4193. [Google Scholar] [CrossRef]
- Want, E.J.; Nordstrom, A.; Morita, H.; Siuzdak, G. From exogenous to endogenous: The inevitable imprint of mass spectrometry in metabolomics. J. Proteom. Res. 2007, 6, 459–468. [Google Scholar] [CrossRef]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016. [Google Scholar] [CrossRef]
- Fuhrer, T.; Heer, D.; Begemann, B.; Zamboni, N. High-Throughput, Accurate Mass Metabolome Profiling of Cellular Extracts by Flow Injection–Time-of-Flight Mass Spectrometry. Anal. Chem. 2011, 83, 7074–7080. [Google Scholar] [CrossRef]
- Zamboni, N.; Saghatelian, A.; Patti, G.J. Defining the Metabolome: Size, Flux, and Regulation. Mol. Cell 2015, 58, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, C.; Han, X. Tutorial on lipidomics. Anal. Chim. Acta 2019, 1061, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Duan, Q.; Han, X. Strategies to Improve/Eliminate the Limitations in Shotgun Lipidomics. Proteomics 2019. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Smilowitz, J.T.; Fiehn, O. Validating Quantitative Untargeted Lipidomics Across Nine Liquid Chromatography-High-Resolution Mass Spectrometry Platforms. Anal. Chem. 2017, 89, 12360–12368. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.J. Separation strategies for untargeted metabolomics. J. Sep. Sci. 2011, 34, 3460–3469. [Google Scholar] [CrossRef]
- Benton, H.P.; Ivanisevic, J.; Mahieu, N.G.; Kurczy, M.E.; Johnson, C.H.; Franco, L.; Rinehart, D.; Valentine, E.; Gowda, H.; Ubhi, B.K.; et al. Autonomous Metabolomics for Rapid Metabolite Identification in Global Profiling. Anal. Chem. 2015, 87, 884–891. [Google Scholar] [CrossRef]
- Ivanisevic, J.; Zhu, Z.-J.; Plate, L.; Tautenhahn, R.; Chen, S.; O’Brien, P.J.; Johnson, C.H.; Marletta, M.A.; Patti, G.J.; Siuzdak, G. Toward Omic Scale Metabolite Profiling: A Dual Separation—Mass Spectrometry Approach for Coverage of Lipid and Central Carbon Metabolism. Anal. Chem. 2013, 85, 6876–6884. [Google Scholar] [CrossRef]
- Yanes, O.; Tautenhahn, R.; Patti, G.J.; Siuzdak, G. Expanding Coverage of the Metabolome for Global Metabolite Profiling. Anal. Chem. 2011, 83, 2152–2161. [Google Scholar] [CrossRef]
- Lu, W.; Clasquin, M.F.; Melamud, E.; Amador-Noguez, D.; Caudy, A.A.; Rabinowitz, J.D. Metabolomic Analysis via Reversed-Phase Ion-Pairing Liquid Chromatography Coupled to a Stand Alone Orbitrap Mass Spectrometer. Anal. Chem. 2010, 82, 3212–3221. [Google Scholar] [CrossRef]
- Gallart-Ayala, H.; Konz, I.; Mehl, F.; Teav, T.; Oikonomidi, A.; Peyratout, G.; van der Velpen, V.; Popp, J.; Ivanisevic, J. A global HILIC-MS approach to measure polar human cerebrospinal fluid metabolome: Exploring gender-associated variation in a cohort of elderly cognitively healthy subjects. Anal. Chim. Acta 2018, 1037, 327–337. [Google Scholar] [CrossRef]
- Wernisch, S.; Pennathur, S. Evaluation of coverage, retention patterns, and selectivity of seven liquid chromatographic methods for metabolomics. Anal. Bioanal. Chem. 2016, 408, 6079–6091. [Google Scholar] [CrossRef]
- Naz, S.; Gallart-Ayala, H.; Reinke, S.N.; Mathon, C.; Blankley, R.; Chaleckis, R.; Wheelock, C.E. Development of a Liquid Chromatography-High Resolution Mass Spectrometry Metabolomics Method with High Specificity for Metabolite Identification Using All Ion Fragmentation Acquisition. Anal. Chem. 2017, 89, 7933–7942. [Google Scholar] [CrossRef]
- Cífková, E.; Holčapek, M.; Lísa, M.; Ovčačíková, M.; Lyčka, A.; Lynen, F.; Sandra, P. Nontargeted Quantitation of Lipid Classes Using Hydrophilic Interaction Liquid Chromatography–Electrospray Ionization Mass Spectrometry with Single Internal Standard and Response Factor Approach. Anal. Chem. 2012, 84, 10064–10070. [Google Scholar] [CrossRef]
