
Machine Learning-Driven Insights in Cancer Metabolomics: From Subtyping to Biomarker Discovery and Prognostic Modeling



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Overview of Metabolomics
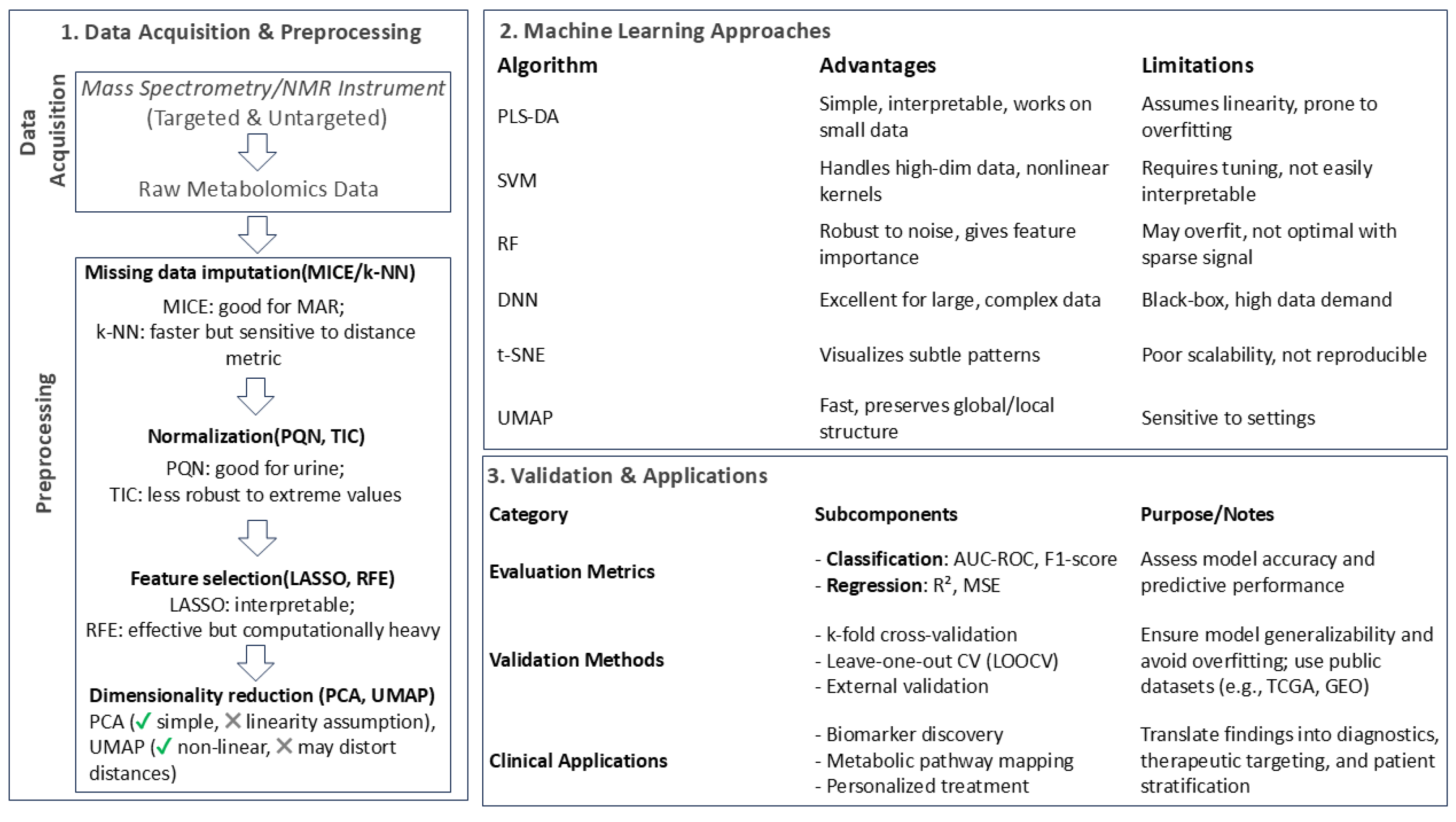
3. Overview of Machine Learning Analysis in Cancer Metabolomics
3.1. Preprocessing Analysis Approaches and Techniques
3.2. Common Machine Learning Approaches in Cancer Metabolomics
3.3. Model Evaluation and Validation Approaches
3.4. Strengths and Limitations of ML Methods in Cancer Metabolomics
4. Machine Learning-Driven Insights in Breast Cancer: From Subtyping to Biomarker Discovery and Prognostic Modeling
4.1. Subtyping and Classification of Breast Cancer
4.2. Biomarker Discovery and Treatment Response Prediction
4.3. Prognostic Modeling and Racial Disparities in Breast Cancer
5. Metabolomics-Based Machine Learning for Colorectal Cancer Detection
5.1. Colorectal Cancer Detection Using Metabolomics and Machine Learning
5.2. Multi-Omics Approaches for Colorectal Cancer Prediction
5.3. Alternative Biological Fluids for Colorectal Cancer Detection
6. Machine Learning Approaches for Ovarian Cancer Diagnosis Using Metabolomics
6.1. Plasma and Serum Metabolomics for Ovarian Cancer Classification
6.2. Multi-Sample Metabolomics Analysis in Ovarian Cancer
7. Bladder Cancer Prognostic and Diagnostic Models
8. Machine Learning Approaches for Lung Cancer Diagnosis, Subtyping, and Treatment Response Using Metabolomics
8.1. Metabolomics and Machine Learning in Lung Cancer Diagnosis and Subtyping
8.2. Metabolomics Biomarkers for Lung Cancer Detection and Treatment Response
9. Metabolomics and Machine Learning in Diagnosing Pancreatic and Related Diseases
10. Metabolomics-Based Machine Learning for Thyroid Cancer Diagnosis
11. Metabolomics and Machine Learning for Prostate Cancer Diagnosis and Classification
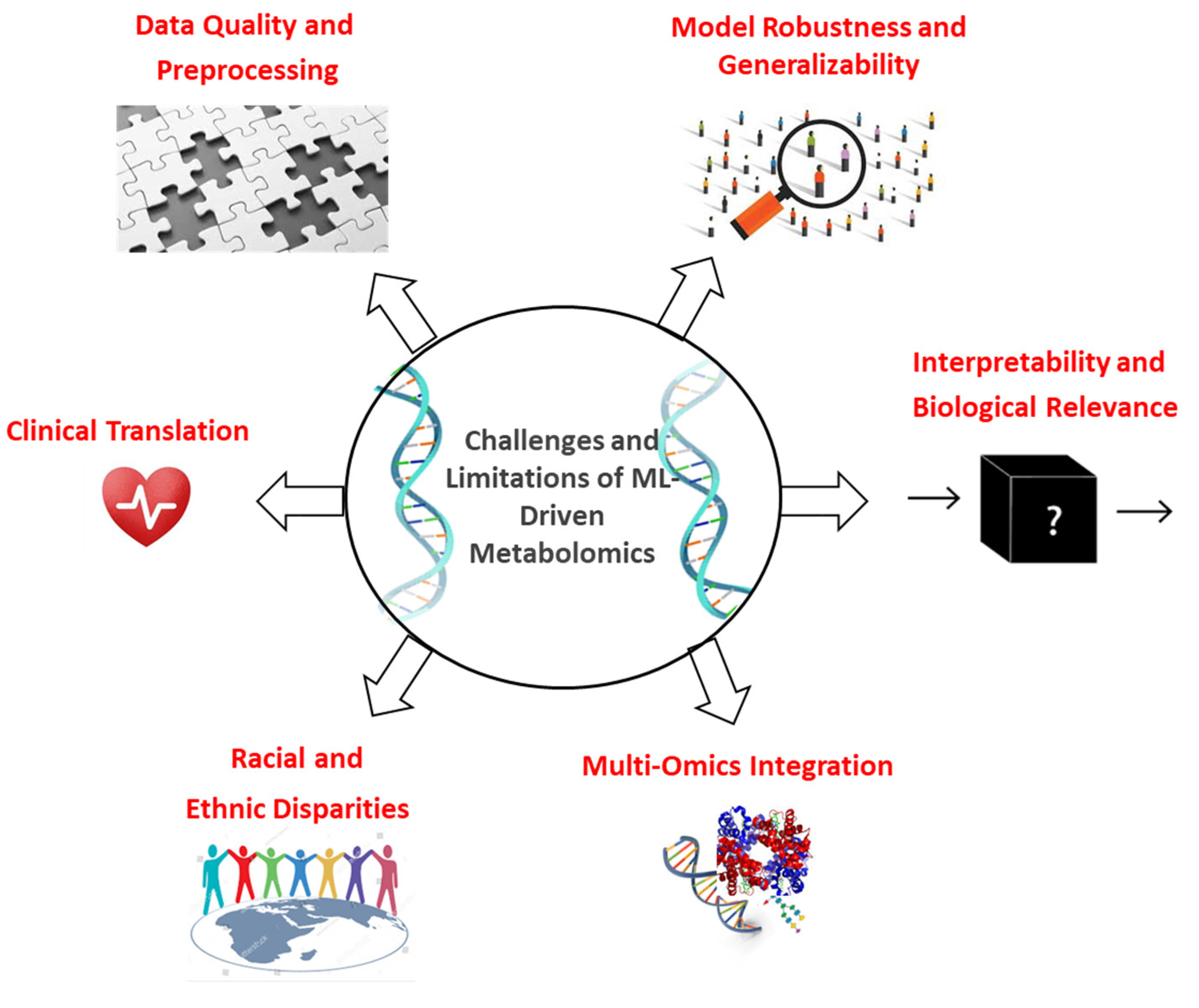
12. Limitations, Challenges, and Future Perspectives of Integrating Machine Learning (ML) with Cancer Metabolomics
12.1. Data Quality and Preprocessing Challenges: The Foundation That Shapes All Outcomes
12.2. Model Overfitting and Generalizability: A Barrier to Translation
12.3. Interpretability and Biological Relevance: Bridging Prediction and Insight
12.4. Multi-Omics Integration: A Valuable Yet Underutilized Approach
12.5. Racial and Ethnic Disparities: A Scientific and Ethical Priority
12.6. Clinical Translation: From Promise to Practice
13. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MS | Mass Spectrometry; |
| LC-MS | Liquid Chromatography–Mass Spectrometry; |
| GC-MS | Gas Chromatography–Mass Spectrometry; |
| NMR | Nuclear Magnetic Resonance; |
| DDA | Data-Dependent Acquisition; |
| DIA | Data-Independent Acquisition; |
| SWATH | Sequential Windowed Acquisition of All Theoretical Fragment Ions; |
| ML | Machine Learning; |
| k-NN | k-Nearest Neighbor; |
| MICE | Multiple Imputation by Chained Equation; |
| PQN | Probabilistic Quotient Normalization; |
| MDFC | Maximal Density Fold Change; |
| RFE | Recursive Feature Elimination; |
| PCA | Principal Component Analysis; |
| t-SNE | t-Distributed Stochastic Neighbor Embedding; |
| UMAP | Uniform Manifold Approximation and Projection; |
| SNF | Similarity Network Fusion; |
| LASSO | Adaptive Least Absolute Shrinkage and Selection Operator; |
| SVM | Support Vector Machine; |
| NB | Naive Bayes; |
