1. Introduction
In the post genomics era where the plant biology community is facing the challenge of identifying the functionalities of genes of unknown function (GUFs), metabolomics offers a link between biochemical phenotype and gene function [
1]. However, the use of metabolomics for the prediction of the function of plant genes faces technical challenges due to the large size (between 100,000 to 200,000 biocompounds) [
2,
3,
4], the chemical complexity and the different abundance levels of a plant metabolic pool. These challenges are currently being tackled by combining targeted and non-targeted metabolic analyses to characterize and compare changes in metabolic networks [
1,
2,
5,
6,
7]. Those combined strategies are a partial solution to the lack of universality of a single analytical technique, as they exploit the power of current separation technologies and the various dynamic ranges and sensitivities offered by the arsenal of commercially available analytical detectors to cover a larger portion of the metabolome than any single platform alone [
2,
5,
6,
7,
8].
Currently, combined metabolomics technologies are being tested as functional genomic tools for the annotation of
Arabidopsis thaliana GUFs [
1,
7,
9]. Usually, high throughput biochemical screening methods are employed to first identify previously uncharacterized
Arabidopsis mutants affecting a variety of metabolic pathways. The screening is carried out by targeted analysis of specific groups of compounds or metabolic subsets (glucosinolates, fatty acids, phytosterols, isoprenoids, amino acids, among others) across a large population of mutagenized
Arabidopsis lines. Once new loci involved in plant metabolism are identified further work is performed in those particular mutants using non-targeted analysis in order to characterize metabolite changes more broadly. Identification of metabolites that are discriminatory between the knockout plant compared to the wild-type help fill up the gaps in our understanding of plant-specific regulatory and biosynthetic pathways and determine the function of the GUFs [
1,
7,
9].
Because of the central role that amino acids play in plant biochemistry, screening methods that quantify free levels of this class of metabolites in plant tissue are in demand. Despite the numerous methods available for amino acid analysis, many lack the suitability for metabolomic studies. Three aspects are vital in developing an effective targeted metabolite analysis platform for large-scale mutant screening: (i) reduction of sample preparation and analysis time, (ii) collection of high-quality data, and (iii) broad dynamic range [
10].
Chromatographic separation methods (gas chromatography, GC, and liquid chromatography, LC) combined with tandem mass spectrometric (MS/MS) detection are dominating the field of metabolomics. Although considerable work has been done in the development of LC-MS methods for analysis of both underivatized and derivatized amino acids in complex matrices, the former are being particularly implemented in metabolomic research and employ the ion-pairing (IP) reversed-phase (RP) LC [
10,
11] or hydrophilic interaction chromatography (HILIC) alternatives [
12,
13]. Although these methodologies are very attractive due to the elimination of the sample derivatization step, they suffer of several problems.
In IPRPLC, for example, the occurrence of system peaks during the gradient elution disturbs the quantitation of amino acids. Systems peaks are caused by low volatility of ion-pairing reagents (example, pentadecafluorooctanoic acid, PDFOA) and their adsorption on the column support surface [
14,
15]. In addition, long equilibration times (t
e) between runs and column regeneration after few injections are needed in order to avoid degradation in chromatography and retention time drift for amino acids due to accumulation of the ion-pairing reagent on the column surface. Equilibration times from 9 to 105 min [
15,
16,
17,
18] and column flushing from 3 to 30 min are reported in the literature [
14,
15,
19,
20]. Another drawback associated with the use of ion-pairing reagents in LC-ESI-MS analysis is the decrease in ionization efficiency of amino acids due to interference by these easy-ionized mobile phase modifiers [
21]. The occurrence of undesirable reactions between ion-pairing reagents and salts present in biological samples can also contribute to this problem. Armstrong
et al. [
20] reported the formation of a sodium adduct of tridecafluoroheptanoate (TDFHA) during the analysis of 25 physiological amino acids and one peptide in plasma samples by IPRPLC coupled to time-of-flight (TOF) MS which caused significant signal suppression of alanyl-glutamine dipeptide and valine. A cation-exchange cleanup step had to be added to the sample preparation in order to decrease the abundance of the TDFHA adduct and improve the accuracy and precision of the analysis [
20]. Last but not least, surfactant impurities can make the eluent particularly noisy at the m/z range corresponding to underivatized amino acids, affecting the sensitivity of the analysis [
15,
22].
Alternatively, when HILIC separation mode is used instead of a reversed-phase system underivatized amino acids are retained without any mobile phase modifier and the above mentioned drawbacks associated to the use of iron-pairing reagents can be avoided. Despite of that, column care (
i.e., installation of in-line filter and guard column [
23]) and long equilibration times (usually about 10 min in order to ensure retention time repeatability [
23]) are essential in HILIC analysis. Furthermore, HILIC columns suffer of poor separation efficiency compared to the RPLC technique [
24,
25].
