Construction of a Genome-Scale Kinetic Model of Mycobacterium Tuberculosis Using Generic Rate Equations

Abstract
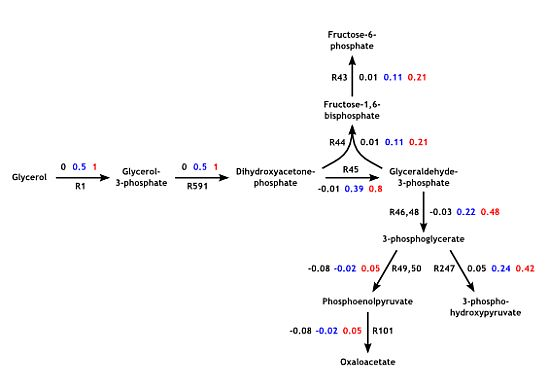
:
1. Introduction
2. Methods
2.1. Enzyme Kinetics and Rate Equations
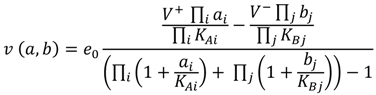 (1)
(1) 2.2. Parameter Estimation
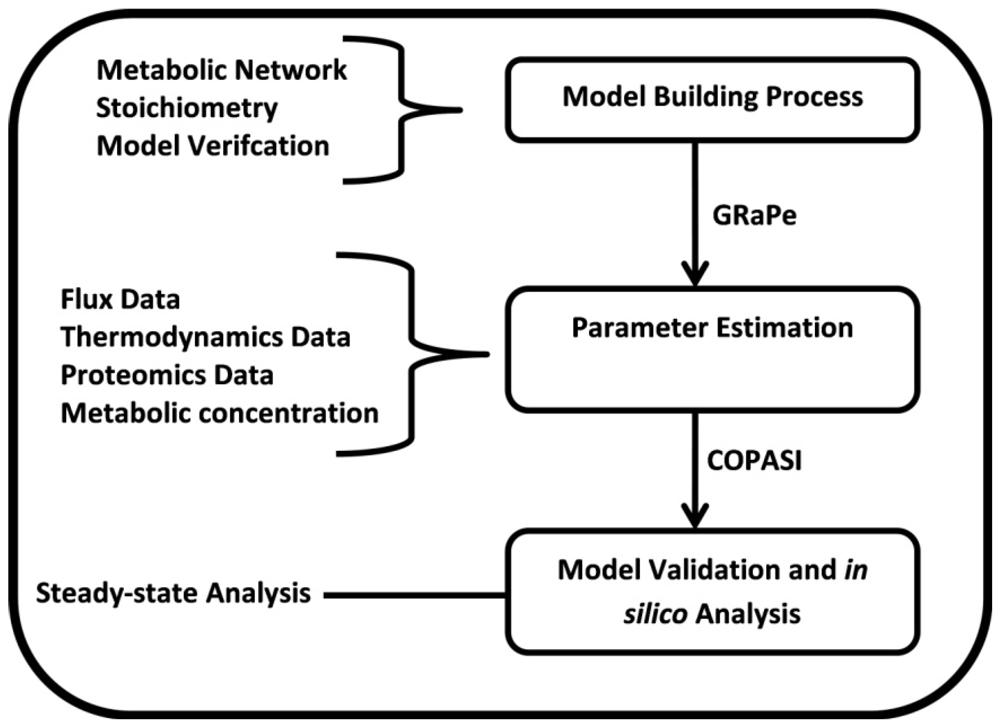
2.3. Parameter Variability Analysis (PVA)
3. Results
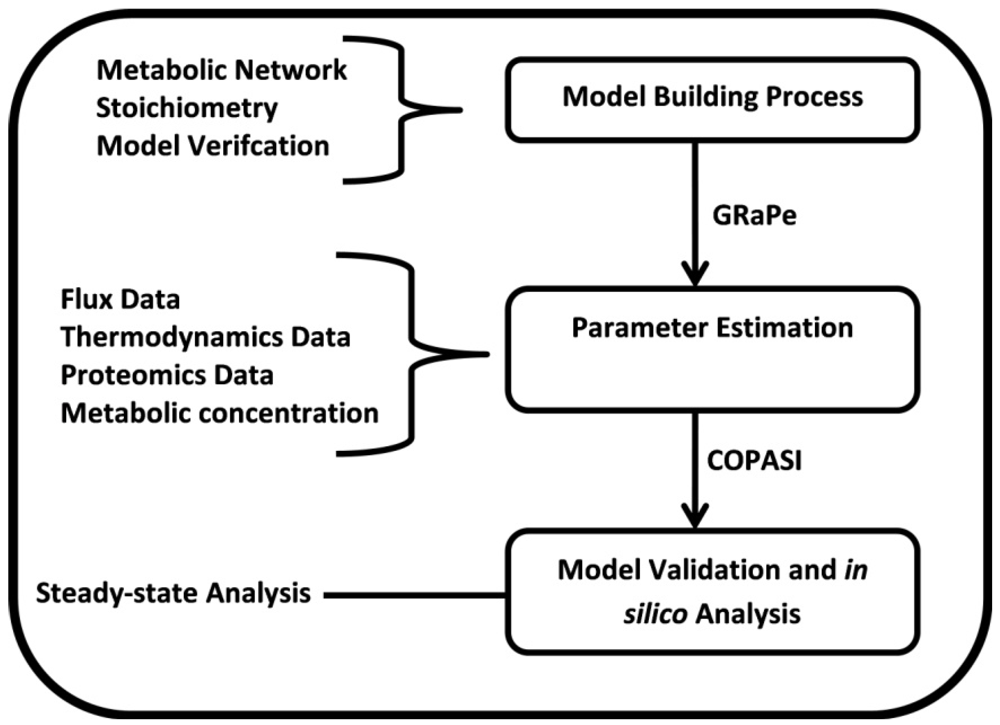
3.1. The Genome-Scale Kinetic Model of Mycobacterium Tuberculosis
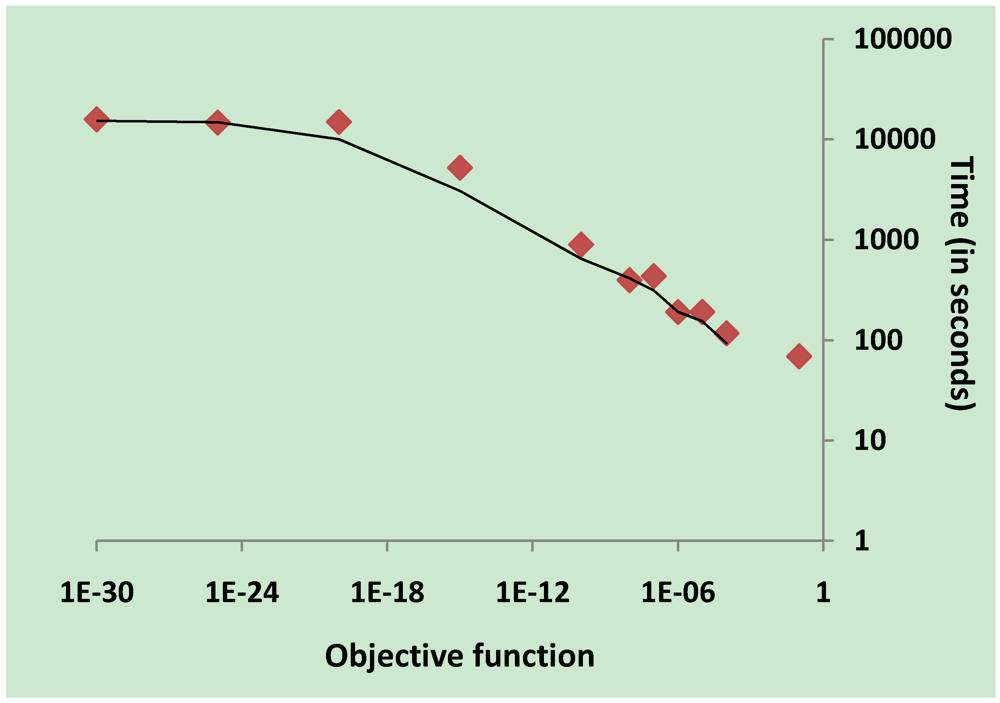
3.2. Parameter Estimation
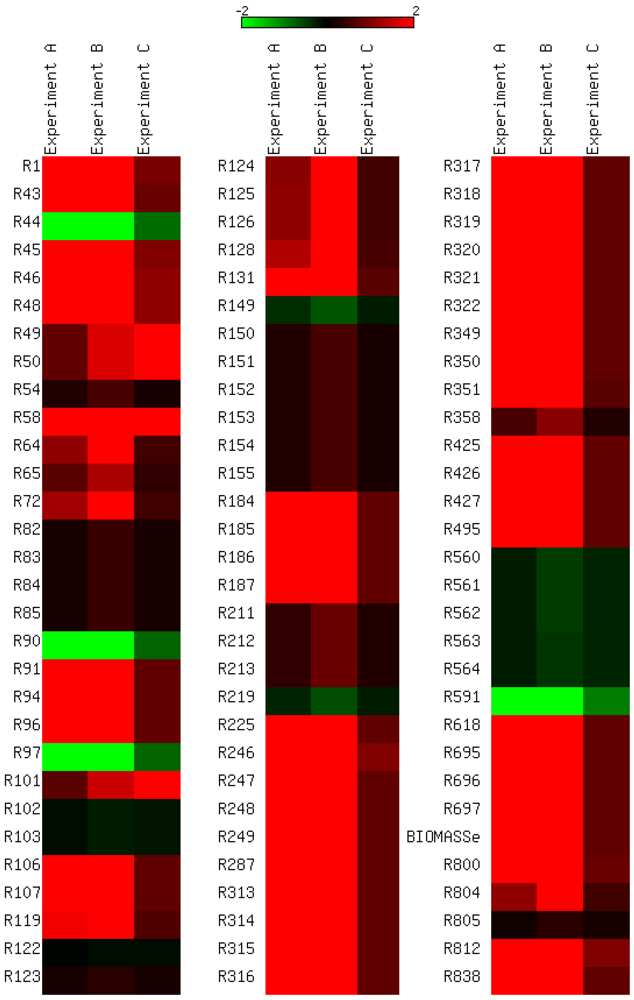
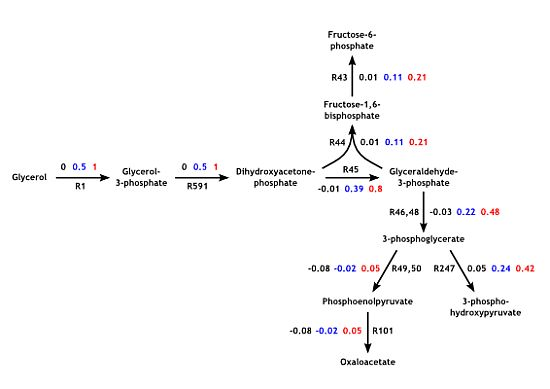
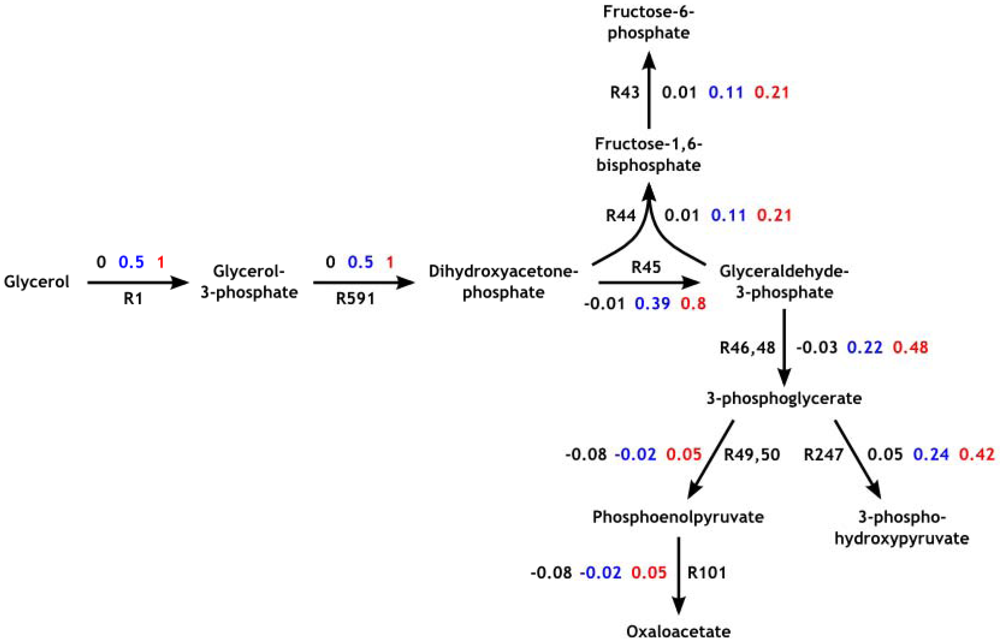
3.3. Model Validation
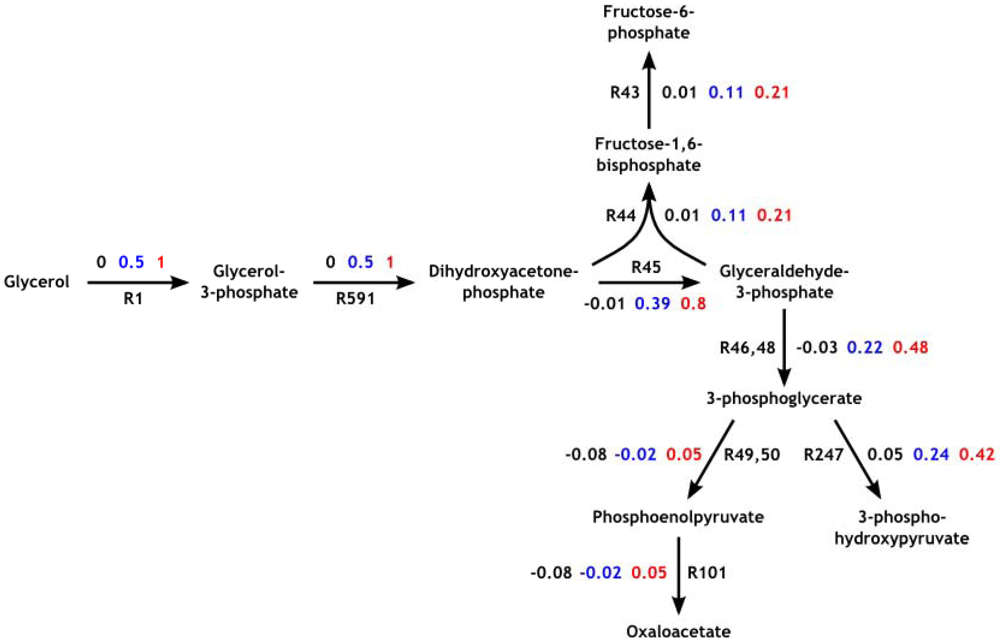
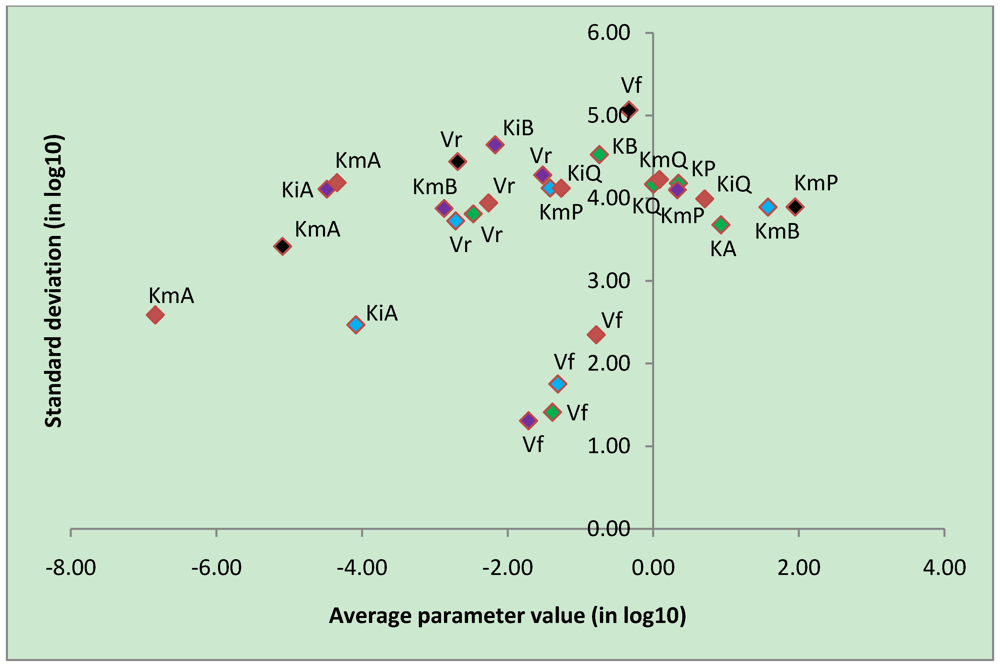
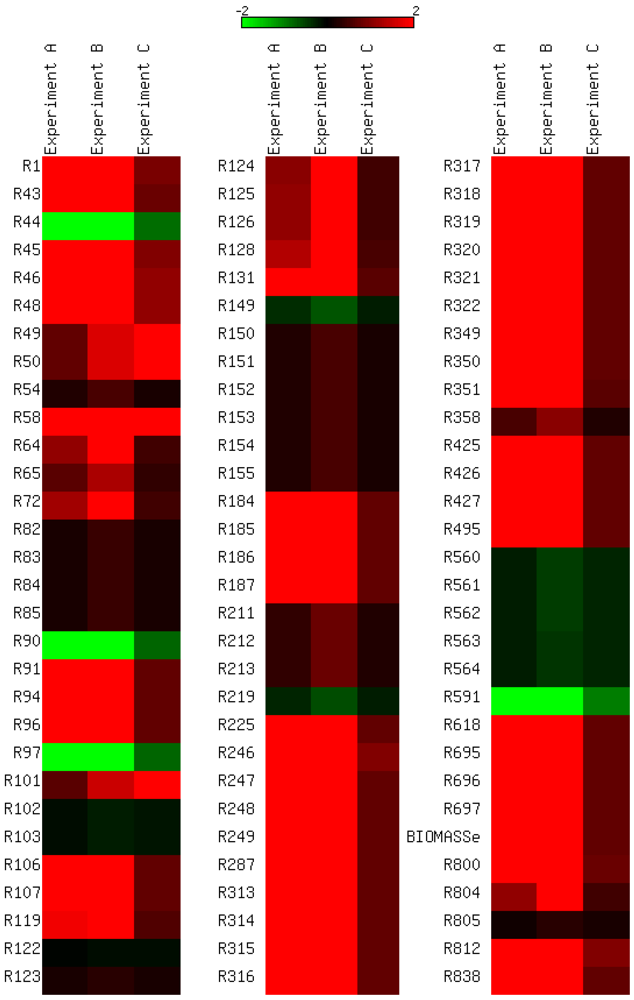
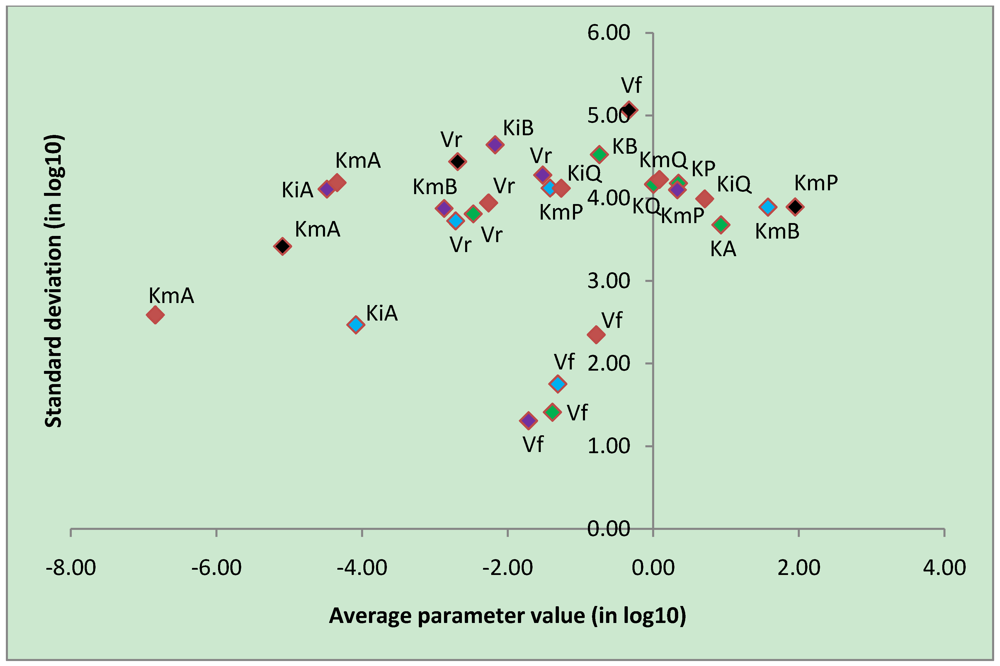
3.4. Parameter Variability Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Most constrained parameters | |||
|---|---|---|---|
| Parameter | Reaction type | Average | Stdev |
| Vf | CK | -0.78 | 2.35 |
| Vf | uni-bi | -1.39 | 1.41 |
| Vf | bi-uni | -1.71 | 1.31 |
