
Unravelling Shared Pathways Linking Metabolic Syndrome, Mild Cognitive Impairment, Dementia, and Sarcopenia

Abstract
1. Introduction
2. Methods
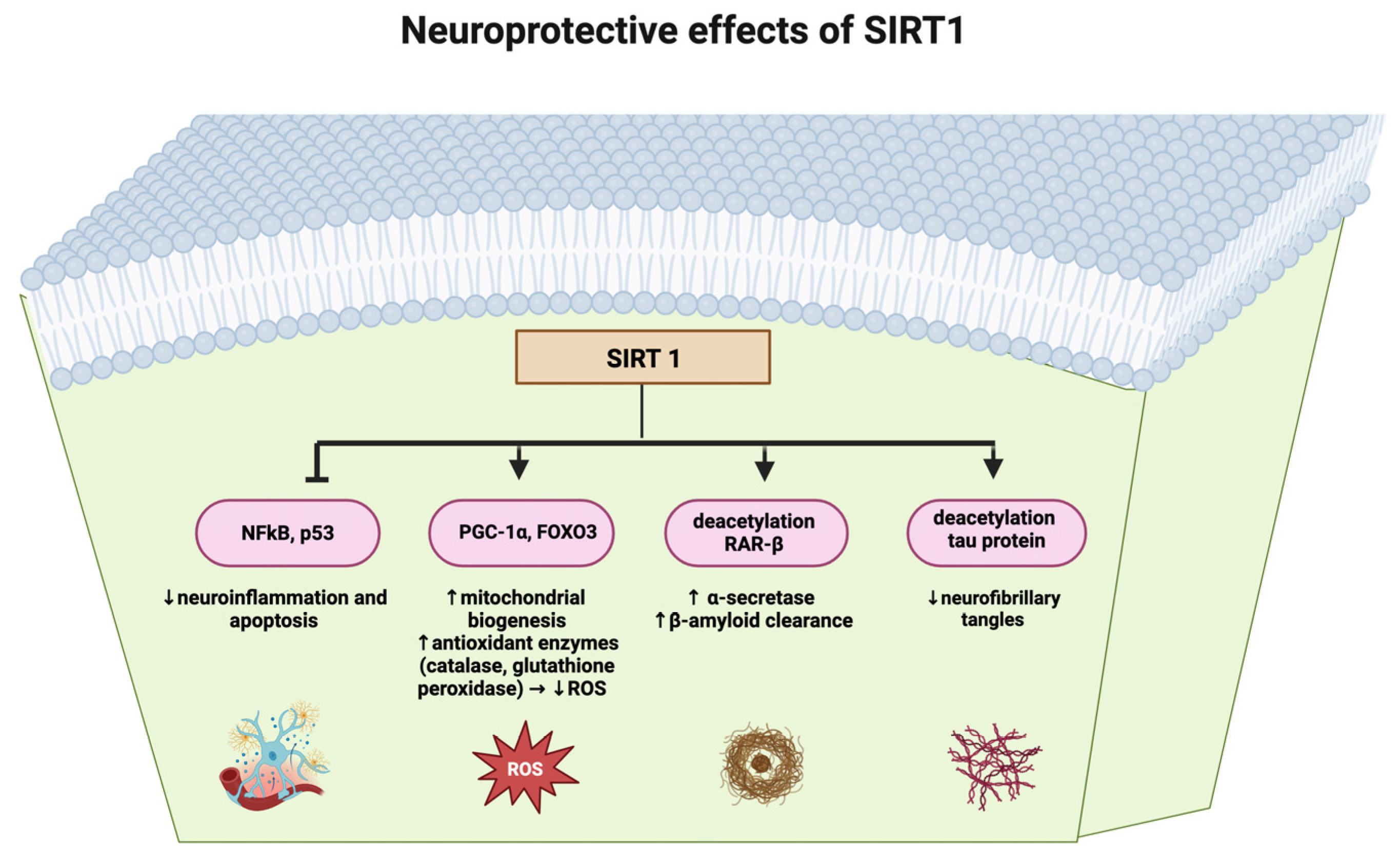
3. Discussion
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| GLP-1 | Glucagon-Like Peptide 1 |
| SCD | Subjective Cognitive Decline |
| IRS-1 | Insulin Receptor Substrate 1 |
| AD | Alzheimer’s Disease |
| BDGF | Brain-Derived Growth Factor |
| PCG | Peroxisome Proliferator-Activated Receptor Gamma Coactivator |
| NAD | Nicotinamide Adenine Dinucleotide |
| NF kB | Nuclear Factor Kappa Light-Chain Enhancer of Activated B Cells |
| FOXO | Fork Head Box O |
| PPAR | Peroxisome Proliferator-Activated Receptor |
| PTP1B | Protein Tyrosine Phosphatase 1B |
| AMPK | AMP-Activated Protein Kinase |
| ROS | Reactive Oxygen Species |
| MCI | Mild Cognitive Impairment |
| MPO | Myeloperoxidase |
| HOMA IR | Homeostatic Model Assessment of Insulin Resistance |
| FDG | Fluorodeoxyglucose |
| IGF-1 | Insulin-Like Growth Factor 1 |
References
- Dybjer, E.; Nilsson, P.M.; Engström, G.; Helmer, C.; Nägga, K. Pre-diabetes and diabetes are independently associated with adverse cognitive test results: A cross-sectional, population-based study. BMC Endocr. Disord. 2018, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.D.; Lee, J.; Hansen, L.; Yuan, M.; Shoelson, S.E. Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J. Biol. Chem. 2004, 279, 35298–35305. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Arumugam, T.V. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Norman, E.D.; Lee, K.; Cutler, R.G.; Telljohann, R.S.; Egan, J.M.; Mattson, M.P. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 2008, 18, 1085–1088. [Google Scholar] [CrossRef]
- Penna, E.; Pizzella, A.; Cimmino, F.; Trinchese, G.; Cavaliere, G.; Catapano, A.; Allocca, I.; Chun, J.T.; Campanozzi, A.; Messina, G.; et al. Neurodevelopmental Disorders: Effect of High-Fat Diet on Synaptic Plasticity and Mitochondrial Functions. Brain Sci. 2020, 10, 805. [Google Scholar] [CrossRef]
- Nencioni, A.; Caffa, I.; Cortellino, S.; Longo, V.D. Fasting and cancer: Molecular mechanisms and clinical application. Nat. Rev. Cancer 2018, 18, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Chandramowlishwaran, P.; Vijay, A.; Abraham, D.; Li, G.; Mwangi, S.M.; Srinivasan, S. Role of Sirtuins in Modulating Neurodegeneration of the Enteric Nervous System and Central Nervous System. Front. Neurosci. 2020, 14, 614331. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. Sirtuins, aging, and metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Li, X. SIRT1 and energy metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; dos Santos, S.M.; Rodrigues, C.A.; Gonçalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Piragine, E.; Brogi, S.; Camodeca, C.; Fucci, R.; Calderone, V.; Nencetti, S.; Martelli, A.; Orlandini, E. Resveratrol-like Compounds as SIRT1 Activators. Int. J. Mol. Sci. 2022, 23, 15105. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wu, Z.; Zhao, P. The protective effects of activating Sirt1/NF-κB pathway for neurological disorders. Rev. Neurosci. 2021, 33, 427–438. [Google Scholar] [CrossRef]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-Pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef]