| RF | Random Forest; |
| PLS-DA | Partial Least Squares Discriminant Analysis; |
| CNNs | Convolutional Neural Networks; |
| RNNs | Recurrent Neural Networks; |
| AUC-ROC | Area Under the Receiver Operating Characteristic Curve; |
| MSE | Mean Squared Error; |
| LOOCV | Leave-one-out cross-validation; |
| TCGA | The Cancer Genome Atlas; |
| GEO | Gene Expression Omnibus; |
| SHAP | SHapley Additive exPlanations; |
| LIME | Local Interpretable Model-Agnostic Explanations; |
| SMOTE | Synthetic Minority Oversampling; |
| MCC | Mathews Correlation Coefficient; |
| TNBC | Triple-Negative Breast Cancer; |
| ER+ | Estrogen Receptor-Positive; |
| BLIS | Basal-Like Immune-Suppressed; |
| BRCA | Breast Invasive Carcinoma; |
| CRC | Colorectal cancer; |
| PTC | Papillary Thyroid Carcinomas; |
| PDAC | Pancreatic Ductal Adenocarcinoma; |
| S1P | Sphingosine-1-Phosphate; |
| NAAG | N-Acetyl-Aspartyl-Glutamate; |
| RFS | Relapse-Free Survival; |
| PNT | Precancerous Normal Tissue; |
| SAH | S-adenosylhomocysteine; |
| PC | Phosphatidylcholine; |
| PE | Phosphatidylethanolamine; |
| LOWESS | Locally Weighted Scatterplot Smoothing; |
| OPLS-DA | Orthogonal Partial Least Squares Discriminant Analysis. |
References
- Liu, S.; Zhang, X.; Wang, W.; Li, X.; Sun, X.; Zhao, Y.; Wang, Q.; Li, Y.; Hu, F.; Ren, H. Metabolic reprogramming and therapeutic resistance in primary and metastatic breast cancer. Mol. Cancer 2024, 23, 261. [Google Scholar] [CrossRef]
- Nong, S.; Han, X.; Xiang, Y.; Qian, Y.; Wei, Y.; Zhang, T.; Tian, K.; Shen, K.; Yang, J.; Ma, X. Metabolic reprogramming in cancer: Mechanisms and therapeutics. MedComm 2023, 4, e218. [Google Scholar] [CrossRef]
- Vo, D.K.; Trinh, K.T.L. Emerging Biomarkers in Metabolomics: Advancements in Precision Health and Disease Diagnosis. Int. J. Mol. Sci. 2024, 25, 13190. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Wilson, C.M.; Fridley, B.L.; Li, Q. Statistics and Machine Learning in Mass Spectrometry-Based Metabolomics Analysis. Methods Mol. Biol. 2023, 2629, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ma, D.; Yang, Y.S.; Yang, F.; Ding, J.H.; Gong, Y.; Jiang, L.; Ge, L.P.; Wu, S.Y.; Yu, Q.; et al. Comprehensive metabolomics expands precision medicine for triple-negative breast cancer. Cell Res. 2022, 32, 477–490. [Google Scholar] [CrossRef]
- Anh, N.K.; Lee, A.; Phat, N.K.; Yen, N.T.H.; Thu, N.Q.; Tien, N.T.N.; Kim, H.S.; Kim, T.H.; Kim, D.H.; Kim, H.Y.; et al. Combining metabolomics and machine learning to discover biomarkers for early-stage breast cancer diagnosis. PLoS ONE 2024, 19, e0311810. [Google Scholar] [CrossRef]
- Yagin, F.H.; Alkhateeb, A.; Colak, C.; Azzeh, M.; Yagin, B.; Rueda, L. A Fecal-Microbial-Extracellular-Vesicles-Based Metabolomics Machine Learning Framework and Biomarker Discovery for Predicting Colorectal Cancer Patients. Metabolites 2023, 13, 589. [Google Scholar] [CrossRef]
- Santaliz-Casiano, A.; Mehta, D.; Danciu, O.C.; Patel, H.; Banks, L.; Zaidi, A.; Buckley, J.; Rauscher, G.H.; Schulte, L.; Weller, L.R.; et al. Identification of metabolic pathways contributing to ER(+) breast cancer disparities using a machine-learning pipeline. Sci. Rep. 2023, 13, 12136. [Google Scholar] [CrossRef]
- Sumon, M.S.I.; Malluhi, M.; Anan, N.; AbuHaweeleh, M.N.; Krzyslak, H.; Vranic, S.; Chowdhury, M.E.H.; Pedersen, S. Integrative Stacking Machine Learning Model for Small Cell Lung Cancer Prediction Using Metabolomics Profiling. Cancers 2024, 16, 4225. [Google Scholar] [CrossRef]
