Statistical Analysis of NMR Metabolic Fingerprints: Established Methods and Recent Advances



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Preprocessing
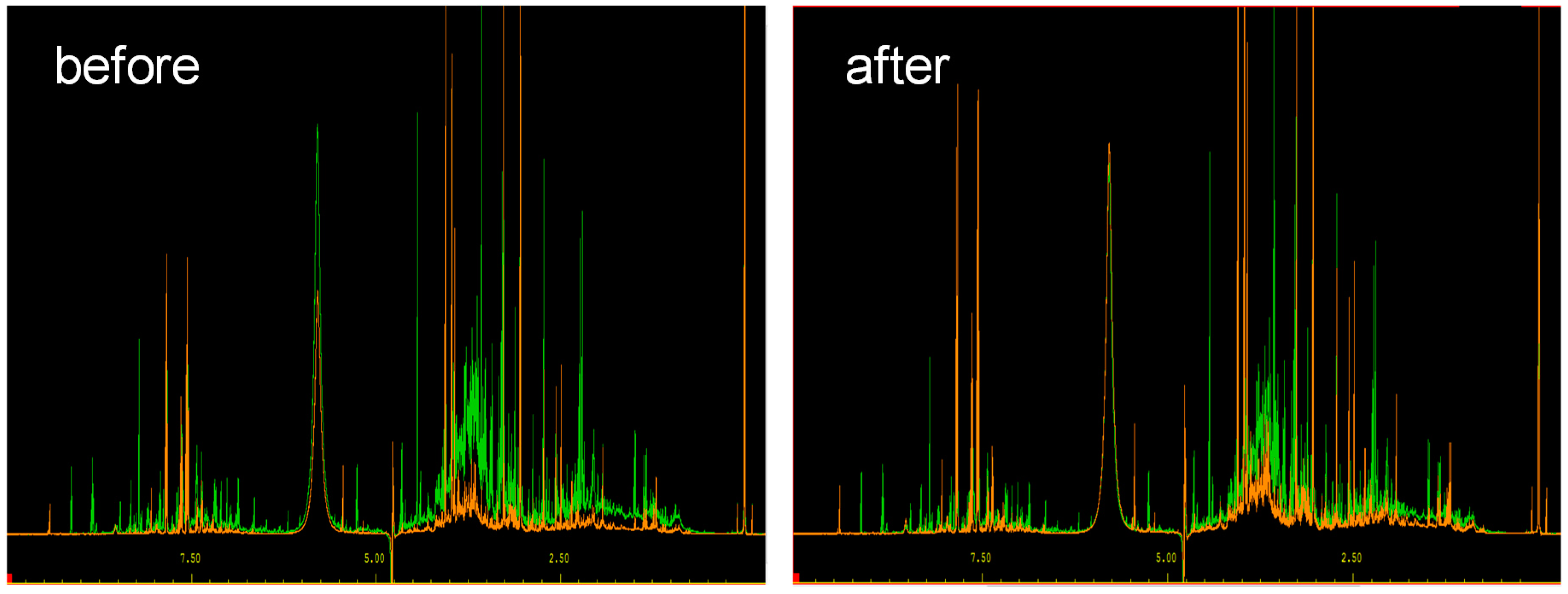
2.1. Data Extraction
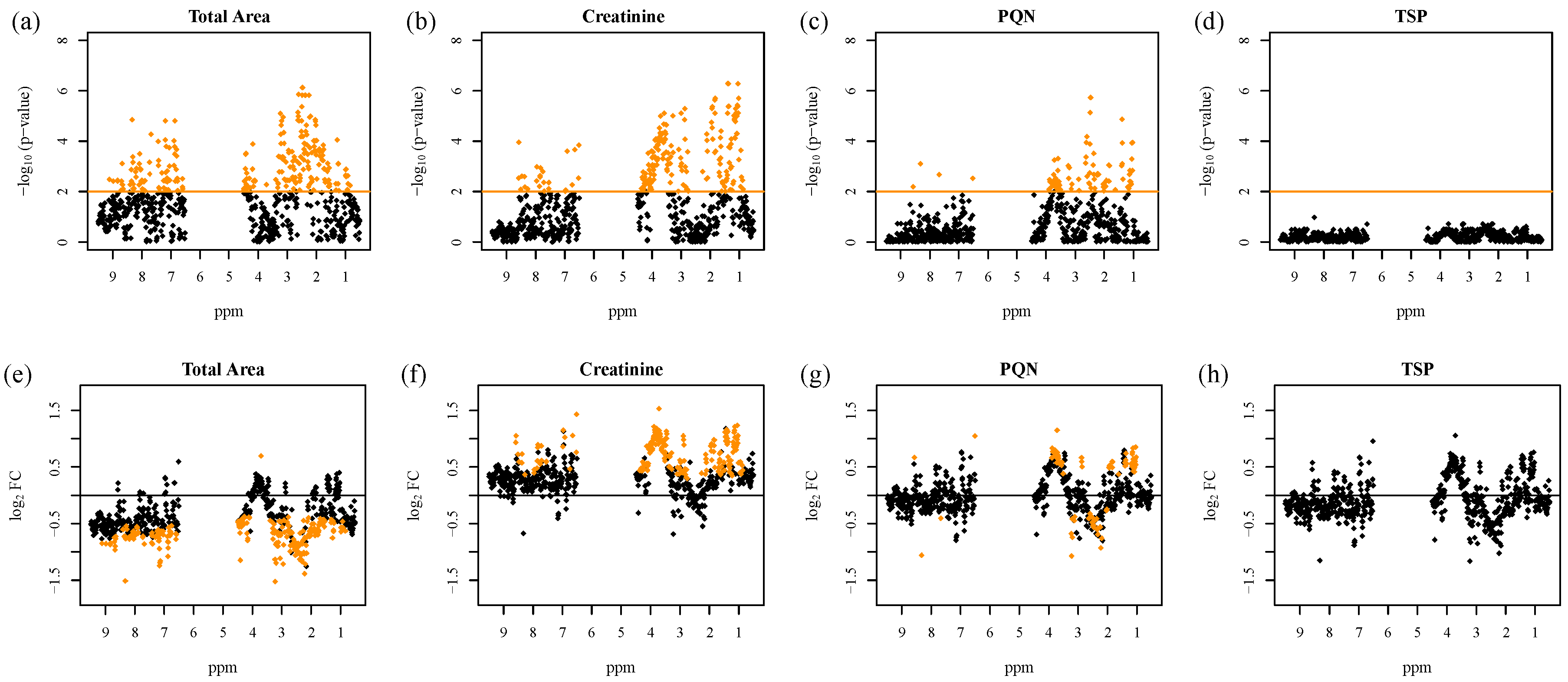
2.2. Normalization
2.3. Additional Data Transformation
3. Statistical Data Analysis Strategies
3.1. Unsupervised Machine Learning Methods
3.2. Hypothesis Testing
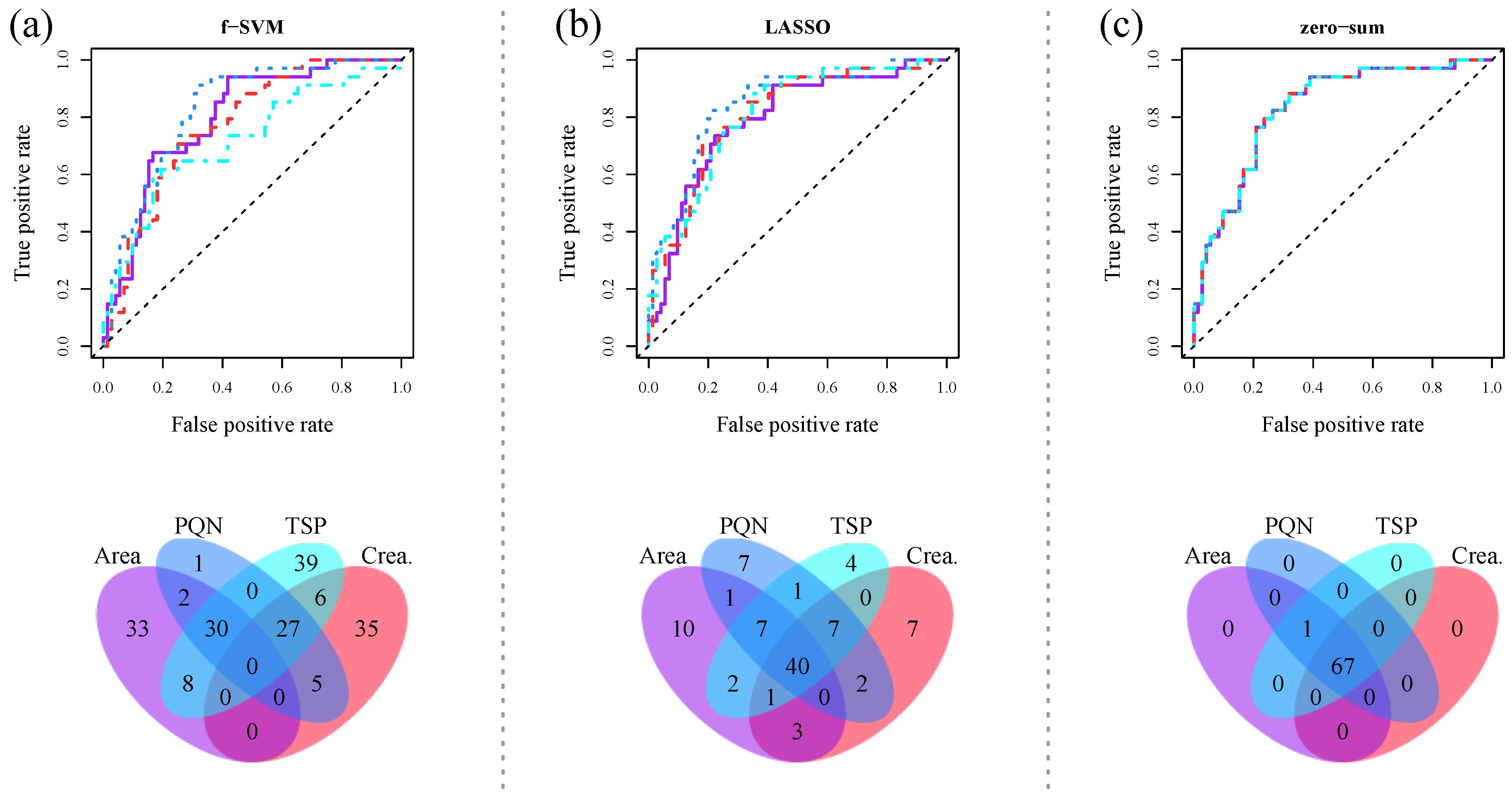
3.3. Supervised Machine Learning Methods
4. Zero-Sum Regression
5. Available Software for Metabolomics Data Preprocessing and Statistical Analysis
6. Conclusions
Funding
Conflicts of Interest
References
- Klein, M.S.; Buttchereit, N.; Miemczyk, S.P.; Immervoll, A.K.; Louis, C.; Wiedemann, S.; Junge, W.; Thaller, G.; Oefner, P.J.; Gronwald, W. NMR metabolomic analysis of dairy cows reveals milk glycerophosphocholine to phosphocholine ratio as prognostic biomarker for risk of ketosis. J. Proteome Res. 2012, 11, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, H.U.; Schley, G.; Hochrein, J.; Klein, M.S.; Köberle, C.; Eckardt, K.U.; Willam, C.; Oefner, P.J.; Gronwald, W. Analysis of Human Urine Reveals Metabolic Changes Related to the Development of Acute Kidney Injury Following Cardiac Surgery. Metabolomics 2013, 9, 697–707. [Google Scholar] [CrossRef]
- Zacharias, H.U.; Hochrein, J.; Vogl, F.C.; Schley, G.; Mayer, F.; Jeleazcov, C.; Eckardt, K.-U.; Willam, C.; Oefner, P.J.; Gronwald, W. Identification of Plasma Metabolites Prognostic of Acute Kidney Injury after Cardiac Surgery with Cardiopulmonary Bypass. J. Proteome Res. 2015, 14, 2897–2905. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Charlton, A.J.; Godward, J.; Jones, S.A.; Harrison, M.; Wilson, J.C. Adaptive binning: An improved binning method for metabolomics data using the undecimated wavelet transform. Chemom. Intell. Lab. 2007, 85, 144–154. [Google Scholar] [CrossRef]
- Vu, T.N.; Laukens, K. Getting your peaks in line: A review of alignment methods for NMR spectral data. Metabolites 2013, 3, 259–276. [Google Scholar] [CrossRef] [PubMed]