- Cuyàs, E.; Verdura, S.; Llorach-Parés, L.; Fernández-Arroyo, S.; Joven, J.; Martin-Castillo, B.; Bosch-Barrera, J.; Brunet, J.; Nonell-Canals, A.; Sanchez-Martinez, M.; et al. Metformin Is a Direct SIRT1-Activating Compound: Computational Modeling and Experimental Validation. Front. Endocrinol. 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Munemura, C.; Fukui, T.; Fukuda, S.; Murawaki, Y. Influence of Olmesartan on Sirtuin 1 mRNA Expression in 5/6 Nephrectomized Spontaneously Hypertensive Rats. Yonago Acta Med. 2015, 58, 63–68. [Google Scholar]
- Chang, N.; Li, J.; Lin, S.; Zhang, J.; Zeng, W.; Ma, G.; Wang, Y. Emerging roles of SIRT1 activator, SRT2104, in disease treatment. Sci. Rep. 2024, 14, 5521. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Kim, T.H.; Wu, H.; Choi, Y.H.; Kim, S.G. SIRT1 activation by methylene blue, a repurposed drug, leads to AMPK-mediated inhibition of steatosis and steatohepatitis. Eur. J. Pharmacol. 2014, 727, 115–124. [Google Scholar] [CrossRef]
- Yang, Y.; Duan, W.; Lin, Y.; Yi, W.; Liang, Z.; Yan, J.; Wang, N.; Deng, C.; Zhang, S.; Li, Y.; et al. SIRT1 activation by curcumin pretreatment attenuates mitochondrial oxidative damage induced by myocardial ischemia reperfusion injury. Free Radic. Biol. Med. 2013, 65, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ding, H.; Tang, X.; Liang, M.; Li, S.; Zhang, J.; Cao, J. Quercetin induces pro-apoptotic autophagy via SIRT1/AMPK signaling pathway in human lung cancer cell lines A549 and H1299 in vitro. Thorac. Cancer 2021, 12, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Joshi, U.; Evans, J.E.; Pearson, A.; Saltiel, N.; Cseresznye, A.; Darcey, T.; Ojo, J.; Keegan, A.P.; Oberlin, S.; Mouzon, B.; et al. Targeting sirtuin activity with nicotinamide riboside reduces neuroinflammation in a GWI mouse model. Neurotoxicology 2020, 79, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Lu, F.; Zhang, X.; Liu, S.; Mu, P. SIRT1 Is Involved in the Neuroprotection of Pterostilbene Against Amyloid β 25-35-Induced Cognitive Deficits in Mice. Front. Pharmacol. 2022, 13, 877098. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflam. 2017, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef] [PubMed]
- Rangan, P.; Lobo, F.; Parrella, E.; Rochette, N.; Morselli, M.; Stephen, T.-L.; Cremonini, A.L.; Tagliafico, L.; Persia, A.; Caffa, I.; et al. Fasting-mimicking diet cycles reduce neuroinflammation to attenuate cognitive decline in Alzheimer’s models. Cell Rep. 2022, 40, 111417. [Google Scholar] [CrossRef] [PubMed]
- Zabolotny, J.M.; Kim, Y.B. Silencing insulin resistance through SIRT1. Cell Metab. 2007, 6, 247–249. [Google Scholar] [CrossRef]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917–924, Correction in Nat. Med. 2010, 16, 237. [Google Scholar] [CrossRef]
- Elias, A.; Padinjakara, N.; Lautenschlager, N.T. Effects of intermittent fasting on cognitive health and Alzheimer’s disease. Nutr. Rev. 2023, 81, 1225–1233. [Google Scholar] [CrossRef]