- Long, F.; Pu, X.; Wang, X.; Ma, D.; Gao, S.; Shi, J.; Zhong, X.; Ran, R.; Wang, L.; Chen, Z.; et al. A metabolic fingerprint of ovarian cancer: A novel diagnostic strategy employing plasma EV-based metabolomics and machine learning algorithms. J. Ovarian Res. 2025, 18, 26. [Google Scholar] [CrossRef]
- Kuwabara, H.; Katsumata, K.; Iwabuchi, A.; Udo, R.; Tago, T.; Kasahara, K.; Mazaki, J.; Enomoto, M.; Ishizaki, T.; Soya, R.; et al. Salivary metabolomics with machine learning for colorectal cancer detection. Cancer Sci. 2022, 113, 3234–3243. [Google Scholar] [CrossRef]
- Troisi, J.; Tafuro, M.; Lombardi, M.; Scala, G.; Richards, S.M.; Symes, S.J.K.; Ascierto, P.A.; Delrio, P.; Tatangelo, F.; Buonerba, C.; et al. A Metabolomics-Based Screening Proposal for Colorectal Cancer. Metabolites 2022, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Penney, K.L.; Tyekucheva, S.; Rosenthal, J.; El Fandy, H.; Carelli, R.; Borgstein, S.; Zadra, G.; Fanelli, G.N.; Stefanizzi, L.; Giunchi, F.; et al. Metabolomics of Prostate Cancer Gleason Score in Tumor Tissue and Serum. Mol. Cancer Res. MCR 2021, 19, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Kuang, A.; Kouznetsova, V.L.; Kesari, S.; Tsigelny, I.F. Diagnostics of Thyroid Cancer Using Machine Learning and Metabolomics. Metabolites 2023, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Iwano, T.; Yoshimura, K.; Watanabe, G.; Saito, R.; Kiritani, S.; Kawaida, H.; Moriguchi, T.; Murata, T.; Ogata, K.; Ichikawa, D.; et al. High-performance Collective Biomarker from Liquid Biopsy for Diagnosis of Pancreatic Cancer Based on Mass Spectrometry and Machine Learning. J. Cancer 2021, 12, 7477–7487. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef]
- Baharum, S.N.; Azizan, K.A. Metabolomics in Systems Biology. Adv. Exp. Med. Biol. 2018, 1102, 51–68. [Google Scholar] [CrossRef]
- Chacko, S.; Haseeb, Y.B.; Haseeb, S. Metabolomics Work Flow and Analytics in Systems Biology. Curr. Mol. Med. 2022, 22, 870–881. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Xu, H.; Qiu, S.; Wang, X. Cell metabolomics. Omics A J. Integr. Biol. 2013, 17, 495–501. [Google Scholar] [CrossRef]
- Pan, Z.; Raftery, D. Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Anal. Bioanal. Chem. 2007, 387, 525–527. [Google Scholar] [CrossRef]
- Kopka, J. Current challenges and developments in GC-MS based metabolite profiling technology. J. Biotechnol. 2006, 124, 312–322. [Google Scholar] [CrossRef]
- Ten-Domenech, I.; Martinez-Sena, T.; Moreno-Torres, M.; Sanjuan-Herraez, J.D.; Castell, J.V.; Parra-Llorca, A.; Vento, M.; Quintas, G.; Kuligowski, J. Comparing Targeted vs. Untargeted MS(2) Data-Dependent Acquisition for Peak Annotation in LC-MS Metabolomics. Metabolites 2020, 10, 126. [Google Scholar] [CrossRef]
- Zhou, J.; Yin, Y. Strategies for large-scale targeted metabolomics quantification by liquid chromatography-mass spectrometry. Anal. 2016, 141, 6362–6373. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, H.; Liu, Y.; Liu, J.; Zhao, X.; Yin, Y. Development and Evaluation of a Parallel Reaction Monitoring Strategy for Large-Scale Targeted Metabolomics Quantification. Anal. Chem. 2016, 88, 4478–4486. [Google Scholar] [CrossRef] [PubMed]