- Savorani, F.; Tomasi, G.; Engelsen, S.B. Icoshift: A versatile Tool for the Rapid Alignment of 1D NMR Spectra. J. Magn. Reson. 2010, 202, 190–202. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, T.; Sinnaeve, D.; van Gasse, B.; Rietzschel, E.R.; de Buyzere, M.L.; Langlois, M.R.; Bekaert, S.; Martins, J.C.; van Criekinge, W. Evaluation of Standard and Advanced Preprocessing Methods for the Univariate Analysis of Blood Serum 1H-NMR Spectra. Anal. Bioanal. Chem. 2010, 398, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.E.; Reo, N.V.; DelRaso, N.J.; Doom, T.E.; Raymer, M.L. Gaussian binning: A new kernel-based method for processing NMR spectroscopic data for metabolomics. Metabolomics 2008, 4, 261–272. [Google Scholar] [CrossRef]
- Sousa, S.; Magalhães, A.; Ferreira, M.M.C. Optimized bucketing for NMR spectra: Three case studies. Chemom. Intell. Lab. 2013, 122, 93–102. [Google Scholar] [CrossRef]
- Craig, A.; Cloarec, O.; Holmes, E.; Nicholson, J.K.; Lindon, J.C. Scaling and Normalization Effects in NMR Spectroscopic Metabolomic Data Sets. Anal. Chem. 2006, 78, 2262–2267. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.; Robards, K.; Prenzler, P.D.; Kendall, M. Recent and potential developments in the analysis of urine: A review. Anal. Chim. Acta 2011, 684, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Lindon, J.C.; Nicholson, J.K.; Holmes, E. (Eds.) The Handbook of Metabonomics and Metabolomics. NMR Spectroscopy Techniques for Application to Metabonomics; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Waikar, S.S.; Sabbisetti, V.S.; Bonventre, J.V. Normalization of Urinary Biomarkers to Creatinine during Changes in Glomerular Filtration Rate. Kidney Int. 2010, 78, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Curhan, G. Cystatin C: A Marker for Renal Function of Something More? Clin. Chem. 2005, 51, 293–294. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.A.; Levey, A.S. Measured GFR as a confirmatory test for estimated GFR. J. Am. Soc. Nephrol. 