Interlaboratory Coverage Test on Plant Food Bioactive Compounds and Their Metabolites by Mass Spectrometry-Based Untargeted Metabolomics

 , , ,
, , ,  ,
,  , ,
, ,  , ,
, ,  ,
,  add
Show full author list
add
Show full author list
Abstract
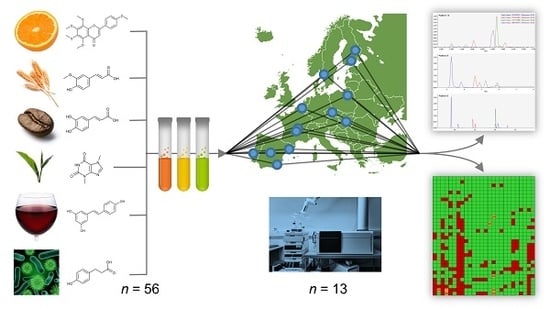
1. Introduction
2. Results and Discussion
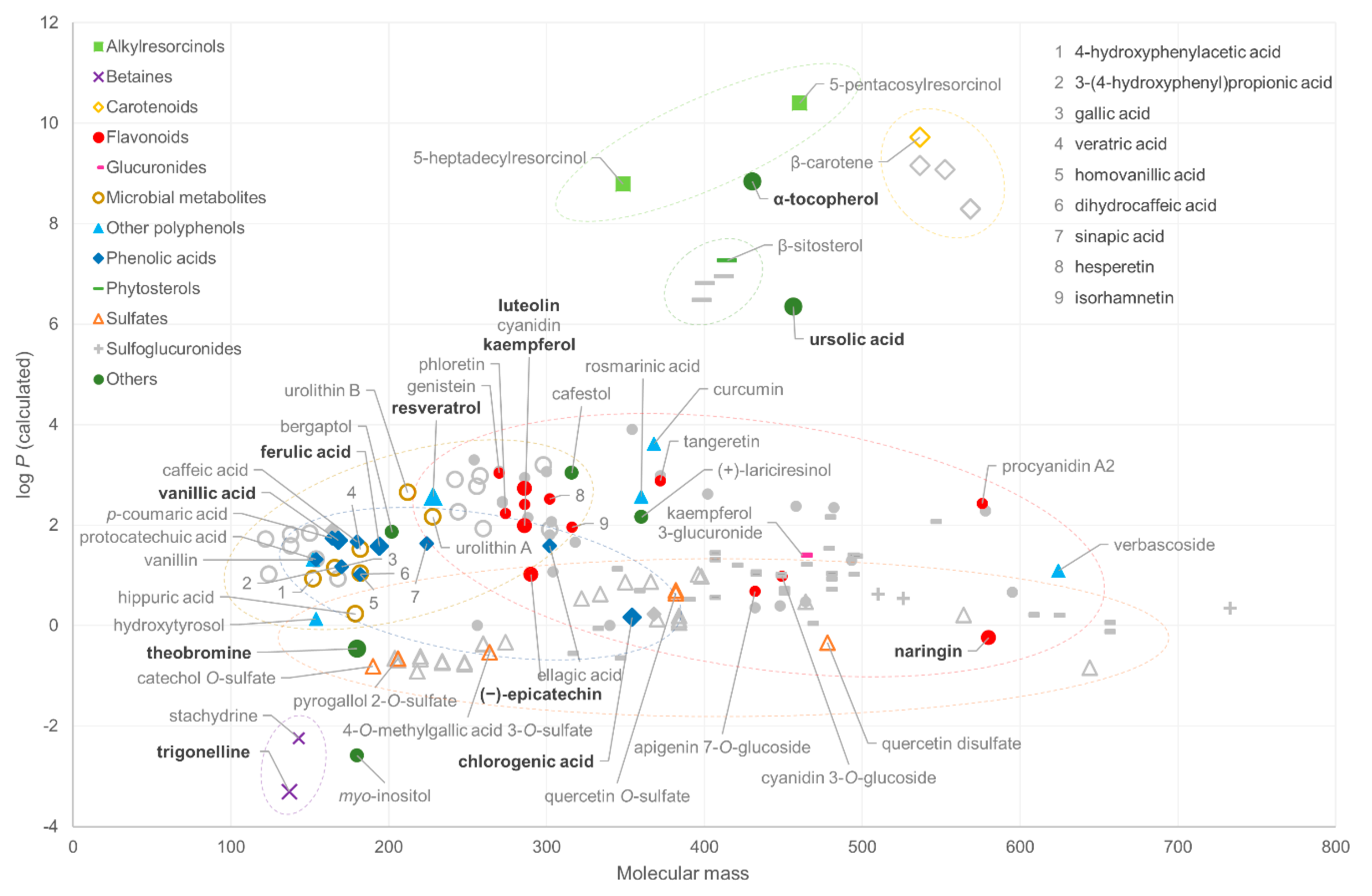
2.1. Chemical Space
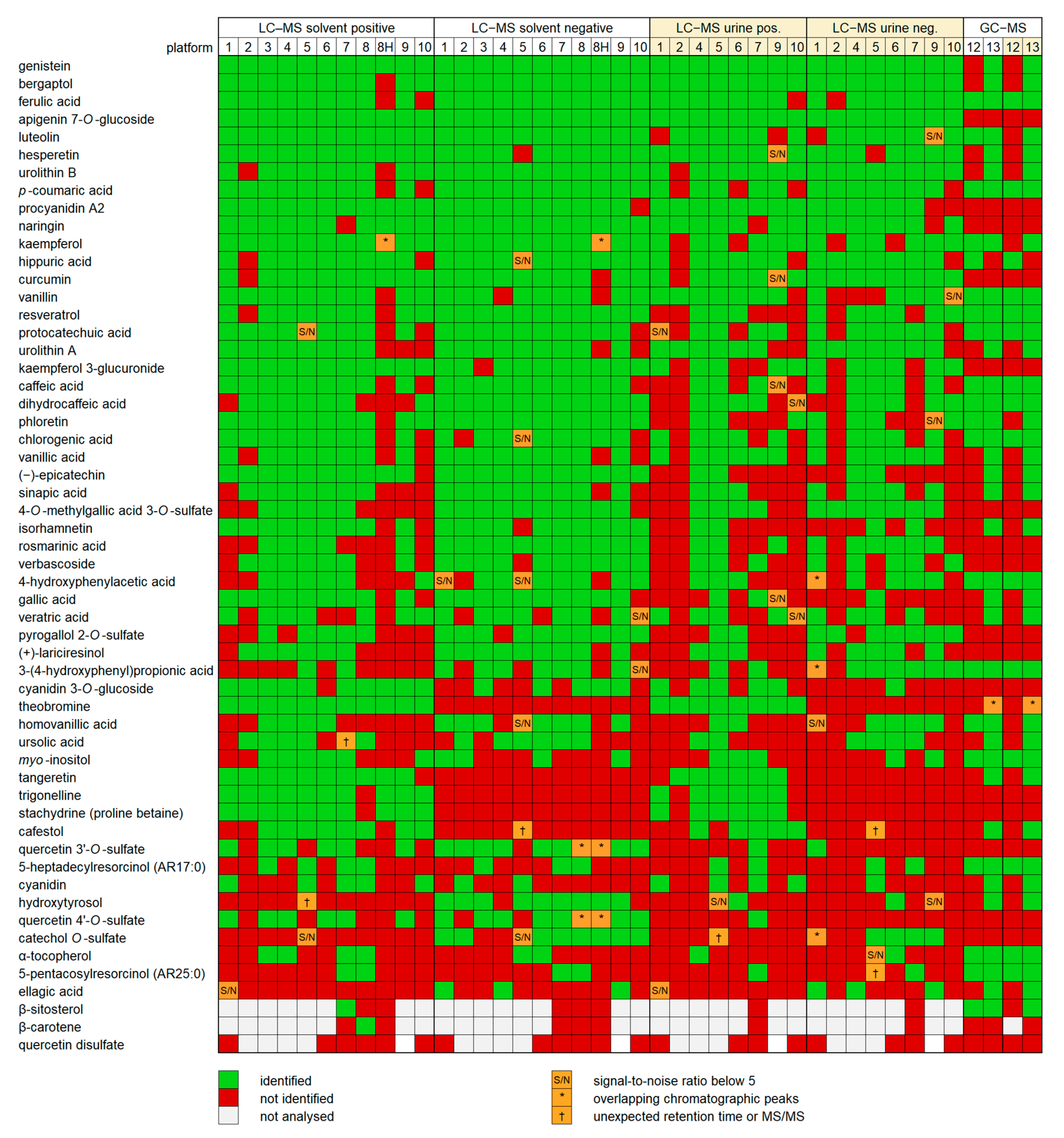
2.2. Detection of Compounds
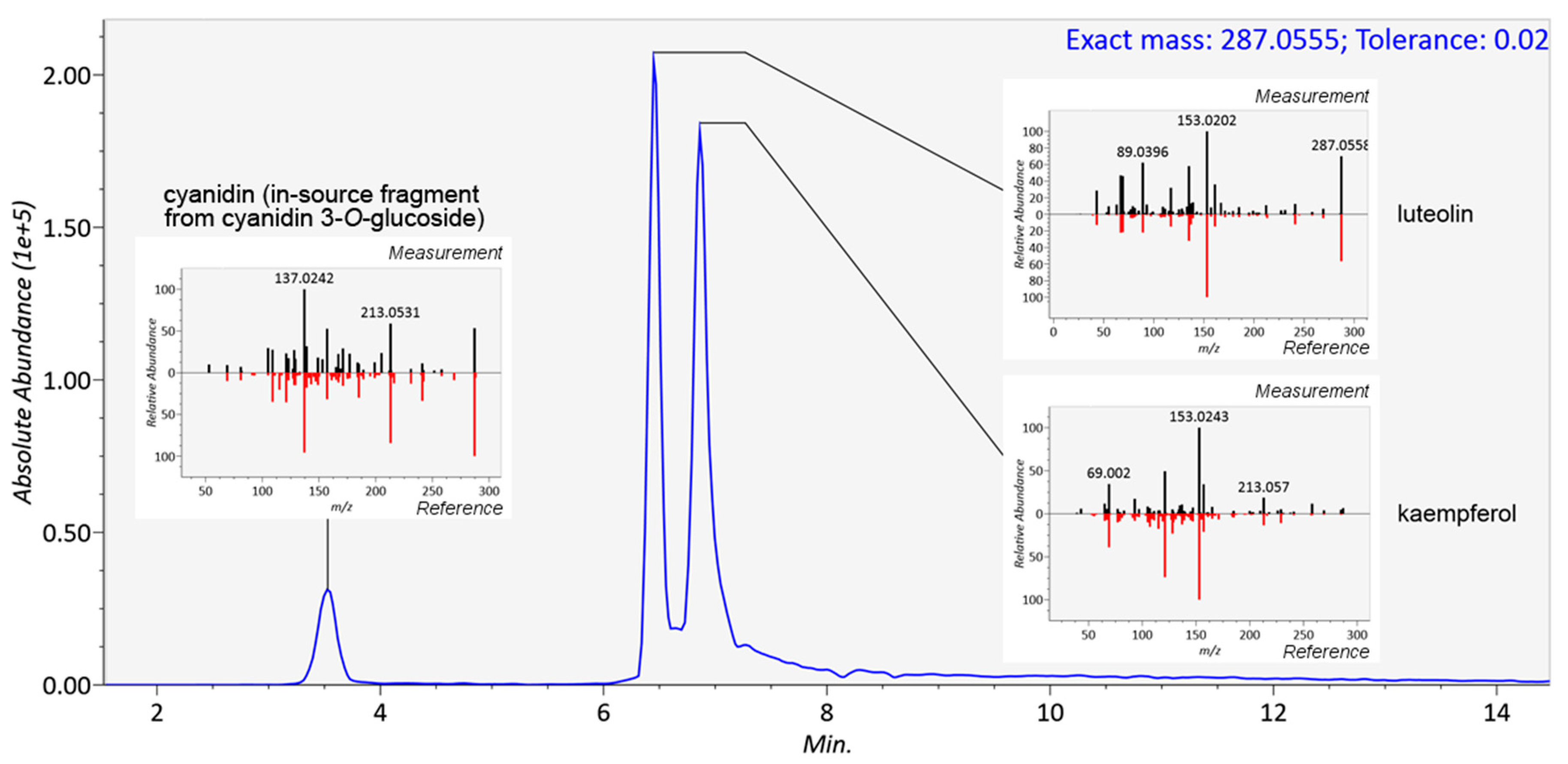
2.3. Reliability of Identifications