- Schwaiger-Haber, M.; Stancliffe, E.; Arends, V.; Thyagarajan, B.; Sindelar, M.; Patti, G.J. A Workflow to Perform Targeted Metabolomics at the Untargeted Scale on a Triple Quadrupole Mass Spectrometer. ACS Meas. Sci. Au 2021, 1, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huan, T. Comparison of Full-Scan, Data-Dependent, and Data-Independent Acquisition Modes in Liquid Chromatography-Mass Spectrometry Based Untargeted Metabolomics. Anal. Chem. 2020, 92, 8072–8080. [Google Scholar] [CrossRef]
- Defossez, E.; Bourquin, J.; von Reuss, S.; Rasmann, S.; Glauser, G. Eight key rules for successful data-dependent acquisition in mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2023, 42, 131–143. [Google Scholar] [CrossRef]
- Stincone, P.; Pakkir Shah, A.K.; Schmid, R.; Graves, L.G.; Lambidis, S.P.; Torres, R.R.; Xia, S.N.; Minda, V.; Aron, A.T.; Wang, M.; et al. Evaluation of Data-Dependent MS/MS Acquisition Parameters for Non-Targeted Metabolomics and Molecular Networking of Environmental Samples: Focus on the Q Exactive Platform. Anal. Chem. 2023, 95, 12673–12682. [Google Scholar] [CrossRef]
- Wang, R.; Yin, Y.; Zhu, Z.J. Advancing untargeted metabolomics using data-independent acquisition mass spectrometry technology. Anal. Bioanal. Chem. 2019, 411, 4349–4357. [Google Scholar] [CrossRef]
- Pandey, R.; Collins, M.; Lu, X.; Sweeney, S.R.; Chiou, J.; Lodi, A.; Tiziani, S. Novel Strategy for Untargeted Chiral Metabolomics using Liquid Chromatography-High Resolution Tandem Mass Spectrometry. Anal. Chem. 2021, 93, 5805–5814. [Google Scholar] [CrossRef]
- Rahnenfuhrer, J.; De Bin, R.; Benner, A.; Ambrogi, F.; Lusa, L.; Boulesteix, A.L.; Migliavacca, E.; Binder, H.; Michiels, S.; Sauerbrei, W.; et al. Statistical analysis of high-dimensional biomedical data: A gentle introduction to analytical goals, common approaches and challenges. BMC Med. 2023, 21, 182. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, S. Deeply integrating latent consistent representations in high-noise multi-omics data for cancer subtyping. Brief. Bioinform. 2024, 25, bbae061. [Google Scholar] [CrossRef]
- Yao, J.Z.; Tsigelny, I.F.; Kesari, S.; Kouznetsova, V.L. Diagnostics of ovarian cancer via metabolite analysis and machine learning. Integr. Biol. Quant. Biosci. Nano Macro 2023, 15, zyad005. [Google Scholar] [CrossRef]
- Cardoso, M.R.; Silva, A.A.R.; Talarico, M.C.R.; Sanches, P.H.G.; Sforca, M.L.; Rocco, S.A.; Rezende, L.M.; Quintero, M.; Costa, T.; Viana, L.R.; et al. Metabolomics by NMR Combined with Machine Learning to Predict Neoadjuvant Chemotherapy Response for Breast Cancer. Cancers 2022, 14, 5055. [Google Scholar] [CrossRef]
- Liu, X.; Wang, W.; Zhang, X.; Liang, J.; Feng, D.; Li, Y.; Xue, M.; Ling, B. Metabolism pathway-based subtyping in endometrial cancer: An integrated study by multi-omics analysis and machine learning algorithms. Mol. Ther. Nucleic Acids 2024, 35, 102155. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Allhorn, A.; Biessmann, F. A Benchmark for Data Imputation Methods. Front. Big Data 2021, 4, 693674. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Ellis, J.L.; Adams, S.; Sahar, M.; Jacobs, M.; Tulpan, D. Comparison of imputation methods for missing production data of dairy cattle. Anim. Int. J. Anim. Biosci. 2023, 17 (Suppl. 5), 100921. [Google Scholar] [CrossRef] [PubMed]
