
The Metabolic Implications of Glucocorticoids in a High-Fat Diet Setting and the Counter-Effects of Exercise

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of GCs in Health and Disease
3. Metabolic Actions of GCs within the Skeletal Muscle, Liver, and Adipose Tissue
3.1. GCs Cause Dyslipidemia and Inhibit Insulin Signaling Proteins within the Skeletal Muscle
3.2. GCs Cause Hepatic Insulin Resistance and Dyslipidemia
3.3. GCs Increase Adipose Tissue Proliferation and Lipolysis
4. GCs and Food Intake
Impact of GCs on Brain and Feeding Behaviour
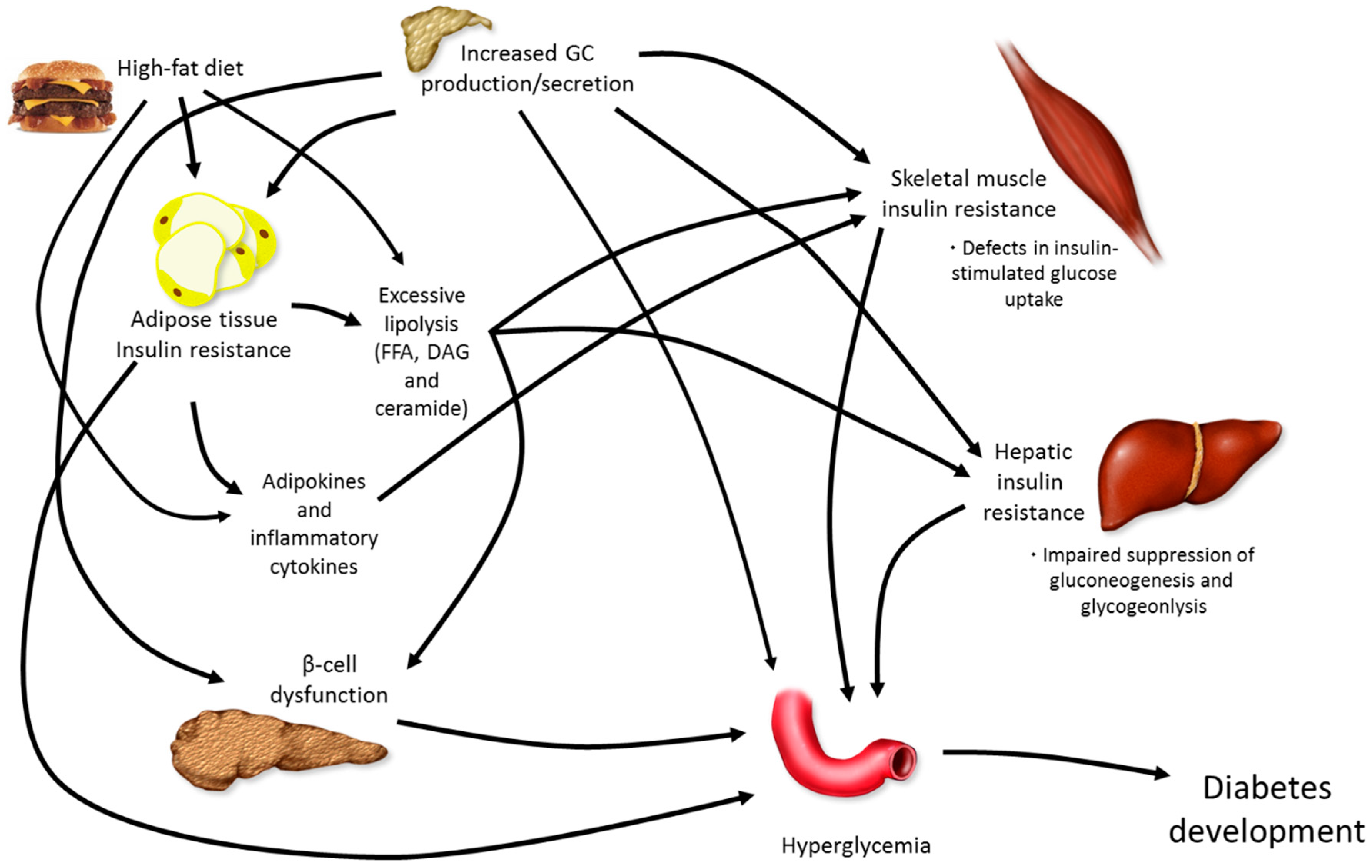
5. Combining Elevations in GCs with High-Fat Feeding
5.1. Dexamethasone or Corticosterone Exogenous Treatment and High-Fat Feeding
5.2. Chronic Stress and High-Fat Feeding
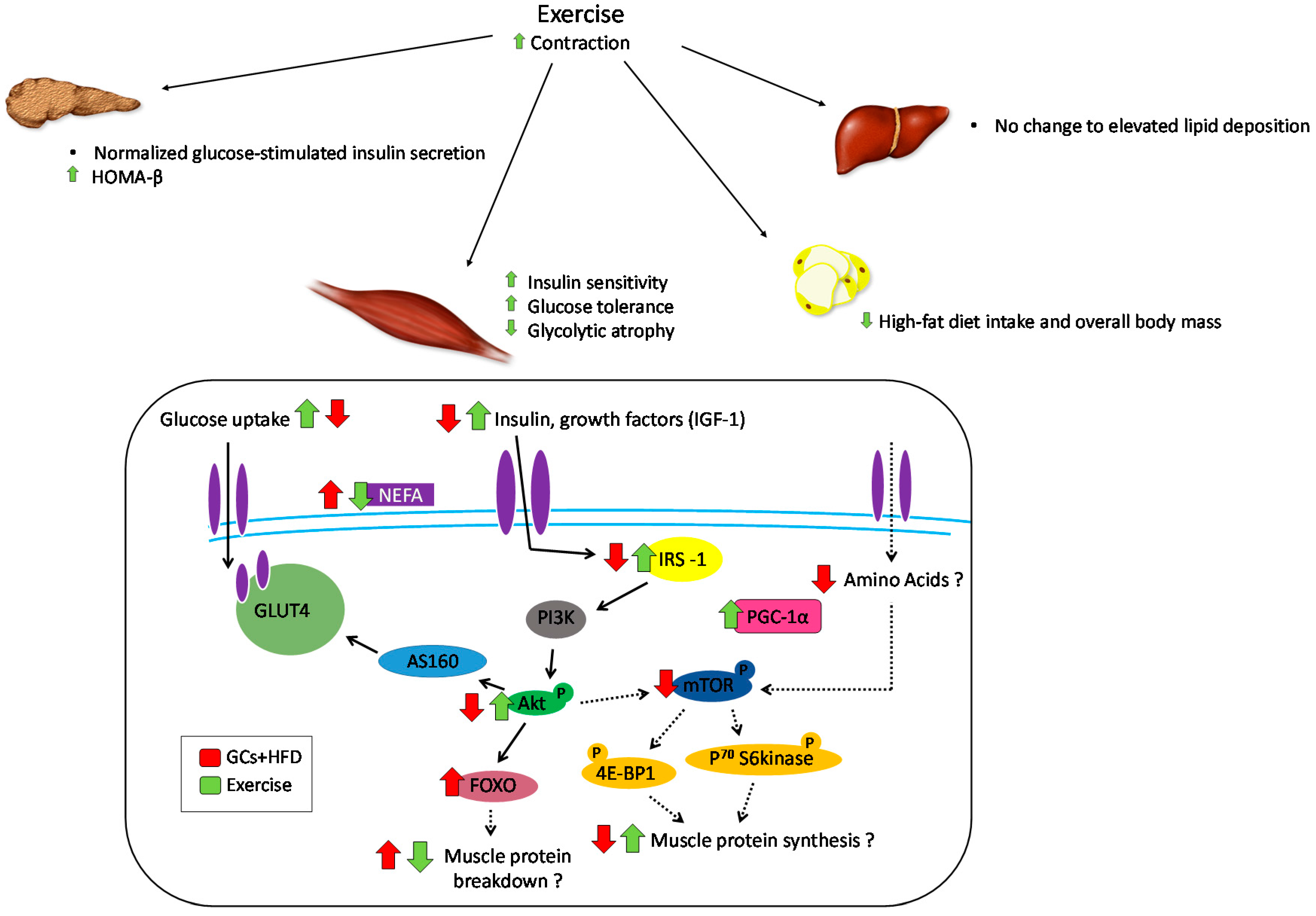
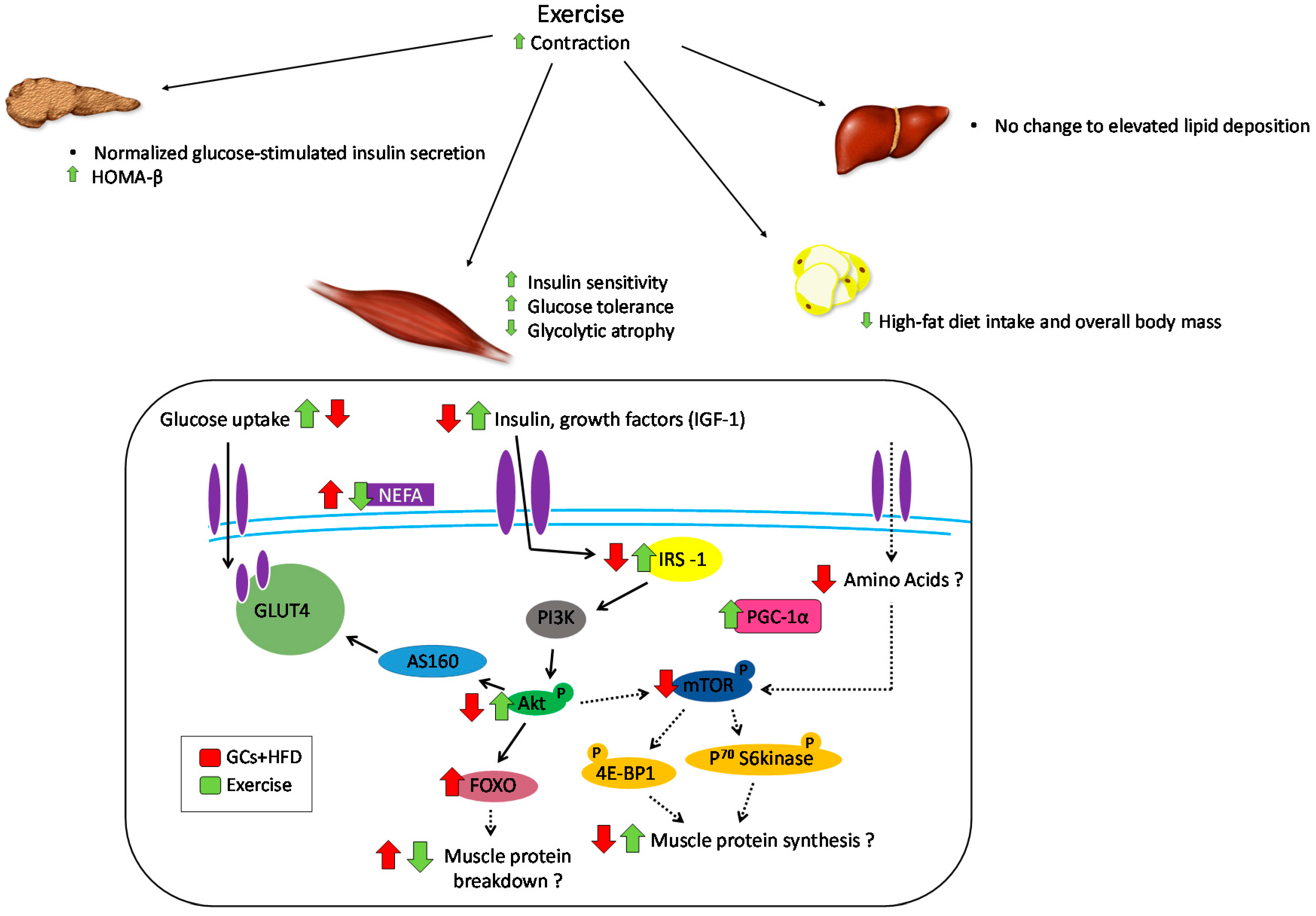
6. Role of Exercise in Offsetting the Metabolic Implications of Both Elevated GCs and High-Fat Feeding
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walsh, D.; Avashia, J. Gcs in clinical oncology. Clevel. Clin. J. Med. 1992, 59, 505–515. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Anti-inflammatory action of glucocorticoids—new mechanisms for old drugs. New Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Caporali, R.; Scire, C.A.; Todoerti, M.; Montecucco, C. The role of low dose glucocorticoids for rheumatoid arthritis in the biologic era. Clin. Exp. Rheumatol. 2013, 31, S9–S13. [Google Scholar] [PubMed]
- Overman, R.A.; Yeh, J.Y.; Deal, C.L. Prevalence of oral glucocorticoid usage in the united states: A general population perspective. Arthritis Care Res. 2013, 65, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.J.; Wong, C.A.; Pringle, M.; Tattersfield, A.E. Use of oral corticosteroids in the community and the prevention of secondary osteoporosis: A cross sectional study. Br. Med. J. 1996, 313, 344–346. [Google Scholar] [CrossRef]
- Van Staa, T.P.; Leufkens, H.G.M.; Abenhaim, L.; Begaud, B.; Zhang, B.; Cooper, C. Use of oral corticosteroids in the United Kingdom. QJM 2000, 93, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Gudbjornsson, B.; Juliusson, U.I.; Gudjonsson, F.V. Prevalence of long term steroid treatment and the frequency of decision making to prevent steroid induced osteoporosis in daily clinical practice. Ann. Rheum. Dis. 2002, 61, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, J.; Jasinska, M.; Orszulak-Michalak, D. Drug-induced myopathies. An overview of the possible mechanisms. Pharmacol. Rep. 2005, 57, 23–34. [Google Scholar] [PubMed]
- Stojanovska, L.; Rosella, G.; Proietto, J. Evolution of dexamethasone-induced insulin resistance in rats. Am. J. Physiol. Endocrinol. Metab. 1990, 258, E748–E756. [Google Scholar]
- Asensio, C.; Muzzin, P.; Rohner-Jeanrenaud, F. Role of glucocorticoids in the physiopathology of excessive fat deposition and insulin resistance. Int. J. Obes. Relat. Metab. Disord. 2004, 28 (Suppl. 4), S45–S52. [Google Scholar] [CrossRef] [PubMed]
- Pivonello, R.; De Leo, M.; Vitale, P.; Cozzolino, A.; Simeoli, C.; De Martino, M.C.; Lombardi, G.; Colao, A. Pathophysiology of diabetes mellitus in cushing’s syndrome. Neuroendocrinology 2010, 92 (Suppl. 1), 77–81. [Google Scholar] [CrossRef] [PubMed]
