
Impact of Glucocorticoid Excess on Glucose Tolerance: Clinical and Preclinical Evidence


Abstract
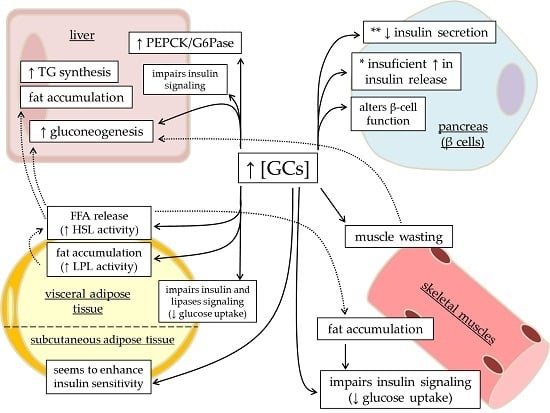
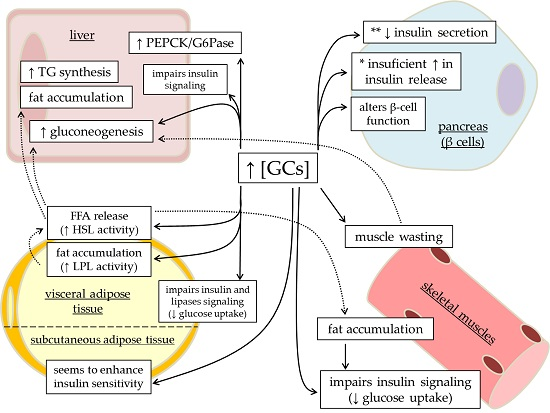
:
1. Introduction
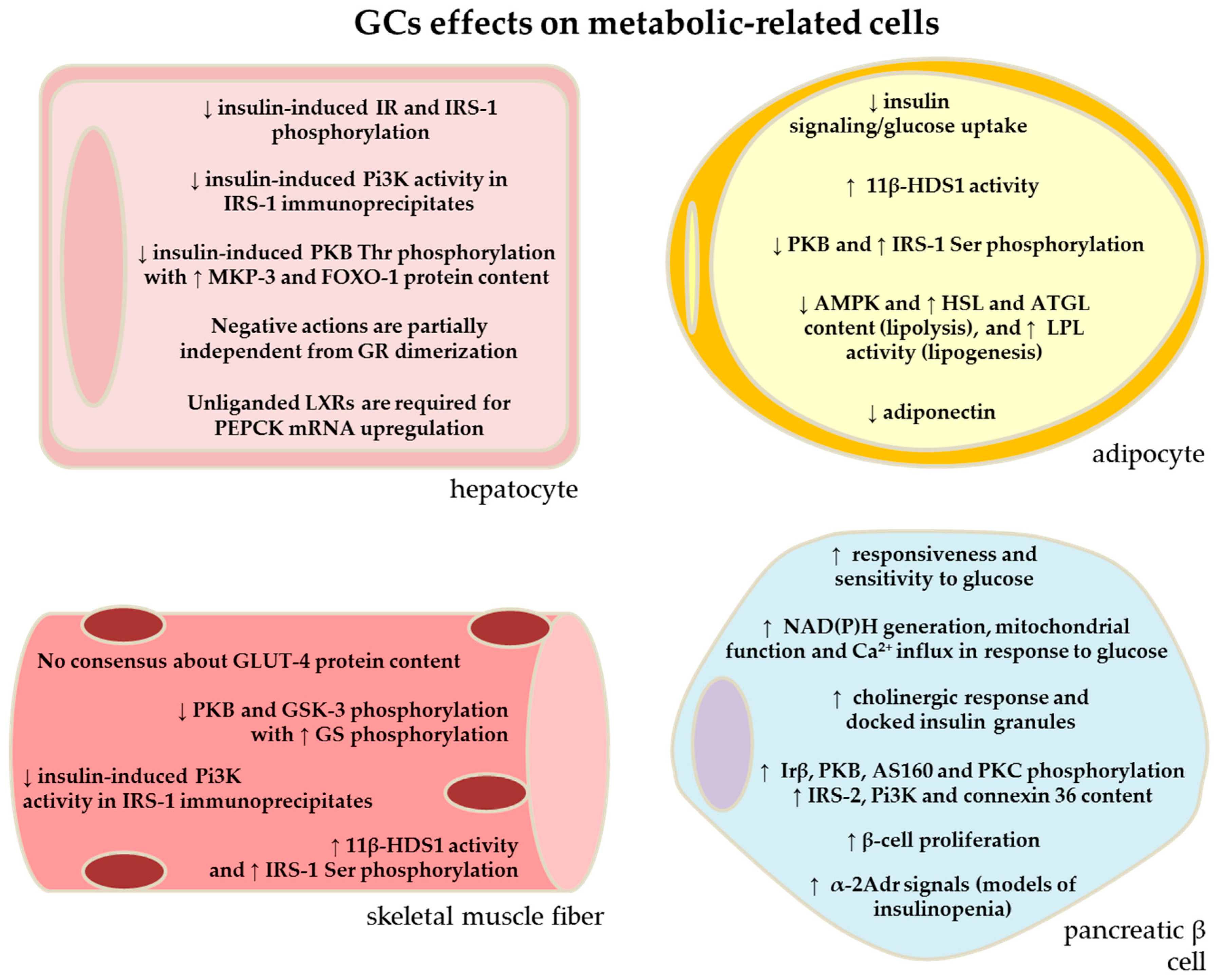
2. Effects of GC Excess in the Liver: Contribution of Increased HGO to GC-Induced Glucose Intolerance
3. Effects of GC Excess in the Skeletal Muscle: Contribution of Reduced Glucose Uptake to GC-Induced Glucose Intolerance
4. GC Excess and Adipose Tissue: Contribution of Increased Adipose Tissue Lipolysis for GC-Related Glucose Intolerance
5. Effects of GC Excess in the Pancreatic Islets: Contribution of Reduced β-Cell Function for GC-Induced Glucose Intolerance
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AMPK | 5′ Adenosine Monophosphate-Activated Kinase |
| cAMP | cyclic Adenosine Monophosphate |
| DAG | Diacylglycerol |
| DEX | Dexamethasone |
| EGP | Endogenous Glucose Production |
| FFA | Free Fatty Acids |
| FoxO-1 | Forkhead Box O-1 |
| GC | Glucocorticoid |
| GLUT-4 | Glucose Transporter 4 |
| GR | Glucocorticoid Receptor |
| GRdim | Genetic Engineered Mice Model of GR Dimerization |
| GS | Glycogen Synthase |
| GSIS | Glucose-Stimulated insulin Secretion |
| GSK-3 | Glycogen Synthase Kinase 3 |
| G6Pase | Glucose-6-Phosphatase |
| HGO | Hepatic Glucose Output |
| HGP | Hepatic Glucose Production |
| IGF-1 | Insulin-Like Growth Factor 1 |
| IL | Interleukine |
| IMTG | increased intramuscular triglycerides |
| IR | Insulin Resistance |
| IRS-1 | Insulin Receptor Substrate 1 |
| IRS-2 | Insulin Receptor Substrate 2 |
| KK | mouse model of metabolic syndrome |
| LPL | Lipoprotein Lipase |
| LXR | Liver X Receptors |
| MKP-3 | MAP kinase phosphatase 3 |
| mRNA | messenger Ribonucleic Acid |
| oGTT | oral Glucose Tolerance Test |
| PEPCK | Phosphoenolpyruvate-Carboxykinase |
| Pi3K | Phosphoinositide 3-Kinase |
| PKA | Protein Kinase A |
| PKB | Protein Kinase B |
| PKC | Protein Kinase C |
| PP-1 | Protein Phosphatase 1 |
| p-PKB | Phosphorylated Protein Kinase B |
| PRED | Prednisolone |
| RA | Rheumatoid Arthritis |
| RXR | Retinoid X Receptors |
| TNF-α | Tumor Necrosis Factor α |
| T2DM | Type 2 Diabetes Mellitus |
| 2-[3H]DG | 2-[3H] Deoxyglucose |
| 11β-HSD-1 | 11β Hydroxysteroid Dehydrogenase Type 1 |
| 11-DHC | 11 Dehydrocorticosterone |
References
- Andrews, R.C.; Walker, B.R. Glucocorticoids and insulin resistance: Old hormones, new targets. Clin. Sci. 1999, 96, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Schäcke, H.; Döcke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- De Bosscher, K.; Haegeman, G.; Elewaut, D. Targeting inflammation using selective glucocorticoid receptor modulators. Curr. Opin. Pharmacol. 2010, 10, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, A.; Boschero, A.C.; Ortsäter, H. Functional and Molecular Aspects of Glucocorticoids in the Endocrine Pancreas and Glucose Homeostasis. In State of the Art of Therapeutic Endocrinology; Magdeldin, S., Ed.; InTech: Rijeka, Croatia, 2012; pp. 121–152. [Google Scholar] [CrossRef]
- Kwon, S.; Hermayer, K.L. Glucocorticoid-induced hyperglycemia. Am. J. Med. Sci. 2013, 345, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Mokuda, O.; Sakamoto, Y.; Ikeda, T.; Mashiba, H. Sensitivity and responsiveness of glucose output to insulin in isolated perfused liver from dexamethasone-treated rats. Horm. Metab. Res. 1991, 23, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, N.; Lim, J.; Bouch, C.; Marciano, D.; Davidson, M.B. Dexamethasone-induced impairment in skeletal muscle glucose transport is not reversed by inhibition of free fatty acid oxidation. Metabolism 1996, 45, 92–100. [Google Scholar] [CrossRef]
- Kimura, M.; Daimon, M.; Tominaga, M.; Manaka, H.; Sasaki, H.; Kato, T. Thiazolidinediones exert different effects on insulin resistance between dexamethasone-treated rats and wistar fatty rats. Endocr. J. 2000, 47, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Nicod, N.; Giusti, V.; Besse, C.; Tappy, L. Metabolic adaptations to dexamethasone-induced insulin resistance in healthy volunteers. Obes. Res. 2003, 11, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Van Raalte, D.H.; Brands, M.; van der Zijl, N.J.; Muskiet, M.H.; Pouwels, P.J.; Ackermans, M.T.; Sauerwein, H.P.; Serlie, M.J.; Diamant, M. Low-dose glucocorticoid treatment affects multiple aspects of intermediary metabolism in healthy humans: A randomized controlled trial. Diabetologia 2011, 54, 2103–2112. [Google Scholar] [CrossRef] [PubMed]
