
Distinguishing Benign from Malignant Pancreatic and Periampullary Lesions Using Combined Use of 1H-NMR Spectroscopy and Gas Chromatography–Mass Spectrometry

 ,
,  ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Serum Samples
2.2. Patient Data
2.3. Metabolomic Analysis
2.4. Data Pre-Processing
2.5. Multivariate Projection Modeling
2.6. Metabolic Pathway Analysis
3. Results
3.1. Demographic and Technical Factors
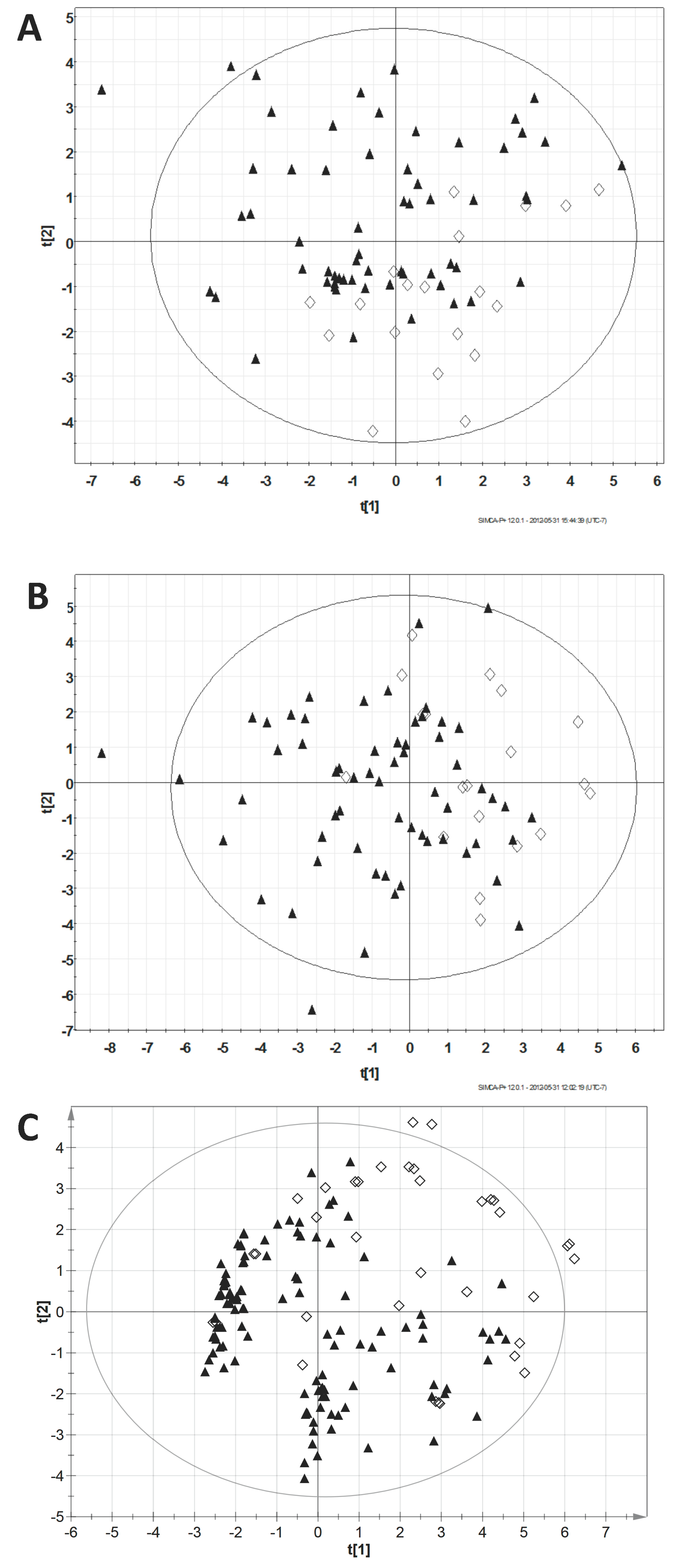
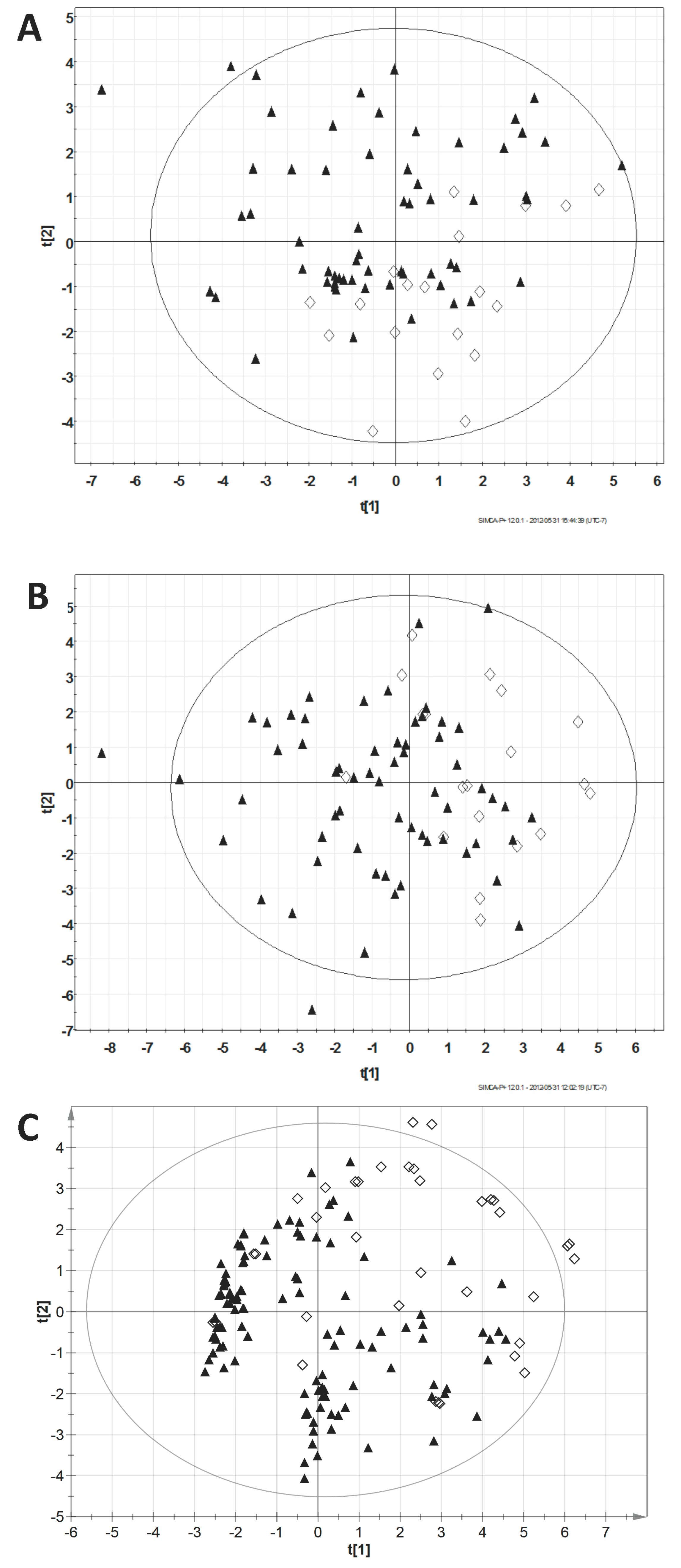
3.2. Principal Component Analysis
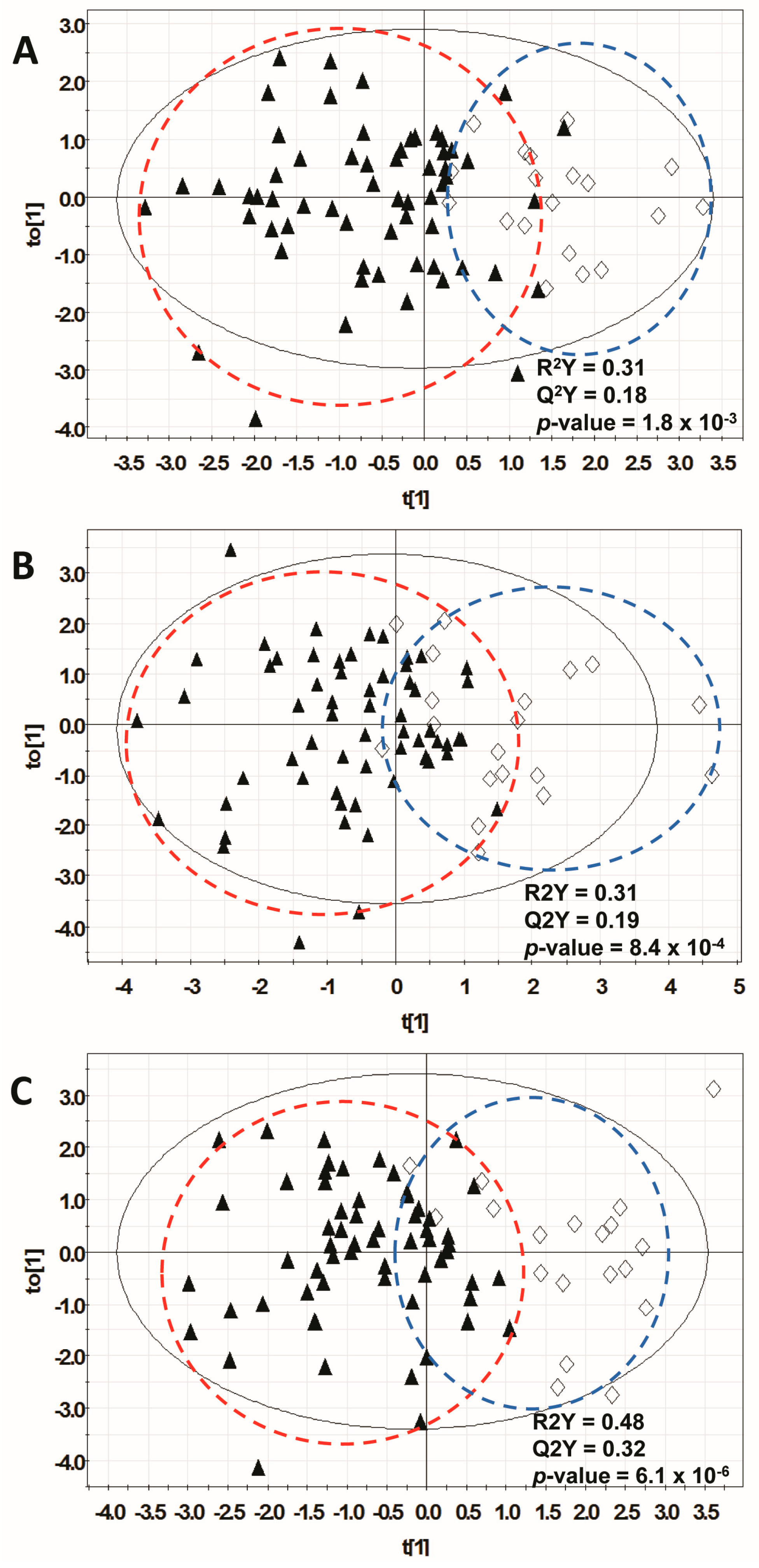
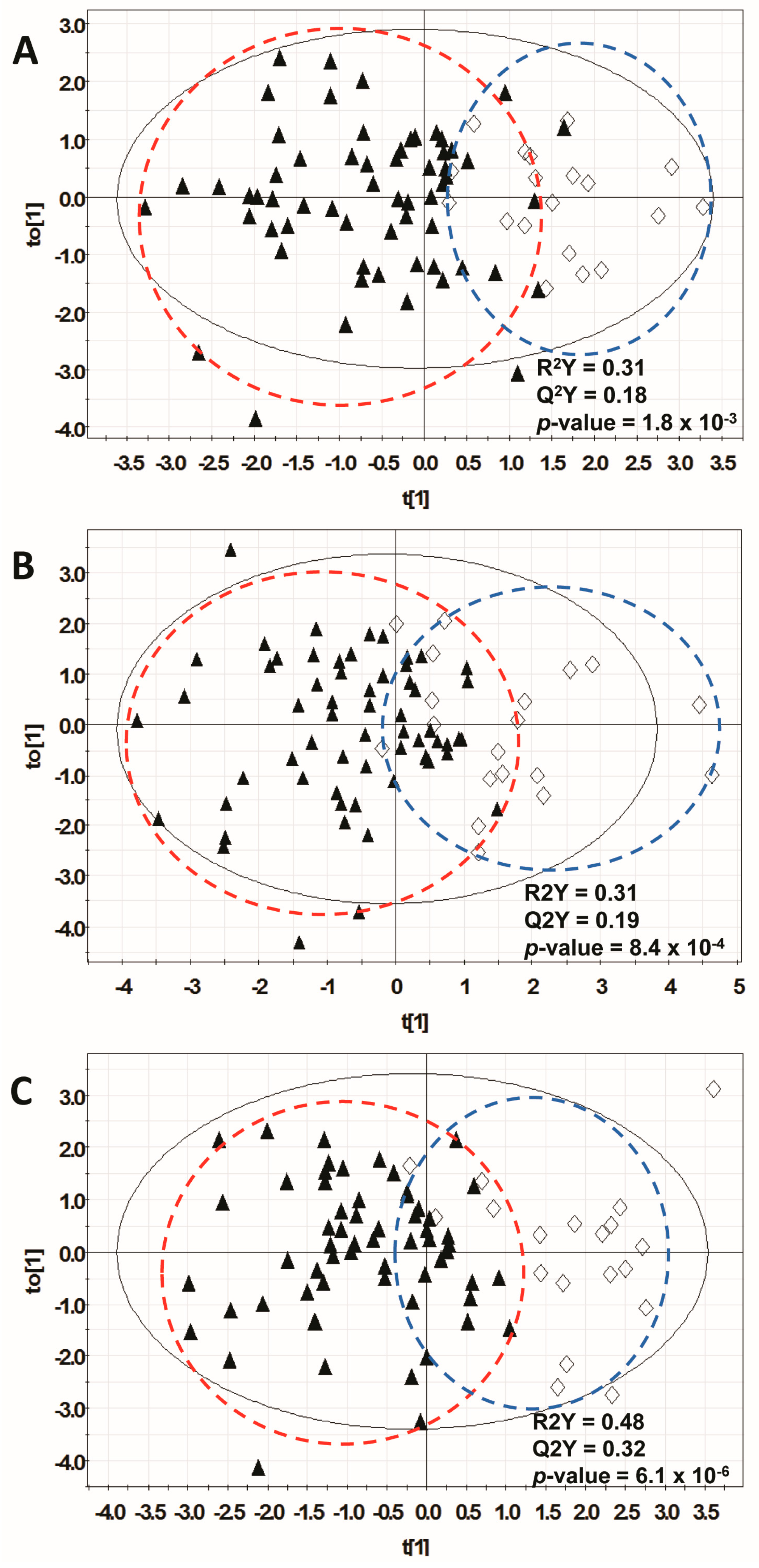
3.3. Orthogonal Multivariate Projection Modeling
3.4. Metabolic Pathway Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goonetilleke, K.S.; Siriwardena, A.K. Systematic review of carbohydrate antigen (CA 19-9) as a biochemical marker in the diagnosis of pancreatic cancer. Eur. J. Surg. Oncol. 2007, 33, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Kinney, T. Evidence-based imaging of pancreatic malignancies. Surg. Clin. N. Am. 2010, 90, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Savides, T.J.; Donohue, M.; Hunt, G.; Al-Haddad, M.; Aslanian, H.; Ben-Menachem, T.; Chen, V.K.; Coyle, W.; Deutsch, J.; DeWitt, J.; et al. EUS-guided FNA diagnostic yield of malignancy in solid pancreatic masses: A benchmark for quality performance measurement. Gastrointest. Endosc. 2007, 66, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Fogel, E.L.; deBellis, M.; McHenry, L.; Watkins, J.L.; Chappo, J.; Cramer, H.; Schmidt, S.; Lazzell-Pannell, L.; Sherman, S.; Lehman, G.A. Effectiveness of a new long cytology brush in the evaluation of malignant biliary obstruction: A prospective study. Gastrointest. Endosc. 2006, 63, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.C.; Wilentz, R.E.; Yeo, C.J.; Sohn, T.A.; Cameron, J.L.; Boitnott, J.K.; Hruban, R.H. Pancreaticoduodenectomy (Whipple resections) in patients without malignancy: are they all ‘chronic pancreatitis’? Am. J. Surg. Pathol. 