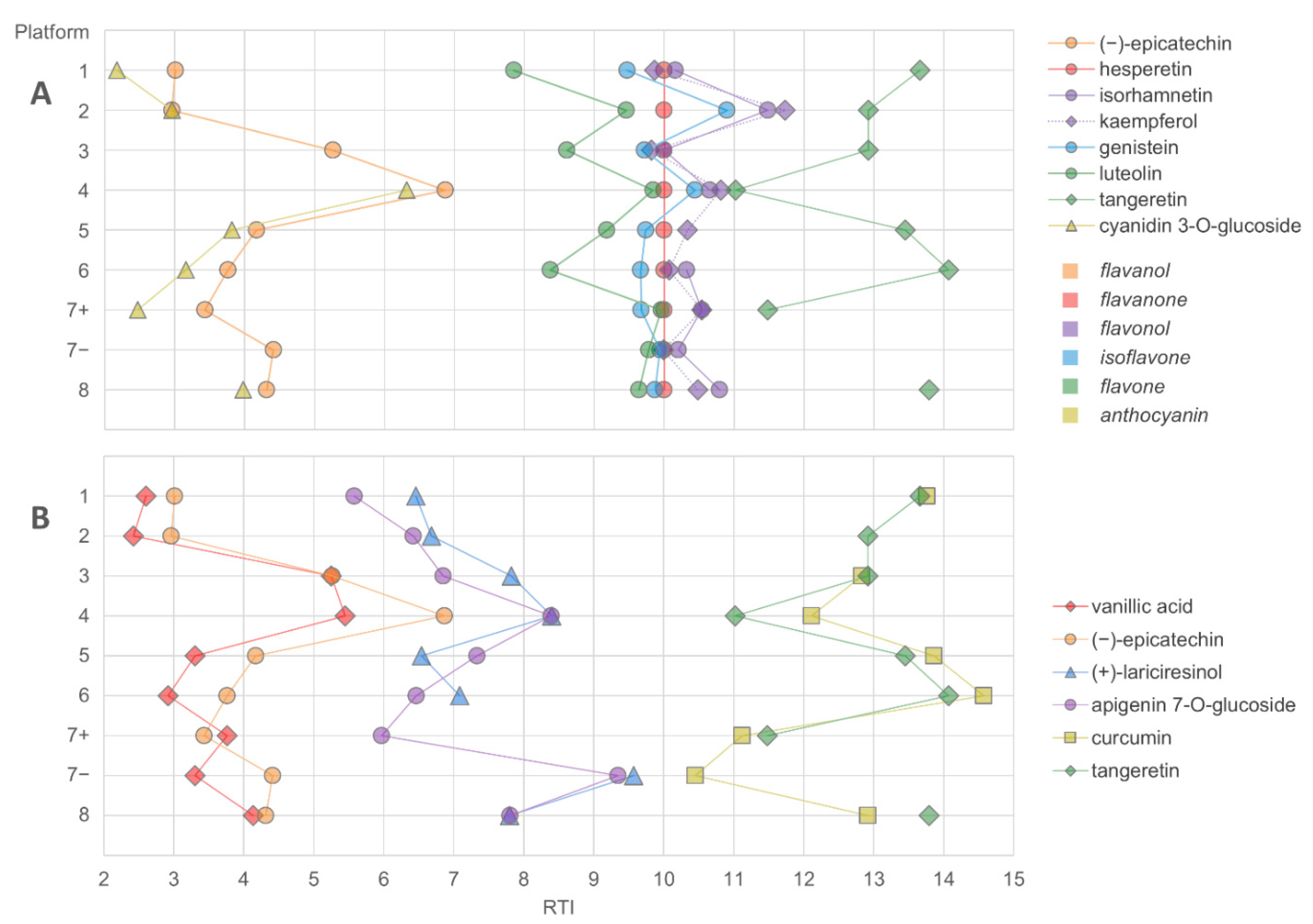
2.4. Considerations on the Optimal Coverage of Plant Bioactives
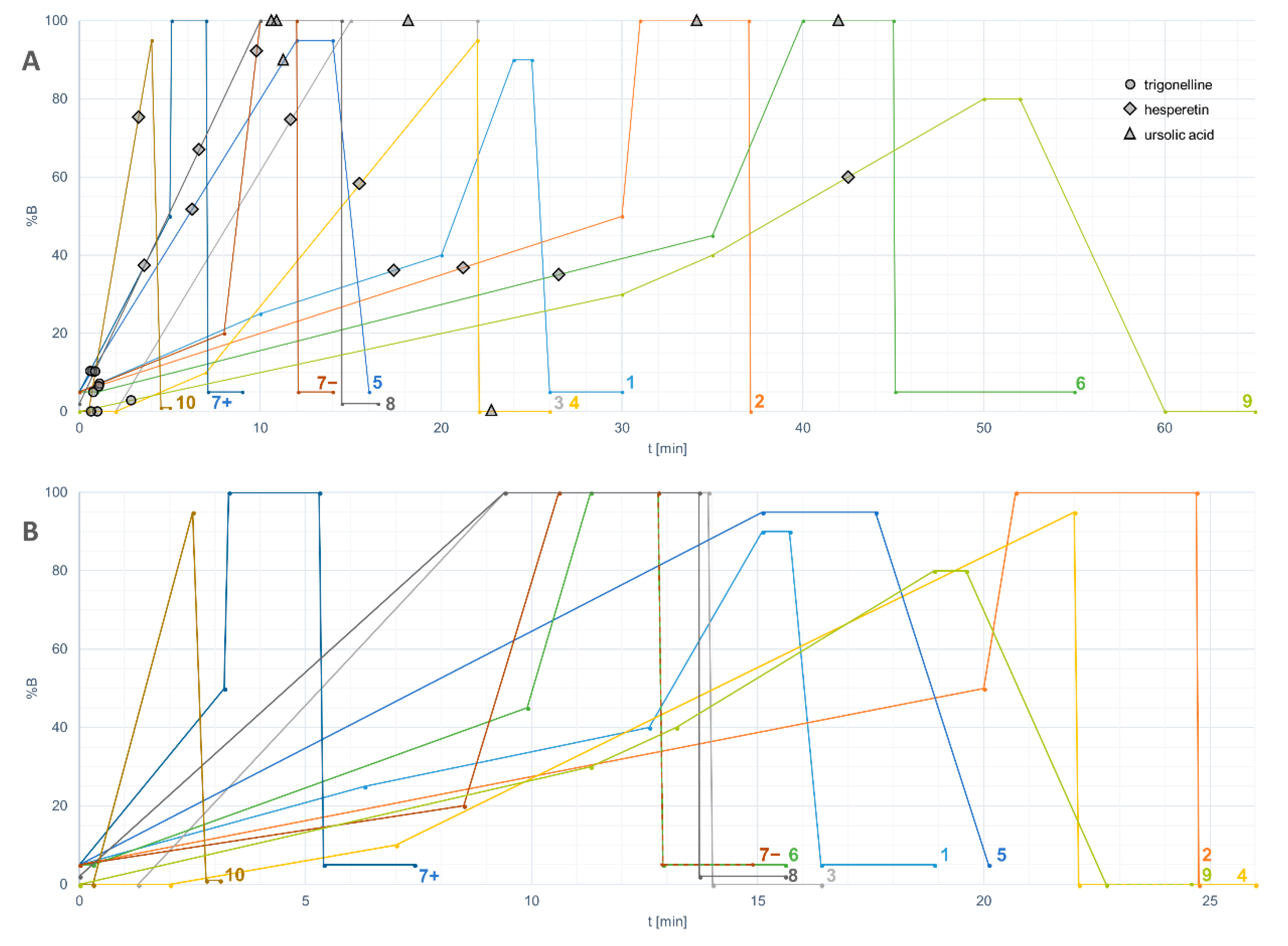
- The retention capability of the method for highly polar compounds can be tested by compounds such as theobromine and trigonelline. The suitability of the initial solvent composition and injection volume of the HPLC method should be optimized based on the results of these test runs. In an optimal setting, these two hydrophilic compounds (or at least theobromine) should elute after the solvent peak.