- Paquot, N.; Schneiter, P.; Jéquier, E.; Tappy, L. Effects of glucocorticoids and sympathomimetic agents on basal and insulin-stimulated glucose metabolism. Clin. Physiol. 1995, 15, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Ruzzin, J.; Wagman, A.S.; Jensen, J. Glucocorticoid-induced insulin resistance in skeletal muscles: Defects in insulin signalling and the effects of a selective glycogen synthase kinase-3 inhibitor. Diabetologia 2005, 48, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Burén, J.; Lai, Y.C.; Lundgren, M.; Eriksson, J.W.; Jensen, J. Insulin action and signalling in fat and muscle from dexamethasone-treated rats. Arch. Biochem. Biophys. 2008, 474, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Delarue, J.; Allain-Jeannic, G.; Guillerm, S.; Cruciani-Guglielmacci, C.; Magnan, C.; Moineau, M.P.; Le Guen, V. Interaction of low dose of fish oil and glucocorticoids on insulin sensitivity and lipolysis in healthy humans: A randomized controlled study. Mol. Nutr. Food Res. 2016, 60, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Schneiter, P.; Tappy, L. Kinetics of dexamethasone-induced alterations of glucose metabolism in healthy humans. Am. J. Physiol. 1998, 275, E806–E813. [Google Scholar] [PubMed]
- Rafacho, A.; Giozzet, V.A.; Boschero, A.C.; Bosqueiro, J.R. Functional alterations in endocrine pancreas of rats with different degrees of dexamethasone-induced insulin resistance. Pancreas 2008, 36, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Van Raalte, D.H.; Nofrate, V.; Bunck, M.C.; van Iersel, T.; Elassaiss Schaap, J.; Nässander, U.K.; Heine, R.J.; Mari, A.; Dokter, W.H.; Diamant, M. Acute and 2-week exposure to prednisolone impair different aspects of beta-cell function in healthy men. Eur. J. Endocrinol. 2010, 162, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, A.; Abrantes, J.L.; Ribeiro, D.L.; Paula, F.M.; Pinto, M.E.; Boschero, A.C.; Bosqueiro, J.R. Morphofunctional alterations in endocrine pancreas of short- and long-term dexamethasone-treated rats. Horm. Metab. Res. 2011, 43, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Van Raalte, D.H.; van Genugten, R.E.; Linssen, M.M.; Ouwens, D.M.; Diamant, M. Glucagon-like peptide-1 receptor agonist treatment prevents glucocorticoid-induced glucose intolerance and islet-cell dysfunction in humans. Diabetes Care. 2011, 34, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Van Raalte, D.H.; Kwa, K.A.; van Genugten, R.E.; Tushuizen, M.E.; Holst, J.J.; Deacon, C.F.; Karemaker, J.M.; Heine, R.J.; Mari, A.; Diamant, M. Islet-cell dysfunction induced by glucocorticoid treatment: Potential role for altered sympathovagal balance? Metabolism 2013, 62, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Nosadini, R.; Del Prato, S.; Tiengo, A.; Valerio, A.; Muggeo, M.; Opocher, G.; Mantero, F.; Duner, E.; Marescotti, C.; Mollo, F.; et al. Insulin resistance in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1983, 57, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Jitrapakdee, S. Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int. J. Biochem. Cell Biol. 2012, 44, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Margolis, R.N.; Curnow, R.T. Effects of dexamethasone administration on hepatic glycogen synthesis and accumulation in adrenalectomized fasted rats. Endocrinology 1984, 115, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Wajngot, A.; Khan, A.; Giacca, A.; Vranic, M.; Efendic, S. Dexamethasone increases glucose cycling, but not glucose production, in healthy subjects. Am. J. Physiol. 1990, 259, E626–E632. [Google Scholar] [PubMed]
- Wajngot, A.; Giacca, A.; Grill, V.; Vranic, M.; Efendic, S. The diabetogenic effects of glucocorticoids are more pronounced in low- than in high-insulin responders. Proc. Natl. Acad. Sci. USA 1992, 89, 6035–6039. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.; Folli, F.; Kahn, J.A.; Kahn, C.R. Modulation of insulin receptor, insulin receptor substrate-1, and phosphatidylinositol 3-kinase in liver and muscle of dexamethasone-treated rats. J. Clin. Investig. 1993, 92, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Nader, N.; Ng, S.S.; Wang, Y.; Abel, B.S.; Chrousos, G.P.; Kino, T. Liver X receptors regulate the transcriptional activity of the glucocorticoid receptor: Implications for the carbohydrate metabolism. PLoS ONE 2012, 7, e26751. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Johnson, J.; Liu, F.; Jen, P.; Reaven, G.M. The effects of acute and chronic dexamethasone administration on insulin binding to isolated rat hepatocytes and adipocytes. Metabolism 1975, 24, 517–527. [Google Scholar] [CrossRef]