- Fei, F.; Bowdish, D.M.; McCarry, B.E. Comprehensive and simultaneous coverage of lipid and polar metabolites for endogenous cellular metabolomics using HILIC-TOF-MS. Anal. Bioanal. Chem. 2014, 406, 3723–3733. [Google Scholar] [CrossRef]
- Teav, T.; Gallart-Ayala, H.; van der Velpen, V.; Mehl, F.; Henry, H.; Ivanisevic, J. Merged Targeted Quantification and Untargeted Profiling for Comprehensive Assessment of Acylcarnitine and Amino Acid Metabolism. Anal. Chem. 2019, 91, 11757–11769. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. LC-MS-Based Lipidomics and Automated Identification of Lipids Using the LipidBlast In-Silico MS/MS Library. Methods Mol. Biol. 2017, 1609, 149–170. [Google Scholar] [CrossRef]
- Rinehart, D.; Johnson, C.H.; Nguyen, T.; Ivanisevic, J.; Benton, H.P.; Lloyd, J.; Arkin, A.P.; Deutschbauer, A.M.; Patti, G.J.; Siuzdak, G. Metabolomic data streaming for biology-dependent data acquisition. Nat. Biotech. 2014, 32, 524–527. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef]
- Shen, X.; Wang, R.; Xiong, X.; Yin, Y.; Cai, Y.; Ma, Z.; Liu, N.; Zhu, Z.J. Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat. Commun. 2019, 10, 1516. [Google Scholar] [CrossRef]
- Hu, Y.; Cai, B.; Huan, T. Enhancing metabolome coverage in data-dependent LC-MS/MS analysis through an integrated feature extraction strategy. Anal. Chem. 2019, 91, 14433–14441. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Meth. 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Koelmel, J.P.; Kroeger, N.M.; Gill, E.L.; Ulmer, C.Z.; Bowden, J.A.; Patterson, R.E.; Yost, R.A.; Garrett, T.J. Expanding Lipidome Coverage Using LC-MS/MS Data-Dependent Acquisition with Automated Exclusion List Generation. J. Am. Soc. Mass Spectrom. 2017, 28, 908–917. [Google Scholar] [CrossRef]
- Wang, Y.; Feng, R.; Wang, R.; Yang, F.; Li, P.; Wan, J.B. Enhanced MS/MS coverage for metabolite identification in LC-MS-based untargeted metabolomics by target-directed data dependent acquisition with time-staggered precursor ion list. Anal. Chim. Acta 2017, 992, 67–75. [Google Scholar] [CrossRef]
- Li, H.; Cai, Y.; Guo, Y.; Chen, F.; Zhu, Z.J. MetDIA: Targeted Metabolite Extraction of Multiplexed MS/MS Spectra Generated by Data-Independent Acquisition. Anal. Chem. 2016, 88, 8757–8764. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, R.; Cai, Y.; Wang, Z.; Zhu, Z.J. DecoMetDIA: Deconvolution of Multiplexed MS/MS Spectra for Metabolite Identification in SWATH-MS-Based Untargeted Metabolomics. Anal. Chem. 2019, 91, 11897–11904. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828. [Google Scholar] [CrossRef]
- Aguilar-Mogas, A.; Sales-Pardo, M.; Navarro, M.; Guimera, R.; Yanes, O. iMet: A Network-Based Computational Tool To Assist in the Annotation of Metabolites from Tandem Mass Spectra. Anal. Chem. 2017, 89, 3474–3482. [Google Scholar] [CrossRef]
- Kind, T.; Liu, K.H.; Lee, D.Y.; DeFelice, B.; Meissen, J.K.; Fiehn, O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat. Methods 2013, 10, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Duhrkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Bocker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef]
- Paglia, G.; Astarita, G. Metabolomics and lipidomics using traveling-wave ion mobility mass spectrometry. Nat. Protoc. 2017, 12, 797–813. [Google Scholar] [CrossRef]
- Blazenovic, I.; Kind, T.; Ji, J.; Fiehn, O. Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 2018, 8, 31. [Google Scholar] [CrossRef]