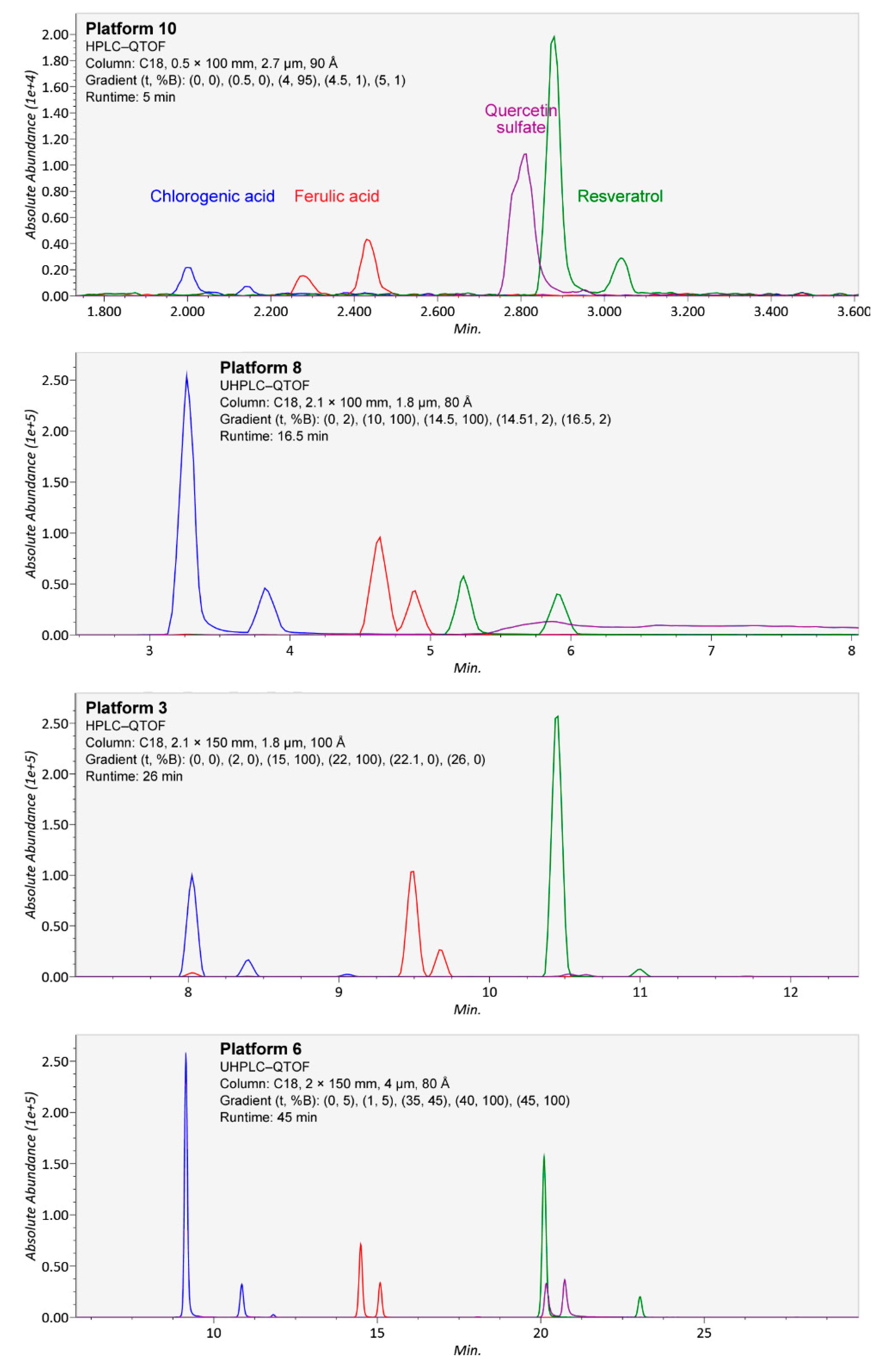
- The chromatographic resolution of the method should be challenged by analyzing critical peak pairs. For instance, vanillic acid and (−)-epicatechin are good candidates to test the chromatographic resolution in the initial separation phase. Kaempferol and luteolin are good choices for the mid-polarity range. Stricter evaluation can be performed by using the cis and trans epimers of ferulic acid or resveratrol.
- The performance of the method for highly non-polar compounds can be tested by ursolic acid and α-tocopherol. These two lipophilic compounds should elute before the solvent gradient with the highest %B (percentage of the organic solvent) reaches the column end.