2003, 27, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.J.; Cameron, J.L.; Sohn, T.A.; Lillemoe, K.D.; Pitt, H.A.; Talamini, M.A.; Hruban, R.H.; Ord, S.E.; Sauter, P.K.; Coleman, J.; et al. Six hundred fifty consecutive pancreaticoduodenectomies in the 1990s: Pathology, complications, and outcomes. Ann. Surg. 1997, 226, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Camp, E.R.; Vogel, S.B. Blind Whipple resections for periampullary and pancreatic lesions. Am. Surg. 2004, 70, 6–12. [Google Scholar] [PubMed]
- McLean, S.R.; Karsanji, D.; Wilson, J.; Dixon, E.; Sutherland, F.R.; Pasieka, J.; Ball, C.; Bathe, O.F. The effect of wait times on oncological outcomes from periampullary adenocarcinomas. J. Surg. Oncol. 2013, 107, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Simunovic, M.; Urbach, D.; Major, D.; Sutradhar, R.; Baxter, N.; To, T.; Brown, A.; Davis, D.; Levine, M.N. Assessing the volume-outcome hypothesis and region-level quality improvement interventions: Pancreas cancer surgery in two Canadian Provinces. Ann. Surg. Oncol. 2010, 17, 2537–2544. [Google Scholar] [CrossRef] [PubMed]
- Simons, J.P.; Shah, S.A.; Ng, S.C.; Whalen, G.F.; Tseng, J.F. National complication rates after pancreatectomy: Beyond mere mortality. J. Gastrointest. Surg. 2009, 13, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Bathe, O.F.; Shaykhutdinov, R.; Kopciuk, K.; Weljie, A.M.; McKay, A.; Sutherland, F.R.; Dixon, E.; Dunse, N.; Sotiropoulos, D.; Vogel, H.J. Feasibility of identifying pancreatic cancer based on serum metabolomics. Cancer Epidemiol. Biomark. Prev. 2011, 20, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.I.; Atherton, H.J.; Goodacre, R.; Griffin, J.L. Systems level studies of mammalian metabolomes: The roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; He, X.; Deng, H.; Chen, Q.; Lu, J.; Spraul, M.; Yu, Y. Discrimination of metabolic profiles of pancreatic cancer from chronic pancreatitis by high-resolution magic angle spinning 1H nuclear magnetic resonance and principal components analysis. Cancer Sci. 2007, 98, 1678–1682. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nishiumi, S.; Ikeda, A.; Yoshie, T.; Sakai, A.; Matsubara, A.; Izumi, Y.; Tsumura, H.; Tsuda, M.; Nishisaki, H.; et al. A novel serum metabolomics-based diagnostic approach to pancreatic cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Leichtle, A.B.; Ceglarek, U.; Weinert, P.; Nakas, C.T.; Nuoffer, J.M.; Kase, J.; Conrad, T.; Witzigmann, H.; Thiery, J.; Fiedler, G.M. Pancreatic carcinoma, pancreatitis, and healthy controls: Metabolite models in a three-class diagnostic dilemma. Metabolomics 2013, 9, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Davis, V.W.; Schiller, D.E.; Eurich, D.; Bathe, O.F.; Sawyer, M.B. Pancreatic Ductal Adenocarcinoma is Associated with a Distinct Urinary Metabolomic Signature. Ann. Surg. Oncol. 2013, 20, 415–423. [Google Scholar] [CrossRef] [PubMed]
- American Joint Committee on Cancer. Exocrine and endocrine pancreas. In AJCC Cancer Staging Manual; Edge, S.B., Byrd, D.R., Compton, C.C., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 241–249. [Google Scholar]
- Farshidfar, F.; Weljie, A.M.; Kopciuk, K.; Buie, W.D.; Maclean, A.; Dixon, E.; Sutherland, F.R.; Molckovsky, A.; Vogel, H.J.; Bathe, O.F. Serum metabolomic profile as a means to distinguish stage of colorectal cancer. Genome Med. 2012, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef] [PubMed]
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted profiling: Quantitative analysis of 1H-NMR metabolomics data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef] [PubMed]
- Hiller, K.; Hangebrauk, J.; Jager, C.; Spura, J.; Schreiber, K.; Schomburg, D. MetaboliteDetector: Comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Anal. Chem. 2009, 81, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Kopka, J.; Schauer, N.; Krueger, S.; Birkemeyer, C.; Usadel, B.; Bergmuller, E.; Dörmann, P.; Weckwerth, W.; Gibon, Y.; Stitt, M.; et al. GMD@CSB.DB: The Golm Metabolome Database. Bioinformatics 2005, 21, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Veselkov, K.A.; Vingara, L.K.; Masson, P.; Robinette, S.L.; Want, E.; Li, J.V.; Barton, R.H.; Boursier-Neyret, C.; Walther, B.; Ebbels, T.M.; et al. Optimized preprocessing of ultra-performance liquid chromatography/mass spectrometry urinary metabolic profiles for improved information recovery. Anal. Chem. 2011, 83, 5864–5872. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Booth, S.C.; Workentine, M.L.; Wen, J.; Shaykhutdinov, R.; Vogel, H.J.; Ceri, H.; Turner, R.J.; Weljie, A.M. Differences in metabolism between the biofilm and planktonic response to metal stress. J. Proteome Res. 2011, 10, 3190–3199. [Google Scholar] [CrossRef] [PubMed]
- Trygg, J.; Holmes, E.; Lundstedt, T. Chemometrics in metabonomics. J. Proteome Res. 2007, 6, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. MSEA: A web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010, 38, W71–W77. [Google Scholar] [CrossRef] [PubMed]
- Fiehn, O.; Garvey, W.T.; Newman, J.W.; Lok, K.H.; Hoppel, C.L.; Adams, S.H. Plasma metabolomic profiles reflective of glucose homeostasis in non-diabetic and type 2 diabetic obese African-American women. PLoS ONE 2010, 5, e15234. [Google Scholar] [CrossRef] [PubMed]
- Urayama, S.; Zou, W.; Brooks, K.; Tolstikov, V. Comprehensive mass spectrometry based metabolic profiling of blood plasma reveals potent discriminatory classifiers of pancreatic cancer. Rapid Commun. Mass Spectrom. 2010, 24, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Gu, X.; Zhou, J.; Yan, C. Metabolomic profiling of human plasma in pancreatic cancer using pressurized capillary electrochromatography. Electrophoresis 2011, 32, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Nishiumi, S.; Shinohara, M.; Ikeda, A.; Yoshie, T.; Hatano, N.; Kakuyama, S.; Mizuno, S.; Sanuki, T.; Kutsumi, H.; Fukusaki, E.; et al. Serum metabolomics as a novel diagnostic approach for pancreatic cancer. Metabolomics 2010, 6, 518–528. [Google Scholar] [CrossRef]