- Mattson, M.P.; Moehl, K.; Ghena, N.; Schmaedick, M.; Cheng, A. Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 2018, 19, 63–80, Corrected in Nat. Rev. Neurosci. 2020, 21, 445. [Google Scholar] [CrossRef]
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem. 2016, 139, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Yang, Y.; Zhou, Y.; Maharana, C.; Lu, D.; Peng, W.; Liu, Y.; Wan, R.; Marosi, K.; Misiak, M.; et al. Mitochondrial SIRT3 Mediates Adaptive Responses of Neurons to Exercise and Metabolic and Excitatory Challenges. Cell Metab. 2016, 23, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Norwitz, N.G.; Hu, M.T.; Clarke, K. The Mechanisms by Which the Ketone Body D-β-Hydroxybutyrate May Improve the Multiple Cellular Pathologies of Parkinson’s Disease. Front. Nutr. 2019, 6, 63. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Demine, S.; Renard, P.; Arnould, T. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8, 795. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Frangou, E.; Love, S.B.; Busza, G.; Holmes, C.; Ritchie, C.; Lawrence, R.; McFarlane, B.; Tadros, G.; Ridha, B.H.; et al. Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: Study protocol for a randomised controlled trial (ELAD study). Trials 2019, 20, 191, Correction in Trials 2020, 21, 660. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-T.; Chen, J.-H.; Hu, C.-J. Role of glucagon-like peptide-1 receptor agonists in Alzheimer’s disease and Parkinson’s disease. J. Biomed. Sci. 2024, 31, 102. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Q.; Qi, X.; Gurney, M.; Perry, G.; Volkow, N.D.; Davis, P.B.; Kaelber, D.C.; Xu, R. Associations of semaglutide with first-time diagnosis of Alzheimer’s disease in patients with type 2 diabetes: Target trial emulation using nationwide real-world data in the US. Alzheimer’s Dement. 2024, 20, 8661–8672. [Google Scholar] [CrossRef]
- Tahmi, M.; Luchsinger, J.A. Metformin in the Prevention of Alzheimer’s Disease and Alzheimer’s Disease Related Dementias. J. Prev. Alzheimer’s Dis. 2023, 10, 706–717. [Google Scholar] [CrossRef] [PubMed]
- Atti, A.R.; Valente, S.; Iodice, A.; Caramella, I.; Ferrari, B.; Albert, U.; Mandelli, L.; De Ronchi, D. Metabolic syndrome, mild cognitive impairment, and dementia: A meta-analysis of longitudinal studies. Am. J. Geriatr. Psychiatry 2019, 27, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Ford, E.S.; Meng, Y.X.; Mokdad, A.H.; Reaven, G.M. Does the association of the triglyceride to high-density lipoprotein cholesterol ratio with fasting serum insulin differ by race/ethnicity? Cardiovasc. Diabetol. 2008, 7, 4. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bernath, M.M.; Bhattacharyya, S.; Nho, K.; Barupal, D.K.; Fiehn, O.; Baillie, R.; Risacher, S.L.; Arnold, M.; Jacobson, T.; Trojanowski, J.Q.; et al. Serum triglycerides in Alzheimer disease: Relation to neuroimaging and CSF biomarkers. Neurology 2020, 94, e2088–e2098. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Choi, H.J.; Byun, M.S.; Yi, D.; Choe, Y.M.; Sohn, B.K.; Baek, H.W.; Lee, J.H.; Kim, H.J.; Han, J.Y.; Yoon, E.J.; et al. Association between serum triglycerides and cerebral amyloidosis in cognitively normal elderly. Am. J. Geriatr. Psychiatry 2016, 24, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Nagga, K.; Gustavsson, A.M.; Stomrud, E.; Lindqvist, D.; van Westen, D.; Blennow, K.; Zetterberg, H.; Melander, O.; Hansson, O. Increased midlife triglycerides predict brain beta-amyloid and tau pathology 20 years later. Neurology 2018, 90, e73–e81. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pillai, J.A.; Bena, J.; Bekris, L.; Kodur, N.; Kasumov, T.; Leverenz, J.B.; Kashyap, S.R. Metabolic syndrome biomarkers relate to rate of cognitive decline in MCI and dementia stages of Alzheimer’s disease. Alzheimer’s Res. Ther. 2023, 15, 54. [Google Scholar] [CrossRef]
- Reed, B.; Villeneuve, S.; Mack, W.; DeCarli, C.; Chui, H.C.; Jagust, W. Associations Between Serum Cholesterol Levels and Cerebral Amyloidosis. JAMA Neurol. 2014, 71, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Galloway, S.; Pallebage-Gamarallage, M.M.; Takechi, R.; Jian, L.; Johnsen, R.D.; Dhaliwal, S.S.; Mamo, J.C. Synergistic effects of high fat feeding and apolipoprotein E deletion on enterocytic amyloid-beta abundance. Lipids Health Dis. 2008, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Petek, B.; Häbel, H.; Xu, H.; Villa-Lopez, M.; Kalar, I.; Hoang, M.T.; Maioli, S.; Pereira, J.B.; Mostafaei, S.; Winblad, B.; et al. Statins and cognitive decline in patients with Alzheimer’s and mixed dementia: A longitudinal registry-based cohort study. Alzheimer’s Res. Ther. 2023, 15, 220. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.B.; Lin, V.W.; Boudreau, D.; Devine, E.B. Statins in the prevention of dementia and Alzheimer’s disease: A meta-analysis of observational studies and an assessment of confounding. Pharmacoepidemiol. Drug Saf. 2013, 22, 345–358. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, B.; Craig, D.; Bullock, R.; Passmore, P. Statins for the prevention of dementia. Cochrane Database Syst. Rev. 2016, 2016, CD003160. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Willette, A.A.; Modanlo, N.; Kapogiannis, D.; Alzheimer’s Disease Neuroimaging Initiative. Insulin resistance predicts medial temporal hypermetabolism in mild cognitive impairment conversion to Alzheimer disease. Diabetes 2015, 64, 1933–1940. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, Y.L.; Ou, Y.N.; Ma, Y.H.; Tan, L.; Yu, J.T. Characteristics of Subjective Cognitive Decline Associated with Alzheimer’s Disease Amyloid Pathology: Findings from The CABLE Study. J. Alzheimer’s Dis. 2023, 92, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, J.; Chen, J.; Luo, M.; Xie, Q.; Rong, Y.; Wu, Y.; Cao, Z.; Liu, Y. High-resolution NMR metabolomics of patients with subjective cognitive decline plus: Perturbations in the metabolism of glucose and branched-chain amino acids. Neurobiol. Dis. 2022, 171, 105782. [Google Scholar] [CrossRef] [PubMed]
- Zeydan, B.; Kantarci, K. Decreased glutamine and glutamate: An early biomarker of neurodegeneration. Int. Psychogeriatr. 2021, 33, 1–2. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Haughey, N.J.; Bandaru, V.V.; Bae, M.; Mattson, M.P. Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim. Biophys. Acta 2010, 1801, 878–886. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.-I.; Mook-Jung, I. A Breakdown in Metabolic Reprogramming Causes Microglia Dysfunction in Alzheimer’s Disease. Cell Metab. 2019, 30, 493–507.e6. [Google Scholar] [CrossRef]
