
Detection and Quantification of Ammonia as the Ammonium Cation in Human Saliva by 1H NMR: A Promising Probe for Health Status Monitoring, with Special Reference to Cancer



Abstract
1. Introduction
2. Materials and Methods
2.1. Saliva Sample Collection and Preliminary Laboratory Processing
2.2. Preparation of WMSS Samples for 1H NMR Analysis
2.3. Acquisition of 1H NMR Spectra
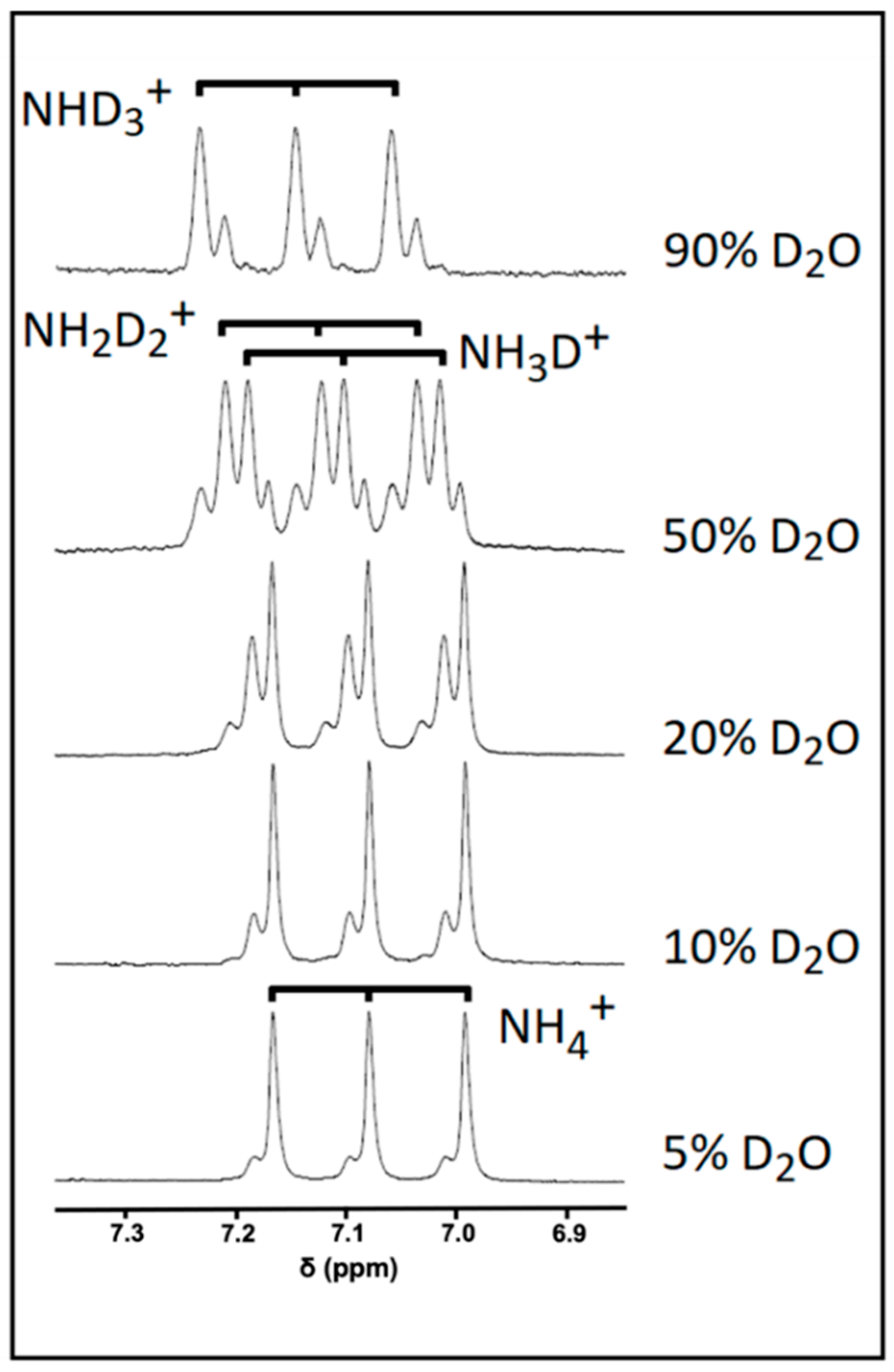
2.4. Investigations of the Influence of Increasing Solution D2O Contents on 1H NMR Spectral Profiles
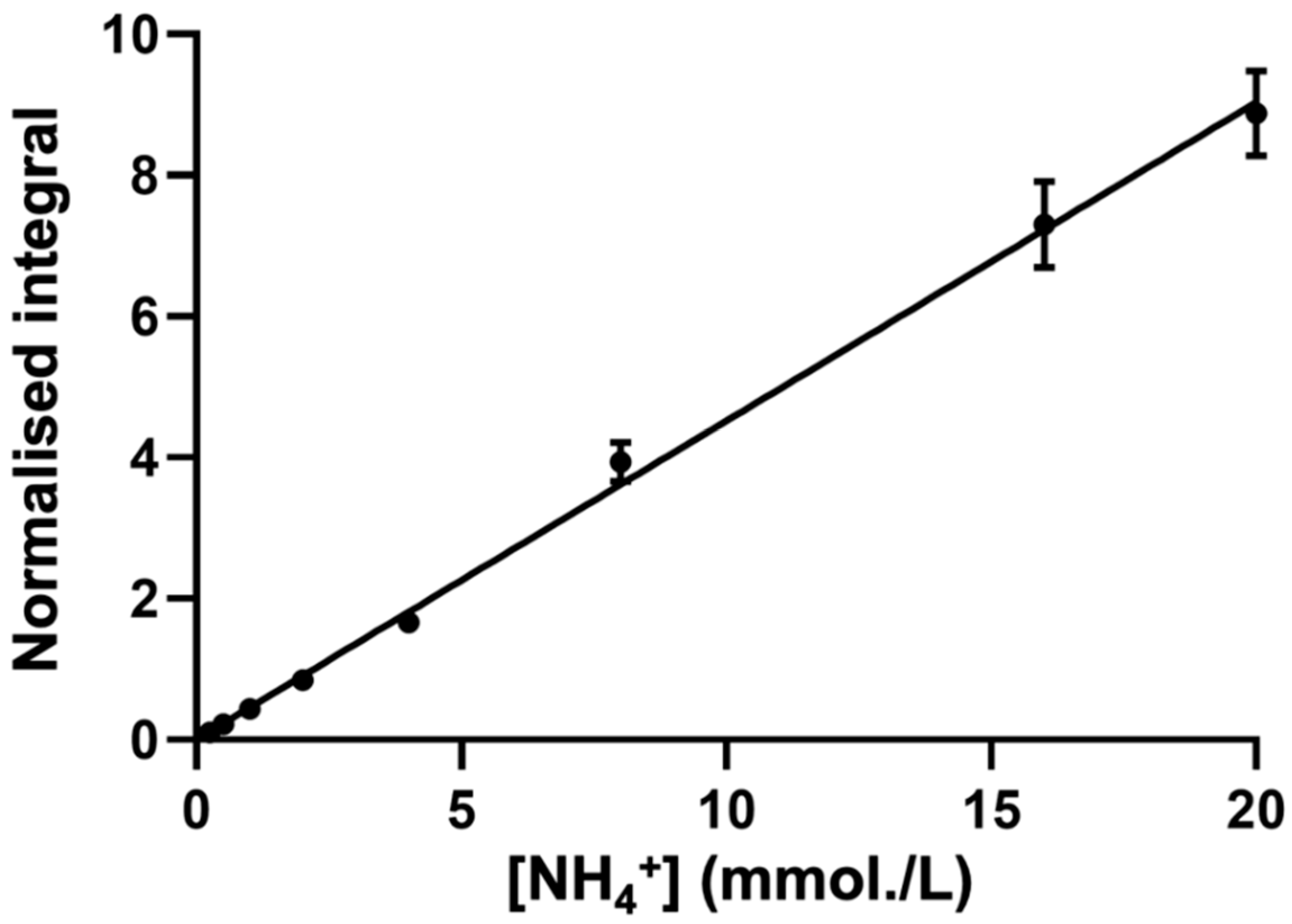
2.5. Generation of Calibration Curves
2.6. Standard Addition Method (SAM) for Salivary NH4+ ion Determinations: Sample Preparation
2.7. Time-Dependent Determinations of Salivary Ammonia/Ammonium Ion Concentration following WMS Sample Collection and Laboratory Processing
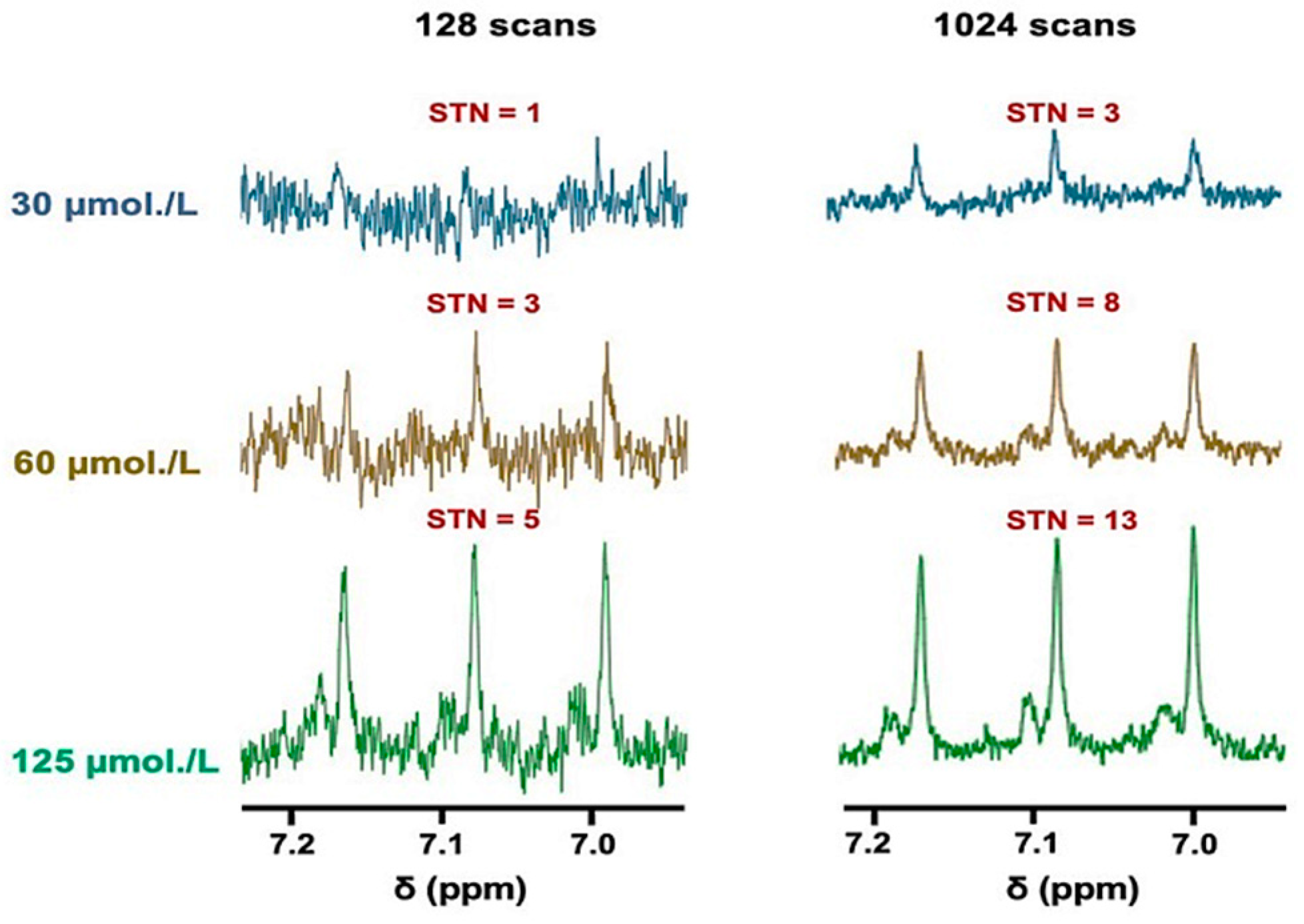
2.8. Sensitivity of 1H NMR Analysis of Salivary NH4+ Ion, and Computation of Signal-to-Noise (STN) Ratios and Lower Limit of Detection and Quantification Values (LLOD and LLOQ Respectively)
2.9. Statistical Analysis of Experimental Data
3. Results
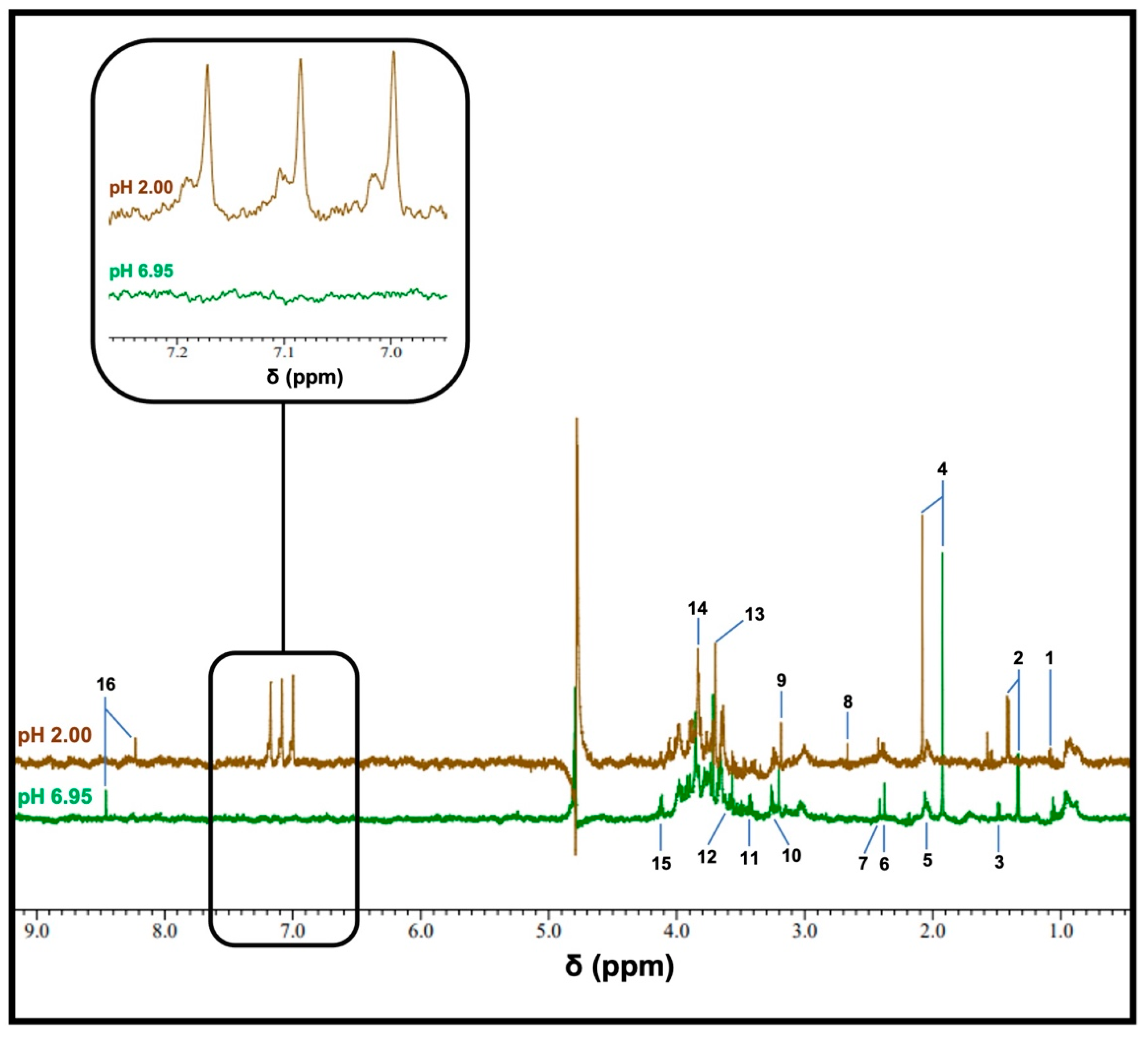
3.1. Detection of Ammonia as Ammonium Ion (NH4+) in WMSS Samples

3.2. Sensitivity of the 1H NMR Analysis of NH4+ in WMSS Samples at pH 2
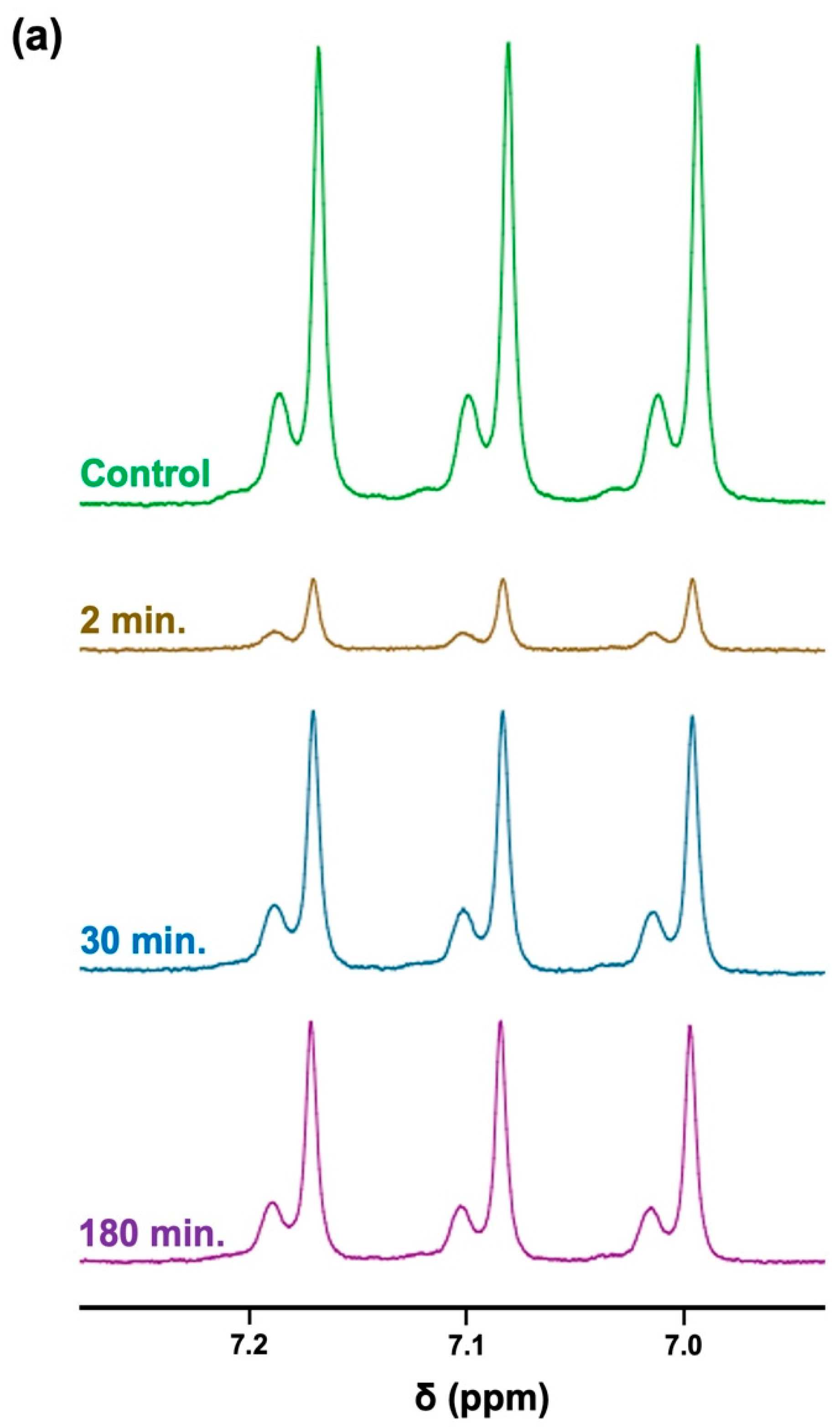
3.3. Investigations of Hydrogen-Deuterium Exchange and Quadrupolar Splitting of the NH4+ Ion 1H NMR Resonance
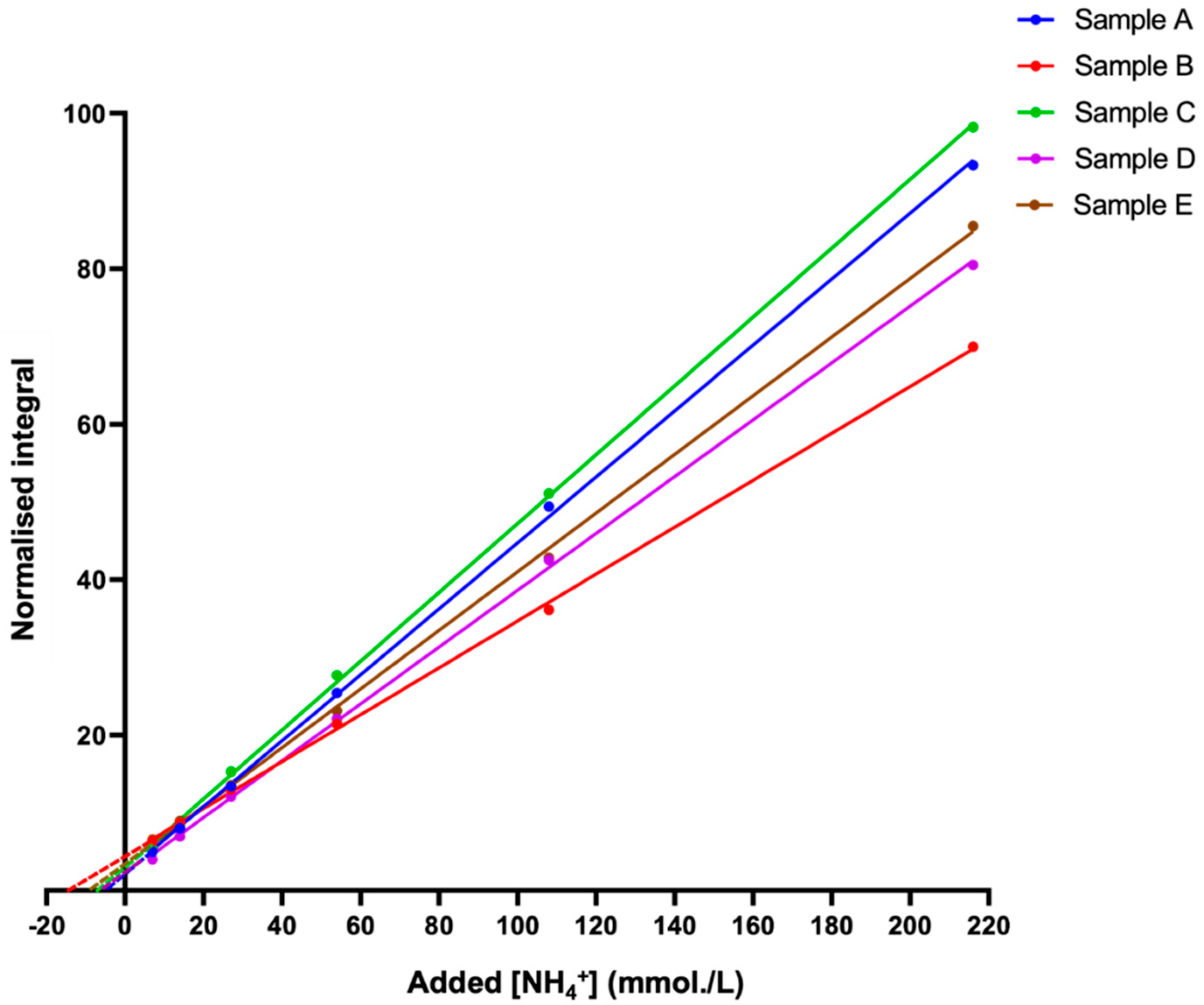
3.4. Establishment of a Reliable Standard Calibration Curve Method to Determine NH4+ Ion Concentrations in WMSS Samples
3.5. Application of the Standard Addition Method (SAM) to Confirm Estimated Salivary NH4+ Ion Concentrations
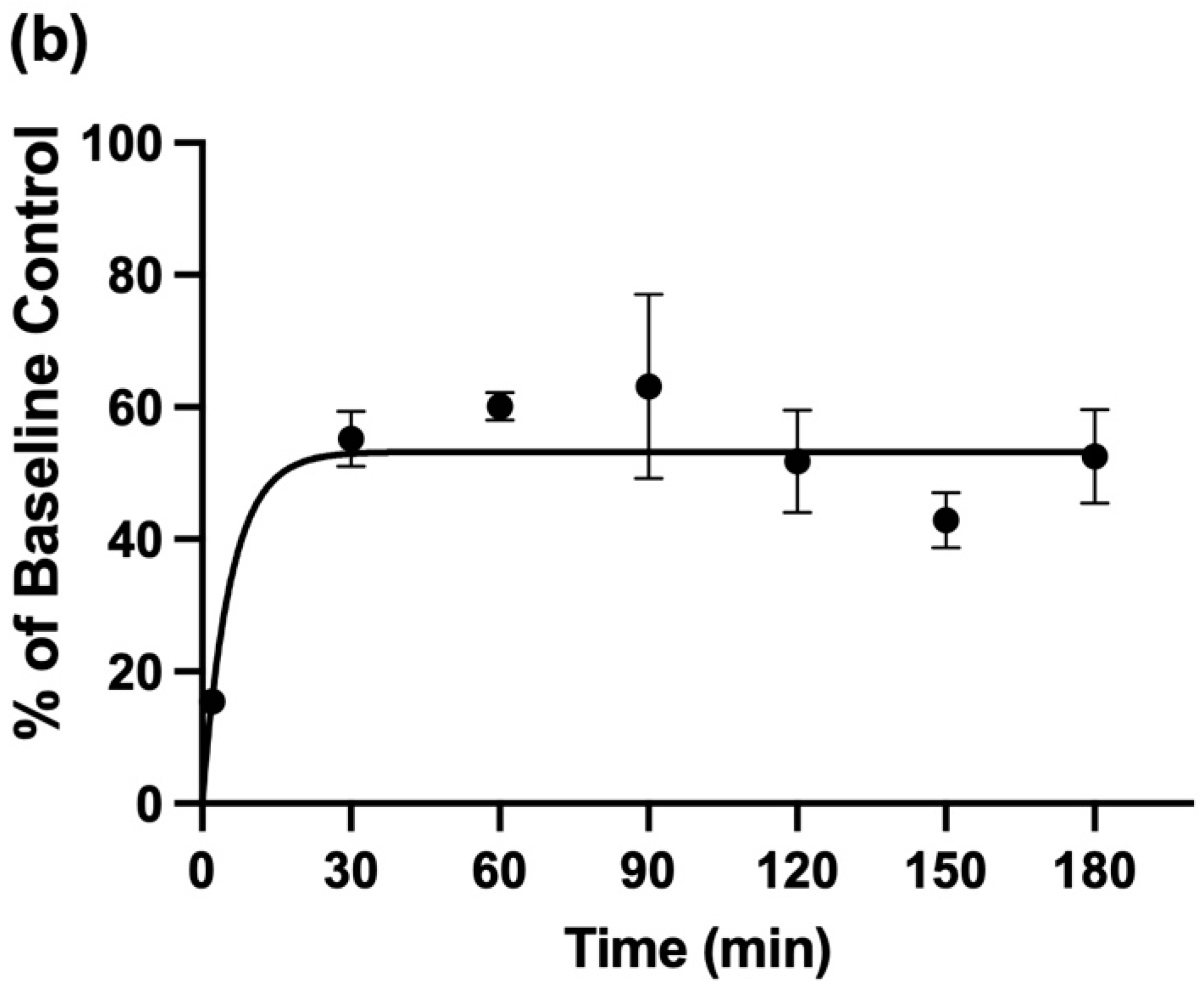
3.6. Attenuation of Salivary NH4+ Concentration by the Pre-Sampling Administration of an Aqueous oral Rinse to Participants, and Its Recovery Therefrom
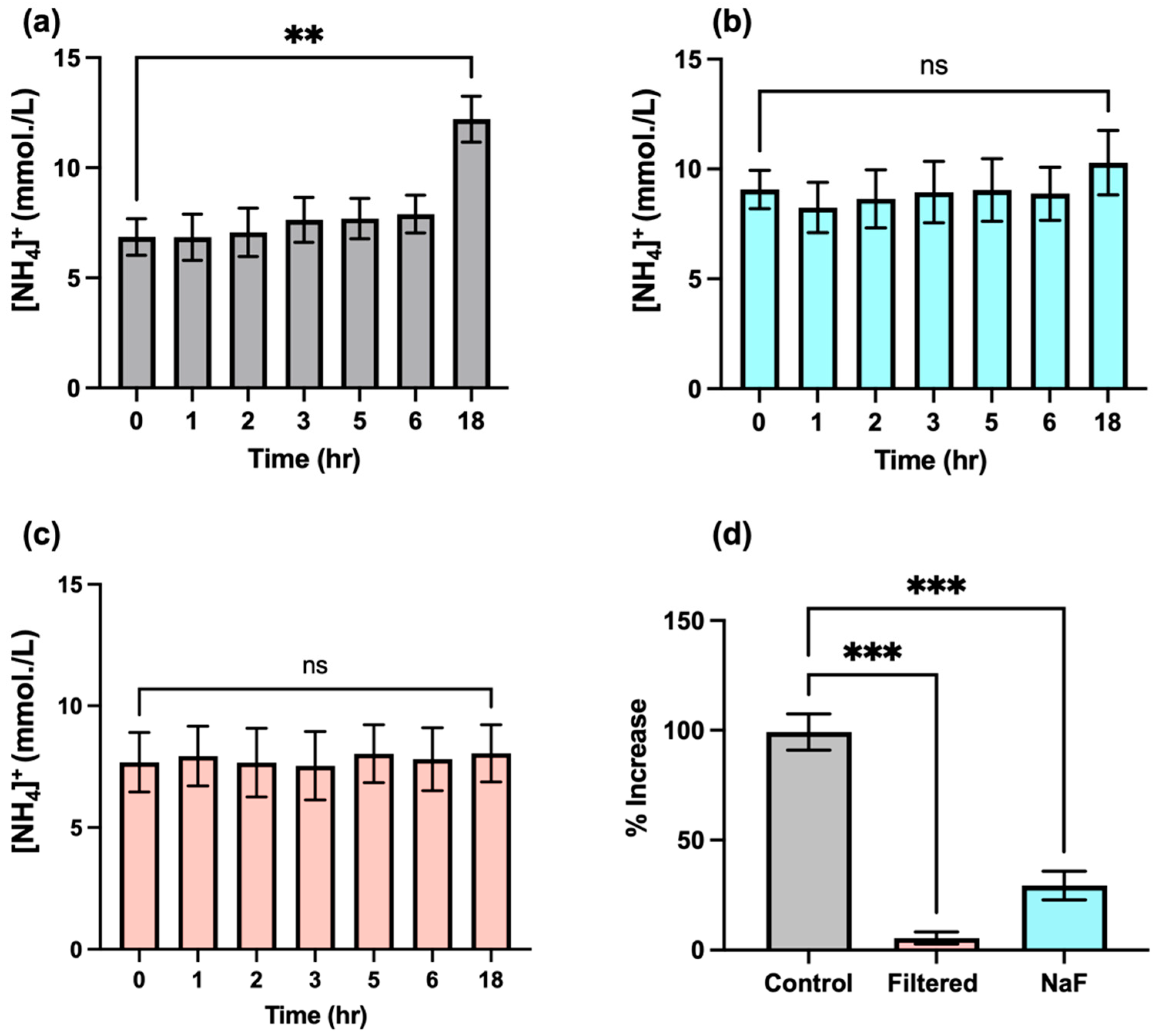
3.7. Time-Dependence of Salivary NH4+ Ion Concentration in WMSS Samples following Sample Collection and Laboratory Processing: Influence and Suppression of Urease Activity
4. Discussion