- Shamoon, H.; Soman, V.; Sherwin, R.S. The influence of acute physiological increments of cortisol on fuel metabolism and insulin binding to monocytes in normal humans. J. Clin. Endocrinol. Metab. 1980, 50, 495–501, Retraction in: Felig, P.; Sherwin, R.S. J. Clin. Endocrinol. Metab. 1980, 51, 1201. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, A.; Ortsäter, H.; Nadal, A.; Quesada, I. Glucocorticoid treatment and endocrine pancreas function: Implications for glucose homeostasis, insulin resistance and diabetes. J. Endocrinol. 2014, 223, R49–R62. [Google Scholar] [CrossRef] [PubMed]
- Ahrén, B. Evidence that autonomic mechanisms contribute to the adaptive increase in insulin secretion during dexamethasone-induced insulin resistance in humans. Diabetologia 2008, 51, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Peckett, A.J.; D’souza, A.M.; Hawke, T.J.; Riddell, M.C. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am. J. Physiol. Cell Physiol. 2011, 300, C198–C209. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, A.; Cestari, T.M.; Taboga, S.R.; Boschero, A.C.; Bosqueiro, J.R. High doses of dexamethasone induce increased beta-cell proliferation in pancreatic rat islets. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E681–E689. [Google Scholar] [CrossRef] [PubMed]
- Petersons, C.J.; Mangelsdorf, B.L.; Jenkins, A.B.; Poljak, A.; Smith, M.D.; Greenfield, J.R.; Thompson, C.H.; Burt, M.G. Effects of low-dose prednisolone on hepatic and peripheral insulin sensitivity, insulin secretion, and abdominal adiposity in patients with inflammatory rheumatologic disease. Diabetes Care 2013, 36, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Binnert, C.; Ruchat, S.; Nicod, N.; Tappy, L. Dexamethasone-induced insulin resistance shows no gender difference in healthy humans. Diabetes Metab. 2004, 30, 321–326. [Google Scholar] [CrossRef]
- Den Uyl, D.; van Raalte, D.H.; Nurmohamed, M.T.; Lems, W.F.; Bijlsma, J.W.; Hoes, J.N.; Dijkmans, B.A.; Diamant, M. Metabolic effects of high-dose prednisolone treatment in early rheumatoid arthritis: Balance between diabetogenic effects and inflammation reduction. Arthritis Rheum. 2012, 64, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martínez, M.U.; Martínez-Lozano, J.; Abud-Mendoza, C. Hyperglycemia and methylprednisolone: Comment on the article by den Uyl et al. Arthritis Rheum. 2012, 64, 3822–3833. [Google Scholar] [CrossRef] [PubMed]
- Frijters, R.; Fleuren, W.; Toonen, E.J.; Tuckermann, J.P.; Reichardt, H.M.; van der Maaden, H.; van Elsas, A.; van Lierop, M.J.; Dokter, W.; de Vlieg, J.; et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genomics 2010, 5, 259. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; He, Q.; Xu, H. FOXO1-dependent up-regulation of MAP kinase phosphatase 3 (MKP-3) mediates glucocorticoid-induced hepatic lipid accumulation in mice. Mol. Cell. Endocrinol. 2014, 393, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, A.; Gonçalves-Neto, L.M.; Santos-Silva, J.C.; Alonso-Magdalena, P.; Merino, B.; Taboga, S.R.; Carneiro, E.M.; Boschero, A.C.; Nadal, A.; Quesada, I. Pancreatic alpha-cell dysfunction contributes to the disruption of glucose homeostasis and compensatory insulin hypersecretion in glucocorticoid-treated rats. PLoS ONE 2014, 9, e93531. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, M.; Smith, T.J.; Krichevsky, A.; Bilezikian, J.P. Glucocorticoids enhance glucose uptake and affect differentiation and beta-adrenergic responsiveness in muscle cell cultures. Cell Differ. 1981, 10, 101–107. [Google Scholar] [CrossRef]
- Haber, R.S.; Weinstein, S.P. Role of glucose transporters in glucocorticoid-induced insulin resistance. GLUT4 isoform in rat skeletal muscle is not decreased by dexamethasone. Diabetes 1992, 41, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.; Elayan, H.; Ziegler, M.G. Glucocorticoid induction of epinephrine synthesizing enzyme in rat skeletal muscle and insulin resistance. J. Clin. Investig. 1993, 92, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.P.; Paquin, T.; Pritsker, A.; Haber, R.S. Glucocorticoid-induced insulin resistance: Dexamethasone inhibits the activation of glucose transport in rat skeletal muscle by both insulin- and non-insulin-related stimuli. Diabetes 1995, 44, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, S.P.; Wilson, C.M.; Pritsker, A.; Cushman, S.W. Dexamethasone inhibits insulin-stimulated recruitment of GLUT4 to the cell surface in rat skeletal muscle. Metabolism 1998, 47, 3–6. [Google Scholar] [CrossRef]