- Zha, H.; Cai, Y.; Yin, Y.; Wang, Z.; Li, K.; Zhu, Z.J. SWATHtoMRM: Development of High-Coverage Targeted Metabolomics Method Using SWATH Technology for Biomarker Discovery. Anal. Chem. 2018, 90, 4062–4070. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Ivanisevic, J.; Thomas, A.; Sidibé, J.; Teav, T.; Guijas, C.; Aisporna, A.E.; Rinehart, D.; Hoang, L.; et al. XCMS-MRM and METLIN-MRM: A cloud library and public resource for targeted analysis of small molecules. Nat. Methods 2018, 15, 681–684. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Bottcher, C.; Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Gowda, H.; Ivanisevic, J.; Johnson, C.H.; Kurczy, M.E.; Benton, H.P.; Rinehart, D.; Nguyen, T.; Ray, J.; Kuehl, J.; Arevalo, B.; et al. Interactive XCMS Online: Simplifying Advanced Metabolomic Data Processing and Subsequent Statistical Analyses. Anal. Chem. 2014, 86, 6931–6939. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [PubMed]
- Tugizimana, F.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. A Conversation on Data Mining Strategies in LC-MS Untargeted Metabolomics: Pre-Processing and Pre-Treatment Steps. Metabolites 2016, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Gross, T.; Mapstone, M.; Miramontes, R.; Padilla, R.; Cheema, A.K.; Macciardi, F.; Federoff, H.J.; Fiandaca, M.S. Toward Reproducible Results from Targeted Metabolomic Studies: Perspectives for Data Pre-processing and a Basis for Analytic Pipeline Development. Curr. Top. Med. Chem. 2018, 18, 883–895. [Google Scholar] [CrossRef]
- Myers, O.D.; Sumner, S.J.; Li, S.; Barnes, S.; Du, X. Detailed Investigation and Comparison of the XCMS and MZmine 2 Chromatogram Construction and Chromatographic Peak Detection Methods for Preprocessing Mass Spectrometry Metabolomics Data. Anal. Chem. 2017, 89, 8689–8695. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Benton, H.P.; Siuzdak, G. Annotation: A Computational Solution for Streamlining Metabolomics Analysis. Anal. Chem. 2018, 90, 480–489. [Google Scholar] [CrossRef]
- Domingo-Almenara, X.; Montenegro-Burke, J.R.; Guijas, C.; Majumder, E.L.W.; Benton, H.P.; Siuzdak, G. Autonomous METLIN-Guided In-source Fragment Annotation for Untargeted Metabolomics. Anal. Chem. 2019, 91, 3246–3253. [Google Scholar] [CrossRef]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2011, 84, 283–289. [Google Scholar] [CrossRef]
- Alonso, A.; Julia, A.; Beltran, A.; Vinaixa, M.; Diaz, M.; Ibanez, L.; Correig, X.; Marsal, S. AStream: An R package for annotating LC/MS metabolomic data. Bioinformatics 2011, 27, 1339–1340. [Google Scholar] [CrossRef]
- Broeckling, C.D.; Afsar, F.A.; Neumann, S.; Ben-Hur, A.; Prenni, J.E. RAMClust: A novel feature clustering method enables spectral-matching-based annotation for metabolomics data. Anal. Chem. 2014, 86, 6812–6817. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Sangster, T.; Major, H.; Plumb, R.; Wilson, A.J.; Wilson, I.D. A pragmatic and readily implemented quality control strategy for HPLC-MS and GC-MS-based metabonomic analysis. Analyst 2006, 131, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Dunn, W.B.; Bandukwala, A.; Bethan, B.; Broadhurst, D.; Clish, C.B.; Dasari, S.; Derr, L.; Evans, A.; Fischer, S.; et al. Towards quality assurance and quality control in untargeted metabolomics studies. Metabolomics 2019, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Dudzik, D.; Barbas-Bernardos, C.; Garcia, A.; Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. a review. J. Pharm. Biomed. Anal. 2018, 147, 149–173. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef]
- Worley, B.; Powers, R. Multivariate Analysis in Metabolomics. Curr Metab. 2013, 1, 92–107. [Google Scholar] [CrossRef]
- Liland, K.H. Multivariate methods in metabolomics—From pre-processing to dimension reduction and statistical analysis. TrAC Trends Anal. Chem. 2011, 30, 827–841. [Google Scholar] [CrossRef]