4.1. Evaluation of the 1H NMR Method Developed for the Determination of Salivary NH4+ Ions and Its Potential Limitations
4.2. Application of 1H NMR Techniques to the Analysis of Alternative, Non-Ammoniacal Biomolecules with Exchangeable Protons
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nascimento, M.M.; Gordan, V.V.; Garvan, C.W.; Browngardt, C.M.; Burne, R.A. Correlations of Oral Bacterial Arginine and Urea Catabolism with Caries Experience. Oral Microbiol. Immunol. 2009, 24, 89–95. [Google Scholar] [CrossRef]
- Griswold, A.R.; Chen, Y.Y.M.; Burne, R.A. Analysis of an Agmatine Deiminase Gene Cluster in Streptococcus Mutans UA159. J. Bacteriol. 2004, 186, 1902–1904. [Google Scholar] [CrossRef]
- Liu, J.; Lkhagva, E.; Chung, H.J.; Kim, H.J.; Hong, S.T. The Pharmabiotic Approach to Treat Hyperammonemia. Nutrients 2018, 10, 140. [Google Scholar] [CrossRef]
- Aldridge, D.R.; Tranah, E.J.; Shawcross, D.L. Pathogenesis of Hepatic Encephalopathy: Role of Ammonia and Systemic Inflammation. J. Clin. Exp. Hepatol. 2015, 5, 7–20. [Google Scholar] [CrossRef]
- Azagra, M.; Pose, E.; De Chiara, F.; Perez, M.; Avitabile, E.; Servitja, J.M.; Brugnara, L.; Azcón, J.; Marco-Rius, I. Ammonium Quantification in Human Plasma by Proton Nuclear Magnetic Resonance for Staging of Liver Fibrosis in Alcohol-Related Liver Disease and Nonalcoholic Fatty Liver Disease. NMR Biomed. 2022, 35, nbm.4745. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic Recycling of Ammonia via Glutamate Dehydrogenase Supports Breast Cancer Biomass. Science 2017, 358, 941–946. [Google Scholar] [CrossRef]
- Kopstein, J.; Wrong, O.M. The Origin and Fate of Salivary Urea and Ammonia in Man. Clin. Sci. Molec. Med. 1977, 52, 9–17. [Google Scholar] [CrossRef]
- Grootveld, M.; Page, G.; Bhogadia, M.; Edgar, M. Updates and Original Case Studies Focused on the NMR-Linked Metabolomics Analysis of Human Oral Fluids Part I: Emerging Platforms and Perspectives. Appl. Sci. 2022, 12, 1235. [Google Scholar] [CrossRef]