Due to the above, it is necessary to explore the possibility of implementing LC-MS methods for the analysis of derivatized amino acids to large-scale mutant screening in metabolomic studies. It is undeniable that derivatization brings several advantages to the LC-MS amino acid analysis in complex biological samples. First, derivatization of amino acids improves chromatographic properties (symmetric peak shape, better retention and resolution) in RPLC techniques [
22]. In addition, if the amino acid derivatives are amenable for electrospray ionization-tandem mass spectrometry (ESI-MS/MS), better ionization efficiency and increased detection sensitivity can be obtained in their analysis due to the shift of the molecular ion masses and enhanced hydrophobicity caused by derivatization [
22]. Despite these advantages it is recognized that not all of the typical amino acid derivatization methods are amenable for “omic”-scale projects.
Typical pre-column derivatization reagents for RPLC amino acid analysis include
o-phthalaldehyde (OPA), phenylisothiocyanate (PITC), 5-dimethylamino-1-naphthalenesulphonyl-chloride (Dansyl), 9-fluorenylmethyl chloroformate (FMOC), propyl chloroformate (PrCl), butanol, among others. Several disadvantages are associated with these pre-column derivatization methods and the analysis of their derivatives by LC-MS and LC-MS/MS: (i) long derivatization reaction time (Dansyl, 35–50 min [
26], PITC, 20 min [
27], FMOC, 1 hr [
22], Butanol, 1 hr [
22]), (ii) complex sample preparation (PITC [
27]), (iii) inability to derivatize secondary amino acids (OPA [
26]), (iv) derivative instability (OPA [
26,
28,
29]; PITC [
30]), (v) photosensitive adducts (Dansyl [
28]), (vi) inconsistent production of derivatives (Dansyl [
28]), (vii) extraction of excess reagent must be performed to stop derivatization and avoid spontaneous hydrolysis of adducts (FMOC [
26,
31]), (viii) removal of excess reagent is necessary to avoid rapid RPLC column deterioration (OPA [
32], PITC [
26,
27]) and (ix) long analysis time of amino acid derivatives by LC-MS and LC-MS/MS (20–45 min [
22,
31,
32,
33,
34]). These disadvantages render these derivatization methods impractical for metabolomics analysis since they introduce errors which can compromise the quality of the data.
The aforementioned shortcomings have urged the development of additional pre-column derivatization reagents. This new generation of reagents has the additional advantage of rendering amino acid adducts with desirable features for LC-MS/MS analysis. These reagents include
N-hydroxysuccinimide-activated
N-alkylnicotinic acid esters (Cn-NA-NHS) [
35],
p-N,N,N-trimethyl- -ammonioanilyl
N’-hydroxysuccinimidyl carbamate iodide (TAHS) [
36], 3-aminopyridyl-
N-hydroxysuccinimidyl carbamate (APDS) [
37,
38], (5-
N-succinimidoxy-5-oxopentyl)- triphenylphosphonium bromide (SPTPP) [
25], and iTRAQ (isobaric tag for relative and absolute quantitation) [
39,
40]. Although highly sensitive and selective detection of amino acids is attained by LC-MS/MS when employing these new generation of reagents, unfortunately the reagents are not commercially available (iTRAQ being the exception but it is prohibitively expensive) and some derivatization procedures are still complex and time-consuming. Advantages and shortcomings of these pre-column derivatization methods can be found in the literature [