- Ruzzin, J.; Jensen, J. Contraction activates glucose uptake and glycogen synthase normally in muscles from dexamethasone-treated rats. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E241–E250. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.; Lew, M.J.; Mayba, O.; Harris, C.A.; Speed, T.P.; Wang, J.C. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 11160–11165. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, T.; Yamamoto, M.; Nagashima, T.; Kajita, K.; Taniguchi, O.; Yasuda, K.; Miura, K. Effect of dexamethasone and prednisolone on insulin-induced activation of protein kinase C in rat adipocytes and soleus muscles. Metabolism 1995, 44, 298–306. [Google Scholar] [CrossRef]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.A.; Sherlock, M.; Gathercole, L.L.; Lavery, G.G.; Lenaghan, C.; Bujalska, I.J.; Laber, D.; Yu, A.; Convey, G.; Mayers, R.; et al. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 2009, 58, 2506–2515. [Google Scholar] [CrossRef] [PubMed]
- Arnaldi, G.; Scandali, V.M.; Trementino, L.; Cardinaletti, M.; Appolloni, G.; Boscaro, M. Pathophysiology of dyslipidemia in Cushing’s syndrome. Neuroendocrinology 2010, 92 (Suppl. 1), 86–90. [Google Scholar] [CrossRef] [PubMed]
- Geer, E.B.; Shen, W.; Gallagher, D.; Punyanitya, M.; Looker, H.C.; Post, K.D.; Freda, P.U. MRI assessment of lean and adipose tissue distribution in female patients with Cushing’s disease. Clin. Endocrinol. 2010, 73, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Urbanet, R.; Pilon, C.; Calcagno, A.; Peschechera, A.; Hubert, E.-L.; Giacchetti, G.; Gomez-Sanchez, C.; Mulatero, P.; Toffanin, M.; Sonino, N.; et al. Analysis of insulin sensitivity in adipose tissue of patients with primary aldosteronism. J. Clin. Endocrinol. Metab. 2010, 95, 4037–4042. [Google Scholar] [CrossRef] [PubMed]
- Carey, D.G.; Jenkins, A.B.; Campbell, L.V.; Freund, J.; Chisholm, D.J. Abdominal fat and insulin resistance in normal and overweight women: Direct measurements reveal a strong relationship in subjects at both low and high risk of NIDDM. Diabetes 1996, 45, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Han, T.S.; Lean, M.E. A clinical perspective of obesity, metabolic syndrome and cardiovascular disease. JRSM Cardiovasc. Dis. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Djurhuus, C.B.; Gravholt, C.H.; Nielsen, S.; Pedersen, S.B.; Møller, N.; Schmitz, O. Additive effects of cortisol and growth hormone on regional and systemic lipolysis in humans. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E488–E494. [Google Scholar] [CrossRef] [PubMed]
- Divertie, G.D.; Jensen, M.D.; Miles, J.M. Stimulation of lipolysis in humans by physiological hypercortisolemia. Diabetes 1991, 40, 1228–1232. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; He, J.; Jiang, H.; Zu, L.; Zhai, W.; Pu, S.; Xu, G. Direct Effect of Glucocorticoids on Lipolysis in Adipocytes. Mol. Endocrinol. 2009, 23, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Christ-Crain, M.; Kola, B.; Lolli, F.; Fekete, C.; Seboek, D.; Wittmann, G.; Feltrin, D.; Igreja, S.C.; Ajodha, S.; Harvey-White, J.; et al. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: A novel mechanism in Cushing’s syndrome. FASEB J. 2008, 22, 1672–1683. [Google Scholar] [CrossRef] [PubMed]
- Fried, S.K.; Russell, C.D.; Grauso, N.L.; Brolin, R.E. Lipoprotein lipase regulation by insulin and glucocorticoid in subcutaneous and omental adipose tissues of obese women and men. J. Clin. Investig. 1993, 92, 2191–2198. [Google Scholar] [CrossRef] [PubMed]
- Appel, B.; Fried, S.K. Effects of insulin and dexamethasone on lipoprotein lipase in human adipose tissue. Am. J. Physiol. 1992, 262, E695–E699. [Google Scholar] [PubMed]
- Peckett, A.J.; Wright, D.C.; Riddell, M.C. The effects of glucocorticoids on adipose tissue lipid metabolism. Metabolism 2011, 60, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Pramyothin, P.; Karastergiou, K.; Fried, S.K. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim. Biophys. Acta 2014, 1842, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Bikman, B.T.; Summers, S.A. Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Investig. 2011, 121, 4222–4230. [Google Scholar] [CrossRef] [PubMed]