- Grootveld, M.; Percival, B.C.; Page, G.; Hunwin, K.; Bhogadia, M.; Chan, W.; Edgar, M. Updates and Original Case Studies Focused on the NMR-Linked Metabolomics Analysis of Human Oral Fluids Part II: Applications to the Diagnosis and Prognostic Monitoring of Oral and Systemic Cancers. Metabolites 2022, 12, 778. [Google Scholar] [CrossRef]
- Grootveld, M.; Page, G.; Bhogadia, M.; Hunwin, K.; Edgar, M. Updates and Original Case Studies Focused on the NMR-Linked Metabolomics Analysis of Human Oral Fluids Part III: Implementations for the Diagnosis of Non-Cancerous Disorders, Both Oral and Systemic. Metabolites 2023, 13, 66. [Google Scholar] [CrossRef]
- Evelyn, R.; Javier, M.; Gustavo, M.; Miguel, N.; Patricia, P.; Valeria, G.; Juan, E.O.; Ismael, Y. Caries-Free Subjects Have High Levels of Urease and Arginine Deiminase Activity. J. Appl. Oral Sci. 2014, 22, 235–240. [Google Scholar] [CrossRef]
- Lasisi, T.J.; Raji, Y.R.; Salako, B.L. Salivary Creatinine and Urea Analysis in Patients with Chronic Kidney Disease: A Case Control Study. BMC Nephrol. 2016, 17, 10. [Google Scholar] [CrossRef]
- Huizenga, J.R.; Tangerman, A.; Gips, C.H. Determination of Ammonia in Biological Fluids. Ann. Clin. Biochem. 1994, 31, 529–543. [Google Scholar] [CrossRef]
- Wang, C.; Sahay, P. Breath Analysis Using Laser Spectroscopic Techniques: Breath Biomarkers, Spectral Fingerprints, and Detection Limits. Sensors 2009, 9, 8230–8262. [Google Scholar] [CrossRef]
- Dumitras, D.C.; Dutu, D.C.; Matei, C.; Cernat, R.; Banita, S.; Patachia, M.; Bratu, A.M.; Petrus, M.; Popa, C. Evaluation of Ammonia Absorption Coefficients by Photoacoustic Spectroscopy for Detection of Ammonia Levels in Human Breath. Laser Phys. 2011, 21, 796–800. [Google Scholar] [CrossRef]
- Hibbard, T.; Killard, A.J. Breath Ammonia Levels in a Normal Human Population Study as Determined by Photoacoustic Laser Spectroscopy. J. Breath Res. 2011, 5, 037101. [Google Scholar] [CrossRef]
- Chan, M.-J.; Li, Y.-J.; Wu, C.-C.; Lee, Y.-C.; Zan, H.-W.; Meng, H.-F.; Hsieh, M.-H.; Lai, C.-S.; Tian, Y.-C. Breath Ammonia Is a Useful Biomarker Predicting Kidney Function in Chronic Kidney Disease Patients. Biomedicines 2020, 8, 468. [Google Scholar] [CrossRef]