- Zakrzewska, K.E.; Cusin, I.; Stricker-Krongrad, A.; Boss, O.; Ricquier, D.; Jeanrenaud, B.; Rohner-Jeanrenaud, F. Induction of obesity and hyperleptinemia by central glucocorticoid infusion in the rat. Diabetes 1999, 48, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Dallman, M.F. Stress-induced obesity and the emotional nervous system. Trends Endocrinol. Metab. 2010, 21, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Dallman, M.F.; Pecoraro, N.C.; la Fleur, S.E. Chronic stress and comfort foods: Self-medication and abdominal obesity. Brain Behav. Immun. 2005, 19, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Stewart, P.M. Modulation of glucocorticoid action and the treatment of type-2 diabetes. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Dallman, M.F.; Warne, J.P.; Foster, M.T.; Pecoraro, N.C. Glucocorticoids and insulin both modulate caloric intake through actions on the brain. J. Physiol. 2007, 583, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Stimson, R.H.; Walker, B.R. The role and regulation of 11β-hydroxysteroid dehydrogenase type 1 in obesity and the metabolic syndrome. Horm. Mol. Biol. Clin. Investig. 2013, 15, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Björntorp, P.; Holm, G.; Rosmond, R. Hypothalamic arousal, insulin resistance and type 2 diabetes mellitus. Diabet. Med. 1999, 16, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11β-hsd1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. USA 2014, 111, E2482–E2491. [Google Scholar] [CrossRef] [PubMed]
- Anagnostis, P.; Katsiki, N.; Adamidou, F.; Athyros, V.G.; Karagiannis, A.; Kita, M.; Mikhailidis, D.P. 11beta-hydroxysteroid dehydrogenase type 1 inhibitors: Novel agents for the treatment of metabolic syndrome and obesity-related disorders? Metab. Clin. Exp. 2013, 62, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.C.; Chong, L.E.; Riddle, M.C. Influence of glucocorticoids and growth hormone on insulin sensitivity in humans. Diabet. Med. 2013, 30, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Stellato, C. Post-transcriptional and nongenomic effects of glucocorticoids. Proc. Am. Thorac. Soc. 2004, 1, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Peckett, A.J.; Wright, D.C.; Riddell, M.C. The effects of glucocorticoids on adipose tissue lipid metabolism. Metab. Clin. Exp. 2011, 60, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- La Fleur, S.E.; Akana, S.F.; Manalo, S.L.; Dallman, M.F. Interaction between corticosterone and insulin in obesity: Regulation of lard intake and fat stores. Endocrinology 2004, 145, 2174–2185. [Google Scholar] [CrossRef] [PubMed]
- Koyama, K.; Chen, G.; Lee, Y.; Unger, R.H. Tissue triglycerides, insulin resistance, and insulin production: Implications for hyperinsulinemia of obesity. Am. J. Physiol. 1997, 273, E708–E713. [Google Scholar] [PubMed]
- Pan, D.A.; Lillioja, S.; Kriketos, A.D.; Milner, M.R.; Baur, L.A.; Bogardus, C.; Jenkins, A.B.; Storlien, L.H. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 1997, 46, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.J.; Vegiopoulos, A.; Herzig, S. Role of glucocorticoids and the glucocorticoid receptor in metabolism: Insights from genetic manipulations. J. Steroid Biochem. Mol. Biol. 2010, 122, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Asrih, M.; Jornayvaz, F.R. Diets and nonalcoholic fatty liver disease: The good and the bad. Clin. Nutr. 2014, 33, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.S.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 1 and its role in the hypothalamus-pituitary-adrenal axis, metabolic syndrome, and inflammation. J. Clin. Endocrinol. Metab. 2009, 94, 4645–4654. [Google Scholar] [CrossRef] [PubMed]