- Maher, A.D.; Cysique, L.A.; Brew, B.J.; Rae, C.D. Statistical integration of 1H-NMR and MRS data from different biofluids and tissues enhances recovery of biological information from individuals with HIV-1 infection. J. Proteome Res. 2011, 10, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Pan, Z.; Xi, B.; Asiago, V.; Musselman, B.; Raftery, D. Principal component directed partial least squares analysis for combining nuclear magnetic resonance and mass spectrometry data in metabolomics: application to the detection of breast cancer. Anal. Chim. Acta. 2011, 686, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Weljie, A.M.; Jirik, F.R. Hypoxia-induced metabolic shifts in cancer cells: Moving beyond the Warburg effect. Int. J. Biochem. Cell Biol. 2011, 43, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Morvan, D.; Demidem, A. Metabolomics by proton nuclear magnetic resonance spectroscopy of the response to chloroethylnitrosourea reveals drug efficacy and tumor adaptive metabolic pathways. Cancer Res. 2007, 67, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Allocation A | Allocation B | Allocation C | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Training n = 80 | Test n = 77 | p * | Training n = 80 | Test n = 77 | p | Training n = 80 | Test n = 77 | p * | ||
| Age | <60 yrs | 24 | 24 | 0.87 | 27 | 21 | 0.38 | 22 | 26 | 0.40 |
| ≥60 yrs | 56 | 53 | 53 | 56 | 58 | 51 | ||||
| Gender | Male | 45 | 37 | 0.31 | 46 | 36 | 0.18 | 40 | 42 | 0.57 |
| Female | 35 | 40 | 34 | 41 | 40 | 35 | ||||
| Lesion Location | Head/Uncinate | 52 | 58 | 0.31 | 54 | 56 | 0.59 | 55 | 55 | 0.46 |
| Body/Tail | 20 | 15 | 19 | 16 | 20 | 15 | ||||
| Lesion Type | Mass | 58 | 53 | 0.37 | 59 | 52 | 0.57 | 54 | 57 | 0.12 |
| Stricture | 8 | 11 | 7 | 12 | 13 | 6 | ||||
| Cyst | 9 | 11 | 11 | 9 | 12 | 8 | ||||
| Diagnosis | Malignant | 61 | 61 | 0.61 | 61 | 61 | 0.61 | 61 | 61 | 0.61 |
| Benign | 19 | 16 | 19 | 16 | 19 | 16 | ||||
| Stage (for Malignant Lesions Only) | I | 11 | 8 | 0.39 | 5 | 14 | 0.24 | 12 | 7 | 0.25 |
| II | 27 | 28 | 30 | 25 | 27 | 28 | ||||
| III | 16 | 13 | 18 | 11 | 14 | 15 | ||||
| IV | 7 | 12 | 8 | 11 | 8 | 11 | ||||
| Surgically Resected | Yes | 48 | 44 | 0.72 | 43 | 49 | 0.21 | 49 | 43 | 0.49 |
| No | 32 | 33 | 37 | 28 | 31 | 34 | ||||
| Jaundice | Yes | 13 | 18 | 0.26 | 14 | 17 | 0.47 | 15 | 16 | 0.75 |
| No | 67 | 59 | 66 | 60 | 65 | 61 | ||||