- Chen, W.; Laiho, S.; Vaittinen, O.; Halonen, L.; Ortiz, F.; Forsblom, C.; Groop, P.-H.; Lehto, M.; Metsälä, M. Biochemical Pathways of Breath Ammonia (NH3) Generation in Patients with End-stage Renal Disease undergoing Hemodialysis. J. Breath Res. 2016, 10, 036011. [Google Scholar] [CrossRef]
- Chen, C.-C.; Hsieh, J.-C.; Chao, C.-H.; Yang, W.-S.; Cheng, H.-T.; Chan, C.-K.; Lu, C.-J.; Hsin, F.; Meng, H.-F.; Zan, H.-W. Correlation Between Breath Ammonia and Blood Urea Nitrogen Levels in Chronic Kidney Disease and Dialysis Patients. J. Breath Res. 2020, 14, 036002. [Google Scholar] [CrossRef]
- Takaesu, Y.; Suzuki, N.; Naito, M.; Watanabe, T.; Shimazu, A.; Yatabe, N.; Yoneda, M.; Hirofuji, T.; Hanioka, T. Novel Oral Biomarkers Predicting Oral Malodor. Oral Surg. Oral Med. Oral Path. Oral Radiol. 2020, 130, 667–674. [Google Scholar] [CrossRef]
- Preti, G.; Labows, J.N.; Kostelc, J.G.; Aldinger, S.; Daniele, R. Analysis of Lung Air from Patients with Bronchogenic Carcinoma and Controls using Gas Chromatography-Mass Spectrometry. J. Chromatograph. B Biomed. Sci. Appl. 1988, 432, 1–11. [Google Scholar] [CrossRef]
- Das, R.; Bej, S.; Chandra, M.N.; Banerjee, P. Selective Recognition of Ammonia and Aliphatic Amines by C-N Fused Phenazine Derivative: A Hydrogel-Based Smartphone Assisted ‘Opto-Electronic Nose’ for Food Spoilage Evaluation with Potent Anti-Counterfeiting Activity and a Potential Prostate Cancer Biomarker Sensor. Anal. Chim. Acta 2002, 1202, 339597. [Google Scholar] [CrossRef]
- Changsen, B.; Hailong, W.; Dong, D.; Tong, L.; Zhi, Y.; Junfei, G.; Wei, Z.; Ding, L.; Ruochen, Y.; Liyan, W.; et al. Urea as a By-Product of Ammonia Metabolism Can Be a Potential Serum Biomarker of Hepatocellular Carcinoma. Front. Cell Develop. Biol. 2021, 9. Available online: https://www.frontiersin.org/articles/10.3389/fcell.2021.650748 (accessed on 12 April 2023).
- Bell, H.N.; Huber, A.K.; Singhal, R.; Korimerla, N.; Rebernick, R.J.; Kumar, R.; El-derany, M.O.; Sajjakulnukit, P.; Das, N.K.; Kerk, S.A.; et al. Microenvironmental Ammonia Enhances T Cell Exhaustion in Colorectal Cancer. Cell Metab. 2023, 35, 134.e6–149.e6. [Google Scholar] [CrossRef]