| Vf | bi-bi | -1.31 | 1.75 |
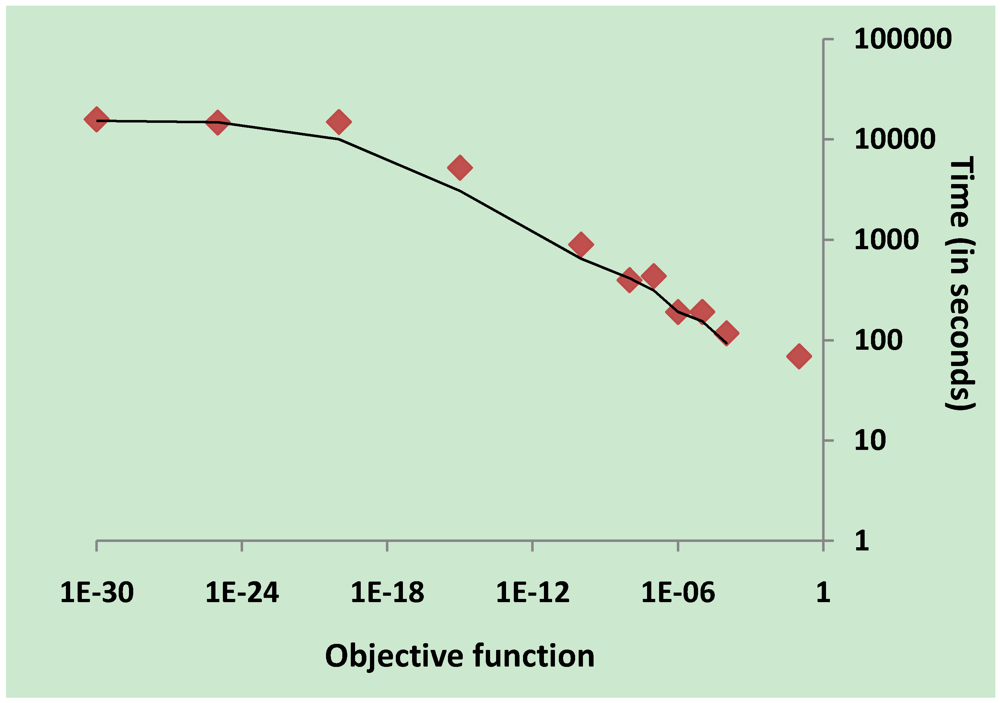
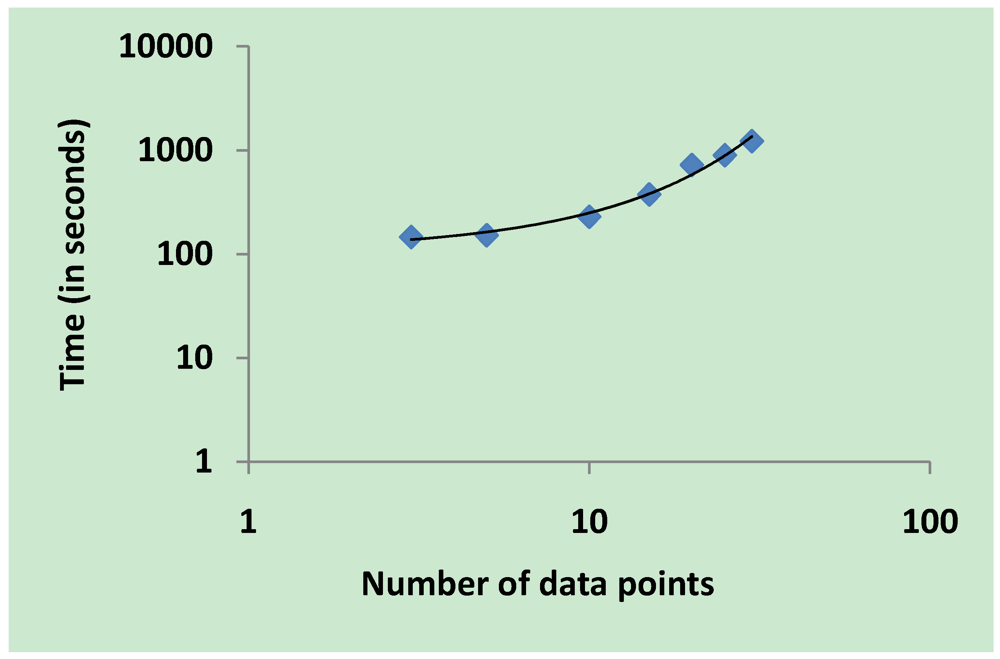
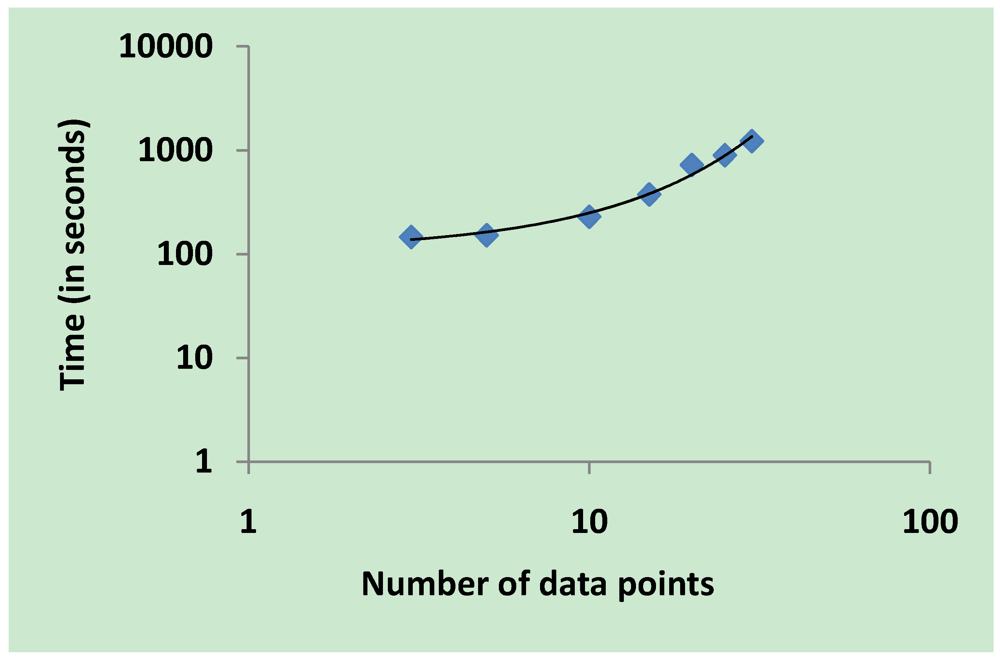
3.5. Validation on Model Integrity
4. Discussion
4. Conclusions
Supplementary files
Acknowledgments
Conflict of Interest
References
- Oberhardt, M.A.; Palsson, B.Ø.; Papin, J.A. Applications of Genome-scale Metabolic Reconstructions. Mol. Syst. Biol. 2009, 5, 320:1–320:15. [Google Scholar]
- Covert, M.W.; Famili, I.; Palsson, B.Ø. Identifying Constraints that Govern Cell Behavior: a Key to Converting Conceptual to Computational Models in Biology? Biotechnol. Bioeng. 2003, 84, 763–772. [Google Scholar] [CrossRef]
- Durot, M.; Bourguignon, P.Y.; Schachter, V. Genome-scale Models of Bacterial Metabolism: Reconstruction and Applications. FEMS Microbiol. Rev. 2009, 33, 164–190. [Google Scholar] [CrossRef]
- Borodina, I.; Nielsen, J. From Genomes to in Silico Cells via Metabolic Networks. Curr. Opin. Biotechnol. 2005, 16, 350–355. [Google Scholar] [CrossRef]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating Quantitative Proteomics and Metabolomics with a Genome-scale Metabolic Network Model. Bioinformatics 2010, 26, i255–i260. [Google Scholar]