- Kjeldahl, K.; Bro, R. Some common misunderstandings in chemometrics. J. Chemom. 2010, 24, 558–564. [Google Scholar] [CrossRef]
- Eriksson, L.; Trygg, J.; Wold, S. CV-ANOVA for significance testing of PLS and OPLS® models. J. Chemom. 2008, 22, 594–600. [Google Scholar] [CrossRef]
- Rubingh, C.M.; Bijlsma, S.; Derks, E.P.; Bobeldijk, I.; Verheij, E.R.; Kochhar, S.; Smilde, A.K. Assessing the performance of statistical validation tools for megavariate metabolomics data. Metabolomics 2006, 2, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Do, K.T.; Wahl, S.; Raffler, J.; Molnos, S.; Laimighofer, M.; Adamski, J.; Suhre, K.; Strauch, K.; Peters, A.; Gieger, C.; et al. Characterization of missing values in untargeted MS-based metabolomics data and evaluation of missing data handling strategies. Metabolomics 2018, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, M.M.W.B.; Eeuwijk, F.A.V.; Jellema, R.H.; Westerhuis, J.A.; Reijmers, T.H.; Hoefsloot, H.C.J.; Smilde, A.K. Data-processing strategies for metabolomics studies. TrAC Trends Anal. Chem. 2011, 30, 1685–1698. [Google Scholar] [CrossRef]
- Tzoulaki, I.; Ebbels, T.M.; Valdes, A.; Elliott, P.; Ioannidis, J.P. Design and analysis of metabolomics studies in epidemiologic research: A primer on -omic technologies. Am. J. Epidemiol. 2014, 180, 129–139. [Google Scholar] [CrossRef]
- Vinaixa, M.; Samino, S.; Saez, I.; Duran, J.; Guinovart, J.J.; Yanes, O. A Guideline to Univariate Statistical Analysis for LC/MS-Based Untargeted Metabolomics-Derived Data. Metabolites 2012, 2, 775–795. [Google Scholar] [CrossRef]
- Da Silva, R.R.; Dorrestein, P.C.; Quinn, R.A. Illuminating the dark matter in metabolomics. Proc. Natl. Acad. Sci. USA 2015, 112, 12549–12550. [Google Scholar] [CrossRef]
- Dias, D.A.; Jones, O.A.; Beale, D.J.; Boughton, B.A.; Benheim, D.; Kouremenos, K.A.; Wolfender, J.L.; Wishart, D.S. Current and Future Perspectives on the Structural Identification of Small Molecules in Biological Systems. Metabolites 2016, 6, 46. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Cho, K.; Uritboonthai, W.; Zhu, Z.; Patti, G.J.; Siuzdak, G. An accelerated workflow for untargeted metabolomics using the METLIN database. Nat. Biotechnol. 2012, 30, 826–828. [Google Scholar] [CrossRef]
- Vinaixa, M.; Schymanski, E.L.; Neumann, S.; Navarro, M.; Salek, R.M.; Yanes, O. Mass spectral databases for LC/MS- and GC/MS-based metabolomics: State of the field and future prospects. TrAC Trends Anal. Chem. 2016, 78, 23–35. [Google Scholar] [CrossRef]
- Mylonas, R.; Mauron, Y.; Masselot, A.; Binz, P.A.; Budin, N.; Fathi, M.; Viette, V.; Hochstrasser, D.F.; Lisacek, F. X-Rank: A robust algorithm for small molecule identification using tandem mass spectrometry. Anal. Chem. 2009, 81, 7604–7610. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Nguyen, C.H.; Mamitsuka, H. Recent advances and prospects of computational methods for metabolite identification: A review with emphasis on machine learning approaches. Brief. Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Neumann, S.; Posch, S. Improving MetFrag with statistical learning of fragment annotations. BMC Bioinform. 2019, 20, 376. [Google Scholar] [CrossRef] [PubMed]
- Blazenovic, I.; Kind, T.; Sa, M.R.; Ji, J.; Vaniya, A.; Wancewicz, B.; Roberts, B.S.; Torbasinovic, H.; Lee, T.; Mehta, S.S.; et al. Structure Annotation of All Mass Spectra in Untargeted Metabolomics. Anal. Chem. 2019, 91, 2155–2162. [Google Scholar] [CrossRef] [PubMed]
- Van der Hooft, J.J.J.; Wandy, J.; Barrett, M.P.; Burgess, K.E.V.; Rogers, S. Topic modeling for untargeted substructure exploration in metabolomics. Proc. Natl. Acad. Sci. USA 2016, 113, 13738–13743. [Google Scholar] [CrossRef] [PubMed]