- Massart, J.; Zierath, J.R.; Chibalin, A.V. A simple and rapid method to characterize lipid fate in skeletal muscle. BMC Res. Notes 2014, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Silva, K.A.S.; Dong, Y.; Zhang, L. Glucocorticoids increase adipocytes in muscle by affecting IL-4 regulated FAP activity. FASEB J. 2014, 28, 4123–4132. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 13, 785–789. [Google Scholar] [CrossRef]
- Van Raalte, D.H.; Ouwens, D.M.; Diamant, M. Novel insights into glucocorticoid-mediated diabetogenic effects: Towards expansion of therapeutic options? Eur. J. Clin. Investig. 2009, 39, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Müller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [PubMed]
- Shpilberg, Y.; Beaudry, J.L.; D’Souza, A.; Campbell, J.E.; Peckett, A.; Riddell, M.C. A rodent model of rapid-onset diabetes induced by glucocorticoids and high-fat feeding. Dis. Model. Mech. 2012, 5, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Nikkilä, E.A.; Pelkonen, R.; Sane, T. Plasma lipoproteins, lipolytic enzymes, and very low density lipoprotein triglyceride turnover in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1983, 57, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Rockall, A.G.; Sohaib, S.A.; Evans, D.; Kaltsas, G.; Isidori, A.M.; Monson, J.P.; Besser, G.M.; Grossman, A.B.; Reznek, R.H. Hepatic steatosis in Cushing’s syndrome: A radiological assessment using computed tomography. Eur. J. Endocrinol. Eur. Fed. Endocr. Soc. 2003, 149, 543–548. [Google Scholar] [CrossRef]
- D’souza, A.M.; Beaudry, J.L.; Szigiato, A.A.; Trumble, S.J.; Snook, L.A.; Bonen, A.; Giacca, A.; Riddell, M.C. Consumption of a high-fat diet rapidly exacerbates the development of fatty liver disease that occurs with chronically elevated glucocorticoids. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G850–G863. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.P.; Hazlehurst, J.M.; Tomlinson, J.W. Glucocorticoids and non-alcoholic fatty liver disease. J. Steroid Biochem. Mol. Biol. 2015, 154, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Sprangers, F.; Romijn, J.A.; Endert, E.; Ackermans, M.T.; Sauerwein, H.P. The role of free fatty acids (FFA) in the regulation of intrahepatic fluxes of glucose and glycogen metabolism during short-term starvation in healthy volunteers. Clin. Nutr. Edinb. Scotl. 2001, 20, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Allick, G.; Sprangers, F.; Weverling, G.J.; Ackermans, M.T.; Meijer, A.J.; Romijn, J.A.; Endert, E.; Bisschop, P.H.; Sauerwein, H.P. Free fatty acids increase hepatic glycogen content in obese males. Metabolism 2004, 53, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.-X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of Hepatic Insulin Resistance in Non-alcoholic Fatty Liver Disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, M.; Burén, J.; Ruge, T.; Myrnäs, T.; Eriksson, J.W. Glucocorticoids down-regulate glucose uptake capacity and insulin-signaling proteins in omental but not subcutaneous human adipocytes. J. Clin. Endocrinol. Metab. 2004, 89, 2989–2997. [Google Scholar] [CrossRef] [PubMed]
- Motta, K.; Barbosa, A.M.; Bobinski, F.; Boschero, A.C.; Rafacho, A. JNK and IKKβ phosphorylation is reduced by glucocorticoids in adipose tissue from insulin-resistant rats. J. Steroid Biochem. Mol. Biol. 2015, 145, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Munck, A. Glucocorticoid inhibition of glucose uptake by peripheral tissues: Old and new evidence, molecular mechanisms, and physiological significance. Perspect. Biol. Med. 1971, 14, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Armstrong, M.J.; Borrows, S.; Yu, J.; Wagenmakers, A.J.M.; Stewart, P.M.; Tomlinson, J.W. Glucocorticoids fail to cause insulin resistance in human subcutaneous adipose tissue in vivo. J. Clin. Endocrinol. Metab. 2013, 98, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Morton, N.M.; Paterson, J.M.; Masuzaki, H.; Holmes, M.C.; Staels, B.; Fievet, C.; Walker, B.R.; Flier, J.S.; Mullins, J.J.; Seckl, J.R. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 2004, 53, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Veilleux, A.; Rhéaume, C.; Daris, M.; Luu-The, V.; Tchernof, A. Omental adipose tissue type 1 11 beta-hydroxysteroid dehydrogenase oxoreductase activity, body fat distribution, and metabolic alterations in women. J. Clin. Endocrinol. Metab. 2009, 94, 3550–3557. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.E.; Kim, J.M.; Joung, K.H.; Lee, J.H.; You, B.R.; Choi, M.J.; Ryu, M.J.; Ko, Y.B.; Lee, M.A.; Lee, J.; et al. The Roles of Adipokines, Proinflammatory Cytokines, and Adipose Tissue Macrophages in Obesity-Associated Insulin Resistance in Modest Obesity and Early Metabolic Dysfunction. PLoS ONE 2016, 11, e0154003. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Fallo, F.; Scarda, A.; Sonino, N.; Paoletta, A.; Boscaro, M.; Pagano, C.; Federspil, G.; Vettor, R. Effect of glucocorticoids on adiponectin: A study in healthy subjects and in Cushing’s syndrome. Eur. J. Endocrinol. 2004, 150, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Raff, H.; Bruder, E.D. Adiponectin and resistin in the neonatal rat. Endocrine 2006, 29, 341–344. [Google Scholar] [CrossRef]
- Steppan, C.M.; Bailey, S.T.; Bhat, S.; Brown, E.J.; Banerjee, R.R.; Wright, C.M.; Patel, H.R.; Ahima, R.S.; Lazar, M.A. The hormone resistin links obesity to diabetes. Nature 2001, 409, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Krsek, M.; Rosická, M.; Nedvídková, J.; Kvasnicková, H.; Hána, V.; Marek, J.; Haluzík, M.; Lai, E.W.; Pacák, K. Increased lipolysis of subcutaneous abdominal adipose tissue and altered noradrenergic activity in patients with Cushing’s syndrome: An in vivo microdialysis study. Physiol. Res. 2006, 55, 421–428. [Google Scholar] [PubMed]
- Kern, P.A.; Ranganathan, S.; Li, C.; Wood, L.; Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E745–E751. [Google Scholar] [PubMed]
- Vicennati, V.; Vottero, A.; Friedman, C.; Papanicolaou, D.A. Hormonal regulation of interleukin-6 production in human adipocytes. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 905–911. [Google Scholar] [PubMed]
- Zilberfarb, V.; Siquier, K.; Strosberg, A.D.; Issad, T. Effect of dexamethasone on adipocyte differentiation markers and tumour necrosis factor-alpha expression in human PAZ6 cells. Diabetologia 2001, 44, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Prigeon, R.L.; McCulloch, D.K.; Boyko, E.J.; Bergman, R.N.; Schwartz, M.W.; Neifing, J.L.; Ward, W.K.; Beard, J.C.; Palmer, J.P. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 1993, 42, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Karlstad, M.D.; Collier, J.J. Pancreatic Islet Responses to Metabolic Trauma. Shock 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Lambillotte, C.; Gilon, P.; Henquin, J.C. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J. Clin. Investig. 1997, 99, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Jeong, I.K.; Oh, S.H.; Kim, B.J.; Chung, J.H.; Min, Y.K.; Lee, M.S.; Lee, M.K.; Kim, K.W. The effects of dexamethasone on insulin release and biosynthesis are dependent on the dose and duration of treatment. Diabetes Res. Clin. Pract. 2001, 51, 163–171. [Google Scholar] [CrossRef]
- Zawalich, W.S.; Tesz, G.J.; Yamazaki, H.; Zawalich, K.C.; Philbrick, W. Dexamethasone suppresses phospholipase C activation and insulin secretion from isolated rat islets. Metabolism 2006, 55, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.C.; Halter, J.B.; Best, J.D.; Pfeifer, M.A.; Porte, D., Jr. Dexamethasone-induced insulin resistance enhances B cell responsiveness to glucose level in normal men. Am. J. Physiol. 1984, 247, E592–E596. [Google Scholar] [PubMed]
- Hollingdal, M.; Juhl, C.B.; Dall, R.; Sturis, J.; Veldhuis, J.D.; Schmitz, O.; Pørksen, N. Glucocorticoid induced insulin resistance impairs basal but not glucose entrained high-frequency insulin pulsatility in humans. Diabetologia 2002, 45, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Willi, S.M.; Kennedy, A.; Wallace, P.; Ganaway, E.; Rogers, N.L.; Garvey, W.T. Troglitazone antagonizes metabolic effects of glucocorticoids in humans: Effects on glucose tolerance, insulin sensitivity, suppression of free fatty acids, and leptin. Diabetes 2002, 51, 2895–2902. [Google Scholar] [CrossRef] [PubMed]
