
A Potential Role for the Ketogenic Diet in Alzheimer’s Disease Treatment: Exploring Pre-Clinical and Clinical Evidence



Abstract
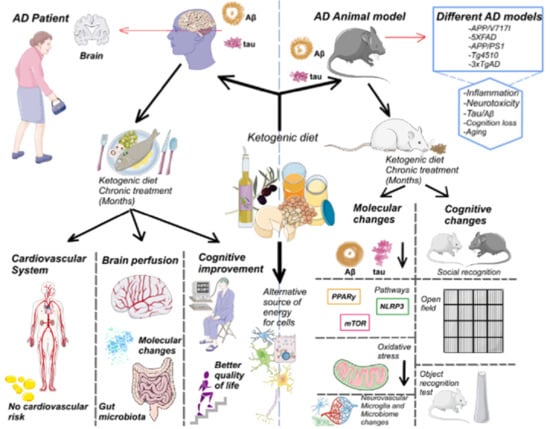
1. Review Design
2. Epidemiology, Neuropathological Insights and Symptoms in Alzheimer ’s Disease
3. Risk Factors for AD
4. Pharmacological Treatment for AD
5. Ketogenic Diet and Ketone Body Biosynthesis
6. Types of KD
7. Possible Risks of KD
8. The Use of the KD in ND
9. The Rationality of KD Use in AD
10. KD Results in Preclinical Models of AD

{kind=link}
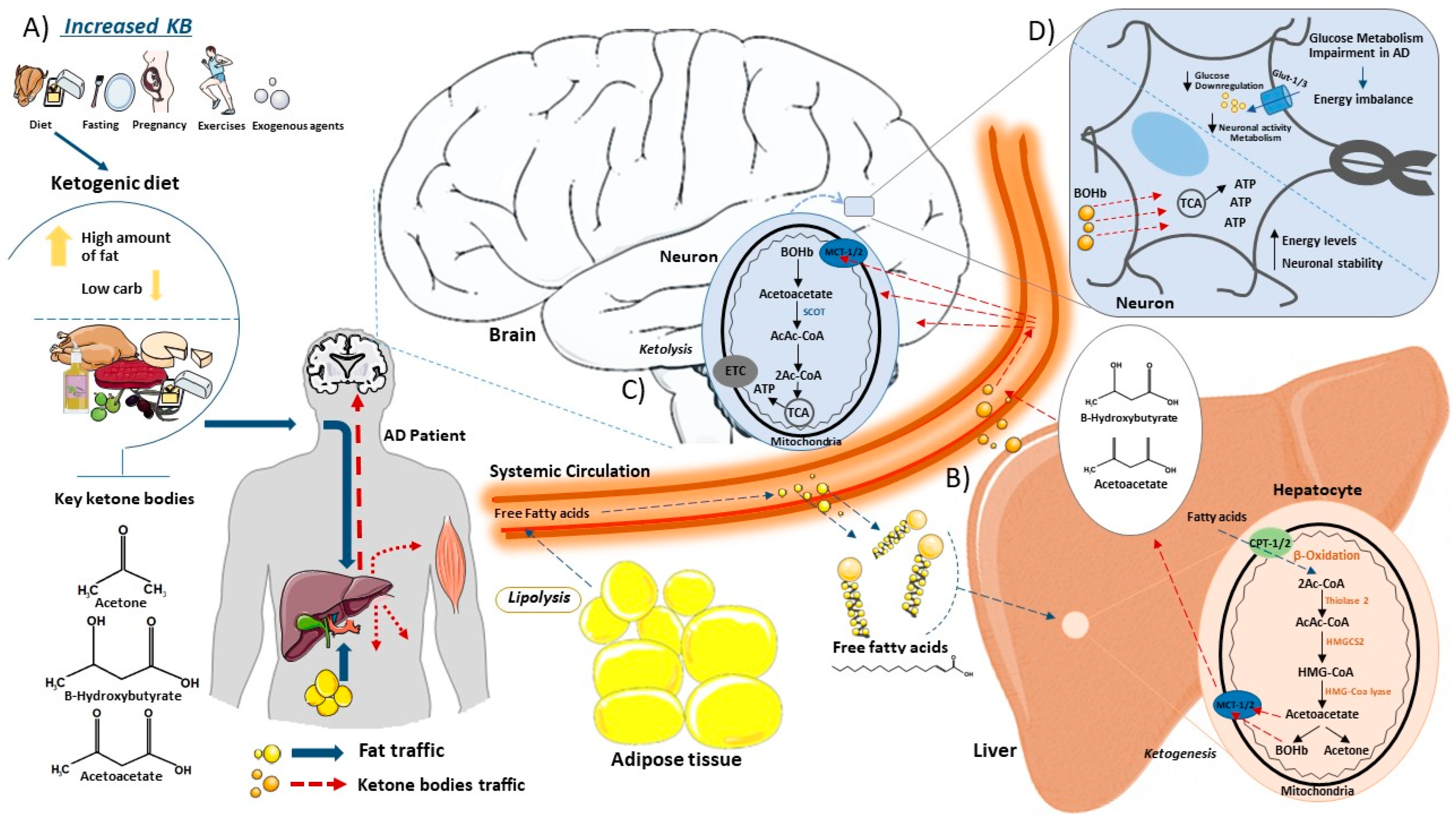{kind=link}
| Author | Species Studied | Experimental Model | Diet/ Supplementation | Duration | Effects |
|---|---|---|---|---|---|
| Yao, J. et al. [120] | Mice (♀) | 3xTgAD | 2-deoxy-D-glucose (2-DG) | 7 weeks | Reduced β-amyloid generation, increased β-amyloid clearance. Enhanced mitochondrial bioenergetic capacity and increased the expression of neurotrophic growth factors. |
| Brownlow, M. L. et al. [125] | Mice | APP/PS1-Tg4510 | Medium-chain triglyceride KD | 16 weeks | Improved motor performance in rotarod test. |
| Xu, Y. et al. [130] | Mice (♂) | 5XFAD | Classic KD | 4 months | Improved spatial learning, spatial memory and working memory. Restored number of neurons and synapses. Reduced neuroinflammation, amyloid plaque deposition and microglial activation. |
| Van der Auwera, I. et al. [131] | Mice (♀) | APP/V717I | Classic KD | 38 days | Reduced levels of Aβ in brain tissue. |
| Liu, H. et al. [155] | Rats (♂) | Sprague-Dawley | KD with or without medium-chain fatty acids | 30 days | Effects on the mTOR pathway and anti-inflammation action. |
| Mohamed, H. E. et al. [168] | Rats (♂) | Obesity induced with HFD | Classic KD | 6 weeks | Improvement of brain oxidative stress responses. Downregulation of brain amyloid protein precursor, apolipoprotein E and caspase-3 mRNA expression. |
| Gzielo, K. et al. [169]. | Rats (♂) | Wistar | Classic KD | 4 months | Morphologically changes in microglial and astroglial cells. |
| Ma, D. et al. [172] | Mice (♂) | C57Bl/6 | Classic KD | 16 weeks | Increased CBF and P-glycoprotein transports on BBB. Reduced mTOR and increased eNOS protein expressions. Enhanced neurovascular functions. Increased the abundance of beneficial gut microbiota. |
| Wang, D. & Mitchell, E. S. [174] | Rats (♂) | Wistar | Medium-chain triglyceride KD | 8 weeks | Increased expression of growth factors, alteration of synaptic markers, transcription factor, protein synthesis and plasticity. Cognitive improvement and difference performance in object and social recognition tests. |
| Pawlosky, R. J. et al. [175] | Mice (♂) | 3xTgAD | Ketone ester (KE) | 8 months | Corrected energy deficiencies in the hippocampus, improved biomarkers and reduced oxidative damage. |
| Kashiwaya, Y. et al. [176] | Mice (♂) | 3xTgAD | Ketone ester (KE) | 8 months | Improved behavioral cognitive function and decreased. Aβ and pTau pathologic changes. |
11. KD Results in Clinical Trials
12. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takada, L.T.; Caramelli, P.; Radanovic, M.; Anghinah, R.; Hartmann, A.P.; Guariglia, C.C.; Bahia, V.S.; Nitrini, R. Prevalence of potentially reversible dementias in a dementia outpatient clinic of a tertiary university-affiliated hospital in Brazil. Arq. Neuropsiquiatr. 2003, 61, 925–929. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Alzheimer’s Disease International. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Leurgans, S.E.; Bennett, D.A. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol. 2009, 66, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Kivipelto, M.; Von Strauss, E. Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward intervention. Dialogues Clin. Neurosci. 2009, 11, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, B.A.; Silva, H.S.; Guimarães, C.; Campino, A.C. Impacto econômico da doença de Alzheimer no Brasil: É possível melhorar a assistência e reduzir custos? Economic impact of Alzheimer’s Disease in Brazil: Is it possible to improve care and minimize costs? Cien. Saude Colet. 2014, 19, 4479–4486. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Corbett, A.; Pickett, J.; Burns, A.; Corcoran, J.; Dunnett, S.B.; Edison, P.; Hagan, J.J.; Holmes, C.; Jones, E.; Katona, C.; et al. Drug repositioning for Alzheimer’s disease. Nat. Rev. Drug Discov. 2012, 11, 833–846. [Google Scholar] [CrossRef]
- Stefanacci, R.G. The costs of Alzheimer’s disease and the value of effective therapies. Am. J. Manag. Care 2011, 17, S356–S362. [Google Scholar]
- Wong, W. Economic burden of Alzheimer disease and managed care considerations. Am. J. Manag. Care. 2020, 26, S177–S183. [Google Scholar]
- Vogt, A.S.; Jennings, G.T.; Mohsen, M.O.; Vogel, M.; Bachmann, M.F. Alzheimer’s Disease: A Brief History of Immunotherapies Targeting Amyloid β. Int. J. Mol. Sci. 2023, 24, 3895. [Google Scholar] [CrossRef]
- Rao, A.K.; Chou, A.; Bursley, B.; Smulofsky, J.; Jezequel, J. Systematic review of the effects of exercise on activities of daily living in people with Alzheimer’s disease. Am. J. Occup. Ther. 2014, 68, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Panegyres, P.K. The Role of Ethnicity in Alzheimer’s Disease: Findings From The C-PATH Online Data Repository. J. Alzheimers Dis. 2016, 51, 515–523. [Google Scholar] [CrossRef]