2009, 20, 2305–2313. [Google Scholar] [CrossRef] [PubMed]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic Quotient Normalization as Robust Method to Account for Dillution of Complex Biological Mixtures. Application to 1H NMR Metabolomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.M.; Klein, M.S.; Hochrein, J.; Oefner, P.J.; Spang, R.; Gronwald, W. State-of-the Art Data Normalization Methods Improve NMR-Based Metabolomic Analysis. Metabolomics 2012, 8, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Variance and Bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Heydebreck, A.V.; Sültmann, H.; Poustka, A.; Vingron, M. Variance Stabilisation Applied to Microarray Data Calibration and to the Quantification of Differential Expression. Bioinformatics 2002, 18, S96–S104. [Google Scholar] [CrossRef] [PubMed]
- Workman, C.; Jensen, L.J.; Jarmer, H.; Berka, R.; Gautier, L.; Nielser, H.B.; Saxild, H.H.; Nielsen, C.; Brunak, S.; Knudsen, S. A New Non-Linear Normalization Method for Reducing Variability in DNA Microarray Experiments. Genome Biol. 2002, 3. [Google Scholar] [CrossRef]
- Hochrein, J.; Zacharias, H.U.; Taruttis, F.; Samol, C.; Engelmann, J.C.; Spang, R.; Oefner, P.J.; Gronwald, W. Data Normalization of 1H NMR Metabolite Fingerprinting Data Sets in the Presence of Unbalanced Metabolite Regulation. J. Proteome Res. 2015, 14, 3217–3228. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, H.U.; Rehberg, T.; Mehrl, S.; Richtmann, D.; Wettig, T.; Oefner, P.J.; Spang, R.; Gronwald, W.; Altenbuchinger, M. Scale-invariant biomarker discovery in urine and plasma metabolite fingerprints. J. Proteome Res. 2017, 16, 3596–3605. [Google Scholar] [CrossRef] [PubMed]
- Gromski, P.S.; Xu, Y.; Hollywood, K.A.; Turner, M.L.; Goodacre, R. The influence of scaling metabolomics data on model classification accuracy. Metabolomics 2015, 11, 684–695. [Google Scholar] [CrossRef]
- Jauhiainen, A.; Madhu, B.; Narita, M.; Narita, M.; Griffiths, J.; Tavaré, S. Normalization of metabolomics data with applications to correlation maps. Bioinformatics 2014, 30, 2155–2161. [Google Scholar] [CrossRef] [PubMed]