25,
35,
36,
37,
38,
39,
40].
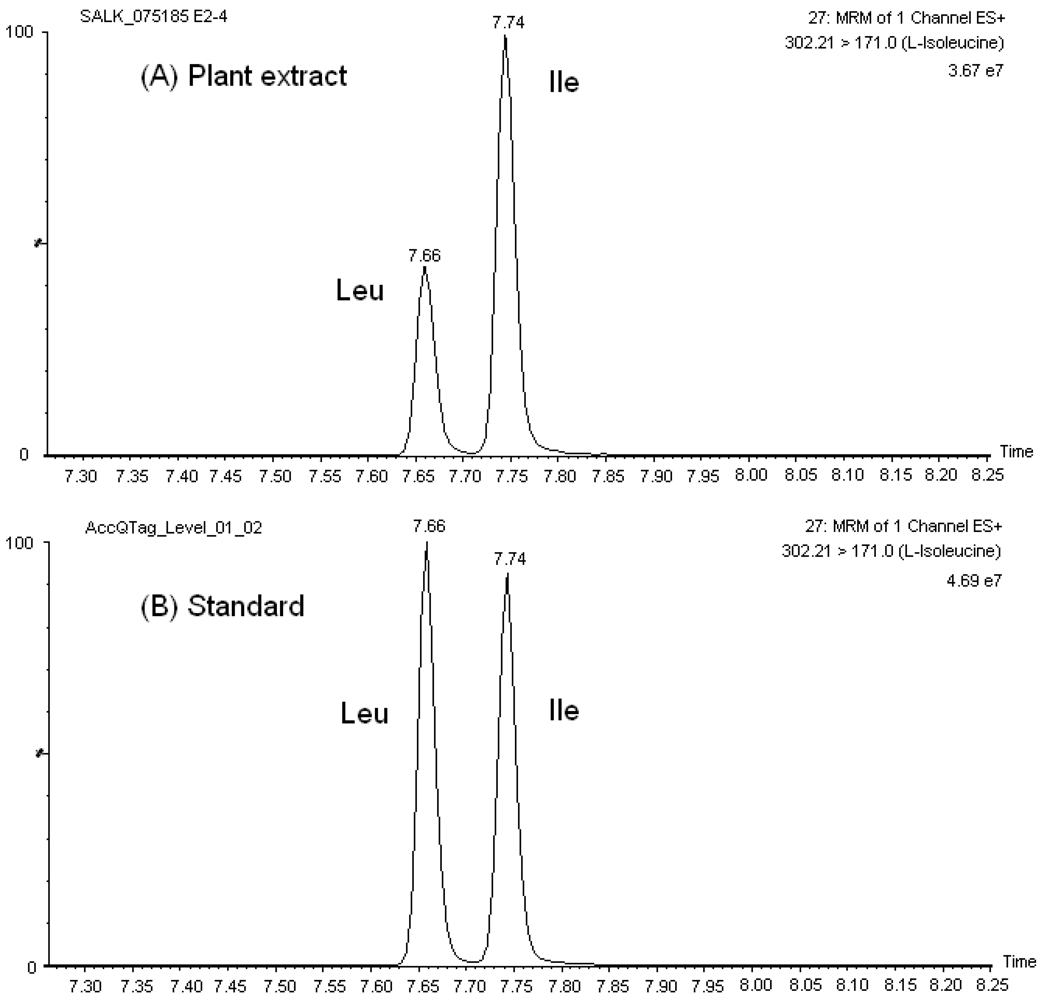
In this study, an analytical platform that combines ultraperformance liquid chromatography with tandem mass spectrometry (UPLC-MS/MS) for targeted amino acid analysis in
Arabidopsis thaliana leaf extracts is presented. Our method uses the commercially available amino acid derivatization reagent 6-aminoquinolyl-
N-hydroxysuccinimidyl carbamate (AQC). Since its introduction as derivatization reagent, AQC has shown interesting features. Reaction of AQC with primary and secondary amino acids is a simple, straightforward process that occurs within seconds and produces stable derivatives; in contrast the hydrolysis of the excess reagent is a much slower reaction [
30,
41]. The only disadvantages reported in the literature are related to the use of HPLC separation with fluorescence or UV detection: long analysis time (25–65 min), low sensitivity (UV only), peak interference by AQC hydrolysis product and intramolecular quenching [
41,
42,
43,
44,
45]. An analytical platform that exploits the greater chromatographic capacity and throughput of UPLC and the sensitivity and selectivity of MS/MS would overcome those drawbacks. The applicability of a UPLC-MS/MS method coupled with AQC precolumn derivatization for targeted amino acid analysis in large-scale metabolomics studies is demonstrated.
3. Experimental Section
3.1. Chemicals and Reagents
The L-amino acids kit (Sigma-Aldrich, Co., St. Louis, MO, USA) was used for direct infusion experiments and a commercial mix of amino acids and related compounds (Sigma-Aldrich, Co., St. Louis, MO, USA) was employed in the preparation of calibration standards. Asparagine, glutamine and homoserine were purchased separately (Sigma-Aldrich, Co., St. Louis, MO, USA) since they are not included in the commercial mix. Stable- isotope-labeled reference compounds (L-asparagine-15-N2; L-serine,2,3,3-d3; L-glutamine-2,3,3,4,4-d5; glycine-d5; D-L-alanine-2,3,3,3-d4; proline-2,5,5-d3; methionine-methyl-d3; tryptophan-2',4',5',6',7'-d5(indole-d5); leucine-d10; valine-d8; L-histidine (ring 2-13C); L-glutamic acid-2,4,4-d3; ornithine-3,3,4,4,5,5-d6; lysine-3,3,4,4,5,5,6,6-d8; phenyl-d5-alanine; 4-hydroxyphenyl-2,6-d2-alanine-2-d1) were used as internal standards and were purchased from Cambridge Isotope Laboratories (Andover, MA, USA) and CDN isotopes (Pointe-Claire, Quebec, Canada). The AccQ•Tag Ultra derivatization kit (AccQ•Tag Ultra borate buffer, AccQ•Tag Ultra reagent powder, and AccQ•Tag Ultra reagent diluent) was obtained from Waters Corporation (Milford, MA, USA). AccQ•Tag Ultra eluents for UPLC-ESI-MS/MS analysis were also from Waters. Ammonium acetate was purchased from Fisher (Fair Lawn, NJ, USA); ammonium hydroxide was supplied by Sigma (St. Louis, MO, USA). LC-MS grade methanol was purchased from J.T. Baker (Phillipsburg, NJ, USA). Ultrapure water (18.2 MΩ-cm) was obtained from a MilliQ Ultrapure water system (Millipore, Bedford, MA, USA). Ultra high purity argon and nitrogen gas for mass spectrometric analysis were purchased from Speciality Gases (Radnor, PA, USA).