- Van der Hooft, J.J.J.; Wandy, J.; Young, F.; Padmanabhan, S.; Gerasimidis, K.; Burgess, K.E.V.; Barrett, M.P.; Rogers, S. Unsupervised Discovery and Comparison of Structural Families Across Multiple Samples in Untargeted Metabolomics. Anal. Chem. 2017, 89, 7569–7577. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Azam, K.; Vadivelu, I.; Burant, C.; Edison, A.; Fiehn, O.; Higashi, R.; Nair, K.S.; et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016, 44, D463–D470. [Google Scholar] [CrossRef]
- Steinbeck, C.; Conesa, P.; Haug, K.; Mahendraker, T.; Williams, M.; Maguire, E.; Rocca-Serra, P.; Sansone, S.A.; Salek, R.M.; Griffin, J.L. MetaboLights: towards a new COSMOS of metabolomics data management. Metabolomics. 2012, 8, 757–760. [Google Scholar] [CrossRef]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef]
- Pirhaji, L.; Milani, P.; Leidl, M.; Curran, T.; Avila-Pacheco, J.; Clish, C.B.; White, F.M.; Saghatelian, A.; Fraenkel, E. Revealing disease-associated pathways by network integration of untargeted metabolomics. Nat. Methods 2016, 13, 770–776. [Google Scholar] [CrossRef]
- Rosato, A.; Tenori, L.; Cascante, M.; De Atauri Carulla, P.R.; Martins dos Santos, V.A.P.; Saccenti, E. From correlation to causation: Analysis of metabolomics data using systems biology approaches. Metabolomics 2018, 14, 37. [Google Scholar] [CrossRef] [PubMed]
- Frainay, C.; Schymanski, E.L.; Neumann, S.; Merlet, B.; Salek, R.M.; Jourdan, F.; Yanes, O. Mind the Gap: Mapping Mass Spectral Databases in Genome-Scale Metabolic Networks Reveals Poorly Covered Areas. Metabolites 2018, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Edison, A.S.; Hall, R.D.; Junot, C.; Karp, P.D.; Kurland, I.J.; Mistrik, R.; Reed, L.K.; Saito, K.; Salek, R.M.; Steinbeck, C.; et al. The Time Is Right to Focus on Model Organism Metabolomes. Metabolites 2016, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.K.; Baer, C.F.; Edison, A.S. Considerations when choosing a genetic model organism for metabolomics studies. Curr. Opin. Chem. Biol. 2017, 36, 7–14. [Google Scholar] [CrossRef]
- Yilmaz, L.S.; Walhout, A.J.M. Metabolic network modeling with model organisms. Curr. Opin. Chem. Biol. 2017, 36, 32–39. [Google Scholar] [CrossRef]
- Milreu, P.V.; Klein, C.C.; Cottret, L.; Acuna, V.; Birmele, E.; Borassi, M.; Junot, C.; Marchetti-Spaccamela, A.; Marino, A.; Stougie, L.; et al. Telling metabolic stories to explore metabolomics data: A case study on the yeast response to cadmium exposure. Bioinformatics 2014, 30, 61–70. [Google Scholar] [CrossRef]
- Stanstrup, J.; Broeckling, C.D.; Helmus, R.; Hoffmann, N.; Mathe, E.; Naake, T.; Nicolotti, L.; Peters, K.; Rainer, J.; Salek, R.M.; et al. The metaRbolomics Toolbox in Bioconductor and beyond. Metabolites 2019, 9, 200. [Google Scholar] [CrossRef]
- Cottret, L.; Frainay, C.; Chazalviel, M.; Cabanettes, F.; Gloaguen, Y.; Camenen, E.; Merlet, B.; Heux, S.; Portais, J.C.; Poupin, N.; et al. MetExplore: Collaborative edition and exploration of metabolic networks. Nucleic Acids Res. 2018, 46, W495–W502. [Google Scholar] [CrossRef]
- Kutmon, M.; van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.R.; Evelo, C.T. PathVisio 3: An extendable pathway analysis toolbox. PLoS Comput. Biol. 2015, 11, e1004085. [Google Scholar] [CrossRef]
- Yamada, T.; Letunic, I.; Okuda, S.; Kanehisa, M.; Bork, P. iPath2.0: Interactive pathway explorer. Nucleic Acids Res. 2011, 39, W412–W415. [Google Scholar] [CrossRef]