- Saccenti, E. Correlation Patterns in Experimental Data Are Affected by Normalization Procedures: Consequences for Data Analysis and Network Inference. J. Proteome Res. 2017, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Viant, M.R.; Lyeth, B.G.; Miller, M.G.; Berman, R.F. An NMR metabolomic investigation of early metabolic disturbances following traumatic brain injury in a mammalian model. NMR Biomed. 2005, 18, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Purohit, P.V.; Rocke, D.M.; Viant, M.R.; Woodruff, D.L. Discrimination models using variance-stabilizing transformation of metabolomic NMR data. Omics 2004, 8, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.; Antti, H.; Gottfries, J.; Holmes, E.; Johansson, E.; Lindgren, F.; Long, I.; Lundstedt, T.; Trygg, J.; Wold, S. Using Chemometrics for Navigating in the Large Data Sets of Genomics, Proteomics, and Metabonomics (gpm). Anal. Bioanal. Chem. 2004, 380, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.E. A User’s Guide to Principal Components; Wiley-Interscience: Hoboken, NJ, USA, 2003. [Google Scholar]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.-H.; Saccenti, E.; Gao, X.; McKay, R.T.; dos Santos, V.A.M.; Roy, R.; Wishart, D.S. Recommended strategies for spectral processing and post-processing of 1D 1H-NMR data of biofluids with a particular focus on urine. Metabolomics 2018, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Gatzek, S.; Sterling, A.; Fiehn, O.; Selbig, J. Metabolite Fingerprinting: Detecting Biological Features by Independent Component Analysis. Bioinformatics 2004, 20, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.S.; Dorn, C.; Saugspier, M.; Hellerbrand, C.; Oefner, P.J.; Gronwald, W. Discrimination of Steatosis and NASH in Mice Using Nuclear Magnetic Resonance Spectroscopy. Metabolomics 2011, 7, 237–246. [Google Scholar] [CrossRef]
- Draisma, H.H.; Reijmers, T.H.; van der Kloet, F.; Bobeldijk-Pastorova, I.; Spies-Faber, E.; Vogels, J.T.; Meulman, J.J.; Boomsma, D.I.; van der Greef, J.; Hankemeier, T. Equating, or correction for between-block effects with application to body fluid LC-MS and NMR metabolomics data sets. Anal. Chem. 2010, 82, 1039–1046. [Google Scholar] [CrossRef] [PubMed]
- Hartigan, J. Clustering Algorithms; John Wiley: New York, NY, USA, 1975. [Google Scholar]