3.2. Plant Material
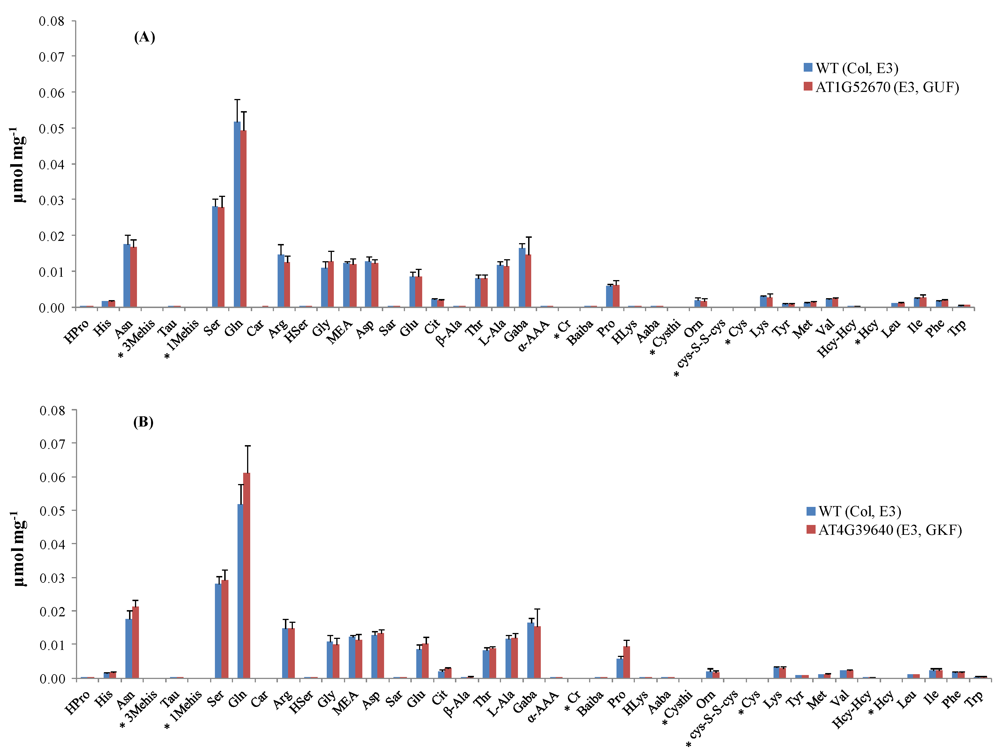
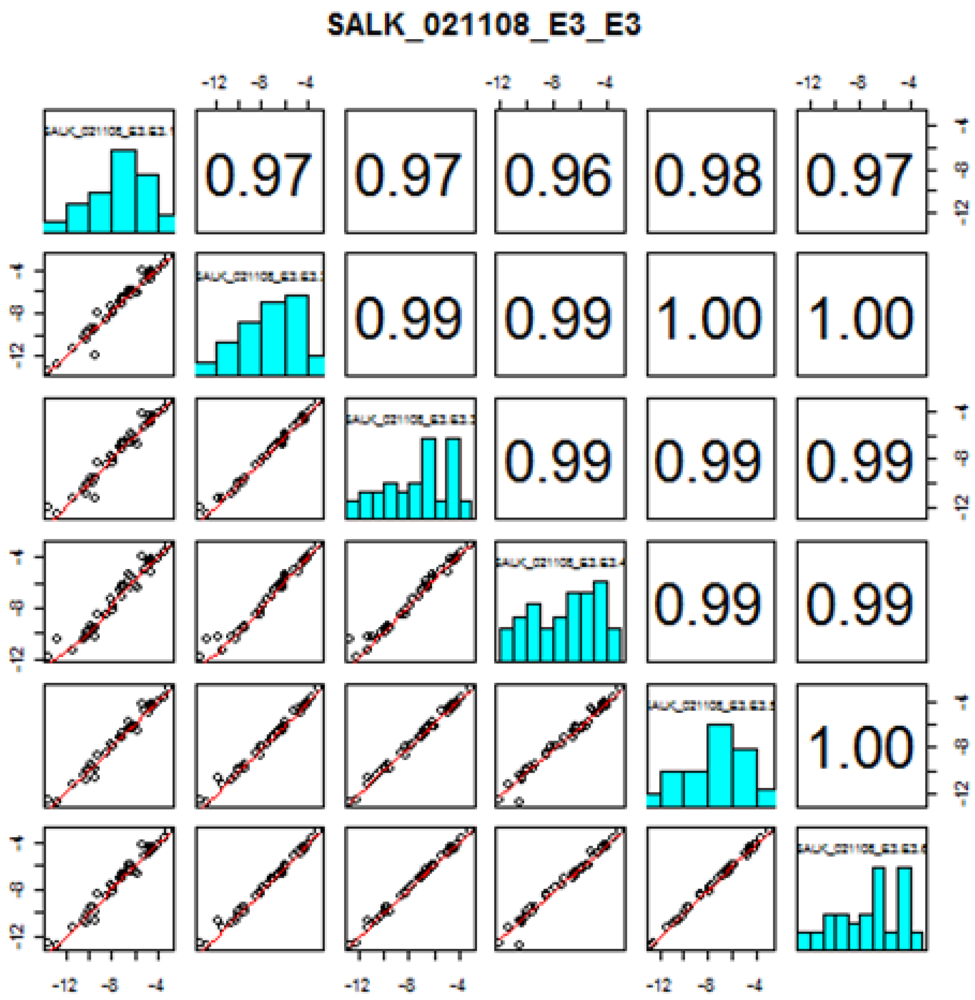
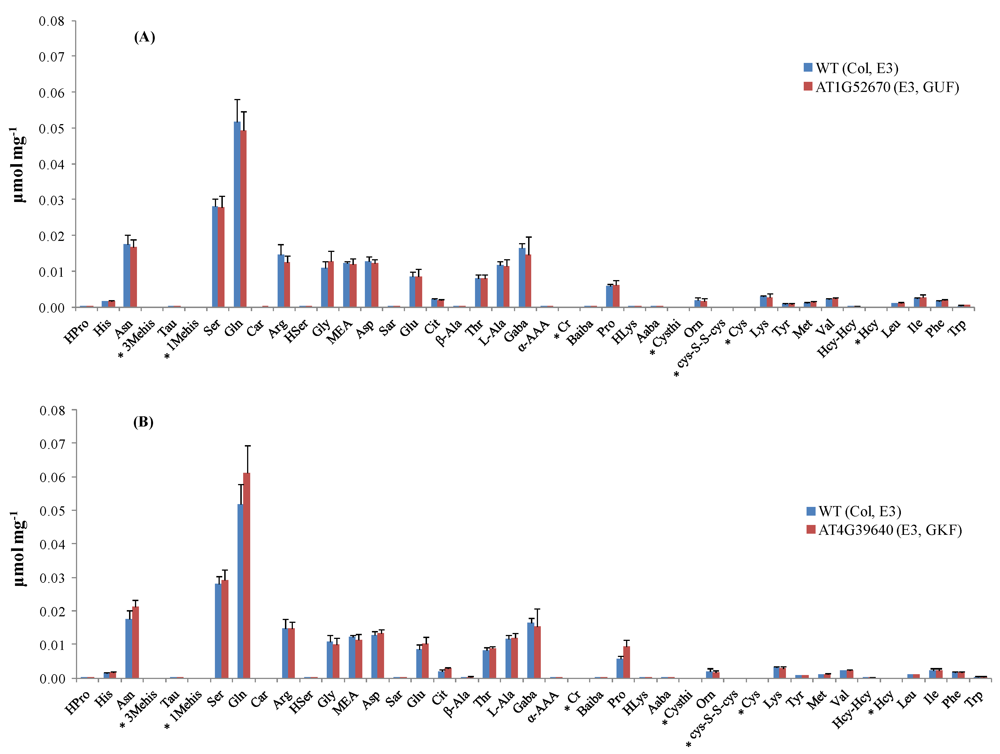
Seed stocks of Arabidopsis thaliana mutants were obtained from ABRC and propagated by the central lab of the Arabidopsis Metabolomics Consortium at Iowa State University. This paper focuses on the results obtained by targeted amino acid analysis on leaf extracts of 69 mutant lines selected for three metabolomic experiments (E1, E2, and E3) designed by the consortium. Six biological replicates of each mutant line were provided along with control samples (Columbia (Col-0) ecotype) for each metabolomic experiment.
The list of T-DNA knock-out mutants, the rationale for their selection, plant growth conditions, and protocol for plant harvesting are published elsewhere [
1,
7] and also available in the project database [
54]. Plant material was stored at −80 °C upon arrival.
3.4. Accq•Tag Ultra Amino Acid Derivatization
The AccQ•Tag Ultra derivatization kit (Waters Corp.) was used in all derivatization procedures, unless otherwise noted. AccQ•Tag Ultra borate buffer was replaced with the ammonium acetate buffer only for direct infusion mass spectrometry experiments. Following the protocol provided by the manufacturer, 10 μL of either a standard amino acid mix solution or an Arabidopsis leaf extract was mixed with 70 μL of AccQ•Tag Ultra borate buffer (pH = 8.8). The derivatization was carried out by adding 20 μL of reconstituted AccQ•Tag Ultra reagent (3 mg/mL of AQC in acetonitrile) to the buffered mixture. The sample was immediately vortexed followed by incubation for 15 min at 55 °C.