- Viña, J.; Lloret, A. Why women have more Alzheimer’s disease than men: Gender and mitochondrial toxicity of amyloid-beta peptide. J. Alzheimers Dis. 2010, 20, S527–S533. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; Van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Canevelli, M.; Quarata, F.; Remiddi, F.; Lucchini, F.; Lacorte, E.; Vanacore, N.; Bruno, G.; Cesari, M. Sex and gender differences in the treatment of Alzheimer’s disease: A systematic review of randomized controlled trials. Pharmacol. Res. 2017, 115, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends. Cell Biol. 1998, 8, 447–453. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998, 21, 428–433. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.G.; Zheng, H.; Van der Ploeg, L.H.; Koo, E.H. The beta-amyloid precursor protein of Alzheimer’s disease enhances neuron viability and modulates neuronal polarity. J. Neurosci. 1997, 17, 9407–9414. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, S.; Wirth, Y.; Möbius, H.J. Effects of memantine on behavioural symptoms in Alzheimer’s disease patients: An analysis of the Neuropsychiatric Inventory (NPI) data of two randomised, controlled studies. Int. J Geriatr. Psychiatry 2005, 20, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Tarawneh, R.; Holtzman, D.M. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Wang, X.; Blanchard, J.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Alzheimer’s disease neurofibrillary degeneration: Pivotal and multifactorial. Biochem. Soc. Trans. 2010, 38, 962–966. [Google Scholar] [CrossRef] [PubMed]
- de la Rubia Ortí, J.E.; Fernández, D.; Platero, F.; García-Pardo, M.P. Can Ketogenic Diet Improve Alzheimer’s Disease? Association With Anxiety, Depression, and Glutamate System. Front. Nutr. 2021, 8, 744398. [Google Scholar] [CrossRef]
- Herrmann, N.; Chau, S.A.; Kircanski, I.; Lanctôt, K.L. Current and emerging drug treatment options for Alzheimer’s disease: A systematic review. Drugs 2011, 71, 2031–2065. [Google Scholar] [CrossRef]
- Corey-Bloom, J. The ABC of Alzheimer’s disease: Cognitive changes and their management in Alzheimer’s disease and related dementias. Int. Psychogeriatr. 2002, 14, 51–75. [Google Scholar] [CrossRef]
- Haaksma, M.L.; Vilela, L.R.; Marengoni, A.; Calderón-Larrañaga, A.; Leoutsakos, J.S.; Olde Rikkert, M.G.M.; Melis, R.J.F. Comorbidity and progression of late onset Alzheimer’s disease: A systematic review. PLoS ONE 2017, 12, e0177044. [Google Scholar] [CrossRef]
- Pinto, A.; Bonucci, A.; Maggi, E.; Corsi, M.; Businaro, R. Anti-Oxidant and Anti-Inflammatory Activity of Ketogenic Diet: New Perspectives for Neuroprotection in Alzheimer’s Disease. Antioxidants 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Verdile, G.; Keane, K.N.; Cruzat, V.F.; Medic, S.; Sabale, M.; Rowles, J.; Wijesekara, N.; Martins, R.N.; Fraser, P.E.; Newsholme, P. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer’s Disease. Mediators Inflamm. 2015, 2015, 105828. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Ehret, M.J.; Chamberlin, K.W. Current Practices in the Treatment of Alzheimer Disease: Where is the Evidence After the Phase III Trials? Clin. Ther. 2015, 37, 1604–1616. [Google Scholar] [CrossRef]
- Farlow, M.R. Etiology and pathogenesis of Alzheimer’s disease. Am. J. Health Syst. Pharm. 1998, 55, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Ahtiluoto, S.; Polvikoski, T.; Peltonen, M.; Solomon, A.; Tuomilehto, J.; Winblad, B.; Sulkava, R.; Kivipelto, M. Diabetes, Alzheimer disease, and vascular dementia: A population-based neuropathologic study. Neurology 2010, 75, 1195–1202. [Google Scholar] [CrossRef]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 630502. [Google Scholar] [CrossRef]
- Norton, S.; Matthews, F.E.; Barnes, D.E.; Yaffe, K.; Brayne, C. Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol. 2014, 13, 788–794. [Google Scholar] [CrossRef]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [PubMed]
- Hersi, M.; Irvine, B.; Gupta, P.; Gomes, J.; Birkett, N.; Krewski, D. Risk factors associated with the onset and progression of Alzheimer’s disease: A systematic review of the evidence. Neurotoxicology 2017, 61, 143–187. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Noble, J.; Tang, M.X.; Schupf, N.; Mayeux, R.; Luchsinger, J.A. Type 2 diabetes and late-onset Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2011, 31, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, X.; Cao, H.; Li, H. Comparison of effects of different dietary interventions on cognitive function in Alzheimer’s disease: Protocol for systematic review and network meta-analysis. BMJ Open 2021, 11, e042997. [Google Scholar] [CrossRef]
- Lloret, A.; Monllor, P.; Esteve, D.; Cervera-Ferri, A.; Lloret, M.A. Obesity as a Risk Factor for Alzheimer’s Disease: Implication of Leptin and Glutamate. Front. Neurosci. 2019, 13, 508. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.B. Stress, inflammation, depression, and dementia associated with phosphate toxicity. Mol. Biol. Rep. 2020, 47, 9921–9929. [Google Scholar] [CrossRef] [PubMed]