- Eckert, A.; Schmitt, K.; Götz, J. Mitochondrial dysfunction—The beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimer’s Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Traxler, L.; Herdy, J.R.; Stefanoni, D.; Eichhorner, S.; Pelucchi, S.; Szücs, A.; Santagostino, A.; Kim, Y.; Agarwal, R.K.; Schlachetzki, J.C.; et al. Warburg-like metabolic transformation underlies neuronal degeneration in sporadic Alzheimer’s disease. Cell Metab. 2022, 34, 1248–1263.e6. [Google Scholar] [CrossRef]
- Mazurek, S.; Boschek, C.B.; Hugo, F.; Eigenbrodt, E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin. Cancer Biol. 2005, 15, 300–308. [Google Scholar] [CrossRef]
- Victor, M.B.; Tsai, L.H. Walking the high wire: How neurons maintain stability in the crossline of neurodegeneration. Cell Metab. 2022, 34, 1227–1229. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Herdy, J.R.; Traxler, L.; Schafer, S.T.; Schlachetzki, J.C.; Böhnke, L.; Reid, D.A.; Lee, H.; Zangwill, D.; Fernandes, D.P.; et al. Age-dependent instability of mature neuronal fate in induced neurons from Alzheimer’s patients. Cell Stem Cell. 2021, 28, 1533–1548.e6. [Google Scholar] [CrossRef] [PubMed]
- Murai, T.; Matsuda, S. Metabolic Reprogramming toward Aerobic Glycolysis and the Gut Microbiota Involved in the Brain Amyloid Pathology. Biology 2023, 12, 1081. [Google Scholar] [CrossRef]
- Isei, M.O.; Girardi, P.A.; Rodwell-Bullock, J.; Nehrke, K.; Johnson, G.V.W. Site-specific phosphorylation of tau impacts mitochondrial function and response to stressors. J. Neurochem. 2024, 168, 1019–1029. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Y.H.; Liu, L.; Gu, Z.; You, Y.; Hao, J.-R.; Sun, N.; Gao, C. Regulation of glycolysis-derived L-lactate production in astrocytes rescues the memory deficits and Aβ burden in early Alzheimer’s disease models. Pharmacol. Res. 2024, 208, 107357. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiao, M.; Leng, L.; Jiang, S.; Feng, L.; Pan, G.; Li, Z.; Wang, Y.; Wang, J.; Wen, Y.; et al. A systematic review and meta-analysis of the prevalence and correlation of mild cognitive impairment in sarcopenia. J. Cachexia Sarcopenia Muscle 2023, 14, 45–56. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bai, A.; Xu, W.; Sun, J.; Liu, j.; Deng, X.; Wu, L.; Zou, X.; Zuo, J.; Zou, L.; Liu, Y.; et al. Associations of sarcopenia and its defining components with cognitive function in community-dwelling oldest old. BMC Geriatr. 2021, 21, 292. [Google Scholar] [CrossRef] [PubMed]
- Oudbier, S.J.; Goh, J.; Looijaard, S.M.L.M.; Reijnierse, E.M.; Meskers, C.G.M.; Maier, A.B. Pathophysiological Mechanisms Explaining the Association Between Low Skeletal Muscle Mass and Cognitive Function. J. Gerontol. Ser. A 2022, 77, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef]