To maintain consistency between the time of extraction and time of analysis due to the large-scale of the project, derivatized samples were prepared and analyzed by UPLC-ESI-MS/MS in daily batches.
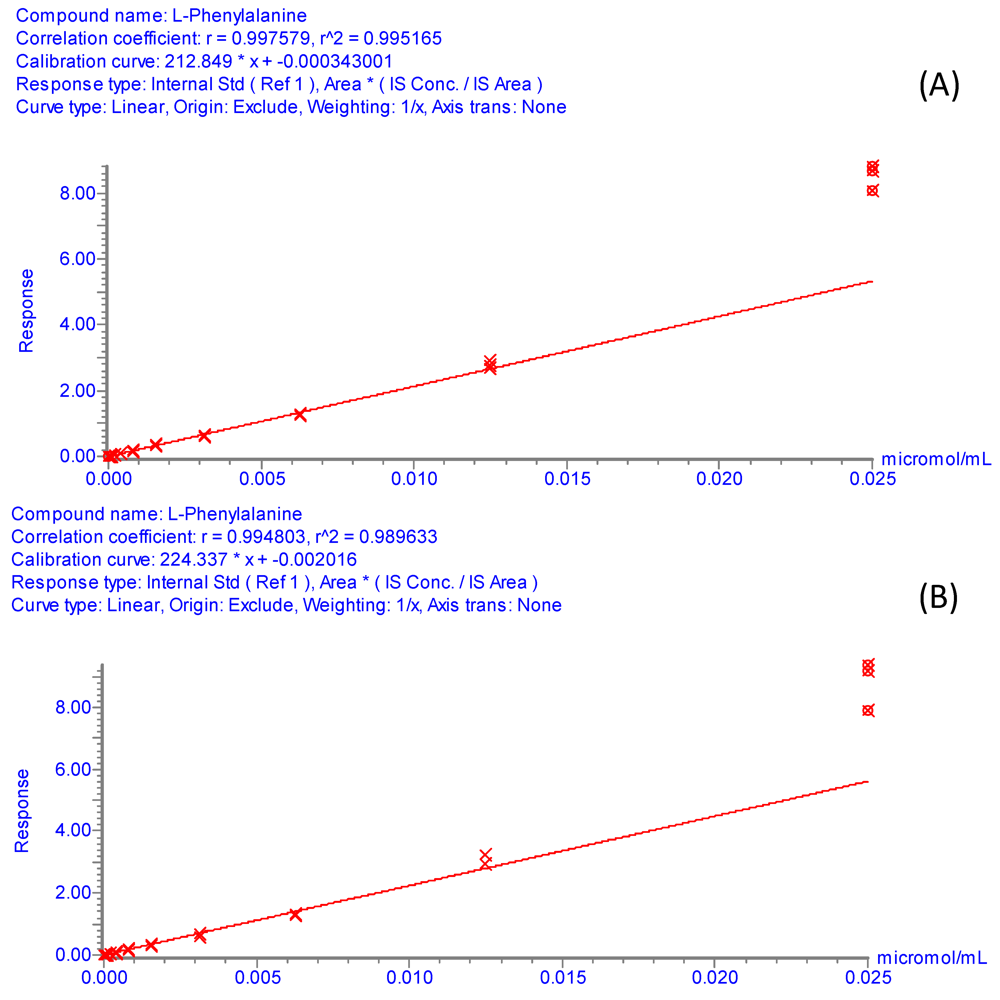
3.6. UPLC-ESI-MS/MS Method Evaluation and Applicability
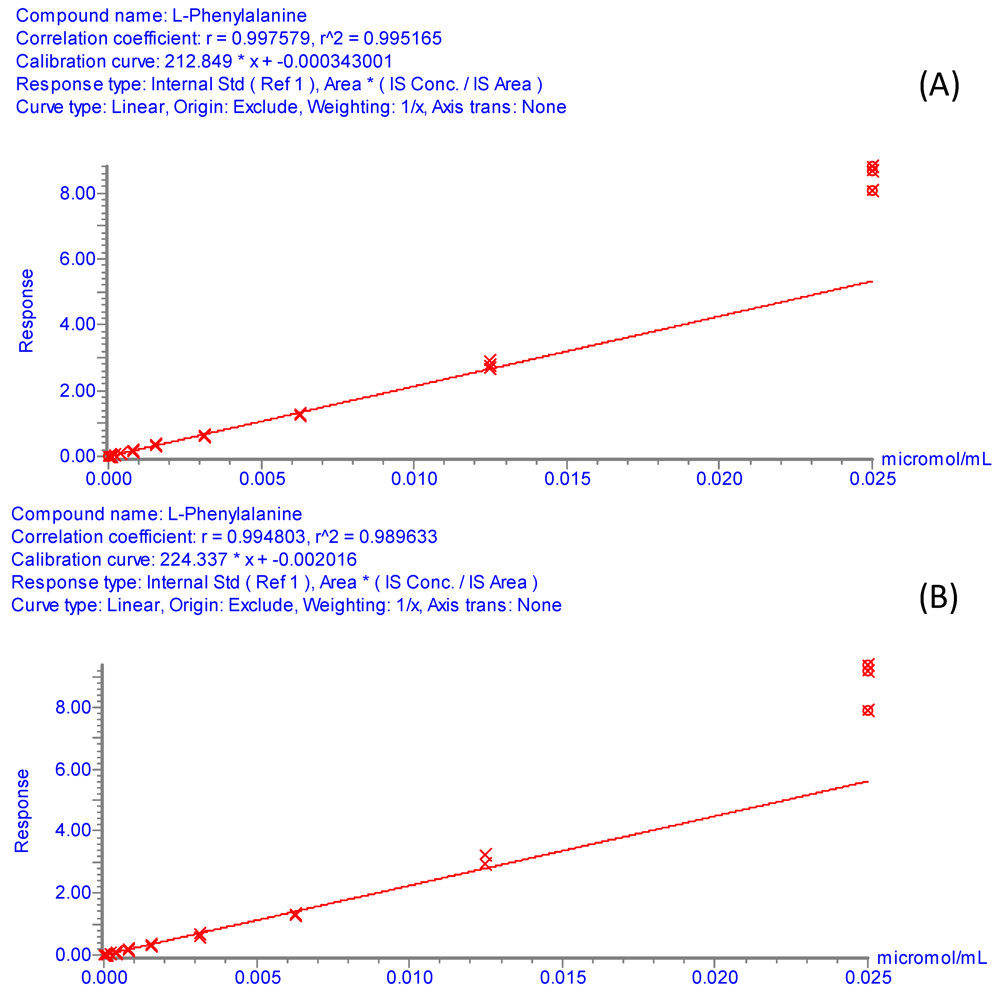
Method evaluation involved the determination of linearity (regression coefficient and dynamic range), sensitivity (detection limits), and reproducibility (relative standard deviations of retention times and peak areas) of the analysis for each amino acid. Working standards with concentration range from 250 μM