- Paradise, M.; Cooper, C.; Livingston, G. Systematic review of the effect of education on survival in Alzheimer’s disease. Int. Psychogeriatr. 2009, 21, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Caamaño-Isorna, F.; Corral, M.; Montes-Martínez, A.; Takkouche, B. Education and dementia: A meta-analytic study. Neuroepidemiology 2006, 26, 226–232. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, N.; Cao, H.; Wei, W.; Ma, L.; Li, H. Different Doses of Pharmacological Treatments for Mild to Moderate Alzheimer’s Disease: A Bayesian Network Meta-Analysis. Front. Pharmacol. 2020, 11, 778. [Google Scholar] [CrossRef]
- May, B.H.; Feng, M.; Hyde, A.J.; Hügel, H.; Chang, S.Y.; Dong, L.; Guo, X.; Zhang, A.L.; Lu, C.; Xue, C.C. Comparisons between traditional medicines and pharmacotherapies for Alzheimer disease: A systematic review and meta-analysis of cognitive outcomes. Int. J. Geriatr. Psychiatry 2018, 33, 449–458. [Google Scholar] [CrossRef]
- Shearer, J.; Green, C.; Ritchie, C.W.; Zajicek, J.P. Health state values for use in the economic evaluation of treatments for Alzheimer’s disease. Drugs Aging 2012, 29, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Ulamek-Koziol, M.; Pluta, R. To treat or not to treat Alzheimer’s disease by the ketogenic diet? That is the question. Neural Regen. Res. 2020, 15, 857–858. [Google Scholar] [PubMed]
- Salek, S.S.; Walker, M.D.; Bayer, A.J. A review of quality of life in Alzheimer’s disease. Part 2: Issues in assessing drug effects. Pharmacoeconomics 1998, 14, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Overshott, R.; Byrne, J.; Burns, A. Nonpharmacological and pharmacological interventions for symptoms in Alzheimer’s disease. Expert Rev. Neurother. 2004, 4, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Sandoz, M.; Démonet, J.F.; Fossard, M. Theory of mind and cognitive processes in aging and Alzheimer type dementia: A systematic review. Aging Ment. Health 2014, 18, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Massoud, F.; Léger, G.C. Pharmacological treatment of Alzheimer disease. Can. J. Psychiatry 2011, 56, 579–588. [Google Scholar] [CrossRef]
- Seibert, M.; Mühlbauer, V.; Holbrook, J.; Voigt-Radloff, S.; Brefka, S.; Dallmeier, D.; Denkinger, M.; Schönfeldt-Lecuona, C.; Klöppel, S.; Von Arnim, C.A.F. Efficacy and safety of pharmacotherapy for Alzheimer’s disease and for behavioural and psychological symptoms of dementia in older patients with moderate and severe functional impairments: A systematic review of controlled trials. Alzheimers Res. Ther. 2021, 13, 131. [Google Scholar] [CrossRef]
- Fink, H.A.; Linskens, E.J.; MacDonald, R.; Silverman, P.C.; McCarten, J.R.; Talley, K.M.C.; Forte, M.L.; Desai, P.J.; Nelson, V.A.; Miller, M.A.; et al. Benefits and Harms of Prescription Drugs and Supplements for Treatment of Clinical Alzheimer-Type Dementia. Ann. Intern. Med. 2020, 172, 656–668. [Google Scholar] [CrossRef]
- Hansen, R.A.; Gartlehner, G.; Lohr, K.N.; Kaufer, D.I. Functional outcomes of drug treatment in Alzheimer’s disease: A systematic review and meta-analysis. Drugs Aging 2007, 24, 155–167. [Google Scholar] [CrossRef]
- Mendiola-Precoma, J.; Berumen, L.C.; Padilla, K.; Garcia-Alcocer, G. Therapies for Prevention and Treatment of Alzheimer’s Disease. BioMed Res. Int. 2016, 2016, 2589276. [Google Scholar] [CrossRef]
- Evans, J.G.; Wilcock, G.; Birks, J. Evidence-based pharmacotherapy of Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2004, 7, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, G.K.; Ballard, C.G.; Cooper, J.A.; Loft, H. Memantine for agitation/aggression and psychosis in moderately severe to severe Alzheimer’s disease: A pooled analysis of 3 studies. J. Clin. Psychiatry 2008, 69, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Olazarán, J.; Reisberg, B.; Clare, L.; Cruz, I.; Peña-Casanova, J.; Del Ser, T.; Woods, B.; Beck, C.; Auer, S.; Lai, C.; et al. Nonpharmacological therapies in Alzheimer’s disease: A systematic review of efficacy. Dement. Geriatr. Cogn. Disord. 2010, 30, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Ruthirakuhan, M.; Luedke, A.C.; Tam, A.; Goel, A.; Kurji, A.; Garcia, A. Use of physical and intellectual activities and socialization in the management of cognitive decline of aging and in dementia: A review. J. Aging Res. 2012, 2012, 384875. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.J.; Kim, S.M.; Han, S.E.; Bae, J.H.; Yu, W.J.; Park, M.Y.; Ku, S.; Yang, Y. Effect of Paper-Based Cognitive Training in Early Stage of Alzheimer’s Dementia. Dement. Neurocogn. Disord. 2019, 18, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Broom, G.M.; Shaw, I.C.; Rucklidge, J.J. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition 2019, 60, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 2006, 17, 431–439. [Google Scholar] [CrossRef]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef]
- Zupec-Kania, B.A.; Spellman, E. An overview of the ketogenic diet for pediatric epilepsy. Nutr. Clin. Pract. 2008, 23, 589–596. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Sethi, S.; Palmer, C.M. Ketogenic diet as a metabolic treatment for mental illness. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 269–274. [Google Scholar] [CrossRef]
- Włodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef] [PubMed]