- Do, K.T.; Wahl, S.; Raffler, J.; Molnos, S.; Laimighofer, M.; Adamski, J.; Suhre, K.; Strauch, K.; Peters, A.; Gieger, C.; et al. Characterization of missing values in untargeted MS-based metabolomics data and evaluation of missing data handling strategies. Metabolomics Off. J. Metabolomic Soc. 2018, 14, 128. [Google Scholar] [CrossRef]
- Altamimi, A.; Alarfaj, A.A.; Umer, M.; Alabdulqader, E.A.; Alsubai, S.; Kim, T.H.; Ashraf, I. An automated approach to predict diabetic patients using KNN imputation and effective data mining techniques. BMC Med. Res. Methodol. 2024, 24, 221. [Google Scholar] [CrossRef]
- Zhang, Z. Multiple imputation with multivariate imputation by chained equation (MICE) package. Ann. Transl. Med. 2016, 4, 30. [Google Scholar] [CrossRef]
- Cinquemani, E.; Laroute, V.; Cocaign-Bousquet, M.; de Jong, H.; Ropers, D. Estimation of time-varying growth, uptake and excretion rates from dynamic metabolomics data. Bioinformatics 2017, 33, i301–i310. [Google Scholar] [CrossRef]
- Deng, K.; Zhang, F.; Tan, Q.; Huang, Y.; Song, W.; Rong, Z.; Zhu, Z.J.; Li, K.; Li, Z. WaveICA: A novel algorithm to remove batch effects for large-scale untargeted metabolomics data based on wavelet analysis. Anal. Chim. Acta 2019, 1061, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Low, B.; Wang, Y.; Zhao, T.; Yu, H.; Huan, T. Closing the Knowledge Gap of Post-Acquisition Sample Normalization in Untargeted Metabolomics. ACS Meas. Sci. Au 2024, 4, 702–711. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xia, Y. Pretreating and normalizing metabolomics data for statistical analysis. Genes Dis. 2024, 11, 100979. [Google Scholar] [CrossRef]
- Han, W.; Li, L. Evaluating and minimizing batch effects in metabolomics. Mass Spectrom. Rev. 2022, 41, 421–442. [Google Scholar] [CrossRef]
- Sanchez-Illana, A.; Perez-Guaita, D.; Cuesta-Garcia, D.; Sanjuan-Herraez, J.D.; Vento, M.; Ruiz-Cerda, J.L.; Quintas, G.; Kuligowski, J. Model selection for within-batch effect correction in UPLC-MS metabolomics using quality control—Support vector regression. Anal. Chim. Acta 2018, 1026, 62–68. [Google Scholar] [CrossRef]
- Reisetter, A.C.; Muehlbauer, M.J.; Bain, J.R.; Nodzenski, M.; Stevens, R.D.; Ilkayeva, O.; Metzger, B.E.; Newgard, C.B.; Lowe, W.L., Jr.; Scholtens, D.M. Mixture model normalization for non-targeted gas chromatography/mass spectrometry metabolomics data. BMC Bioinform. 2017, 18, 84. [Google Scholar] [CrossRef]
- Lin, X.; Yang, F.; Zhou, L.; Yin, P.; Kong, H.; Xing, W.; Lu, X.; Jia, L.; Wang, Q.; Xu, G. A support vector machine-recursive feature elimination feature selection method based on artificial contrast variables and mutual information. J. chromatography. B Anal. Technol. Biomed. Life Sci. 2012, 910, 149–155. [Google Scholar] [CrossRef]
- Hu, C.; Fan, Y.; Lin, Z.; Xie, X.; Huang, S.; Hu, Z. Metabolomic landscape of overall and common cancers in the UK Biobank: A prospective cohort study. Int. J. Cancer 2024, 155, 27–39. [Google Scholar] [CrossRef]
- Xu, S.; Bai, C.; Chen, Y.; Yu, L.; Wu, W.; Hu, K. Comparing univariate filtration preceding and succeeding PLS-DA analysis on the differential variables/metabolites identified from untargeted LC-MS metabolomics data. Anal. Chim. Acta 2024, 1287, 342103. [Google Scholar] [CrossRef] [PubMed]
- Debik, J.; Sangermani, M.; Wang, F.; Madssen, T.S.; Giskeodegard, G.F. Multivariate analysis of NMR-based metabolomic data. NMR Biomed. 2022, 35, e4638. [Google Scholar] [CrossRef] [PubMed]