- α-tocopherol
- chlorogenic acid
- (−)-epicatechin
- ferulic acid
- kaempferol
- luteolin
- naringin
- resveratrol
- theobromine
- trigonelline
- ursolic acid
- vanillic acid
3. Materials and Methods
3.1. Reference Standard Mixtures
3.2. Sample Preparation and Analysis with Mass Spectrometry
3.3. Data Analysis and Compound Identification
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wishart, D.S. Metabolomics: Applications to food science and nutrition research. Trends Food Sci. Technol. 2008, 19, 482–493. [Google Scholar] [CrossRef]
- Del-Castillo-Alonso, M.; Castagna, A.; Csepregi, K.; Hideg, E.; Jakab, G.; Jansen, M.A.K.; Jug, T.; Llorens, L.; Matai, A.; Martinez-Luescher, J.; et al. Environmental Factors Correlated with the Metabolite Profile of Vitis vinifera cv. Pinot Noir Berry Skins along a European Latitudinal Gradient. J. Agric. Food Chem. 2016, 64, 8722–8734. [Google Scholar] [CrossRef] [PubMed]
- Creydt, M.; Fischer, M. Omics approaches for food authentication. Electrophoresis 2018, 39, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Dragsted, L.O.; Gao, Q.; Scalbert, A.; Vergères, G.; Kolehmainen, M.; Manach, C.; Brennan, L.; Afman, L.A.; Wishart, D.S.; Andres Lacueva, C.; et al. Validation of biomarkers of food intake-critical assessment of candidate biomarkers. Genes Nutr. 2018, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Allwood, J.W.; Goodacre, R. An introduction to liquid chromatography–mass spectrometry instrumentation applied in plant metabolomic analyses. Phytochem. Anal. 2010, 21, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Kaspar, S.; Peukert, M.; Svatos, A.; Matros, A.; Mock, H. MALDI-imaging mass spectrometry—An emerging technique in plant biology. Proteomics 2011, 11, 1840–1850. [Google Scholar] [CrossRef] [PubMed]
- Kueger, S.; Steinhauser, D.; Willmitzer, L.; Giavalisco, P. High-resolution plant metabolomics: From mass spectral features to metabolites and from whole-cell analysis to subcellular metabolite distributions. Plant J. 2012, 70, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Guo, J.; Chen, S.; Liu, X.; Zhou, Y.; Zhang, X.; Xu, X. Recent developments in qualitative and quantitative analysis of phytochemical constituents and their metabolites using liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2013, 72, 267–291. [Google Scholar] [CrossRef] [PubMed]
- Koistinen, V.M.; Hanhineva, K. Mass Spectrometry-based Analysis of Whole Grain Phytochemicals. Crit. Rev. Food Sci. Nutr. 2015, 57, 1688–1709. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, A.; Venter, P.; Pasch, H. Recent advances and trends in the liquid-chromatography-mass spectrometry analysis of flavonoids. J. Chromatogr. A 2016, 1430, 16–78. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Brennan, L.; Fiehn, O.; Hankemeier, T.; Kristal, B.S.; van Ommen, B.; Pujos-Guillot, E.; Verheij, E.; Wishart, D.; Wopereis, S. Mass-spectrometry-based metabolomics: Limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics 2009, 5, 435–458. [Google Scholar] [CrossRef] [PubMed]
- De Vos, R.C.; Moco, S.; Lommen, A.; Keurentjes, J.J.; Bino, R.J.; Hall, R.D. Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2007, 2, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Motilva, M.; Serra, A.; Macià, A. Analysis of food polyphenols by ultra high-performance liquid chromatography coupled to mass spectrometry: An overview. J. Chromatogr. A 2013, 1292, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Willför, S.M.; Smeds, A.I.; Holmbom, B.R. Chromatographic analysis of lignans. J. Chromatogr. A 2006, 1112, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Feliciano, R.P.; Mecha, E.; Bronze, M.R.; Rodriguez-Mateos, A. Development and validation of a high-throughput micro solid-phase extraction method coupled with ultra-high-performance liquid chromatography-quadrupole time-of-flight mass spectrometry for rapid identification and quantification of phenolic metabolites in human plasma and urine. J. Chromatogr. A 2016, 1464, 21–31. [Google Scholar] [PubMed]
- Abidi, S.L. Chromatographic analysis of plant sterols in foods and vegetable oils. J. Chromatogr. A 2001, 935, 173–201. [Google Scholar] [CrossRef]
- Lerma-García, M.J.; Simó-Alfonso, E.F.; Méndez, A.; Lliberia, J.L.; Herrero-Martínez, J.M. Fast separation and determination of sterols in vegetable oils by ultraperformance liquid chromatography with atmospheric pressure chemical ionization mass spectrometry detection. J. Agric. Food Chem. 2010, 58, 2771–2776. [Google Scholar] [CrossRef] [PubMed]
- Amorim-Carrilho, K.T.; Cepeda, A.; Fente, C.; Regal, P. Review of methods for analysis of carotenoids. Trends Anal. Chem. 2014, 56, 49–73. [Google Scholar] [CrossRef]