- Barry, D.; Ellul, S.; Watters, L.; Lee, D.; Haluska, R., Jr.; White, R. The ketogenic diet in disease and development. Int. J. Dev. Neurosci. 2018, 68, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Zhang, T.; Wu, X.; Qiu, J.Y. Ketone production by ketogenic diet and by intermittent fasting has different effects on the gut microbiota and disease progression in an Alzheimer’s disease rat model. J. Clin. Biochem. Nutr. 2020, 67, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.W.; Lange, K.M.; Makulska-Gertruda, E.; Nakamura, Y.; Reissmann, A.; Kanaya, S.; Hauser, J. Ketogenic diets and Alzheimer’s disease. Food Sci. Hum. Wellness 2017, 6, 1–9. [Google Scholar] [CrossRef]
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Dhamija, R.; Eckert, S.; Wirrell, E. Ketogenic diet. Can. J. Neurol. Sci. 2013, 40, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Mantha, O.L.; Schor, J.; Pascual, A.; Plaçais, P.Y.; Pavlowsky, A.; Preat, T. Glia fuel neurons with locally synthesized ketone bodies to sustain memory under starvation. Nat. Metab. 2022, 4, 213–224. [Google Scholar] [CrossRef]
- Higashino-Matsui, Y.; Shirato, K.; Suzuki, Y.; Kawashima, Y.; Someya, Y.; Sato, S.; Shiraishi, A.; Jinde, M.; Matsumoto, A.; Ideno, H.; et al. Age-related effects of fasting on ketone body production during lipolysis in rats. Environ. Health Prev. Med. 2012, 17, 157–163. [Google Scholar] [CrossRef][Green Version]
- Paoli, A.; Bosco, G.; Camporesi, E.M.; Mangar, D. Ketosis, ketogenic diet and food intake control: A complex relationship. Front. Psychol. 2015, 6, 27. [Google Scholar] [CrossRef]
- Evans, M.; Cogan, K.E.; Egan, B. Metabolism of ketone bodies during exercise and training: Physiological basis for exogenous supplementation. J. Physiol. 2017, 595, 2857–2871. [Google Scholar] [CrossRef]
- Qian, M.; Wu, N.; Li, L.; Yu, W.; Ouyang, H.; Liu, X.; He, Y.; Al-Mureish, A. Effect of Elevated Ketone Body on Maternal and Infant Outcome of Pregnant Women with Abnormal Glucose Metabolism During Pregnancy. Diabetes Metab. Syndr. Obes. 2020, 13, 4581–4588. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Liu, C.L.; Chiu, W.C.; Twu, Y.C.; Liao, Y.J. HMGCS2 Mediates Ketone Production and Regulates the Proliferation and Metastasis of Hepatocellular Carcinoma. Cancers 2019, 11, 1876. [Google Scholar] [CrossRef] [PubMed]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Orii, K.E.; Fukao, T.; Song, X.Q.; Mitchell, G.A.; Kondo, N. Liver-specific silencing of the human gene encoding succinyl-CoA: 3-ketoacid CoA transferase. Tohoku J. Exp. Med. 2008, 215, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Roubergue, A.; Philibert, B.; Gautier, A.; Kuster, A.; Markowicz, K.; Billette de Villemeur, T.; Vuillaumier-Barrot, S.; Nicole, S.; Roze, E.; Doummar, D. Excellent response to a ketogenic diet in a patient with alternating hemiplegia of childhood. JIMD Rep. 2015, 15, 7–12. [Google Scholar]
- Kim, J.A.; Yoon, J.R.; Lee, E.J.; Lee, J.S.; Kim, J.T.; Kim, H.D.; Kang, H.C. Efficacy of the classic ketogenic and the modified Atkins diets in refractory childhood epilepsy. Epilepsia 2016, 57, 51–58. [Google Scholar] [CrossRef]
- Zhu, H.; Bi, D.; Zhang, Y.; Kong, C.; Du, J.; Wu, X.; Wei, Q.; Qin, H. Ketogenic diet for human diseases: The underlying mechanisms and potential for clinical implementations. Signal Transduct. Target Ther. 2022, 7, 11. [Google Scholar] [CrossRef]
- Liu, Y.M.; Wang, H.S. Medium-chain triglyceride ketogenic diet, an effective treatment for drug-resistant epilepsy and a comparison with other ketogenic diets. Biomed. J. 2013, 36, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Croteau, E.; Castellano, C.A.; Richard, M.A.; Fortier, M.; Nugent, S.; Lepage, M.; Duchesne, S.; Whittingstall, K.; Turcotte, É.E.; Bocti, C.; et al. Ketogenic Medium Chain Triglycerides Increase Brain Energy Metabolism in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 64, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Caprio, M.; Infante, M.; Moriconi, E.; Armani, A.; Fabbri, A.; Mantovani, G.; Mariani, S.; Lubrano, C.; Poggiogalle, E.; Migliaccio, S.; et al. Very-low-calorie ketogenic diet (VLCKD) in the management of metabolic diseases: Systematic review and consensus statement from the Italian Society of Endocrinology (SIE). J. Endocrinol. Investig. 2019, 42, 1365–1386. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Greco, T.; Glenn, T.C.; Hovda, D.A.; Prins, M.L. Ketogenic diet decreases oxidative stress and improves mitochondrial respiratory complex activity. J. Cereb. Blood Flow Metab. 2016, 36, 1603–1613. [Google Scholar] [CrossRef]
- Dowis, K.; Banga, S. The Potential Health Benefits of the Ketogenic Diet: A Narrative Review. Nutrients 2021, 13, 1654. [Google Scholar] [CrossRef] [PubMed]
- Gupta, L.; Khandelwal, D.; Kalra, S.; Gupta, P.; Dutta, D.; Aggarwal, S. Ketogenic diet in endocrine disorders: Current perspectives. J. Postgrad. Med. 2017, 63, 242–251. [Google Scholar]
- Charlot, A.; Zoll, J. Beneficial Effects of the Ketogenic Diet in Metabolic Syndrome: A Systematic Review. Diabetology 2022, 3, 292–309. [Google Scholar] [CrossRef]
- Bostock, E.C.S.; Kirkby, K.C.; Taylor, B.V.; Hawrelak, J.A. Consumer Reports of “Keto Flu” Associated With the Ketogenic Diet. Front. Nutr. 2020, 7, 20. [Google Scholar] [CrossRef]