- Navaneethan, U.; Spencer, C.; Zhu, X.; Vargo, J.J.; Grove, D.; Dweik, R.A. Volatile Organic Compounds in Bile can Distinguish Pancreatic Cancer from Chronic Pancreatitis: A Prospective Observational Study. Endoscopy 2021, 53, 732–736. [Google Scholar] [CrossRef]
- Silwood, C.; Grootveld, M.; Lynch, E. 1H NMR Investigations of the Molecular Nature of Low-Molecular-Mass Calcium Ions in Biofluids. J. Biol. Inorg. Chem. 2002, 7, 46–57. [Google Scholar] [CrossRef]
- Hodgetts, R.Y.; Kiryutin, A.S.; Nichols, P.; Du, H.L.; Bakker, J.M.; MacFarlane, D.R.; Simonov, A.N. Refining Universal Procedures for Ammonium Quantification via Rapid 1H NMR Analysis for Dinitrogen Reduction Studies. ACS Energy Lett. 2020, 5, 736–741. [Google Scholar] [CrossRef]
- Kolen, M.; Smith, W.A.; Mulder, F.M. Accelerating 1H NMR Detection of Aqueous Ammonia. ACS Omeg. 2021, 6, 5698–5704. [Google Scholar] [CrossRef]
- Preece, N.E.; Cerdan, S. Determining 15N to 14N Ratios in Biofluids by Single-Pulse 1H Nuclear Magnetic Resonance. Anal. Biochem. 1993, 215, 180–183. [Google Scholar] [CrossRef]
- Ross, A.; Schlotterbeck, G.; Dieterle, F.; Senn, H. NMR Spectroscopy Techniques for Application to Metabonomics. In The Handbook of Metabonomics and Metabolomics; Elsevier: Amsterdam, The Netherlands, 2007; pp. 55–112. [Google Scholar]
- Aguilar, J.A.; Kenwright, S.J. Robust NMR Water Signal Suppression for Demanding Analytical Applications. Analyst 2016, 141, 236–242. [Google Scholar] [CrossRef]
- Komarowski, S.; Yu, Q. Ammonium Ion Removal from Wastewater Using Australian Natural Zeolite: Batch Equilibrium and Kinetic Studies. Environ. Technol. 1997, 18, 1085–1097. [Google Scholar] [CrossRef]
- Svane, S.; Sigurdarson, J.J.; Finkenwirth, F.; Eitinger, T.; Karring, H. Inhibition of Urease Activity by Different Compounds Provides Insight into the Modulation and Association of Bacterial Nickel Import and Ureolysis. Sci. Rep. 2020, 10, 8503. [Google Scholar] [CrossRef] [PubMed]
- Thepchuay, Y.; Mesquita, R.B.R.; Nacapricha, D.; Rangel, A.O.S. Micro-PAD Card for Measuring Total Ammonia Nitrogen in Saliva. Anal. Bioanal. Chem. 2020, 412, 3167–3176. [Google Scholar] [CrossRef] [PubMed]