- Bosma, M.; Kersten, S.; Hesselink, M.K.; Schrauwen, P. Re-evaluating lipotoxic triggers in skeletal muscle: Relating intramyocellular lipid metabolism to insulin sensitivity. Prog. Lipid Res. 2012, 51, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Buren, J.; Lai, Y.C.; Lundgren, M.; Eriksson, J.W.; Jensen, J. Insulin action and signalling in fat and muscle from dexamethasone-treated rats. Arch. Biochem. Biophys. 2008, 474, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.; Leighton, B.; Parry-Billings, M.; Sasson, S.; Young, M.; Krause, U.; Bevan, S.; Piva, T.; Wegener, G.; Newsholme, E.A. Effects of gc excess on the sensitivity of glucose transport and metabolism to insulin in rat skeletal muscle. J. Biochem. 1997, 321, 707–712. [Google Scholar] [CrossRef]
- Stefanyk, L.E.; Dyck, D.J. The interaction between adipokines, diet and exercise on muscle insulin sensitivity. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 255–259. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; Goodpaster, B.H. Skeletal muscle triglyceride an aspect of regional adiposity and insulin resistance. Diabetes Care 2001, 24, 933–941. [Google Scholar] [CrossRef] [PubMed]
- Arnaldi, G.; Scandali, V.M.; Trementino, L.; Cardinaletti, M.; Appolloni, G.; Boscaro, M. Pathophysiology of dyslipidemia in cushing’s syndrome. Neuroendocrinology 2010, 92, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Rutters, F.; Nieuwenhuizen, A.G.; Lemmens, S.G.T.; Born, J.M.; Westerterp-Plantenga, M.S. Hypothalamic–pituitary–adrenal (hpa) axis functioning in relation to body fat distribution. Clin. Endocrinol. 2010, 72, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: Their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Krisan, A.D.; Crain, A.M.; Collins, D.E.; Yaspelkis, B.B. High-fat diet and leptin treatment alter skeletal muscle insulin-stimulated phosphatidylinositol 3-kinase activity and glucose transport. Metab. Clin. Exp. 2003, 52, 1196–1205. [Google Scholar] [CrossRef]
- Weinstein, S.P.; Wilson, C.M.; Pritsker, A.; Cushman, S.W. Dexamethasone inhibits insulin-stimulated recruitment of glut4 to the cell surface in rat skeletal muscle. Metab. Clin. Exp. 1998, 47, 3–6. [Google Scholar] [CrossRef]
- Perseghin, G.; Petersen, K.; Shulman, G.I. Cellular mechanism of insulin resistance: Potential links with inflammation. Int. J. Obes. Relat. Metab. Disord. 2003, 27 (Suppl. 3), S6–S11. [Google Scholar] [CrossRef] [PubMed]
- Dirks-Naylor, A.J.; Griffiths, C.L. Glucocorticoid-induced apoptosis and cellular mechanisms of myopathy. J. Steroid Biochem. Mol. Biol. 2009, 117, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Knotts, T.A.; Wang, L.P.; Li, G.; Dobrowsky, R.T.; Florant, G.L.; Summers, S.A. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J. Biol. Chem. 2003, 278, 10297–10303. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Barrett, E.J.; Wei, L.; Liu, Z. Adrenalectomy enhances the insulin sensitivity of muscle protein synthesis. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E102–E109. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Brown, P.D.; Badal, S.; Morrison, S.; Ragoobirsingh, D. Acute impairment of insulin signalling by dexamethasone in primary cultured rat skeletal myocytes. Mol. Cell. Biochem. 