- Dashti, H.M.; Mathew, T.C.; Hussein, T.; Asfar, S.K.; Behbahani, A.; Khoursheed, M.A.; Al-Sayer, H.M.; Bo-Abbas, Y.Y.; Al-Zaid, N.S. Long-term effects of a ketogenic diet in obese patients. Exp. Clin. Cardiol. 2004, 9, 200–205. [Google Scholar] [PubMed]
- Batch, J.T.; Lamsal, S.P.; Adkins, M.; Sultan, S.; Ramirez, M.N. Advantages and Disadvantages of the Ketogenic Diet: A Review Article. Cureus 2020, 12, e9639. [Google Scholar] [CrossRef] [PubMed]
- Muscogiuri, G.; El Ghoch, M.; Colao, A.; Hassapidou, M.; Yumuk, V.; Busetto, L.; Obesity Management Task Force (OMTF) of the European Association for the Study of Obesity (EASO). European Guidelines for Obesity Management in Adults with a Very Low-Calorie Ketogenic Diet: A Systematic Review and Meta-Analysis. Obes. Facts 2021, 14, 222–245. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Jackson, C.F.; Levy, R.G.; Cooper, P.N. Ketogenic diet and other dietary treatments for epilepsy. Cochrane Database Syst. Rev. 2016, 9, CD001903. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, Y.; Wang, Y.; Tang, H.; Zhang, F.; Zhang, Y.; Zhao, Y. Ketogenic diet for treatment of intractable epilepsy in adults: A meta-analysis of observational studies. Epilepsia Open 2018, 3, 9–17. [Google Scholar] [CrossRef]
- Vanitallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef]
- Palmer, C.M.; Gilbert-Jaramillo, J.; Westman, E.C. The ketogenic diet and remission of psychotic symptoms in schizophrenia: Two case studies. Schizophr. Res. 2019, 208, 439–440. [Google Scholar] [CrossRef]
- Sarnyai, Z.; Kraeuter, A.K.; Palmer, C.M. Ketogenic diet for schizophrenia: Clinical implication. Curr. Opin. Psychiatry 2019, 32, 394–401. [Google Scholar] [CrossRef]
- Murphy, P.; Likhodii, S.; Nylen, K.; Burnham, W.M. The antidepressant properties of the ketogenic diet. Biol. Psychiatry 2004, 56, 981–983. [Google Scholar] [CrossRef]
- Włodarczyk, A.; Cubała, W.J.; Stawicki, M. Ketogenic diet for depression: A potential dietary regimen to maintain euthymia? Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 109, 110257. [Google Scholar] [CrossRef]
- Phelps, J.R.; Siemers, S.V.; El-Mallakh, R.S. The ketogenic diet for type II bipolar disorder. Neurocase 2013, 19, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Kass, H.R.; Winesett, S.P.; Bessone, S.K.; Turner, Z.; Kossoff, E.H. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure 2016, 35, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.R.; Mielnik, C.A.; Funk, A.; O’Donovan, S.M.; Bentea, E.; Pletnikov, M.; Ramsey, A.J.; Wen, Z.; Rowland, L.M.; McCullumsmith, R.E. Measurement of lactate levels in postmortem brain, iPSCs, and animal models of schizophrenia. Sci. Rep. 2019, 9, 5087. [Google Scholar] [CrossRef] [PubMed]
- Rauchenzauner, M.; Klepper, J.; Leiendecker, B.; Luef, G.; Rostasy, K.; Ebenbichler, C. The ketogenic diet in children with Glut1 deficiency syndrome and epilepsy. J. Pediatr. 2008, 153, 716–718. [Google Scholar] [CrossRef]
- Morris, G.; Puri, B.K.; Maes, M.; Olive, L.; Berk, M.; Carvalho, A.F. The role of microglia in neuroprogressive disorders: Mechanisms and possible neurotherapeutic effects of induced ketosis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 99, 109858. [Google Scholar] [CrossRef] [PubMed]
- Castellano, C.A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G.; Imbeault, H.; Turcotte, É.; Fulop, T.; Cunnane, S.C. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 43, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M.; Brown-Borg, H.M. Does Diet Have a Role in the Treatment of Alzheimer’s Disease? Front. Aging Neurosci. 2020, 12, 617071. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219. [Google Scholar] [CrossRef]
- Yao, J.; Chen, S.; Mao, Z.; Cadenas, E.; Brinton, R.D. 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS ONE 2011, 6, e21788. [Google Scholar] [CrossRef]
- Gough, S.M.; Casella, A.; Ortega, K.J.; Hackam, A.S. Neuroprotection by the Ketogenic Diet: Evidence and Controversies. Front. Nutr. 2021, 8, 782657. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 294–315. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, D.; Giménez-Cassina, A. Ketone Bodies in the Brain Beyond Fuel Metabolism: From Excitability to Gene Expression and Cell Signaling. Front. Mol. Neurosci. 2021, 14, 732120. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T. Ketone bodies as a therapeutic for Alzheimer’s disease. Neurotherapeutics 2008, 5, 470–480. [Google Scholar] [CrossRef]
- Brownlow, M.L.; Benner, L.; D’Agostino, D.; Gordon, M.N.; Morgan, D. Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer’s pathology. PLoS ONE 2013, 8, e75713. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, H.; Chen, Z.; Xu, H.; Bu, G.; Zheng, H. Implications of GABAergic Neurotransmission in Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, D.J.; Hogg, E.L.; Regan, P.; Piers, T.; Narayan, P.; Whitehead, G.; Winters, B.L.; Kim, D.H.; Kim, E.; St George-Hyslop, P.; et al. Intracellular oligomeric amyloid-beta rapidly regulates GluA1 subunit of AMPA receptor in the hippocampus. Sci. Rep. 2015, 5, 10934. [Google Scholar] [CrossRef]