| Diabetes Mellitus | Yes | 20 | 13 | 0.21 | 17 | 16 | 0.94 | 18 | 15 | 0.64 |
| No | 60 | 64 | 63 | 61 | 62 | 62 | ||||
| Bowel Cleansing | Yes | 43 | 43 | 1.0 | 42 | 44 | 0.30 | 47 | 39 | 0.46 |
| No | 25 | 25 | 29 | 21 | 24 | 26 | ||||
| Sampling Year | 2006-8 | 45 | 44 | 0.91 | 45 | 44 | 0.91 | 45 | 44 | 0.91 |
| 2009-10 | 35 | 33 | 35 | 33 | 35 | 33 | ||||
| Sampling Location | Laboratory | 12 | 17 | 0.25 | 17 | 12 | 0.36 | 11 | 18 | 0.12 |
| OR | 68 | 60 | 63 | 65 | 69 | 59 | ||||
| GC-MS Extraction | Day 1/2 | 42 | 38 | 0.69 | 42 | 38 | 0.69 | 42 | 38 | 0.69 |
| Day 3/4 | 38 | 39 | 38 | 39 | 38 | 39 | ||||
| GC-MS Derivatization | Day 1/2 | 40 | 41 | 0.68 | 40 | 41 | 0.68 | 40 | 41 | 0.68 |
| Day 3/4 | 40 | 36 | 40 | 36 | 40 | 36 | ||||
| Dataset | Mean of Training Sets (n = 80 each) | Mean of Test Sets (n = 77 each) | ||||
|---|---|---|---|---|---|---|
| X | R2 | Q2 | p | AUROC | SE | |
| 1H-NMR | 14 | 0.308 | 0.184 | 1.80 × 10−3 | 0.74 | 0.06 |
| GC-MS | 18 | 0.312 | 0.188 | 8.40 × 10−4 | 0.62 | 0.08 |
| Combined * | 20 | 0.478 | 0.324 | 6.14 × 10−6 | 0.66 | 0.08 |
| Metabolite | Datasets | Mean Coeff | Mean SE (Coeff) | Mean VIP | Mean SE (VIP) | p-Value in NMR | p-Value in GC-MS | |
|---|---|---|---|---|---|---|---|---|
| Higher in Malignant | Galactose | G, C | 0.121 | 0.069 | 1.123 | 0.683 | - | 0.001 |
| Unmatched RI:1007.82 QI: 67, 82, 83 | G, C | 0.120 | 0.074 | 1.337 | 0.708 | - | 0.11 | |
| Isopropanol | N, C | 0.114 | 0.042 | 1.001 | 0.382 | 0.01 | - | |
| Phenylalanine | N, G, C | 0.109 | 0.057 | 1.052 | 0.621 | 0.004 | 0.15 | |
| Glutamate | N, G, C | 0.105 | 0.064 | 1.127 | 0.616 | 0.01 | 0.01 | |
| Mannose | N, C | 0.102 | 0.069 | 1.220 | 0.410 | 0.01 | - | |
| Trimethylamine-N-oxide | N | 0.092 | 0.061 | 0.867 | 0.503 | 0.08 | - | |
| Arabitol | G, C | 0.090 | 0.047 | 0.967 | 0.409 | - | 0.16 | |
| Threitol | G, C | 0.088 | 0.080 | 0.889 | 0.816 | - | 0.14 | |
| Succinate | N, C | 0.086 | 0.115 | 0.743 | 0.777 | - | - | |
| Urea | N, G, C | 0.074 | 0.058 | 0.965 | 0.604 | 0.08 | 0.19 | |
| Myo-Inositol | N, G, C | 0.070 | 0.061 | 0.991 | 0.582 | 0.04 | 0.16 | |