- Wishart, D.S.; Li, C.; Marcu, A.; Badran, H.; Pon, A.; Budinski, Z.; Patron, J.; Lipton, D.; Cao, X.; Oler, E.; et al. PathBank: A comprehensive pathway database for model organisms. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, M.R.; Jeucken, A.; Wassenaar, T.A.; van de Lest, C.H.A.; Brouwers, J.F.; Helms, J.B. LION/web: A web-based ontology enrichment tool for lipidomic data analysis. GigaScience 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Forsberg, E.M.; Rinehart, D.; Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Fang, M.; Aisporna, A.; Hilmers, B.; Poole, F.L.; et al. Systems biology guided by XCMS Online metabolomics. Nat. Methods 2017, 14, 461. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, G.; Haldiya, P.K.; Willighagen, E.; Kind, T.; Fiehn, O. The Chemical Translation Service--a web-based tool to improve standardization of metabolomic reports. Bioinformatics 2010, 26, 2647–2648. [Google Scholar] [CrossRef]
- Cottret, L.; Wildridge, D.; Vinson, F.; Barrett, M.P.; Charles, H.; Sagot, M.F.; Jourdan, F. MetExplore: A web server to link metabolomic experiments and genome-scale metabolic networks. Nucleic Acids Res. 2010, 38, W132–W137. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, 25. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Goeman, J.; van de Geer, S.; Kort, F.; van Houwelingen, J. A global test for groups of genes: Testing association with a clinical outcome. Bioinformatics 2004, 20, 93–99. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Fabregat, A.; Korninger, F.; Viteri, G.; Sidiropoulos, K.; Marin-Garcia, P.; Ping, P.; Wu, G.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome graph database: Efficient access to complex pathway data. PLoS Comput. Biol. 2018, 14, e1005968. [Google Scholar] [CrossRef]
- Bohler, A.; Wu, G.; Kutmon, M.; Pradhana, L.A.; Coort, S.L.; Hanspers, K.; Haw, R.; Pico, A.R.; Evelo, C.T. Reactome from a WikiPathways Perspective. PLoS Comput. Biol. 2016, 12, e1004941. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes-a 2019 update. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Noronha, A.; Danielsdottir, A.D.; Gawron, P.; Johannsson, F.; Jonsdottir, S.; Jarlsson, S.; Gunnarsson, J.P.; Brynjolfsson, S.; Schneider, R.; Thiele, I.; et al. ReconMap: An interactive visualization of human metabolism. Bioinformatics 2017, 33, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Daníelsdóttir, A.D.; Krecke, M.; Merten, D.; Haraldsdóttir, H.S.; et al. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 2019, 47, D614–D624. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef]
- Kaza, M.; Karaźniewicz-Łada, M.; Kosicka, K.; Siemiątkowska, A.; Rudzki, P.J. Bioanalytical method validation: new FDA guidance vs. EMA guideline. Better or worse? J. Pharm. Biomed. Anal. 2019, 165, 381–385. [Google Scholar] [CrossRef]
- Egertson, J.D.; MacLean, B.; Johnson, R.; Xuan, Y.; MacCoss, M.J. Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat. Protoc. 2015, 10, 887–903. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS/MS Data Acquisition Mode | Selection of Precursor Ions | Advantage | Pitfall |
|---|---|---|---|
| Selective or targeted MS/MS | Only selected ions specified on an inclusion list will be targeted | Highest quality MS/MS data | A posteriori acquisition, in a separate batch of analyses |
| Data-Dependent Acquisition (DDA) | Ions are selected for MS/MS acquisition in real-time based on threshold intensity: Top «n» ions are «picked» in each scan Preferred list and exclusion list | High-quality MS/MS data and established link between precursor and product ions | High acquisition rates required. Selection of the most highly abundant ions each time, across multiple scans, resulting in low MS/MS coverage |