2007, 297, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Girogino, F.; Almahfouz, A.; Goodyear, L.J.; Smith, R.J. Glucocorticoid regulation of insulin receptor and substrate irs-1 tyrosine phosphorylation in rat skeletal muscle in vivo. J. Clin. Investig. 1993, 91, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.A.; Folli, F.; Khan, J.A.; Kahn, C.R. Modulation of ir, irs-1 and pi3-k in liver and muscle of dexamethasone-treated rats. J. Clin. Investig. 1993, 92, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Buren, J.; Liu, H.X.; Jensen, J.; Eriksson, J.W. Dexamethasone impairs insulin signalling and glucose transport by depletion of irs-1, pi3k and pkb. Eur. J. Endocrinol. 2002, 146, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.-i.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between glucocorticoid receptor and nutritional sensor mtor in skeletal muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pessin, J.E. Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Schakman, O.; Gilson, H.; Thissen, J.P. Mechanisms of glucocorticoid-induced myopathy. J. Endocrinol. 2008, 197, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L.; Tischler, M.; DeMartino, G.; Griffin, G. Hormonal regulation of protein degradation and synthesis in skeletal muscle. Fed. Proc. 1980, 39, 31–36. [Google Scholar] [PubMed]
- Whorwood, C.B.; Donovan, S.J.; Flanagan, D.; Phillips, D.I.W.; Byrne, C.D. Increased glucocorticoid receptor expression in human skeletal muscle cells may contribute to the pathogenesis of the metabolic syndrome. Diabetes 2002, 51, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Barbera, M.; Fierabracci, V.; Novelli, M.; Bombara, M.; Masiello, P.; Bergamini, E.; De Tata, V. Dexamethasone-induced insulin resistance and pancreatic adaptive response in aging rats are not modified by oral vanadyl sulfate treatment. Eur. J. Endocrinol. 2001, 145, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Schmoll, D. Novel concepts in insulin regulation of hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E685–E692. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.C.; Walker, B.R. Glucocorticoids and insulin resistance: Old hormones, new targets. Clin. Sci. 1999, 96, 513–523. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Gerich, J.; Rizza, R. Effects of glucocorticoids on carbohydrate metabolism. Diabetes Metab. Rev. 1988, 4, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Rizza, R.A.; Mandarino, L.J.; Gerich, J.E. Cortisol-induced insulin resistance in man: Impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor defect of insulin action. J. Clin. Endocrinol. Metab. 1982, 54, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Kooi, B.T.V.; Onuma, H.; Oeser, J.K.; Svitek, C.A.; Allen, S.R.; Kooi, C.W.V.; Chazin, W.J.; O’Brien, R.M. The glucose-6-phosphatase catalytic subunit gene promoter contains both positive and negative glucocorticoid response elements. Mol. Endocrinol. 2005, 19, 3001–3022. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.Y.; DuBois, D.C.; Almon, R.R.; Jusko, W.J. Receptor/gene-mediated pharmacodynamic effects of methylprednisolone on phosphoenolpyruvate carboxykinase regulation in rat liver. J. Pharmacol. Exp. Ther. 2004, 309, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Parekh, S.; Anania, F.A. Abnormal lipid and glucose metabolism in obesity: Implications for nonalcoholic fatty liver disease. Gastroenterology 2007, 132, 2191–2207. [Google Scholar] [CrossRef] [PubMed]