| Trehalose-alpha | G, C | 0.059 | 0.053 | 0.624 | 0.572 | - | 0.21 | |
| Higher in Benign | Match RI:2018.25 QI: 191, 217, 305, 318, 507 | G, C | −0.029 | 0.055 | 0.568 | 0.680 | - | 0.79 |
| Tridecanol | G | −0.060 | 0.051 | 0.738 | 0.613 | - | 0.28 | |
| Azelaic acid | G | −0.061 | 0.038 | 0.814 | 0.526 | - | 0.04 | |
| Unmatched RI:2475.33 QI: 73, 375, 376 | G, C | −0.066 | 0.048 | 0.791 | 0.475 | - | 0.01 | |
| Pyroglutamate | N | −0.068 | 0.036 | 0.696 | 0.306 | 0.18 | - | |
| Isoleucine | G | −0.069 | 0.091 | 0.778 | 1.069 | - | 0.05 | |
| Tyrosine | N, G | −0.074 | 0.058 | 0.862 | 0.669 | 0.21 | 0.08 | |
| Arginine | N, C | −0.080 | 0.055 | 0.721 | 0.500 | 0.38 | - | |
| Unmatched RI:1913.88 QI: 156, 174, 317 | G, C | −0.090 | 0.067 | 1.092 | 0.863 | - | 0.01 | |
| Proline | N, G, C | −0.096 | 0.063 | 1.009 | 0.547 | 0.03 | 0.10 | |
| Alanine | N, C | −0.098 | 0.041 | 0.853 | 0.311 | 0.01 | - | |
| Ornithine | N, G, C | −0.104 | 0.068 | 0.997 | 0.687 | 0.06 | 0.07 | |
| Creatine | N, C | −0.107 | 0.041 | 0.952 | 0.267 | 0.06 | - | |
| Glutamine | N, G, C | −0.115 | 0.072 | 1.107 | 0.686 | 0.0002 | 0.0001 | |
| Lysine | N, C | −0.117 | 0.037 | 1.289 | 0.345 | 0.01 | - | |
| Threonine | N, G, C | −0.137 | 0.065 | 1.360 | 0.538 | 0.04 | 0.001 | |
| Unmatched RI:1971.99 QI: 185, 247, 275 | G, C | –0.138 | 0.069 | 1.346 | 0.640 | - | 0.03 |
| Metabolic Pathway | Total Compounds in Pathway | Hits in Current Dataset | p | Impact Factor |
|---|---|---|---|---|
| Arginine and proline metabolism | 77 | 7 | 8.49 × 10−5 | 0.456 |
| Alanine, aspartate, and glutamate metabolism | 24 | 4 | 2.60 × 10−4 | 0.441 |
| Galactose metabolism | 41 | 3 | 8.63 × 10−5 | 0.224 |
| Lysine degradation | 47 | 1 | 4.09 × 10−3 | 0.147 |
| D-Glutamine and D-glutamate metabolism | 11 | 2 | 1.37 × 10−3 | 0.139 |
| Inositol phosphate metabolism | 39 | 1 | 3.00 × 10−2 | 0.137 |
| Phenylalanine metabolism | 45 | 3 | 6.60 × 10−3 | 0.119 |
| Aminoacyl-tRNA biosynthesis | 75 | 10 | 8.90 × 10−7 | 0.113 |
| Lysine biosynthesis | 32 | 1 | 4.09 × 10−3 | 0.100 |