- Kim, D.Y.; Abdelwahab, M.G.; Lee, S.H.; O’Neill, D.; Thompson, R.J.; Duff, H.J.; Sullivan, P.G.; Rho, J.M. Ketones prevent oxidative impairment of hippocampal synaptic integrity through KATP channels. PLoS ONE 2015, 10, e0119316. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, C.; Wu, J.; Liu, P.; Deng, X.; Zhang, Y.; Peng, B.; Zhu, Y. Ketogenic diet ameliorates cognitive impairment and neuroinflammation in a mouse model of Alzheimer’s disease. CNS Neurosci. Ther. 2022, 28, 580–592. [Google Scholar] [CrossRef]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2005, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Harmon, D.M.; Kludtke, E.; Mickow, A.; Simha, V.; Kopecky, S. Dramatic elevation of LDL cholesterol from ketogenic-dieting: A Case Series. Am. J. Prev. Cardiol. 2023, 14, 100495. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.A.; Jeon, B.T.; Shin, H.J.; Kim, N.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 2011, 232, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Ruskin, D.N.; Sturdevant, I.C.; Wyss, L.S.; Masino, S.A. Ketogenic diet effects on inflammatory allodynia and ongoing pain in rodents. Sci. Rep. 2021, 11, 725. [Google Scholar] [CrossRef] [PubMed]
- Ruskin, D.N.; Kawamura, M.; Masino, S.A. Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS ONE 2009, 4, e8349. [Google Scholar] [CrossRef]
- Zou, Y.; Fineberg, S.; Pearlman, A.; Feinman, R.D.; Fine, E.J. The effect of a ketogenic diet and synergy with rapamycin in a mouse model of breast cancer. PLoS ONE 2020, 15, e0233662. [Google Scholar] [CrossRef]
- Cooper, M.A.; Menta, B.W.; Perez-Sanchez, C.; Jack, M.M.; Khan, Z.W.; Ryals, J.M.; Winter, M.; Wright, D.E. A ketogenic diet reduces metabolic syndrome-induced allodynia and promotes peripheral nerve growth in mice. Exp. Neurol. 2018, 306, 149–157. [Google Scholar] [CrossRef]
- Poplawski, M.M.; Mastaitis, J.W.; Isoda, F.; Grosjean, F.; Zheng, F.; Mobbs, C.V. Reversal of diabetic nephropathy by a ketogenic diet. PLoS ONE 2011, 6, e18604. [Google Scholar] [CrossRef]
- Okuda, T.; Morita, N. A very low carbohydrate ketogenic diet prevents the progression of hepatic steatosis caused by hyperglycemia in a juvenile obese mouse model. Nutr. Diabetes 2012, 2, e50. [Google Scholar] [CrossRef]
- Todorova, M.T.; Tandon, P.; Madore, R.A.; Stafstrom, C.E.; Seyfried, T.N. The ketogenic diet inhibits epileptogenesis in EL mice: A genetic model for idiopathic epilepsy. Epilepsia 2000, 41, 933–940. [Google Scholar] [CrossRef]
- Masino, S.A.; Li, T.; Theofilas, P.; Sandau, U.S.; Ruskin, D.N.; Fredholm, B.B.; Geiger, J.D.; Aronica, E.; Boison, D. A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J. Clin. Investig. 2011, 121, 2679–2683. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lu, Y.; Jia, M.; Wang, X.; Zhang, Z.; Hou, Q.; Wang, B. Ketogenic diet attenuates spatial and item memory impairment in pentylenetetrazol-kindled rats. Brain Res. 2016, 1646, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.F.; Huang, G.B.; Xu, M.D.; Gao, F.; Lin, S.; Huang, J.; Wang, J.; Li, Y.Q.; Wu, C.H.; Yao, S.; et al. Anti-depression effects of ketogenic diet are mediated via the restoration of microglial activation and neuronal excitability in the lateral habenula. Brain Behav. Immun. 2020, 88, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, Y.Q.; Wu, C.H.; Zhang, Y.L.; Zhao, S.T.; Chen, Y.J.; Deng, Y.H.; Xuan, A.; Sun, X.D. The effect of ketogenic diet on behaviors and synaptic functions of naive mice. Brain Behav. 2019, 9, e01246. [Google Scholar] [CrossRef] [PubMed]
- Ruskin, D.N.; Ross, J.L.; Kawamura, M., Jr.; Ruiz, T.L.; Geiger, J.D.; Masino, S.A. A ketogenic diet delays weight loss and does not impair working memory or motor function in the R6/2 1J mouse model of Huntington’s disease. Physiol. Behav. 2011, 103, 501–507. [Google Scholar] [CrossRef]
- Shaafi, S.; Najmi, S.; Aliasgharpour, H.; Mahmoudi, J.; Sadigh-Etemad, S.; Farhoudi, M.; Baniasadi, N. The efficacy of the ketogenic diet on motor functions in Parkinson’s disease: A rat model. Iran. J. Neurol. 2016, 15, 63–69. [Google Scholar]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef]
- Yoshikawa, K. Neurotoxicity of β-amyloid. Nature 1993, 361, 122–123. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Hao, J.; Liu, R.; Turner, G.; Shi, F.D.; Rho, J.M. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS ONE 2012, 7, e35476. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Huang, J.; Liu, H.; Li, F.; Peng, Q.; Liu, C. Effects of ketogenic diet containing medium-chain fatty acids on serum inflammatory factor and mTOR signaling pathway in rats. Chem. Biol. Technol. Agric. 2020, 7, 27. [Google Scholar] [CrossRef]
- McDaniel, S.S.; Rensing, N.R.; Thio, L.L.; Yamada, K.A.; Wong, M. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 2011, 52, e7–e11. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhou, Z.; Lin, X.; Liu, F.; Liu, C.; Liu, Z.; Deng, W.; Zhang, X.; Chang, H.; Zhao, C. Ketogenic diet prevents paclitaxel-induced neuropathic nociception through activation of PPARγ signalling pathway and inhibition of neuroinflammation in rat dorsal root ganglion. Eur. J. Neurosci. 2021, 54, 5341–5356. [Google Scholar] [CrossRef] [PubMed]
- Simeone, T.A.; Matthews, S.A.; Samson, K.K.; Simeone, K.A. Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp. Neurol. 2017, 287, 54–64. [Google Scholar] [CrossRef]