| Data-Independent Acquisition (DIA) | All fragment ions for all precursors are acquired simultaneously: All-ion-fragmentation (Q1 transmits the full mass range, 50–1700 Da of precursor ions in the collision cell: AIF, MSE) or with sequential mass windows (Q1 transmits several increments of 20–50 amu across the mass range in the collision cell: SWATH, SONAR, BASIC DIA—see Figure 2) | Improved coverage for low abundant precursor ions | High acquisition rates required. Difficulty of MS/MS data deconvolution to re-establish the link between the precursor and product ions |
| Parameter | Criteria | Outcome | Notes |
|---|---|---|---|
| Coefficient of variation (CV) | Choose threshold of variation, e.g., of metabolite peak area in repeated injections of QC sample | Remove metabolite features, e.g., with CV > 30% in QC samples * | CV cut-off values may be dependent on sample type, chromatography, or instrument parameters |
| Presence in study samples | Metabolite feature/peak must be present in a certain proportion of the study samples (and/or QCs) | Remove metabolite features present in only a low proportion of study samples | Certain peaks may only be present in one class of samples—adjust threshold accordingly |
| Presence in blank samples | Metabolite feature/peak must not be present in study samples/at very low levels | Remove metabolite features present in blank samples | Some metabolite features may be present in blank samples due to carryover—ensure multiple blanks have been run to address this |
| Response to dilution | Metabolite feature/peak must respond to dilution series with r2 > 0.8** | Remove metabolite features with r2 < 0.8 ** | Some metabolite features may be saturated at higher concentrations and so do not behave linearly—check raw data |
| Bottleneck | Cause | Solutions |
|---|---|---|
| Known metabolite (mis)identification | Isomers or metabolites with identical mass (and molecular formula) but different structures |
|
| Isobars or compounds of similar molecular weight produce interferences |
| |
| In-source fragments—due to production of ions (by loss of H2O, CO2, H3PO4) that have the same mass and/or structure as the molecular ions of other metabolites |
| |
| Unknown metabolite identification | “Known unknowns”—metabolites listed in molecular structure databases but without recorded reference MS/MS spectra in spectral libraries |
|
| “Unknown unknowns”—new metabolites not listed in any database |
|
| Tool | Functionalities |
| MeTexplore web server [128] |
|
| Pathvisio [129] |
|
| iPath—Interactive Pathways Explorer [130] |
|
| MetaboAnalyst* web server [91] |
|
| PathBank [131] |
|
| LION/web [132] |
|
| XCMS online* [133] |
|
| Database | Functionalities |
|---|---|
| KEGG database and pathway browser [139] |
|
| Reactome database and pathway browser [140,141] |
|
| Cyc databases (EcoCyc, HumanCyc, MetaCyc, BioCyc) [142] |
|
| Recon database [143,144] Virtual metabolic human |
|
| WikiPathways database [145] |
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanisevic, J.; Want, E.J. From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data. Metabolites 2019, 9, 308. https://doi.org/10.3390/metabo9120308
Ivanisevic J, Want EJ. From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data. Metabolites. 2019; 9(12):308. https://doi.org/10.3390/metabo9120308
Chicago/Turabian StyleIvanisevic, Julijana, and Elizabeth J. Want. 2019. "From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data" Metabolites 9, no. 12: 308. https://doi.org/10.3390/metabo9120308
APA StyleIvanisevic, J., & Want, E. J. (2019). From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data. Metabolites, 9(12), 308. https://doi.org/10.3390/metabo9120308