- Hoes, J.N.; van der Goes, M.C.; van Raalte, D.H.; van der Zijl, N.J.; den Uyl, D.; Lems, W.F.; Lafeber, F.P.; Jacobs, J.W.; Welsing, P.M.; Diamant, M.; et al. Glucose tolerance, insulin sensitivity and β-cell function in patients with rheumatoid arthritis treated with or without low-to-medium dose glucocorticoids. Ann. Rheum. Dis. 2011, 70, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Larsson, H.; Ahrén, B. Insulin resistant subjects lack islet adaptation to short-term dexamethasone-induced reduction in insulin sensitivity. Diabetologia 1999, 42, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Besse, C.; Nicod, N.; Tappy, L. Changes in insulin secretion and glucose metabolism induced by dexamethasone in lean and obese females. Obes. Res. 2005, 13, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Jensen, D.H.; Aaboe, K.; Henriksen, J.E.; Vølund, A.; Holst, J.J.; Madsbad, S.; Krarup, T. Steroid-induced insulin resistance and impaired glucose tolerance are both associated with a progressive decline of incretin effect in first-degree relatives of patients with type 2 diabetes. Diabetologia 2012, 55, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Challis, J.R.; Newnham, J.P.; Sloboda, D.M. Early-life glucocorticoid exposure: The hypothalamic-pituitary-adrenal axis, placental function, and long-term disease risk. Endocr. Rev. 2013, 34, 885–916. [Google Scholar] [CrossRef] [PubMed]
- Moisiadis, V.G.; Matthews, S.G. Glucocorticoids and fetal programming part 2: Mechanisms. Nat. Rev. Endocrinol. 2014, 10, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, S.; Ostlund, B.; Myrsén-Axcrona, U.; Sundler, F.; Ahrén, B. Beta cell adaptation to dexamethasone-induced insulin resistance in rats involves increased glucose responsiveness but not glucose effectiveness. Pancreas 2001, 22, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Fransson, L.; Franzén, S.; Rosengren, V.; Wolbert, P.; Sjöholm, Å.; Ortsäter, H. β-Cell adaptation in a mouse model of glucocorticoid-induced metabolic syndrome. J. Endocrinol. 2013, 219, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Protzek, A.O.; Costa-Júnior, J.M.; Rezende, L.F.; Santos, G.J.; Araújo, T.G.; Vettorazzi, J.F.; Ortis, F.; Carneiro, E.M.; Rafacho, A.; Boschero, A.C. Augmented β-Cell Function and Mass in Glucocorticoid-Treated Rodents Are Associated with Increased Islet Ir-β/AKT/mTOR and Decreased AMPK/ACC and AS160 Signaling. Int. J. Endocrinol. 2014, 2014, 983453. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, A.; Marroquí, L.; Taboga, S.R.; Abrantes, J.L.; Silveira, L.R.; Boschero, A.C.; Carneiro, E.M.; Bosqueiro, J.R.; Nadal, A.; Quesada, I. Glucocorticoids in vivo induce both insulin hypersecretion and enhanced glucose sensitivity of stimulus-secretion coupling in isolated rat islets. Endocrinology 2010, 151, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, A.; Quallio, S.; Ribeiro, D.L.; Taboga, S.R.; Paula, F.M.; Boschero, A.C.; Bosqueiro, J.R. The adaptive compensations in endocrine pancreas from glucocorticoid-treated rats are reversible after the interruption of treatment. Acta Physiol. 2010, 200, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Angelini, N.; Rafacho, A.; Boschero, A.C.; Bosqueiro, J.R. Involvement of the cholinergic pathway in glucocorticoid-induced hyperinsulinemia in rats. Diabetes Res. Clin. Pract. 2010, 87, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Novelli, M.; De Tata, V.; Bombara, M.; Lorenzini, A.; Masini, M.; Pollera, M.; Bergamini, E.; Masiello, P. Insufficient adaptive capability of pancreatic endocrine function in dexamethasone-treated ageing rats. J. Endocrinol. 1999, 162, 425–432. [Google Scholar] [CrossRef] [PubMed]
- De Paula, F.M.; Boschero, A.C.; Carneiro, E.M.; Bosqueiro, J.R.; Rafacho, A. Insulin signaling proteins in pancreatic islets of insulin-resistant rats induced by glucocorticoid. Biol. Res. 2011, 44, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Rafacho, A.; Roma, L.P.; Taboga, S.R.; Boschero, A.C.; Bosqueiro, J.R. Dexamethasone-induced insulin resistance is associated with increased connexin 36 mRNA and protein expression in pancreatic rat islets. Can. J. Physiol. Pharmacol. 2007, 85, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, C.; Ferreira, F.B.; Gonçalves-Neto, L.M.; Taboga, S.R.; Boschero, A.C.; Rafacho, A. Age- and gender-related changes in glucose homeostasis in glucocorticoid-treated rats. Can. J. Physiol. Pharmacol. 2014, 92, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Protzek, A.O.; Rezende, L.F.; Costa-Júnior, J.M.; Ferreira, S.M.; Cappelli, A.P.; de Paula, F.M.; de Souza, J.C.; Kurauti, M.A.; Carneiro, E.M.; Rafacho, A.; et al. Hyperinsulinemia caused by dexamethasone treatment is associated with reduced insulin clearance and lower hepatic activity of insulin-degrading enzyme. J. Steroid Biochem. Mol. Biol. 2016, 155, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Johnson, J.H.; Ohneda, M.; McAllister, C.T.; Inman, L.; Alam, T.; Unger, R.H. Roles of insulin resistance and beta-cell dysfunction in dexamethasone-induced diabetes. J. Clin. Investig. 1992, 90, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ostenson, C.G.; Berggren, P.O.; Efendic, S. Glucocorticoid increases glucose cycling and inhibits insulin release in pancreatic islets of ob/ob mice. Am. J. Physiol. 1992, 263, E663–E666. [Google Scholar] [PubMed]