- Tiniakos, D.G.; Vos, M.B.; Brunt, E.M. Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.M.; Petersenn, S. 3 rationale for treatment and therapeutic options in cushing’s disease. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, S15–S22. [Google Scholar] [CrossRef]
- Abad, V.; Chrousos, G.P.; Reynolds, J.C.; Nieman, L.K.; Hill, S.C.; Weinstein, R.S.; and Leong, G.M. Glucocorticoid excess during adolescence leads to a major persistent deficit in bone mass and an increase in central body fat. J. Bone Miner. Res. 2001, 16, 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; He, J.; Jiang, H.; Zu, L.; Zhai, W.; Pu, S.; Xu, G. Direct effect of glucocorticoids on lipolysis in adipocytes. Mol. Endocrinol. 2009, 23, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Peckett, A.J.; D’souza, A.M.; Hawke, T.J.; Riddell, M.C. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am. J. Physiol. Cell Physiol. 2011, 300, C198–C209. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-J.; Pramyothin, P.; Karastergiou, K.; Fried, S.K. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim. Biophys. Acta 2014, 1842, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, N.; Akiyama, Y.; Kato, H.; Kuroda, T.; Ono, T.; Imagawa, K.; Asakura, K.; Shinosaki, T.; Murayama, T.; Hanasaki, K. Pharmacological evaluation of adipose dysfunction via 11beta-hydroxysteroid dehydrogenase type 1 in the development of diabetes in diet-induced obese mice with cortisone pellet implantation. J. Pharmacol. Exp. Ther. 2014, 349, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Desbriere, R.; Vuaroqueaux, V.; Achard, V.; Boullu-Ciocca, S.; Labuhn, M.; Dutour, A.; Grino, M. 11β-hydroxysteroid dehydrogenase type 1 mrna is increased in both visceral and subcutaneous adipose tissue of obese patients. Obesity 2006, 14, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Bujalska, I.J.; Kumar, S.; Stewart, P.M. Does central obesity reflect “cushing’s disease of the omentum”? Lancet 1997, 349, 1210–1213. [Google Scholar] [CrossRef]
- Rebuffé-Scrive, M.; Brönnegard, M.; Nilsson, A.; Eldh, J.; Gustafsson, J.-A.; Björntorp, P. Steroid hormone receptors in human adipose tissues. J. Clin. Endocrinol. Metab. 1990, 71, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, J.; Weck, M.; Nilsson, A.; Ottosson, M.; Björntorp, P. Glucocorticoid hormone binding to rat adipocytes. Biochim. Biophys. Acta 1994, 1224, 17–21. [Google Scholar] [CrossRef]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, N.; Dallman, M.F.; Warne, J.P.; Ginsberg, A.B.; Laugero, K.D.; la Fleur, S.E.; Houshyar, H.; Gomez, F.; Bhargava, A.; Akana, S.F. From malthus to motive: How the hpa axis engineers the phenotype, yoking needs to wants. Prog. Neurobiol. 2006, 79, 247–340. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.E.; Czarnecka, M.; Kitlinska, J.B.; Tilan, J.U.; Kvetňanský, R.; Zukowska, Z. Chronic stress, combined with a high-fat/high-sugar diet, shifts sympathetic signaling toward neuropeptide y and leads to obesity and the metabolic syndrome. Ann. N. Y. Acad. Sci. 2008, 1148, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.E.; Bhargava, A.; Soriano, L.; Laugero, K.; Akana, S.F.; Dallman, M.F. Sucrose intake and corticosterone interact with cold to modulate ingestive behaviour, energy balance, autonomic outflow and neuroendocrine responses during chronic stress. J. Neuroendocrinol. 2002, 14, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, N.; Reyes, F.; Gomez, F.; Bhargava, A.; Dallman, M.F. Chronic stress promotes palatable feeding, which reduces signs of stress: Feedforward and feedback effects of chronic stress. Endocrinology 2004, 145, 3754–3762. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, T.W. Glucocorticoids as modulators in the control of feeding. Brain Res. Bull. 1991, 27, 423–428. [Google Scholar] [CrossRef]