- Abdelmoula, W.M.; Balluff, B.; Englert, S.; Dijkstra, J.; Reinders, M.J.; Walch, A.; McDonnell, L.A.; Lelieveldt, B.P. Data-driven identification of prognostic tumor subpopulations using spatially mapped t-SNE of mass spectrometry imaging data. Proc. Natl. Acad. Sci. USA 2016, 113, 12244–12249. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Lin, Z.; Wang, X.; Yang, J.; Zhao, W.; Lu, H.; Zhang, Z.; Chen, Y. Pure Ion Chromatograms Combined with Advanced Machine Learning Methods Improve Accuracy of Discriminant Models in LC-MS-Based Untargeted Metabolomics. Molecules 2021, 26, 2715. [Google Scholar] [CrossRef]
- Gupta, R.; Srivastava, D.; Sahu, M.; Tiwari, S.; Ambasta, R.K.; Kumar, P. Artificial intelligence to deep learning: Machine intelligence approach for drug discovery. Mol. Divers. 2021, 25, 1315–1360. [Google Scholar] [CrossRef]
- She, H.; Du, Y.; Du, Y.; Tan, L.; Yang, S.; Luo, X.; Li, Q.; Xiang, X.; Lu, H.; Hu, Y.; et al. Metabolomics and machine learning approaches for diagnostic and prognostic biomarkers screening in sepsis. BMC Anesthesiol. 2023, 23, 367. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, N.; Zhang, C.; Kunzke, T.; Zens, P.; Feuchtinger, A.; Berezowska, S.; Walch, A. Metabolic heterogeneity in tumor cells impacts immunology in lung squamous cell carcinoma. Oncoimmunology 2025, 14, 2457797. [Google Scholar] [CrossRef]
- Van Dieren, L.; Amar, J.Z.; Geurs, N.; Quisenaerts, T.; Gillet, C.; Delforge, B.; D’Heysselaer, L.C.; Filip Thiessen, E.F.; Cetrulo, C.L.; Lellouch, A.G. Unveiling the power of convolutional neural networks in melanoma diagnosis. Eur. J. Dermatol. EJD 2023, 33, 495–505. [Google Scholar] [CrossRef]
- Byra, M.; Dobruch-Sobczak, K.; Piotrzkowska-Wroblewska, H.; Klimonda, Z.; Litniewski, J. Prediction of response to neoadjuvant chemotherapy in breast cancer with recurrent neural networks and raw ultrasound signals. Phys. Med. Biol. 2022, 67, 185007. [Google Scholar] [CrossRef]
- Raschka, S. Model Evaluation, Model Selection, and Algorithm Selection in Machine Learning. arXiv 2020. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Scott, M.; Lundberg, S.-I.L. A unified approach to interpreting model predictions. In Proceedings of the NIPS’17: Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 22 May 2017. [Google Scholar]
- Gromski, P.S.; Muhamadali, H.; Ellis, D.I.; Xu, Y.; Correa, E.; Turner, M.L.; Goodacre, R. A tutorial review: Metabolomics and partial least squares-discriminant analysis--a marriage of convenience or a shotgun wedding. Anal. Chim. Acta 2015, 879, 10–23. [Google Scholar] [CrossRef]
- Xi, B.; Gu, H.; Baniasadi, H.; Raftery, D. Statistical analysis and modeling of mass spectrometry-based metabolomics data. Methods Mol. Biol. 2014, 1198, 333–353. [Google Scholar] [CrossRef]
- Favorov, O.; Macdonald, J.; Kursun, O. SVM-Based Analysis of NMR Spectra in Metabolomics: Development of Procedures. J. Sci. Med. 2019, 1. [Google Scholar]
- Chen, T.; Cao, Y.; Zhang, Y.; Liu, J.; Bao, Y.; Wang, C.; Jia, W.; Zhao, A. Random forest in clinical metabolomics for phenotypic discrimination and biomarker selection. Evid. -Based Complement. Altern. Med. Ecam 2013, 2013, 298183. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Date, Y.; Kikuchi, J. Application of ensemble deep neural network to metabolomics studies. Anal. Chim. Acta 2018, 1037, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Date, Y.; Kikuchi, J. Application of a Deep Neural Network to Metabolomics Studies and Its Performance in Determining Important Variables. Anal. Chem. 2018, 90, 1805–1810. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef]