- Urpi-Sarda, M.; Monagas, M.; Khan, N.; Llorach, R.; Lamuela-Raventós, R.M.; Jáuregui, O.; Estruch, R.; Izquierdo-Pulido, M.; Andrés-Lacueva, C. Targeted metabolic profiling of phenolics in urine and plasma after regular consumption of cocoa by liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2009, 1216, 7258–7267. [Google Scholar] [CrossRef] [PubMed]
- Sang, S.; Lee, M.; Yang, I.; Buckley, B.; Yang, C.S. Human urinary metabolite profile of tea polyphenols analyzed by liquid chromatography/electrospray ionization tandem mass spectrometry with data-dependent acquisition. Rapid Commun. Mass Spectrom. 2008, 22, 1567–1578. [Google Scholar] [CrossRef] [PubMed]
- Hanhineva, K.; Keski-Rahkonen, P.; Lappi, J.; Katina, K.; Pekkinen, J.; Savolainen, O.; Timonen, O.; Paananen, J.; Mykkänen, H.; Poutanen, K. The postprandial plasma rye fingerprint includes benzoxazinoid-derived phenylacetamide sulfates. J. Nutr. 2014, 144, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.B.; Svelander, C.; Savolainen, O.I.; Lind, M.V.; Kirwan, J.P.; Breton, I.; Godin, J.; Sandberg, A. A high-throughput method for liquid chromatography–tandem mass spectrometry determination of plasma alkylresorcinols, biomarkers of whole grain wheat and rye intake. Anal. Biochem. 2016, 499, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Hernández, L.; Castro-Puyana, M.; Luisa Marina, M.; Crego, A.L. Determination of betaines in vegetable oils by capillary electrophoresis tandem mass spectrometry–application to the detection of olive oil adulteration with seed oils. Electrophoresis 2011, 32, 1394–1401. [Google Scholar] [CrossRef] [PubMed]
- Vrhovsek, U.; Masuero, D.; Gasperotti, M.; Franceschi, P.; Caputi, L.; Viola, R.; Mattivi, F. A versatile targeted metabolomics method for the rapid quantification of multiple classes of phenolics in fruits and beverages. J. Agric. Food Chem. 2012, 60, 8831–8840. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Brennan, L.; Manach, C.; Andres-Lacueva, C.; Dragsted, L.O.; Draper, J.; Rappaport, S.M.; van der Hooft, J.J.; Wishart, D.S. The food metabolome: A window over dietary exposure. Am. J. Clin. Nutr. 2014, 99, 1286–1308. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, H.; O’Gorman, A.; Brennan, L. Metabolomics as a tool in nutritional research. Curr. Opin. Lipidol. 2015, 26, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Huerta, O.D.; Gil, A. Nutrimetabolomics: An Update on Analytical Approaches to Investigate the Role of Plant-Based Foods and Their Bioactive Compounds in Non-Communicable Chronic Diseases. Int. J. Mol. Sci. 2016, 17, 2072. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.J.; García-Villalba, R.; Garrido, Y.; Gil, M.I.; Tomás-Barberán, F.A. Untargeted metabolomics approach using UPLC-ESI-QTOF-MS to explore the metabolome of fresh-cut iceberg lettuce. Metabolomics 2016, 12, 138. [Google Scholar] [CrossRef]
- Koistinen, V.M.; Mattila, O.; Katina, K.; Poutanen, K.; Aura, A.M.; Hanhineva, K. Metabolic profiling of sourdough fermented wheat and rye bread. Sci. Rep. 2018, 8, 5684. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Xiao, J.F.; Tuli, L.; Ressom, H.W. LC-MS-based metabolomics. Mol. BioSyst. 2012, 8, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Valentová, K.; Káňová, K.; Di Meo, F.; Pelantová, H.; Chambers, C.S.; Rydlová, L.; Petrásková, L.; Křenková, A.; Cvačka, J.; Trouillas, P. Chemoenzymatic Preparation and Biophysical Properties of Sulfated Quercetin Metabolites. Int. J. Mol. Sci. 2017, 18, 2231. [Google Scholar] [CrossRef] [PubMed]
- De Rijke, E.; Out, P.; Niessen, W.M.; Ariese, F.; Gooijer, C.; Udo, A.T. Analytical separation and detection methods for flavonoids. J. Chromatogr. A 2006, 1112, 31–63. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.; Fiehn, O.; Goodacre, R.; Griffin, J.L. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Clifford, M.N.; Kirkpatrick, J.; Kuhnert, N.; Roozendaal, H.; Salgado, P.R. LC–MS n analysis of the cis isomers of chlorogenic acids. Food Chem. 2008, 106, 379–385. [Google Scholar] [CrossRef]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Tanchuk, V.Y. Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci. 2002, 42, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plat-Form | General Method(s) | HPLC/GC Model | Column (Dimensions, Particle Size, Pore Size) | MS | Ion Source | HPLC Mobile Phase | HPLC Flow | HPLC Gradient (t [min], %B) |
|---|---|---|---|---|---|---|---|---|
| 1 | UHPLC–QTOF | Agilent 1290 | Agilent Poroshell 120 EC-C18 (3 × 100 mm, 2.7 µm, 120 Å) | Agilent 6550 | ESI+/− | A: H2O + 0.1% FA, B: ACN + 0.1% FA | 0.4 mL/min | (0, 5), (10, 25), (20, 40), (24, 90), (25, 90), (26, 5), (30, 5) |