- Leibowitz, S.F.; Sladek, C.; Spencer, L.; Tempel, D. Neuropeptide y, epinephrine and norepinephrine in the paraventricular nucleus: Stimulation of feeding and the release of corticosterone, vasopressin and glucose. Brain Res. Bull. 1988, 21, 905–912. [Google Scholar] [CrossRef]
- Epel, E.; Lapidus, R.; McEwen, B.; Brownell, K. Stress may add bite to appetite in women: A laboratory study of stress-induced cortisol and eating behavior. Psychoneuroendocrinology 2001, 26, 37–49. [Google Scholar] [CrossRef]
- Strack, A.M.; Schwartz, M.W.; Dallman, M.F. Glucocorticoids and insulin: Reciprocal signals for energy balance. Am. J. Physiol. 1995, 268, R141–R149. [Google Scholar]
- Targownik, L.E.; Nugent, Z.; Singh, H.; Bernstein, C.N. Prevalence of and outcomes associated with corticosteroid prescription in inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Deer, J.; Koska, J.; Ozias, M.; Reaven, P. Dietary models of insulin resistance. Metab. Clin. Exp. 2015, 64, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Gounarides, J.S.; Korach-Andre, M.; Killary, K.; Argentieri, G.; Turner, O.; Laurent, D. Effect of dexamethasone on glucose tolerance and fat metabolism in a diet-induced obesity mouse model. Endocrinology 2008, 149, 758–766. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.; de Mattos, A.B.; Biz, C.; Oyama, L.M.; Ribeiro, E.B.; do Nascimento, C.M.O. High-fat diet and glucocorticoid treatment cause hyperglycemia associated with adiponectin receptor alterations. Lipids Health Dis. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.; Iwanaga-Carvalho, C.; Mota, J.F.; Oyama, L.M.; Ribeiro, E.B.; Oller do Nascimento, C.M. Effects of adrenal hormones on the expression of adiponectin and adiponectin receptors in adipose tissue, muscle and liver. Steroids 2011, 76, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Shpilberg, Y.; Beaudry, J.L.; D’Souza, A.; Campbell, J.E.; Peckett, A.; Riddell, M.C. A rodent model of rapid-onset diabetes induced by glucocorticoids and high-fat feeding. Dis. Mod. Mech. 2012, 5, 671–680. [Google Scholar] [CrossRef] [PubMed]
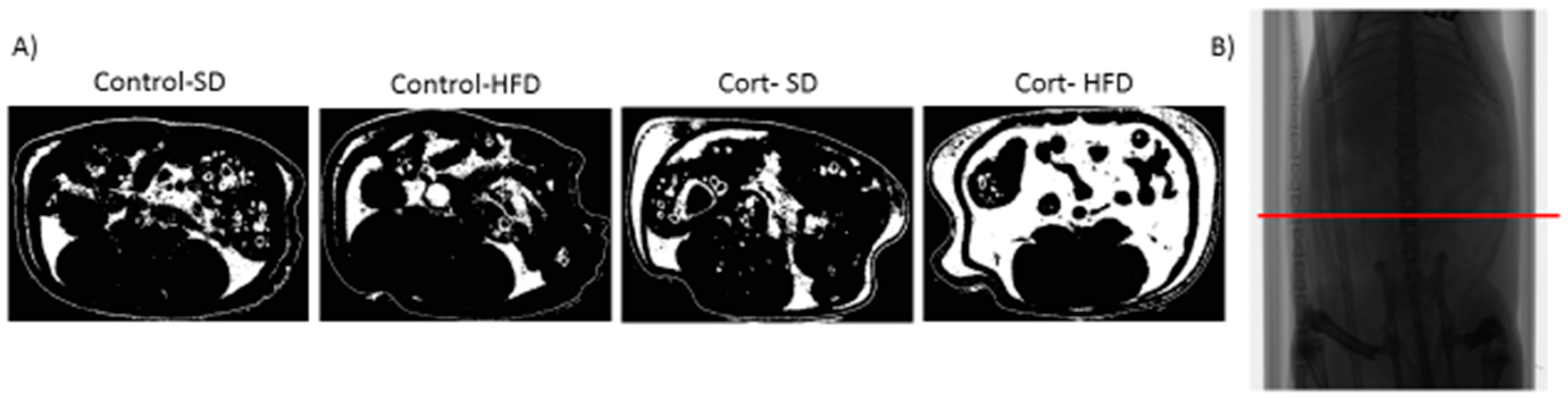
- D’Souza, A.M.; Beaudry, J.L.; Szigiato, A.; Trumble, S.J.; Snook, L.A.; Bonen, A.; Giacca, A.; Riddell, M.C. Consumption of a high-fat diet rapidly exacerbates the development of fatty liver disease that occurs with chronically elevated glucocorticoids. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 302, G850–G863. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, J.L.; D’Souza A, M.; Teich, T.; Tsushima, R.; Riddell, M.C. Exogenous glucocorticoids and a high-fat diet cause severe hyperglycemia and hyperinsulinemia and limit islet glucose responsiveness in young male sprague-dawley rats. Endocrinology 2013, 154, 3197–3208. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, J.L.; Dunford, E.C.; Leclair, E.; Mandel, E.R.; Peckett, A.J.; Haas, T.L.; Riddell, M.C. Voluntary exercise improves metabolic profile in high-fat fed glucocorticoid-treated rats. J. Appl. Physiol. 2015, 118, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, X.; Jiao, H.; Zhao, J.; Lin, H. Glucocorticoids inhibited hypothalamic target of rapamycin in high fat diet-fed chicks. Poult Sci. 2015, 94, 2221–2227. [Google Scholar] [CrossRef] [PubMed]