- Kitano, H. Systems biology: A Brief Overview. Science 2002, 295, 1662–1664. [Google Scholar]
- Adiamah, D.A.; Handl, J.; Schwartz, J.M. Streamlining the Construction of Large-scale Dynamic Models Using Generic Kinetic Equations. Bioinformatics 2010, 26, 1324–1331. [Google Scholar] [CrossRef]
- Smallbone, K.; Simeonidis, E.; Broomhead, D.S.; Kell, D.B. Something from Nothing - Bridging the Gap between Constraint-based and Kinetic Modelling. FEBS J. 2007, 274, 5576–5585. [Google Scholar] [CrossRef]
- Smallbone, K.; Simeonidis, E.; Swainston, N.; Mendes, P. Towards a Genome-scale Kinetic Model of Cellular Metabolism. BMC Syst. Biol. 2010, 4, 6:1–6:9. [Google Scholar]
- Ao, P.; Lee, L.; Lidstrom, M.; Yin, L.; Zhu, X. Towards Kinetic Modeling of Global Metabolic Networks: Methylobacterium extorquens AM1 growth as validation. Chin. J. Biotechnol. 2008, 24, 980–994. [Google Scholar] [CrossRef]
- Lubitz, T.; Schulz, M.; Klipp, E.; Liebermeister, W. Parameter Balancing in Kinetic Models of Cell Metabolism. J. Phys. Chem. B 2010, 114, 16298–16303. [Google Scholar]