- Huizenga, J.R.; Gips, C.H. Determination of Ammonia in Saliva using Indophenol, an Ammonium Electrode and an Enzymatic Method: A Comparative Investigation. J. Clin. Chem. Clin. Biochem. 1982, 20, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Baliga, S.; Muglikar, S.; Kale, R. Salivary pH: A Diagnostic Biomarker. J. Indian Soc. Periodontol. 2013, 17, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Perrin, C.L.; Dwyer, T.L.; Baine, P. Two-dimensional NMR Exchange Spectroscopy Study of Proton Exchange in Aqueous Ammonium Ion: H/D Primary Kinetic Isotope Effect for Direct Nitrogen-to-Nitrogen Proton Transfer. J. Am. Chem. Soc. 1994, 116, 4044–4049. [Google Scholar] [CrossRef]
- Sanders, J.K.M.; Hunter, B.K.; Jameson, C.J.; Romeo, G. Isotope Effects on Proton Chemical Shifts and Coupling Constants in the Ammonium Ions 15,14N4-nDn+. Chem. Phys. Lett. 1988, 143, 471–476. [Google Scholar]
- Frostell, G. A Colourimetric Screening Test for Evaluation of the Buffer Capacity of Saliva. Swed. Dent. J. 1980, 4, 81–86. [Google Scholar]
- Gergov, M.; Nenonen, T.; Ojanperä, I.; Ketola, R.A. Compensation of Matrix Effects in a Standard Addition Method for Metformin in Postmortem Blood Using Liquid Chromatography-Electrospray-Tandem Mass Spectrometry. J. Anal. Toxicol. 2015, 39, 359–364. [Google Scholar] [CrossRef]
- Thepchuay, Y.; Costa, C.F.A.; Mesquita, R.B.R.; Sampaio-Maia, B.; Nacapricha, D.; Rangel, A.O.S.S. Flow-Based Method for the Determination of Biomarkers Urea and Ammoniacal Nitrogen in Saliva. Bioanalysis 2020, 12, 455–465. [Google Scholar] [CrossRef]
- Sissons, C.H.; Hancock, E.M. Urease Activity in Streptococcus Salivarius at Low pH. Arch. Oral Biol. 1993, 38, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Kroupina, K.; Bémeur, C.; Rose, C.F. Amino acids, Ammonia, and Hepatic Encephalopathy. Anal. Biochem. 2022, 649, 114696. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.J. Microdiffusion Analysis and Volumetric Error, 4th ed.; Crosby Lockwood: London, UK, 1957. [Google Scholar]