- Bonen, A.; Jain, S.S.; Snook, L.A.; Han, X.X.; Yoshida, Y.; Buddo, K.H.; Lally, J.S.; Pask, E.D.; Paglialunga, S.; Beaudoin, M.S.; et al. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia 2015, 58, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Balsevich, G.; Uribe, A.; Wagner, K.V.; Hartmann, J.; Santarelli, S.; Labermaier, C.; Schmidt, M.V. Interplay between diet-induced obesity and chronic stress in mice: Potential role of fkbp51. J. Endocrinol. 2014, 222, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bruder-Nascimento, T.; Campos, D.H.S.; Alves, C.; Thomaz, S.; Cicogna, A.C.; Cordellini, S. Effects of chronic stress and hfd on metabolic and nutritional parameters in wistar rats. Arq. Bras. Endocrinol. Metab. 2013, 57, 642–649. [Google Scholar] [CrossRef]
- Campbell, J.E.; Rakhshani, N.; Fediuc, S.; Bruni, S.; Riddell, M.C. Voluntary wheel running initially increases adrenal sensitivity to adrenocorticotrophic hormone, which is attenuated with long-term training. J. Appl. Physiol. 2009, 106, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, A.E.; Campbell, J.E.; Fediuc, S.; Riddell, M.C. Effect of voluntary exercise on peripheral tissue glucocorticoid receptor content and the expression and activity of 11beta-hsd1 in the syrian hamster. J. Appl. Physiol. 2006, 100, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.L.; Kang, H.Y. Effects of endurance exercise and high-fat diet on insulin resistance and ceramide contents of skeletal muscle in sprague-dawley rats. Korean Diabetes J. 2010, 34, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Barel, M.; Perez, O.A.; Giozzet, V.A.; Rafacho, A.; Bosqueiro, J.R.; do Amaral, S.L. Exercise training prevents hyperinsulinemia, muscular glycogen loss and muscle atrophy induced by dexamethasone treatment. Eur. J. Appl. Physiol. 2010, 108, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Hakkinen, A.; Sokka, T.; Kotaniemi, A.; Hannonen, P. A randomized two-year study of the effects of dynamic strength training on muscle strength, disease activity, functional capacity, and bone mineral density in early rheumatoid arthritis. Arthritis Rheum. 2001, 44, 515–522. [Google Scholar] [CrossRef]
- Al-Nassan, S.; Fujita, N.; Kondo, H.; Murakami, S.; Fujino, H. Chronic exercise training down-regulates tnf-alpha and atrogin-1/mafbx in mouse gastrocnemius muscle atrophy induced by hindlimb unloading. Acta Histochem. Cytochem. 2012, 45, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Hickson, R.C.; Davis, J.R. Partial prevention of glucocorticoid-induced muscle atrophy by endurance training. Am. J. Physiol. 1981, 241, E226–E232. [Google Scholar] [PubMed]
- Macedo, A.G.; Krug, A.L.; Herrera, N.A.; Zago, A.S.; Rush, J.W.; Amaral, S.L. Low-intensity resistance training attenuates dexamethasone-induced atrophy in the flexor hallucis longus muscle. J. Steroid Biochem. Mol. Biol. 2014, 143, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Nicastro, H.; Zanchi, N.E.; da Luz, C.R.; de Moraes, W.M.; Ramona, P.; de Siqueira Filho, M.A.; Chaves, D.F.; Medeiros, A.; Brum, P.C.; Dardevet, D.; et al. Effects of leucine supplementation and resistance exercise on dexamethasone-induced muscle atrophy and insulin resistance in rats. Nutrition 2012, 28, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Alberga, A.S.; Prud’homme, D.; Sigal, R.J.; Goldfield, G.S.; Hadjiyannakis, S.; Phillips, P.; Malcolm, J.; Ma, J.; Doucette, S.; Gougeon, R.; et al. Effects of aerobic training, resistance training, or both on cardiorespiratory and musculoskeletal fitness in adolescents with obesity: The hearty trial. Appl. Physiol. Nutr. Metab. 