to 476.8 pM were prepared by serial dilutions of a 500 μM amino acid mix solution spiked with isotopically labeled internal standards at 4 × 10
−3 g/L. The serial dilutions were performed in a Biomek 2000 Beckman Coulter laboratory automation workstation (Fullerton, CA) using a solution containing the internal standards at 4 × 10
−3 g/L in a 50% (v/v) methanol:water mixture in order to keep their concentration constant. After derivatization the concentrations of amino acids were decreased 10-fold and the concentration of all internal standards was maintained constant at 4 × 10
−4 g/L. Calibration curves were obtained by replicate injection of each of the derivatized working standards and were constructed as plots of relative peak area (Area
amino acid/Area
internal standard)
versus amino acid concentration using the TargetLynx software. The assignments of internal standards are given in
Table S4.
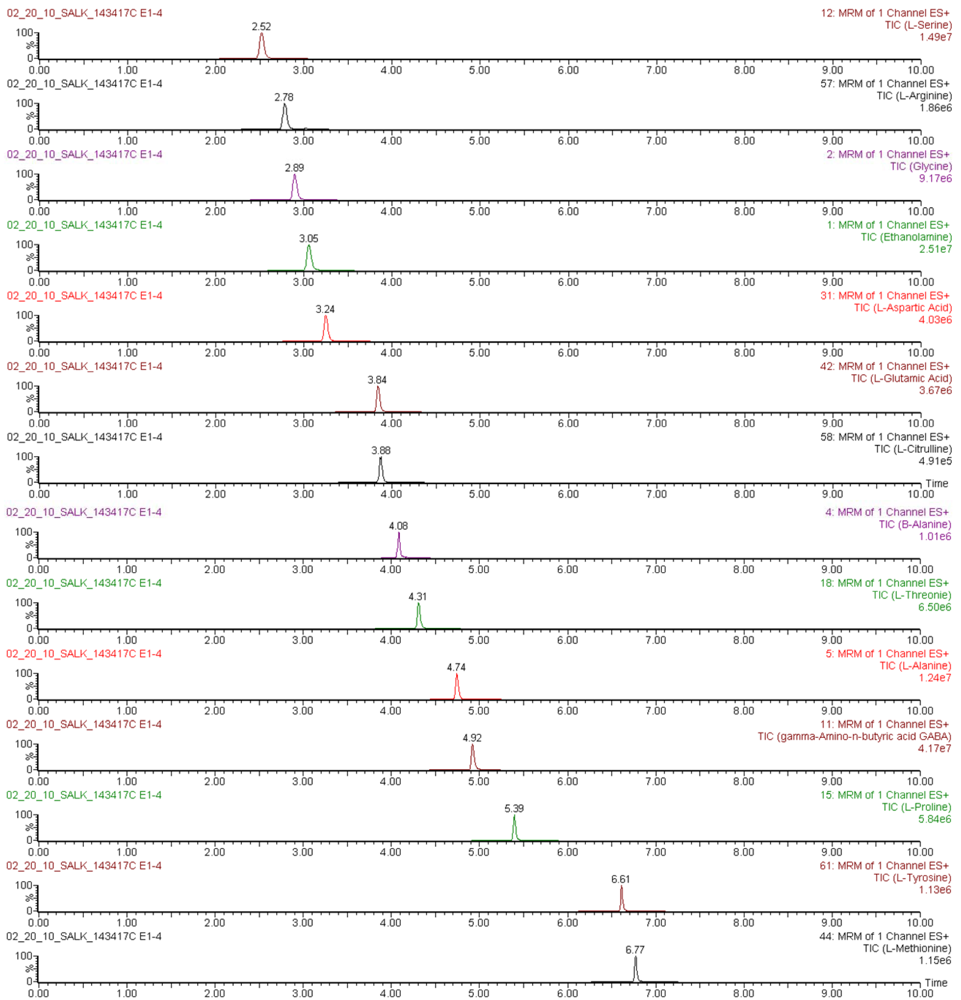
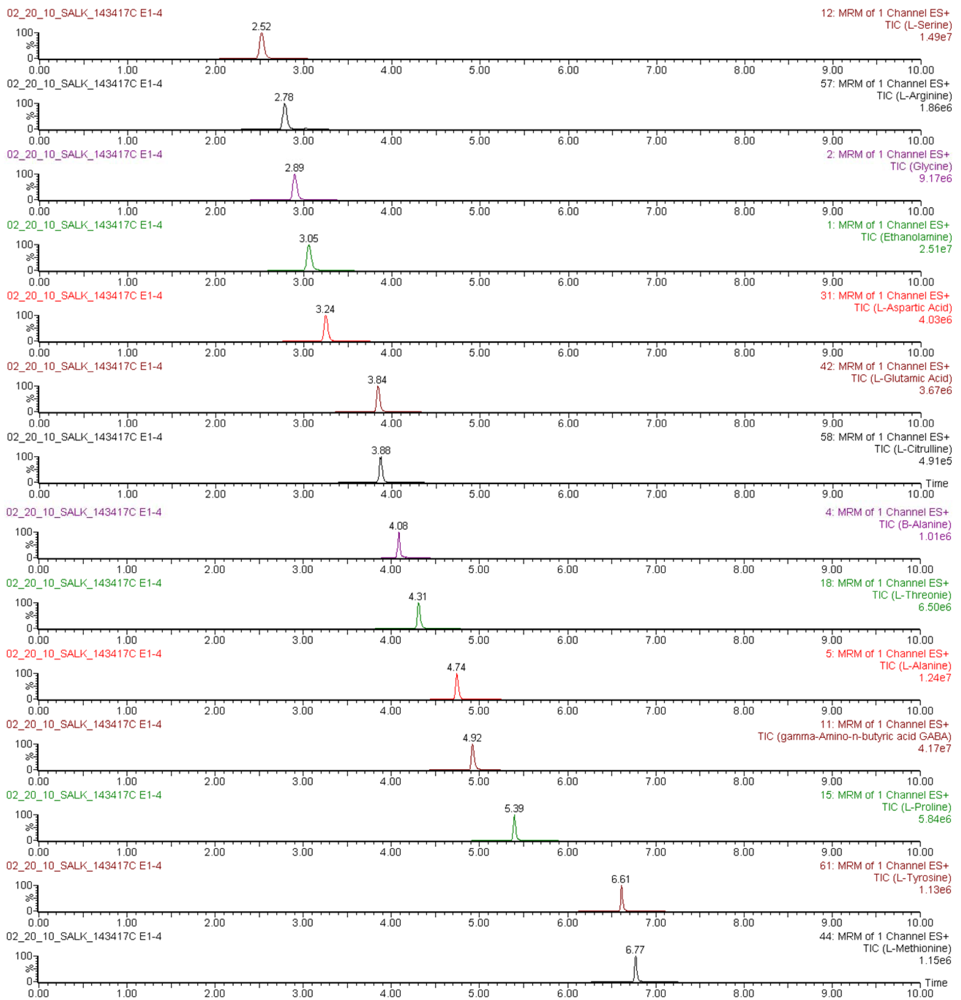
The applicability of the UPLC-ESI-MS/MS method for sensitive throughput analysis of amino acids was evaluated by determination of their concentrations in derivatized Arabidopsis thaliana leaf extracts obtained as described in numeral 3.3 and 3.4.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}