- Martínez-Rivera, J.I.; Xibillé-Friedmann, D.X.; González-Christen, J.; de la Garza-Ramos, M.A.; Carrillo-Vázquez, S.M.; Montiel-Hernández, J.L. Salivary Ammonia Levels and Tannerella forsythia are associated with Rheumatoid Arthritis: A Cross Sectional Study. Clin. Exp. Dent. Res. 2017, 3, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Huizenga, J.; Vissink, A.; Kuipers, E.; Gips, C.H. Helicobacter pylori and Ammonia Concentrations of Whole, Parotid and Submandibular/Sublingual Saliva. Clin. Oral Invest. 1999, 3, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Metsala, M.; Vaittinen, O.; Halonen, L. The Origin of Mouth-Exhaled Ammonia. J. Breath Res. 2014, 8, 036003. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Reference | Disorder | Healthy Control Group Mean [NH3] or [NH4+] in mmol/L (±SD, and Range in Brackets, Where Available) | Disease Group Mean [NH3] or [NH4+] in mmol/L (±SD, and Range in Brackets, Where Available) | Analysis Technique/ Method | Sample Collection Procedure |
|---|---|---|---|---|---|
| [7] | Uraemia | 3.8 * | 13.6 * | Enzyme-based microdiffusion method described by Conway [44]. | Participants chewed gum and rinsed their mouths 10 min prior to sample collection. |
| [41] | CKD | 7.7 | 32.9 | Flow-based NH3-selective electrode. | Saliva samples were collected at least 1 h after eating and tooth-brushing were performed. Mouth rinsing was performed at an unspecified time point prior to sample collection. |
| [45] | Rheumatoid arthritis (RA) | 1.93 ± 0.48 ** | 3.61 ± 0.76 ** (high disease activity group only) | Enzymatic kinetic/ colourimetric assay. | None specified. |
| [46] | Dyspepsia | 2.57 ± 1.64 | 2.49 ± 1.26 | Indophenol spectrophotometric method. | Saliva was collected 5 min after participants rinsed their mouths with a water-based oral solution. Saliva was stimulated via a paraffin pellet-chewing episode. |
| [20] | Oral malodour | 63 (55–74) *** | Physiological oral malodour: 79.5 (73.8–84.3) *** Periodontal disease oral malodour: 76.5 (68.5–84.8) *** | Flow-based NH3-selective electrode. | Each participant was instructed to rinse the oral cavity with 3 mL of distilled water for a period of 10 s before sample collection. |