- Liebermeister, W.; Klipp, E. Bringing Metabolic Networks to Life: Convenience Rate Law and Thermodynamic Constraint. Theor. Biol. Med. Model. 2006, 3, 41:1–41:13. [Google Scholar]
- Banga, R.J. Optimization in Computational Systems Biology. BMC Syst. Biol. 2008, 2, 47:1–47:7. [Google Scholar]
- Price, N.D.; Papin, J.A.; Schilling, C.H.; Palsson, B. Genome-scale Microbial in Silico Models: the Constraints-based Approach. Trends Biotechnol. 2003, 21, 162–169. [Google Scholar]
- Beste, D.; Hooper, T.; Stewart, G.; Bonde, B.; Rossa, C.A.; Bushell, M.; Wheeler, P.; Klamt, S.; Kierzek, A.; McFadden, J. GSNM-TB: A Web-based Genome-scale Network Model of Mycobacterium tuberculosis metabolism. Genome Biol. 2007, 8, R89:1–R89:18. [Google Scholar]
- Chopra, P.; Meena, L.S.; Singh, Y. New Drug Targets for Mycobacterium Tuberculosis. Indian J.Med. Res. 2003, 117, 1–9. [Google Scholar]
- Raman, K.; Yeturu, K.; Chandra, N. TargetTB: A Target Identification Pipeline for Mycobacterium Tuberculosis through an Interactome, Reactome and Genome-scale Structural Analysis. BMC Syst. Biol. 2008, 2, 109:1–109:21. [Google Scholar]