- Mendez, K.M.; Reinke, S.N.; Broadhurst, D.I. A comparative evaluation of the generalised predictive ability of eight machine learning algorithms across ten clinical metabolomics data sets for binary classification. Metabolomics Off. J. Metabolomic Soc. 2019, 15, 150. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, Y.; Xie, S.; Song, X. Screening and diagnosis of triple negative breast cancer based on rapid metabolic fingerprinting by conductive polymer spray ionization mass spectrometry and machine learning. Front. Cell Dev. Biol. 2022, 10, 1075810. [Google Scholar] [CrossRef]
- Xie, N.; Liao, D.; Liu, B.; Zhang, J.; Liu, L.; Huang, G.; Ouyang, Q. Interpretable Machine Learning Algorithms Identify Inetetamab-Mediated Metabolic Signatures and Biomarkers in Treating Breast Cancer. J. Clin. Lab. Anal. 2024, 38, e25124. [Google Scholar] [CrossRef]
- Vaida, M.; Arumalla, K.K.; Tatikonda, P.K.; Popuri, B.; Bux, R.A.; Tappia, P.S.; Huang, G.; Haince, J.F.; Ford, W.R. Identification of a Novel Biomarker Panel for Breast Cancer Screening. Int. J. Mol. Sci. 2024, 25, 11835. [Google Scholar] [CrossRef] [PubMed]
- Cubuk, C.; Loucera, C.; Pena-Chilet, M.; Dopazo, J. Crosstalk between Metabolite Production and Signaling Activity in Breast Cancer. Int. J. Mol. Sci. 2023, 24, 7450. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Tsigelny, I.F.; Kesari, S.; Kouznetsova, V.L. Colorectal Cancer Detection via Metabolites and Machine Learning. Curr. Issues Mol. Biol. 2024, 46, 4133–4146. [Google Scholar] [CrossRef] [PubMed]
- Murovec, B.; Deutsch, L.; Stres, B. Predictive modeling of colorectal cancer using exhaustive analysis of microbiome information layers available from public metagenomic data. Front. Microbiol. 2024, 15, 1426407. [Google Scholar] [CrossRef]
- Zhan, Y.; Weng, M.; Guo, Y.; Lv, D.; Zhao, F.; Yan, Z.; Jiang, J.; Xiao, Y.; Yao, L. Identification and validation of the nicotine metabolism-related signature of bladder cancer by bioinformatics and machine learning. Front. Immunol. 2024, 15, 1465638. [Google Scholar] [CrossRef]
- Shao, C.H.; Chen, C.L.; Lin, J.Y.; Chen, C.J.; Fu, S.H.; Chen, Y.T.; Chang, Y.S.; Yu, J.S.; Tsui, K.H.; Juo, C.G.; et al. Metabolite marker discovery for the detection of bladder cancer by comparative metabolomics. Oncotarget 2017, 8, 38802–38810. [Google Scholar] [CrossRef]
- Choudhary, A.; Yu, J.; Kouznetsova, V.L.; Kesari, S.; Tsigelny, I.F. Two-Stage Deep-Learning Classifier for Diagnostics of Lung Cancer Using Metabolites. Metabolites 2023, 13, 1055. [Google Scholar] [CrossRef]
- Kajiwara, N.; Kakihana, M.; Maeda, J.; Kaneko, M.; Ota, S.; Enomoto, A.; Ikeda, N.; Sugimoto, M. Salivary metabolomic biomarkers for non-invasive lung cancer detection. Cancer Sci. 2024, 115, 1695–1705. [Google Scholar] [CrossRef]
- Sun, R.; Fei, F.; Wang, M.; Jiang, J.; Yang, G.; Yang, N.; Jin, D.; Xu, Z.; Cao, B.; Li, J. Integration of metabolomics and machine learning revealed tryptophan metabolites are sensitive biomarkers of pemetrexed efficacy in non-small cell lung cancer. Cancer Med. 2023, 12, 19245–19259. [Google Scholar] [CrossRef]