| [18] | End-stage renal disease patients undergoing haemodialysis | n/a | 3.5 ± 0.3 | Indophenol spectrophotometric method. | Saliva samples were stimulated via chewing a paraffin pellet prior to collection. |
| [47] | Healthy control only | 2.2 ± 0.2 | n/a | Indophenol spectrophotometric method. | Samples were collected ≥ 2 h after a participant’s last meal. Stimulated samples were collected via chewing a plastic paraffin film for 1 min |
| [34] | Healthy control only | 2.48 | n/a | Micro-PAD card technique. | Samples were collected at least 1 h after an oral mouth-rinse was performed with water following a meal. |
| [35] | Healthy control only | 4.40 (1.1–12.1) | n/a | Indophenol spectrophotometric method. | Sample collection was performed following use of chewing gum, and thorough mouth-rinsing with water for an unspecified period of time. |
| n/a | Current study | 11.4 ± 4.5 | n/a | 1H NMR analysis. | As described herein. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhogadia, M.; Edgar, M.; Hunwin, K.; Page, G.; Grootveld, M. Detection and Quantification of Ammonia as the Ammonium Cation in Human Saliva by 1H NMR: A Promising Probe for Health Status Monitoring, with Special Reference to Cancer. Metabolites 2023, 13, 792. https://doi.org/10.3390/metabo13070792
Bhogadia M, Edgar M, Hunwin K, Page G, Grootveld M. Detection and Quantification of Ammonia as the Ammonium Cation in Human Saliva by 1H NMR: A Promising Probe for Health Status Monitoring, with Special Reference to Cancer. Metabolites. 2023; 13(7):792. https://doi.org/10.3390/metabo13070792
Chicago/Turabian StyleBhogadia, Mohammed, Mark Edgar, Kayleigh Hunwin, Georgina Page, and Martin Grootveld. 2023. "Detection and Quantification of Ammonia as the Ammonium Cation in Human Saliva by 1H NMR: A Promising Probe for Health Status Monitoring, with Special Reference to Cancer" Metabolites 13, no. 7: 792. https://doi.org/10.3390/metabo13070792
APA StyleBhogadia, M., Edgar, M., Hunwin, K., Page, G., & Grootveld, M. (2023). Detection and Quantification of Ammonia as the Ammonium Cation in Human Saliva by 1H NMR: A Promising Probe for Health Status Monitoring, with Special Reference to Cancer. Metabolites, 13(7), 792. https://doi.org/10.3390/metabo13070792