- Chandrasekaran, S.; Price, N.D. Probabilistic Integrative Modelling of Genome-scale Metabolic and Regulatory Networks in Escherichia Coli and Mycobacterium Tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef]
- Bordbar, A.; Lewis, N.E.; Schellenberger, J.; Palsson, B.Ø.; Jamshidi, N. Insight into Human Alveolar Macrophage and M. Tuberculosis Interactions via Metabolic Reconstructions. Mol. Syst. Biol. 2010, 6, 422:1–422:14. [Google Scholar]
- King, E.L.; Altman, C. A Schematic Method of Deriving the Rate Laws for Enzyme-catalyzed Reactions. J. Phys. Chem. 1956, 60, 1375–1378. [Google Scholar] [CrossRef]
- Jamshidi, N.; Palsson, B.Ø. Formulating Genome-scale Kinetic Models in the Post-genome Era. Mol. Syst. Biol. 2008, 4, 171:1–171:10. [Google Scholar]
- Teusink, B.; Passarge, J.; Reijenga, C.A.; Esgalhado, E.; van der Weijden, C.C.; Schepper, M.; Walsh, M.C.; Bakker, B.M.; van Dam, K.; Westerhoff, H.V.; Snoep, J.L. Can Yeast Glycolysis be Understood in Terms of in Vitro Kinetics of the Constituent Enzymes? Testing Biochemistry. Eur. J. Biochem. 2000, 267, 5313–5329. [Google Scholar]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI: A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar]