- Frey, B.J.; Dueck, D. Clustering by passing messages between data points. Science 2007, 315, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.K.; Sandeep, K.; Dow, E.R. Self-organizing Maps for the Analysis of NMR Spectra. Biosilico 2004, 2, 157–163. [Google Scholar] [CrossRef]
- Zacharias, H.U.; Hochrein, J.; Klein, M.S.; Samol, C.; Oefner, P.J.; Gronwald, W. Current Experimental, Bioinformatic and Statistical Methods used in NMR Based Metabolomics. Curr. Metabol. 2013, 1, 253–268. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar]
- Salkind, N.J. (Ed.) Bonferroni and Sidak Corrections for Multiple Comparisons. In Encyclopedia of Measurement and Statistics; Sage: Thousand Oaks, CA, USA, 2007. [Google Scholar]
- Barker, M.; Rayens, W. Partial Least Squares for Discrimination. J. Chemom. 2003, 17, 166–173. [Google Scholar] [CrossRef]
- Trygg, J.; Wold, S. Orthogonal Projections to Latent Structures. J. Chemom. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Burges, C.J.C. A Tutorial on Support Vector Machines for Pattern Recognition. Data Min. Knowl. Discov. 1998, 2, 121–167. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. B 1996, 58, 267–288. [Google Scholar]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and Variable Selection via the Elastic Net. J. R. Stat. Soc. B 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Hochrein, J.; Klein, M.S.; Zacharias, H.U.; Li, J.; Wijffels, G.; Schirra, H.J.; Spang, R.; Oefner, P.J.; Gronwald, W. Performance Evaluation of Algorithms for the Classification of Metabolic 1H-NMR Fingerprints. J. Proteome Res. 2012, 11, 6242–6251. [Google Scholar] [CrossRef] [PubMed]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis—A marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Hinzman, A.A.; Kang, E.L.; Szczesniak, R.D.; Lu, L.J. Computational and statistical analysis of metabolomics data. Metabolomics 2015, 11, 1492–1513. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M. Machine Learning Methods for Analysis of Metabolic Data and Metabolic Pathway Modeling. Metabolites 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Shi, P.; Feng, R.; Li, H. Variable selection in regression with compositional covariates. Biometrika 2014, 101, 785–797. [Google Scholar] [CrossRef]