- Khan, A.; Hong-Lie, C.; Landau, B.R. Glucose-6-phosphatase activity in islets from ob/ob and lean mice and the effect of dexamethasone. Endocrinology 1995, 136, 1934–1938. [Google Scholar] [PubMed]
- Davani, B.; Portwood, N.; Bryzgalova, G.; Reimer, M.K.; Heiden, T.; Ostenson, C.G.; Okret, S.; Ahren, B.; Efendic, S.; Khan, A. Aged transgenic mice with increased glucocorticoid sensitivity in pancreatic beta-cells develop diabetes. Diabetes 2004, 53 (Suppl. 1), S51–S59. [Google Scholar] [CrossRef] [PubMed]
- Wise, J.K.; Hendler, R.; Felig, P. Influence of glucocorticoids on glucagon secretion and plasma amino acid concentrations in man. J. Clin. Investig. 1973, 52, 2774–2782. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.P.; Bremer, A.A.; Kieffer, T.J.; D’Alessio, D.; Havel, P.J. Investigation of the mechanisms contributing to the compensatory increase in insulin secretion during dexamethasone-induced insulin resistance in rhesus macaques. J. Endocrinol. 2013, 216, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, J.L.; D’souza, A.M.; Teich, T.; Tsushima, R.; Riddell, M.C. Exogenous glucocorticoids and a high-fat diet cause severe hyperglycemia and hyperinsulinemia and limit islet glucose responsiveness in young male Sprague-Dawley rats. Endocrinology 2013, 154, 3197–3208. [Google Scholar] [CrossRef] [PubMed]
- Bowles, N.P.; Karatsoreos, I.N.; Li, X.; Vemuri, V.K.; Wood, J.A.; Li, Z.; Tamashiro, K.L.; Schwartz, G.J.; Makriyannis, A.M.; Kunos, G.; et al. A peripheral endocannabinoid mechanism contributes to glucocorticoid-mediated metabolic syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 285–290. [Google Scholar] [CrossRef] [PubMed]

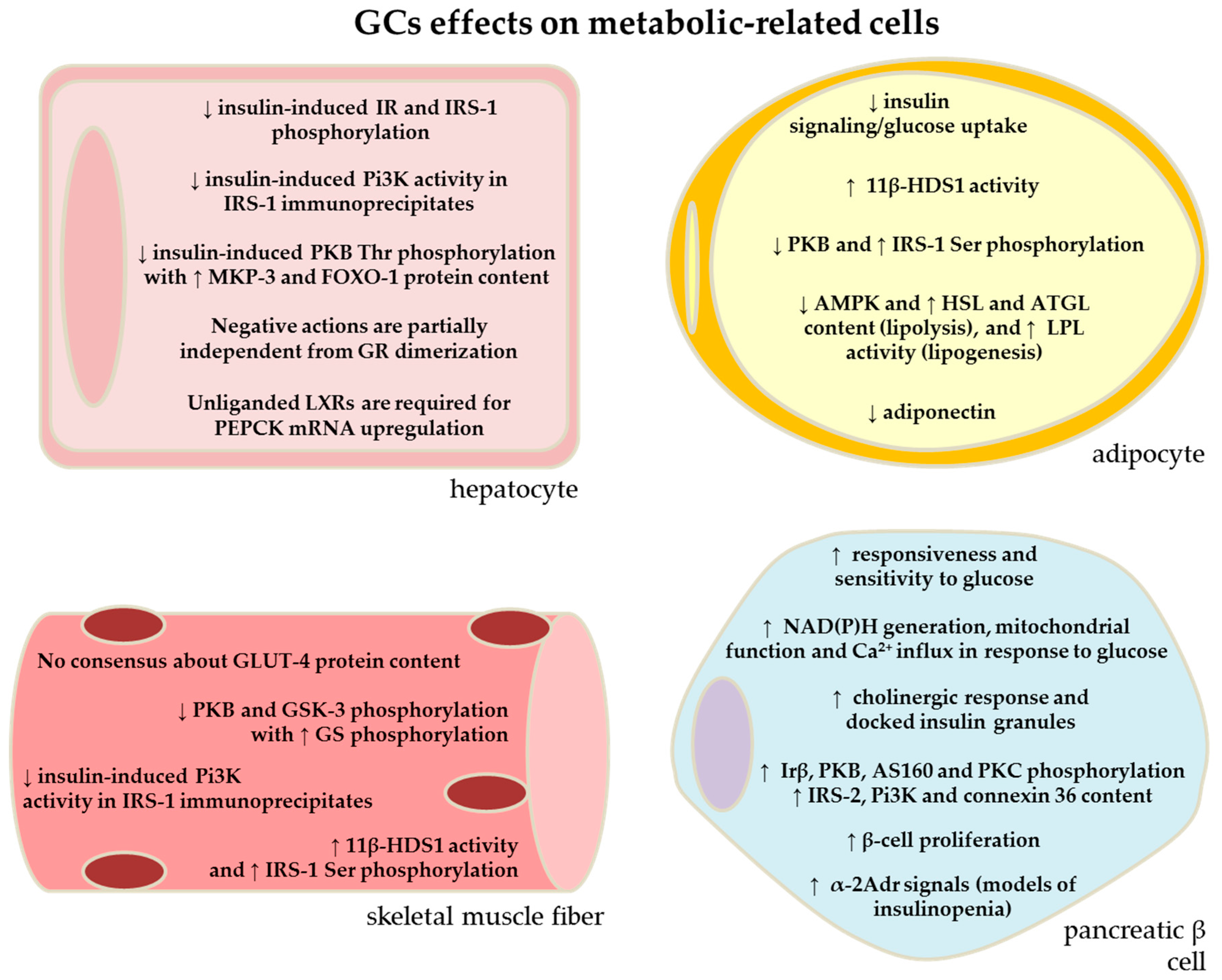

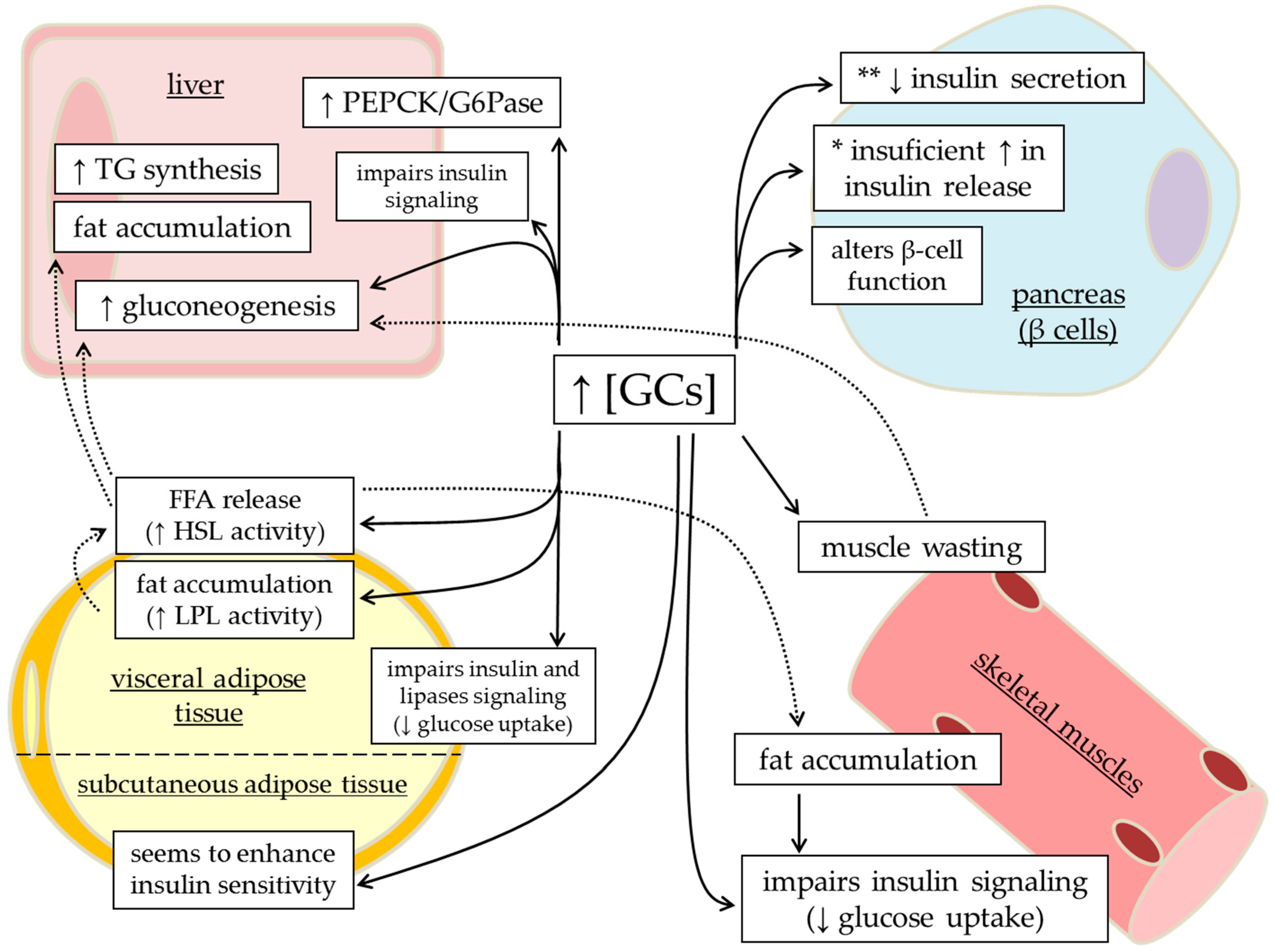
{kind=link}
{kind=link}
{kind=link}
| Study Design | Hepatic Glucose Output | Skeletal Muscle Glucose Uptake | Adipose Tissue Lipolysis | Glycemia and Insulinemia | β-cell Function | Ref. | |
|---|---|---|---|---|---|---|---|
| HUMAN | DEX (4 × 0.5 mg) for 2 days – women | Increased | Unaltered | Unaltered | Normal glycemia and increased insulinemia | Increased | [9] |
| PRED (7.5 and 30 mg) for 14 days – men | Increased (both doses) | Unaltered | Increased (both doses) | Increased glycemia (both doses) and insulinaemia (higher dose) | [10] | ||
| DEX (2 mg) for 2 days – both genders | Unaltered | Reduced | Probably increased | Increased insulinaemia, but not glycemia | [11] | ||
| DEX (4 × 0.5 mg) for 2 days – men | Increased | Reduced | Increased | [14] | |||
| DEX (4 × 0.5 mg) for 2 days – both genders | Unaltered | Probably reduced | Unaltered glycemia Increased insulinemia | [15] | |||
| PRED (75 mg) for 1 day or (30 mg) for 15 days – men | Reduced for 75 mg, but unaltered for 30 mg treatment (based on plasma C-peptide) | [17] | |||||
| DEX (15 mg) over 3 days - women | Unaltered glycemia Increased insulinemia | Increased | [31] | ||||
| RAT | DEX (1 mg) for 7 days – male Wistar rats | Increased | Increased glycemia and insulinemia | [6] | |||
| DEX (0.5 mg) for 7 days – male Wistar rats | Increased | Reduced | Increased glycemia and insulinaemia | [8] | |||
| DEX (1 mg) for 11 days – male Wistar rats | Reduced | Increased | Unaltered glycemia | [13] | |||
| CORT (300 MG) wax pellets for 10 days – male SD rats | Increased | Unaltered insulinemia | [32] | ||||
| DEX (1 mg) for 5 days – male Wistar rats | Increased glycemia and insulinemia | [18,33] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasieka, A.M.; Rafacho, A. Impact of Glucocorticoid Excess on Glucose Tolerance: Clinical and Preclinical Evidence. Metabolites 2016, 6, 24. https://doi.org/10.3390/metabo6030024
Pasieka AM, Rafacho A. Impact of Glucocorticoid Excess on Glucose Tolerance: Clinical and Preclinical Evidence. Metabolites. 2016; 6(3):24. https://doi.org/10.3390/metabo6030024
Chicago/Turabian StylePasieka, Aoibhe M., and Alex Rafacho. 2016. "Impact of Glucocorticoid Excess on Glucose Tolerance: Clinical and Preclinical Evidence" Metabolites 6, no. 3: 24. https://doi.org/10.3390/metabo6030024
APA StylePasieka, A. M., & Rafacho, A. (2016). Impact of Glucocorticoid Excess on Glucose Tolerance: Clinical and Preclinical Evidence. Metabolites, 6(3), 24. https://doi.org/10.3390/metabo6030024