2015, 41, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Colberg, S.R.; Sigal, R.J. Moving beyond cardio: The value of resistance training, balance training, and other forms of exercise in the management of diabetes. Diabetes Spectr. 2015, 28, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; Macedo, A.; Zago, A.; Rush, J.; Santos, C.; Amaral, S. High-intensity resistance training attenuates dexamethasone-induced muscle atrophy. Muscle Nerv. 2015, 53, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Holten, M.K.; Zacho, M.; Gastor, M.; Juel, C.; Wojtaszewski, J.F.P.; Dela, F. Strength training increases insulin-mediated glucose uptake, glut4 content and insulin signaling in skeletal muscle in patients with t2dm. Diabetes 2004, 53, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Dube, J.J.; Amati, F.; Toledo, F.G.; Stefanovic-Racic, M.; Rossi, A.; Coen, P.; Goodpaster, B.H. Effects of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia 2011, 54, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Amati, F.; Dube, J.J.; Alvarez-Carnero, E.; Edreira, M.M.; Chomentowski, P.; Coen, P.M.; Switzer, G.E.; Bickel, P.E.; Stefanovic-Racic, M.; Toledo, F.G.; et al. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance: Another paradox in endurance-trained athletes? Diabetes 2011, 60, 2588–2597. [Google Scholar] [CrossRef] [PubMed]
- Dionísio, T.; Louzada, J.; Viscelli, B.; Dionísio, E.; Martuscelli, A.; Barel, M.; Perez, O.; Bosqueiro, J.R.; Brozoski, D.; Santos, C. Aerobic training prevents dexamethasone-induced peripheral insulin resistance. Horm. Metab. Res. 2014, 46, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhai, X.; Li, H.; Ji, L. Depression-like behaviors in mice subjected to co-treatment of high-fat diet and corticosterone are ameliorated by aicar and exercise. J. Affect. Disord. 2014, 156, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Boersma, G.J.; Tamashiro, K.L.; Moran, T.H.; Liang, N.-C. Corticosterone administration in drinking water decreases high-fat diet intake but not preference in male rats. Am. J. Physiol. 2016, 310, R733–R743. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dunford, E.C.; Riddell, M.C. The Metabolic Implications of Glucocorticoids in a High-Fat Diet Setting and the Counter-Effects of Exercise. Metabolites 2016, 6, 44. https://doi.org/10.3390/metabo6040044
Dunford EC, Riddell MC. The Metabolic Implications of Glucocorticoids in a High-Fat Diet Setting and the Counter-Effects of Exercise. Metabolites. 2016; 6(4):44. https://doi.org/10.3390/metabo6040044
Chicago/Turabian StyleDunford, Emily C., and Michael C. Riddell. 2016. "The Metabolic Implications of Glucocorticoids in a High-Fat Diet Setting and the Counter-Effects of Exercise" Metabolites 6, no. 4: 44. https://doi.org/10.3390/metabo6040044
APA StyleDunford, E. C., & Riddell, M. C. (2016). The Metabolic Implications of Glucocorticoids in a High-Fat Diet Setting and the Counter-Effects of Exercise. Metabolites, 6(4), 44. https://doi.org/10.3390/metabo6040044