- Yan, S.; Peng, Y.; Wu, Z.; Cheng, L.; Li, H.; Xu, H.; Huang, Y.; Zhang, W.; Li, Y. Distinct metabolic biomarkers to distinguish IgG4-related disease from Sjogren’s syndrome and pancreatic cancer and predict disease prognosis. BMC Med. 2022, 20, 497. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, G.; Jing, L.; Peyrottes, I.; Guigonis, J.M.; Graslin, F.; Lindenthal, S.; Sanglier, J.; Gimenez, I.; Haudebourg, J.; Vandersteen, C.; et al. Pilot Study on the Use of Untargeted Metabolomic Fingerprinting of Liquid-Cytology Fluids as a Diagnostic Tool of Malignancy for Thyroid Nodules. Metabolites 2023, 13, 782. [Google Scholar] [CrossRef]
- Zang, X.; Jones, C.M.; Long, T.Q.; Monge, M.E.; Zhou, M.; Walker, L.D.; Mezencev, R.; Gray, A.; McDonald, J.F.; Fernandez, F.M. Feasibility of detecting prostate cancer by ultraperformance liquid chromatography-mass spectrometry serum metabolomics. J. Proteome Res. 2014, 13, 3444–3454. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing Value Imputation Approach for Mass Spectrometry-based Metabolomics Data. Sci. Rep. 2018, 8, 663. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Yu, Y.; Mai, Y.; Zheng, Y.; Shi, L. Assessing and mitigating batch effects in large-scale omics studies. Genome Biol. 2024, 25, 254. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in 1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Vabalas, A.; Gowen, E.; Poliakoff, E.; Casson, A.J. Machine learning algorithm validation with a limited sample size. PLoS ONE 2019, 14, e0224365. [Google Scholar] [CrossRef]
- Cabitza, F.; Campagner, A.; Soares, F.; Garcia de Guadiana-Romualdo, L.; Challa, F.; Sulejmani, A.; Seghezzi, M.; Carobene, A. The importance of being external. methodological insights for the external validation of machine learning models in medicine. Comput. Methods Programs Biomed. 2021, 208, 106288. [Google Scholar] [CrossRef]
- Sampath, P.; Elangovan, G.; Ravichandran, K.; Shanmuganathan, V.; Pasupathi, S.; Chakrabarti, T.; Chakrabarti, P.; Margala, M. Robust diabetic prediction using ensemble machine learning models with synthetic minority over-sampling technique. Sci. Rep. 2024, 14, 28984. [Google Scholar] [CrossRef] [PubMed]
- Armitage, E.G.; Barbas, C. Metabolomics in cancer biomarker discovery: Current trends and future perspectives. J. Pharm. Biomed. Anal. 2014, 87, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. iScience 2022, 25, 103798. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elguoshy, A.; Zedan, H.; Saito, S. Machine Learning-Driven Insights in Cancer Metabolomics: From Subtyping to Biomarker Discovery and Prognostic Modeling. Metabolites 2025, 15, 514. https://doi.org/10.3390/metabo15080514
Elguoshy A, Zedan H, Saito S. Machine Learning-Driven Insights in Cancer Metabolomics: From Subtyping to Biomarker Discovery and Prognostic Modeling. Metabolites. 2025; 15(8):514. https://doi.org/10.3390/metabo15080514
Chicago/Turabian StyleElguoshy, Amr, Hend Zedan, and Suguru Saito. 2025. "Machine Learning-Driven Insights in Cancer Metabolomics: From Subtyping to Biomarker Discovery and Prognostic Modeling" Metabolites 15, no. 8: 514. https://doi.org/10.3390/metabo15080514
APA StyleElguoshy, A., Zedan, H., & Saito, S. (2025). Machine Learning-Driven Insights in Cancer Metabolomics: From Subtyping to Biomarker Discovery and Prognostic Modeling. Metabolites, 15(8), 514. https://doi.org/10.3390/metabo15080514