- Guo, M.; Wang, X.; Zhao, Y.; Yang, Q.; Ding, H.; Dong, Q.; Chen, X.; Cui, M. Ketogenic Diet Improves Brain Ischemic Tolerance and Inhibits NLRP3 Inflammasome Activation by Preventing Drp1-Mediated Mitochondrial Fission and Endoplasmic Reticulum Stress. Front. Mol. Neurosci. 2018, 11, 86. [Google Scholar] [CrossRef]
- Lin, J.; Huang, Z.; Liu, J.; Huang, Z.; Liu, Y.; Liu, Q.; Yang, Z.; Li, R.; Wu, X.; Shi, Z.; et al. Neuroprotective Effect of Ketone Metabolism on Inhibiting Inflammatory Response by Regulating Macrophage Polarization After Acute Cervical Spinal Cord Injury in Rats. Front. Neurosci. 2020, 14, 583611. [Google Scholar] [CrossRef]
- Haskó, G.; Cronstein, B. Regulation of inflammation by adenosine. Front. Immunol. 2013, 4, 85. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.; Dupuis, N.; Auvin, S. Ketogenic diet and Neuroinflammation. Epilepsy Res. 2020, 167, 106454. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Park, S.W.; Brooks, N.; Lang, B.T.; Vemuganti, R. PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res. 2008, 1244, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARγ and the Innate Immune System Mediate the Resolution of Inflammation. PPAR Res. 2015, 14, 549691. [Google Scholar]
- Bernardo, A.; Minghetti, L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr. Pharm. Des. 2006, 12, 93–109. [Google Scholar] [CrossRef]
- Chawla, A. Control of macrophage activation and function by PPARs. Circ. Res. 2010, 106, 1559–1569. [Google Scholar] [CrossRef]
- Carvalho, M.V.; Gonçalves-de-Albuquerque, C.F.; Silva, A.R. PPAR Gamma: From Definition to Molecular Targets and Therapy of Lung Diseases. Int. J. Mol. Sci. 2021, 22, 805. [Google Scholar] [CrossRef]
- Mohamed, H.E.; El-Swefy, S.E.; Rashed, L.A.; Abd El-Latif, S.K. Biochemical effect of a ketogenic diet on the brains of obese adult rats. J. Clin. Neurosci. 2010, 17, 899–904. [Google Scholar] [CrossRef]
- Gzielo, K.; Soltys, Z.; Rajfur, Z.; Setkowicz, Z.K. The Impact of the Ketogenic Diet on Glial Cells Morphology. A Quantitative Morphological Analysis. Neuroscience 2019, 413, 239–251. [Google Scholar] [CrossRef]
- Attaye, I.; Van Oppenraaij, S.; Warmbrunn, M.V.; Nieuwdorp, M. The Role of the Gut Microbiota on the Beneficial Effects of Ketogenic Diets. Nutrients 2021, 14, 191. [Google Scholar] [CrossRef]
- Ang, Q.Y.; Alexander, M.; Newman, J.C.; Tian, Y.; Cai, J.; Upadhyay, V.; Turnbaugh, J.A.; Verdin, E.; Hall, K.D.; Leibel, R.L.; et al. Ketogenic Diets Alter the Gut Microbiome Resulting in Decreased Intestinal Th17 Cells. Cell 2020, 181, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wang, A.C.; Parikh, I.; Green, S.J.; Hoffman, J.D.; Chlipala, G.; Murphy, M.P.; Sokola, B.S.; Bauer, B.; Hartz, A.M.S.; et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci. Rep. 2018, 8, 6670. [Google Scholar] [CrossRef] [PubMed]
- Crosby, L.; Davis, B.; Joshi, S.; Jardine, M.; Paul, J.; Neola, M.; Barnard, N.D. Ketogenic Diets and Chronic Disease: Weighing the Benefits Against the Risks. Front. Nutr. 2021, 8, 702802. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mitchell, E.S. Cognition and Synaptic-Plasticity Related Changes in Aged Rats Supplemented with 8- and 10-Carbon Medium Chain Triglycerides. PLoS ONE 2016, 11, e0160159. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef]
- Kovács, Z.; D’Agostino, D.P.; Diamond, D.; Kindy, M.S.; Rogers, C.; Ari, C. Therapeutic Potential of Exogenous Ketone Supplement Induced Ketosis in the Treatment of Psychiatric Disorders: Review of Current Literature. Front. Psychiatry 2019, 10, 363. [Google Scholar] [CrossRef]
- Tabaie, E.A.; Reddy, A.J.; Brahmbhatt, H. A narrative review on the effects of a ketogenic diet on patients with Alzheimer’s disease. AIMS Public Health 2021, 9, 185–193. [Google Scholar] [CrossRef]
- Brandt, J.; Buchholz, A.; Henry-Barron, B.; Vizthum, D.; Avramopoulos, D.; Cervenka, M.C. Preliminary Report on the Feasibility and Efficacy of the Modified Atkins Diet for Treatment of Mild Cognitive Impairment and Early Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 969–981. [Google Scholar] [CrossRef]
- Phillips, M.C.L.; Deprez, L.M.; Mortimer, G.M.N.; Murtagh, D.K.J.; McCoy, S.; Mylchreest, R.; Gilbertson, L.J.; Clark, K.M.; Simpson, P.V.; McManus, E.J.; et al. Randomized crossover trial of a modified ketogenic diet in Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 51. [Google Scholar] [CrossRef]
- Neth, B.J.; Mintz, A.; Whitlow, C.; Jung, Y.; Solingapuram Sai, K.; Register, T.C.; Kellar, D.; Lockhart, S.N.; Hoscheidt, S.; Maldjian, J.; et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: A pilot study. Neurobiol. Aging 2020, 86, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement. 2017, 4, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Matsuo, J.; Ishida, I.; Takano, H.; Yokoi, Y.; Hori, H.; Yoshida, S.; Ashida, K.; Nakamura, K.; Takahashi, T.; et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci. Lett. 2019, 690, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. BBA Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Toda, A.; Kimoto, A.; Takebayashi, Y.; Higashiyama, R.; Tagata, Y.; Ito, M.; Ota, T.; Shibata, N.; Arai, H. Benefits of use, and tolerance of, medium-chain triglyceride medical food in the management of Japanese patients with Alzheimer’s disease: A prospective, open-label pilot study. Clin. Interv. Aging 2016, 11, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Torosyan, N.; Sethanandha, C.; Grill, J.D.; Dilley, M.L.; Lee, J.; Cummings, J.L.; Ossinalde, C.; Silverman, D.H. Changes in regional cerebral blood flow associated with a 45 day course of the ketogenic agent, caprylidene, in patients with mild to moderate Alzheimer’s disease: Results of a randomized, double-blinded, pilot study. Exp. Gerontol. 2018, 111, 118–121. [Google Scholar] [CrossRef]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Fortier, M.; Castellano, C.A.; Croteau, E.; Langlois, F.; Bocti, C.; St-Pierre, V.; Vandenberghe, C.; Bernier, M.; Roy, M.; Descoteaux, M.; et al. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 2019, 5, 625–634. [Google Scholar] [CrossRef]