- Altenbuchinger, M.; Rehberg, T.; Zacharias, H.U.; Stämmler, F.; Dettmer, K.; Weber, D.; Hiergeist, A.; Gessner, A.; Holler, E.; Oefner, P.J.; et al. Reference point insensitive molecular data analysis. Bioinformatics 2017, 33, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Development Core Team, R. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2009. [Google Scholar]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A Web Server for Metabolomic Data Analysis and Interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [PubMed]
- Worley, B.; Powers, R. MVAPACK: A complete data handling package for NMR metabolomics. ACS Chem. Biol. 2014, 9, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Giacomoni, F.; Le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- De Livera, A.M.; Olshansky, G.; Simpson, J.A.; Creek, D.J. NormalizeMets: Assessing, selecting and implementing statistical methods for normalizing metabolomics data. Metabolomics 2018, 14, 1048. [Google Scholar] [CrossRef]
- Li, B.; Tang, J.; Yang, Q.; Cui, X.; Li, S.; Chen, S.; Cao, Q.; Xue, W.; Chen, N.; Zhu, F. Performance Evaluation and Online Realization of Data-driven Normalization Methods Used in LC/MS based Untargeted Metabolomics Analysis. Sci. Rep. 2016, 6, 38881. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zacharias, H.U.; Altenbuchinger, M.; Gronwald, W. Statistical Analysis of NMR Metabolic Fingerprints: Established Methods and Recent Advances. Metabolites 2018, 8, 47. https://doi.org/10.3390/metabo8030047
Zacharias HU, Altenbuchinger M, Gronwald W. Statistical Analysis of NMR Metabolic Fingerprints: Established Methods and Recent Advances. Metabolites. 2018; 8(3):47. https://doi.org/10.3390/metabo8030047
Chicago/Turabian StyleZacharias, Helena U., Michael Altenbuchinger, and Wolfram Gronwald. 2018. "Statistical Analysis of NMR Metabolic Fingerprints: Established Methods and Recent Advances" Metabolites 8, no. 3: 47. https://doi.org/10.3390/metabo8030047
APA StyleZacharias, H. U., Altenbuchinger, M., & Gronwald, W. (2018). Statistical Analysis of NMR Metabolic Fingerprints: Established Methods and Recent Advances. Metabolites, 8(3), 47. https://doi.org/10.3390/metabo8030047