| 2 | UHPLC–TOF | Acquity H-class | Acquity UPLC BEH Shield RP18 (2.1 × 150 mm, 1.7 µm, 130 Å) | Synapt G2 S | ESI+/− | A: H2O + 0.1% FA, B: ACN + 0.1% FA | 0.35 mL/min | (0, 5), (30, 50), (31, 100), (37, 100), (37.1, 0) |
| 3 | HPLC–QTOF | Thermo U3000 | Acquity HSS T3 (2.1 × 150 mm, 1.8 µm, 100 Å) | Bruker Impact HD2 | ESI+/− | A: H2O + 0.1% FA, B: ACN + 0.1% FA | 0.4 mL/min | (0, 0), (2, 0), (15, 100), (22, 100), (22.1, 0), (26, 0) |
| 4 | HPLC–QTOF | Thermo U3000 | Acquity UPLC BEH Shield RP18 (2.1 × 100 mm, 1.7 µm, 130 Å) | Bruker Impact HD2 | ESI+/− | A: H2O + 0.1% FA, B: ACN + 0.1% FA | 0.4 mL/min | (0, 0), (2, 0), (7, 10), (22, 95), (22.1, 0), (26, 0) |
| 5 | UHPLC–QTOF | Eksigent nanoLC | Eksigent HALO C18 (0.5 × 50 mm, 2.7 µm, 90 Å) | Sciex Triple-TOF 6600 | ESI+/− | A: H2O + 0.1% FA, B: ACN + 0.1% FA | 10 µL/min | (0, 5), (12, 95), (14, 95), (16, 5) |
| 6 | UHPLC–QTOF | Agilent 1260 | Phenomenex Synergi Hydro-RP (2 × 150 mm, 4 µm, 80 Å) | Agilent 6530 | ESI+/− | A: H2O + 0.1% FA, B: ACN + 0.1% FA | 0.5 mL/min | (0, 5), (1, 5), (35, 45), (40, 100), (45, 100) |
| 7+ | UHPLC−QTRAP | Agilent 1290 | Luna Omega Polar C18 (2.1 × 100 mm, 1.6 µm, 100 Å) | Sciex 6500 | ESI+ | A: H2O + 0.5% FA, B: ACN + 0.5% FA | 0.5 mL/min | (0, 5), (5, 50), (5.1, 100), (7, 100), (7.1, 5), (9, 5) |
| 7− | UHPLC−QTRAP | Agilent 1290 | Luna Omega Polar C18 (2.1 × 100 mm, 1.6 µm, 100 Å) | Sciex 6500 | ESI− | A: H2O + 0.1% FA + 10 mM NH4COOH, B: ACN | “ | (0, 5), (8, 20), (10, 100), (12, 100), (12.1, 5), (14, 5) |
| 8 | UHPLC–QTOF | Agilent 1290 | Agilent Zorbax Eclipse XDB-C18 (2.1 × 100 mm, 1.8 µm, 80 Å) | Agilent 6540 | ESI+/− | A: H2O + 0.1% FA, B: MeOH + 0.1% FA | 0.4 mL/min | (0, 2), (10, 100), (14.5, 100), (14.51, 2), (16.5, 2) |
| 8H * | UHPLC–QTOF | Agilent 1290 | Waters Aqcuity UPLC BEH Amide (2.1 × 100 mm, 1.7 µm, 130 Å) | Agilent 6540 | ESI+/− | A: 50% ACN + 20 mM NH4COOH, B: 90% ACN + 20 mM NH4COOH; pH = 3 | 0.6 mL/min | (0, 100), (2.5, 100), (10, 0), (10.1, 100), (14, 100) |
| 9 | HPLC–QTOF | Agilent 1200 | Luna C18 (4.6 × 150 mm, 3 µm, 100 Å) | Agilent G6530A | ESI+/− | A: H2O + 0.1% FA, B: ACN + 0.1% FA | 0.5 mL/min | (0, 0), (30, 30), (35, 40), (50, 80), (52, 80), (60, 0), (65, 0) |
| 10 | HPLC–QTOF | AB Sciex MicroLC 200 | Eksigent HALO C18 (0.5 × 100 mm, 2.7 µm, 90 Å) | AB Sciex 5600+ | ESI+/− | A: H2O + 0.9% FA, B: ACN + 0.9% FA | 15 µL/min | (0, 0), (0.5, 0), (4, 95), (4.5, 1), (5, 1) |
| 11 | HPLC–MS | Shimadzu Prominence | Kinetex PFP (4.6 × 150 mm, 5 µm, 100 Å) | Shimadzu LCMS 2020 | ESI+/– | A: H2O + 0.1% TFAB: MeOH | 0.4 mL/min | (0, 40), (25, 80) |
| 12 | GC–MS | Shimadzu 2010 Plus | Restek Rxi-5 ms (15 m, 0.25 mm, 0.25 µm) | Shimadzu TQ-8030 | EI | - | - | - |
| 13 | GC–MS | Agilent 7890A | DB-5 ms (30 m, 0.25 mm, 0.25 µm) | Agilent 5975C MSD | EI | - | - | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koistinen, V.M.; Bento da Silva, A.; Abrankó, L.; Low, D.; Garcia Villalba, R.; Tomás Barberán, F.; Landberg, R.; Savolainen, O.; Alvarez-Acero, I.; De Pascual-Teresa, S.; et al. Interlaboratory Coverage Test on Plant Food Bioactive Compounds and Their Metabolites by Mass Spectrometry-Based Untargeted Metabolomics. Metabolites 2018, 8, 46. https://doi.org/10.3390/metabo8030046
Koistinen VM, Bento da Silva A, Abrankó L, Low D, Garcia Villalba R, Tomás Barberán F, Landberg R, Savolainen O, Alvarez-Acero I, De Pascual-Teresa S, et al. Interlaboratory Coverage Test on Plant Food Bioactive Compounds and Their Metabolites by Mass Spectrometry-Based Untargeted Metabolomics. Metabolites. 2018; 8(3):46. https://doi.org/10.3390/metabo8030046
Chicago/Turabian StyleKoistinen, Ville Mikael, Andreia Bento da Silva, László Abrankó, Dorrain Low, Rocio Garcia Villalba, Francisco Tomás Barberán, Rikard Landberg, Otto Savolainen, Inmaculada Alvarez-Acero, Sonia De Pascual-Teresa, and et al. 2018. "Interlaboratory Coverage Test on Plant Food Bioactive Compounds and Their Metabolites by Mass Spectrometry-Based Untargeted Metabolomics" Metabolites 8, no. 3: 46. https://doi.org/10.3390/metabo8030046
APA StyleKoistinen, V. M., Bento da Silva, A., Abrankó, L., Low, D., Garcia Villalba, R., Tomás Barberán, F., Landberg, R., Savolainen, O., Alvarez-Acero, I., De Pascual-Teresa, S., Van Poucke, C., Almeida, C., Petrásková, L., Valentová, K., Durand, S., Wiczkowski, W., Szawara-Nowak, D., González-Domínguez, R., Llorach, R., ... Bronze, M. R. (2018). Interlaboratory Coverage Test on Plant Food Bioactive Compounds and Their Metabolites by Mass Spectrometry-Based Untargeted Metabolomics. Metabolites, 8(3), 46. https://doi.org/10.3390/metabo8030046