- Li, D.J.; Li, Y.H.; Yuan, H.B.; Qu, L.F.; Wang, P. The novel exercise-induced hormone irisin protects against neuronal injury via activation of the Akt and ERK1/2 signaling pathways and contributes to the neuroprotection of physical exercise in cerebral ischemia. Metabolism 2017, 68, 31–42. [Google Scholar] [CrossRef]
- Chen, W.; Wang, L.; You, W.; Shan, T. Myokines mediate the cross talk between skeletal muscle and other organs. J. Cell Physiol. 2021, 236, 2393–2412. [Google Scholar] [CrossRef] [PubMed]
- Burtscher, J.; Millet, G.P.; Place, N.; Kayser, B.; Zanou, N. The Muscle-Brain Axis and Neurodegenerative Diseases: The Key Role of Mitochondria in Exercise-Induced Neuroprotection. Int. J. Mol. Sci. 2021, 22, 6479. [Google Scholar] [CrossRef]

{kind=link}
| Compound | Mechanism of Action | Effects |
|---|---|---|
| Resveratrol [11,12] | Activates SIRT1. | Improves cognitive function; protects against neurodegeneration. |
| Metformin [15] | Activates SIRT1 by increasing NAD+ levels; modulates AMPK enhancing SIRT1 activity indirectly. | Promotes mitochondrial biogenesis; improves insulin sensitivity. |
| Olmesartan [16] | Increases SIRT1 mRNA by inhibiting the angiotensin II type 1 receptor. | Reduces oxidative stress and inflammation. |
| SRT2104 [16] | Binds to an allosteric site on SIRT1, enhancing affinity for acetylated lysine residues on targets like p53, NF-kB, and PGC-1α. | Promotes deacetylation of substrates; improves metabolic and inflammatory responses without affecting NAD+ levels. |
| Curcumin [19] | Indirectly activates SIRT1 by scavenging ROS and inhibiting NF kB; binds directly to the SIRT1 active site. | Protects mitochondria; enhances oxidative phosphorylation. |
| Quercetin [20] | Activates SIRT1 through the AMPK; reduces ROS; inhibits mTOR; and increases NAD+ levels. | Promotes mitochondrial biogenesis; prevents β-amyloid-induced microglia death; reduces oxidative stress. |
| Pterostilbene [22] | Directly binds to SIRT1, enhancing deacetylase activity; increases NAD+ levels; and inhibits NAD+-consuming enzymes (CD38). | Reduces tau protein acetylation; prevents β-amyloid aggregation; supports cognitive function. |
| Nicotinamide Riboside [25,26,27] | Precursor for NAD+ biosynthesis. | Enhances NAD+ levels; promotes cellular energy metabolism and repair. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccarelli Ceccarelli, D.; Solerte, S.B. Unravelling Shared Pathways Linking Metabolic Syndrome, Mild Cognitive Impairment, Dementia, and Sarcopenia. Metabolites 2025, 15, 159. https://doi.org/10.3390/metabo15030159
Ceccarelli Ceccarelli D, Solerte SB. Unravelling Shared Pathways Linking Metabolic Syndrome, Mild Cognitive Impairment, Dementia, and Sarcopenia. Metabolites. 2025; 15(3):159. https://doi.org/10.3390/metabo15030159
Chicago/Turabian StyleCeccarelli Ceccarelli, Daniela, and Sebastiano Bruno Solerte. 2025. "Unravelling Shared Pathways Linking Metabolic Syndrome, Mild Cognitive Impairment, Dementia, and Sarcopenia" Metabolites 15, no. 3: 159. https://doi.org/10.3390/metabo15030159
APA StyleCeccarelli Ceccarelli, D., & Solerte, S. B. (2025). Unravelling Shared Pathways Linking Metabolic Syndrome, Mild Cognitive Impairment, Dementia, and Sarcopenia. Metabolites, 15(3), 159. https://doi.org/10.3390/metabo15030159