- Zi, Z.; Klipp, E. SBML-PET: a Systems Biology Markup Language-based Parameter Estimation Tool. Bioinformatics 2006, 22, 2704–2705. [Google Scholar]
- Goel, G.; Chou, I.C.; Voit, E.O. System Estimation from Metabolic Time Series Data. Bioinformatics 2008, 24, 2505–2511. [Google Scholar]
- Jamshidi, N.; Palsson, B.Ø. Investigating the Metabolic Capabilities of Mycobacterium Tuberculosis H37Rv Using the in Silico Strain iNJ661 and Proposing Alternative Drug Targets. BMC Syst. Biol. 2007, 1, 26:1–26:20. [Google Scholar]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for Representation and Analysis of Molecular Networks Involving Diseases and Drugs. Nucl. Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef]
- van Riel, N.A.W. Dynamic Modelling and Analysis of Biochemical Networks: Mechanism-based Models and Model-based Experiments. Brief. Bioinform. 2006, 7, 364–374. [Google Scholar] [CrossRef]
- Chou, I-C.; Voit, E.O. Recent Developments in Parameter Estimation and Structure Identification of Biochemical and Genomic Systems. Math. Biosci. 2009, 219, 57–83. [Google Scholar] [CrossRef]
- Ishii, N.; Nakahigashi, K.; Baba, T.; Robert, M.; Soga, T.; Kanai, A.; Hirasawa, T.; Naba, M.; Hirai, K.; Hogue, A.; et al. Multiple High-throughput Analyses Monitor the Response of E. Coli to Perturbations. Science 2007, 316, 593–597. [Google Scholar]
- Castrillo, J.; Zeef, L.; Hoyle, D.; Zhang, N.; Hayes, A.; Gardner, D.; Cornell, M.; Petty, J.; Hakes, L.; Wardleworth, L.; et al. Growth Control of the Eukaryote Cell: A Systems Biology Study in Yeast. J. Biol. 2007, 6, 4:1–4:25. [Google Scholar]
- Yus, E.; Maier, T.; Michalodimitrakis, K.; van Noort, V.; Yamada, T.; Chen, W.H.; Wodke, J.A.; Güell, M.; Martínez, S.; Bourgeois, R.; et al. Impact of Genome Reduction on Bacterial Metabolism and Its Regulation. Science 2009, 326, 1263–1268. [Google Scholar]
- Reinker, S.; Altman, R.M.; Timmer, J. Parameter Estimation in Stochastic Biochemical Reactions. Syst. Biol. 2006, 153, 168–178. [Google Scholar] [CrossRef]
- Mendes, P.; Kell, D.B. Non-linear Optimization of Biochemical Pathways: Applications to Metabolic Engineering and Parameter Estimation. Bioinformatics 1998, 14, 869–883. [Google Scholar] [CrossRef]