| Glycine, serine and threonine metabolism | 48 | 2 | 7.07 × 10−4 | 0.097 |
| Tyrosine metabolism | 76 | 2 | 2.77 × 10−2 | 0.047 |
| Taurine and hypotaurine metabolism | 20 | 1 | 8.27 × 10−3 | 0.032 |
| Fructose and mannose metabolism | 48 | 1 | 1.56 × 10−3 | 0.029 |
| Butanoate metabolism | 40 | 2 | 6.28 × 10−3 | 0.018 |
| Valine, leucine, and isoleucine biosynthesis | 27 | 2 | 9.74 × 10−4 | 0.013 |
| Glutathione metabolism | 38 | 3 | 3.35 × 10−3 | 0.013 |
| Phenylalanine, tyrosine, and tryptophan biosynthesis | 27 | 2 | 1.05 × 10−2 | 0.008 |
| Purine metabolism | 92 | 2 | 5.70 × 10−4 | 0.008 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McConnell, Y.J.; Farshidfar, F.; Weljie, A.M.; Kopciuk, K.A.; Dixon, E.; Ball, C.G.; Sutherland, F.R.; Vogel, H.J.; Bathe, O.F. Distinguishing Benign from Malignant Pancreatic and Periampullary Lesions Using Combined Use of 1H-NMR Spectroscopy and Gas Chromatography–Mass Spectrometry. Metabolites 2017, 7, 3. https://doi.org/10.3390/metabo7010003
McConnell YJ, Farshidfar F, Weljie AM, Kopciuk KA, Dixon E, Ball CG, Sutherland FR, Vogel HJ, Bathe OF. Distinguishing Benign from Malignant Pancreatic and Periampullary Lesions Using Combined Use of 1H-NMR Spectroscopy and Gas Chromatography–Mass Spectrometry. Metabolites. 2017; 7(1):3. https://doi.org/10.3390/metabo7010003
Chicago/Turabian StyleMcConnell, Yarrow J., Farshad Farshidfar, Aalim M. Weljie, Karen A. Kopciuk, Elijah Dixon, Chad G. Ball, Francis R. Sutherland, Hans J. Vogel, and Oliver F. Bathe. 2017. "Distinguishing Benign from Malignant Pancreatic and Periampullary Lesions Using Combined Use of 1H-NMR Spectroscopy and Gas Chromatography–Mass Spectrometry" Metabolites 7, no. 1: 3. https://doi.org/10.3390/metabo7010003
APA StyleMcConnell, Y. J., Farshidfar, F., Weljie, A. M., Kopciuk, K. A., Dixon, E., Ball, C. G., Sutherland, F. R., Vogel, H. J., & Bathe, O. F. (2017). Distinguishing Benign from Malignant Pancreatic and Periampullary Lesions Using Combined Use of 1H-NMR Spectroscopy and Gas Chromatography–Mass Spectrometry. Metabolites, 7(1), 3. https://doi.org/10.3390/metabo7010003