| KD Type | Ratio | Carbohydrate Intake per Day on a Diet of 1000 Kcal | Considerations |
|---|---|---|---|
| LCT | 4:1 to 3:1 | 8 g on a 4:1 16 g on a 3:1 | Severe carbohydrate restriction, unpalatable |
| MCTD | Not diet-ratio-related | 48 g | More ketogenic, gastrointestinal side effects |
| MAD | Approximately 1:1 | 40–60 g | No precise weighing, no protein/calorie restrictions |
| LOGI | Approximately 1:1 | 10 g for the first month then 20–30 g | Minimized glycemic increases, liberalized regimen |
| Author | Participants (n) | Primary Diagnosis | Diet/ Supplementation | Duration | Ketosis | Results/Side Effects |
|---|---|---|---|---|---|---|
| Croteau, E. et al. [93] | 15 | Mild-moderate AD | Medium-chain triglyceride KD Suppl. C8C10 and C8 | Two periods of 1 month | C8C10: Blood BOHb (mM) = 0.46 ± 0.19 C8: Blood BOHb (mM) = 0.57 ± 0.27 | Increased total brain energy metabolism. |
| Phillips, M. C. L. et al. [180] | 26 | Alzheimer’s disease | Classic KD | Two periods of 12 weeks | BOHb level = 0.95 ± 0.34 mmol/L 18/21 patients | Improved daily function and quality of life. No significant changes were observed in the lipid profile. Mild adverse effects. |
| Neth, B. J. et al. [181] | 20 | Subjective memory complaints or mild cognitive impairment | Mediterranean KD | Two periods of 6 weeks | BOHb level = 0.23 (0.27) mmol/L | Improvement of peripheral metabolic measures, CSF biomarker profile and increased cerebral perfusion. No serious adverse events occurred. |
| Nagpal, R. et al. [182] | 17 | Mild cognitive impairment or cognitively normal | Mediterranean KD | Two periods of 6 weeks | Not measured | Modulating capacity of MMKD in the gut microbiome. |
| Taylor, M. K. et al. [183] | 15 | Very-mild, mild and moderate AD | Medium-chain triglyceride KD | 3 months | Serum BOHb level = 0.31 mmol/L | Overall cognitive improvement. No serious adverse events occurred. |
| Ota, M. et al. [184] | 20 | Mild-moderate AD | Medium-chain triglyceride KD | 12 months | Plasma BOHb level = 470.9 ± 292.6 μmol/L | Positive effects on verbal memory and processing speed. Diarrhea, most frequently reported side effect of MCT. |
| Rebello, C. J. et al. [185] | 6 | Mild cognitive impairment | Medium-chain triglyceride KD | 24 weeks | ApoE4(−) Serum BOHb level = 0.15 mM ApoE4(+) Serum BOHb level = 0.54 mM | Memory improvement. |
| Ohnuma, T. et al. [186] | 22 | Sporadic, mild-moderate AD | AXONA Dietary Suplment | 3 months | Serum BOHb level = 81.1 ± 79.9 μM | Improvement in cognitive functions. No severe gastrointestinal adverse effects. |
| Henderson, S. T. et al. [187] | 152 | Mild-moderate AD | AC-1202 | 90 days | BOHb level = 0.39 mM | Cognitive improvement. Mild to moderate adverse events restricted to the gastrointestinal system. |
| Torosyan, N. et al. [188] | 16 | Mild-moderate AD | Caprylidene | 45 days | Not measured | Increased blood flow in specific brain regions. |
| Reger, M. A. et al. [189] | 20 | AD or mild cognitive | Emulsified medium-chain triglyceride | Two times of 90 min | ApoE4(−) Serum BOHb level = 0.54 mM ApoE4(+) Serum BOHb level = 0.43 mM | Cognitive improvement in AD patients withoutAPoE4. |
| Fortier, M. et al. [190] | 52 | Mild cognitive impairment | Ketogenic medium-chain triglyceride drink | 6 months | Plasma BOHb level = 401 ± 303 μM | Improved several cognitive outcomes in MCI. No severe adverse events. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, T.P.D.; Morais, A.L.B.; dos Reis, P.L.B.; Palotás, A.; Vieira, L.B. A Potential Role for the Ketogenic Diet in Alzheimer’s Disease Treatment: Exploring Pre-Clinical and Clinical Evidence. Metabolites 2024, 14, 25. https://doi.org/10.3390/metabo14010025
Oliveira TPD, Morais ALB, dos Reis PLB, Palotás A, Vieira LB. A Potential Role for the Ketogenic Diet in Alzheimer’s Disease Treatment: Exploring Pre-Clinical and Clinical Evidence. Metabolites. 2024; 14(1):25. https://doi.org/10.3390/metabo14010025
Chicago/Turabian StyleOliveira, Tadeu P. D., Ana L. B. Morais, Pedro L. B. dos Reis, András Palotás, and Luciene B. Vieira. 2024. "A Potential Role for the Ketogenic Diet in Alzheimer’s Disease Treatment: Exploring Pre-Clinical and Clinical Evidence" Metabolites 14, no. 1: 25. https://doi.org/10.3390/metabo14010025
APA StyleOliveira, T. P. D., Morais, A. L. B., dos Reis, P. L. B., Palotás, A., & Vieira, L. B. (2024). A Potential Role for the Ketogenic Diet in Alzheimer’s Disease Treatment: Exploring Pre-Clinical and Clinical Evidence. Metabolites, 14(1), 25. https://doi.org/10.3390/metabo14010025