- Kell, D.B. Metabolomics, Modelling and Machine Learning in Systems Biology – towards an Understanding of the Languages of Cells. FEBS J. 2006, 273, 873–894. [Google Scholar] [CrossRef]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Adiamah, D.A.; Schwartz, J.-M. Construction of a Genome-Scale Kinetic Model of Mycobacterium Tuberculosis Using Generic Rate Equations. Metabolites 2012, 2, 382-397. https://doi.org/10.3390/metabo2030382
Adiamah DA, Schwartz J-M. Construction of a Genome-Scale Kinetic Model of Mycobacterium Tuberculosis Using Generic Rate Equations. Metabolites. 2012; 2(3):382-397. https://doi.org/10.3390/metabo2030382
Chicago/Turabian StyleAdiamah, Delali A., and Jean-Marc Schwartz. 2012. "Construction of a Genome-Scale Kinetic Model of Mycobacterium Tuberculosis Using Generic Rate Equations" Metabolites 2, no. 3: 382-397. https://doi.org/10.3390/metabo2030382
APA StyleAdiamah, D. A., & Schwartz, J.-M. (2012). Construction of a Genome-Scale Kinetic Model of Mycobacterium Tuberculosis Using Generic Rate Equations. Metabolites, 2(3), 382-397. https://doi.org/10.3390/metabo2030382