
Inborn Errors of Purine Salvage and Catabolism

 , , , , ,
, , , , ,
Abstract
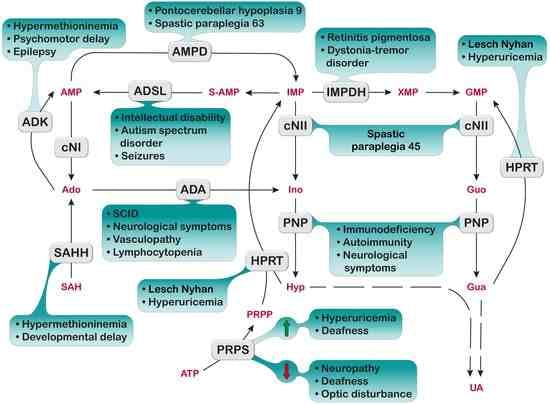
1. Introduction
2. Phosphoribosylpyrophosphate Synthetase
2.1. PRPS Superactivity
2.1.1. Diagnosis and Treatment
2.1.2. Proposed Mechanistic Basis
2.2. PRPS Deficiency
2.2.1. Diagnosis and Treatment
2.2.2. Proposed Mechanistic Basis
3. Hypoxanthine-Guanine Phosphoribosyltransferase
3.1. Diagnosis and Treatment
3.2. Proposed Mechanistic Basis
4. Adenylosuccinate Lyase (ADSL)
4.1. Diagnosis and Treatment
4.2. Proposed Mechanistic Basis
5. Inosine 5′-Monophosphate Dehydrogenase
5.1. Diagnosis and Treatment
5.2. Proposed Mechanistic Basis
6. 5′-Nucleotidases
6.1. Ectosolic 5′-Nucleotidase
Diagnosis and Treatment
6.2. Cytosolic 5′-Nucleotidase I
6.3. Cytosolic 5′-Nucleotidase II
6.3.1. Diagnosis and Treatment
6.3.2. Proposed Mechanistic Basis
7. Adenosine Monophosphate Deaminase
7.1. AMPD1 Deficiency
7.1.1. Diagnosis and Treatment
7.1.2. Proposed Mechanistic Basis
7.2. AMPD2 Deficiency
7.2.1. Diagnosis and Treatment
7.2.2. Proposed Mechanistic Basis
8. Adenosine Kinase
8.1. Diagnosis and Treatment
8.2. Proposed Mechanistic Basis
9. Adenosine Deaminase
9.1. ADA1 Deficiency
9.1.1. Diagnosis and Treatment
9.1.2. Proposed Mechanistic Basis
9.2. ADA2 Deficiency
9.2.1. Diagnosis and Treatment
9.2.2. Proposed Mechanistic Basis
10. S-Adenosylhomocysteine Hydrolase
10.1. Diagnosis and Treatment
10.2. Proposed Mechanistic Basis
11. Purine Nucleoside Phosphorylase
11.1. Diagnosis and Treatment
11.2. Proposed Mechanistic Basis
12. Discussion
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pedley, A.M.; Benkovic, S.J. A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci. 2017, 42, 141–154. [Google Scholar] [CrossRef]
- D’Mello, J.P. Utilization of dietary purines and pyrimidines by non-ruminant animals. Proc. Nutr. Soc. 1982, 41, 301–308. [Google Scholar] [CrossRef]
- Mohamedali, K.A.; Guicherit, O.M.; Kellems, R.E.; Rudolph, F.B. The highest levels of purine catabolic enzymes in mice are present in the proximal small intestine. J. Biol. Chem. 1993, 268, 23728–23733. [Google Scholar] [CrossRef]
- Giannecchini, M.; Matteucci, M.; Pesi, R.; Sgarrella, F.; Tozzi, M.G.; Camici, M. Uptake and utilization of nucleosides for energy repletion. Int. J. Biochem. Cell Biol. 2005, 37, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Fini, M.A.; Elias, A.; Johnson, R.J.; Wright, R.M. Contribution of uric acid to cancer risk, recurrence, and mortality. Clin. Transl. Med. 2012, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Allegrini, S.; Garcia-Gil, M.; Pesi, R.; Camici, M.; Tozzi, M.G. The Good, the Bad and the New about Uric Acid in Cancer. Cancers 2022, 14, 4959. [Google Scholar] [CrossRef] [PubMed]
- Jasinge, E.; Kularatnam, G.A.M.; Dilanthi, H.W.; Vidanapathirana, D.M.; Jayasena, K.; Chandrasiri, N.; Indika, N.L.R.; Ratnayake, P.D.; Gunasekara, V.N.; Fairbanks, L.D.; et al. Uric acid, an important screening tool to detect inborn errors of metabolism: A case series. BMC Res. Notes 2017, 10, 454. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Tylki-Szymanska, A. Inborn errors of purine and pyrimidine metabolism: A guide to diagnosis. Mol. Genet. Metab. 2022, 136, 164–176. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of purine metabolism: Clinical update and therapies. J. Inherit. Metab. Dis. 2014, 37, 669–686. [Google Scholar] [CrossRef]
- Camici, M.; Micheli, V.; Ipata, P.L.; Tozzi, M.G. Pediatric neurological syndromes and inborn errors of purine metabolism. Neurochem. Int. 2010, 56, 367–378. [Google Scholar] [CrossRef]
- Micheli, V.; Camici, M.; Tozzi, M.G.; Ipata, P.L.; Sestini, S.; Bertelli, M.; Pompucci, G. Neurological disorders of purine and pyrimidine metabolism. Curr. Top. Med. Chem. 2011, 11, 923–947. [Google Scholar] [CrossRef]
- Sebesta, I.; Stiburkova, B.; Krijt, J. Hereditary xanthinuria is not so rare disorder of purine metabolism. Nucleosides Nucleotides Nucleic Acids 2018, 37, 324–328. [Google Scholar] [CrossRef]
- Sahota, A.S.; Tischfield, J.A.; Kamatani, N.; Anne Simmonds, H. Adenine Phosphoribosyltransferase Deficiency and 2,8-Dihydroxyadenine Lithiasis. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Cohen, A.; Gudas, L.J.; Ammann, A.J.; Staal, G.E.; Martin, D.W., Jr. Deoxyguanosine triphosphate as a possible toxic metabolite in the immunodeficiency associated with purine nucleoside phosphorylase deficiency. J. Clin. Investg. 1978, 61, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.A. Phosphoribosylpyrophosphate synthetase and the regulation of phosphoribosylpyrophosphate production in human cells. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 69, 115–148. [Google Scholar] [CrossRef] [PubMed]
- Hove-Jensen, B.; Andersen, K.R.; Kilstrup, M.; Martinussen, J.; Switzer, R.L.; Willemoes, M. Phosphoribosyl Diphosphate (PRPP): Biosynthesis, Enzymology, Utilization, and Metabolic Significance. Microbiol. Mol. Biol. Rev. 2017, 81, e00040-16. [Google Scholar] [CrossRef]
- Taira, M.; Iizasa, T.; Yamada, K.; Shimada, H.; Tatibana, M. Tissue-differential expression of two distinct genes for phosphoribosyl pyrophosphate synthetase and existence of the testis-specific transcript. Biochim. Biophys. Acta 1989, 1007, 203–208. [Google Scholar] [CrossRef]
- Sperling, O.; Persky-Brosh, S.; Boer, P.; De Vries, A. Human erythrocyte phosphoribosylpyrophosphate synthetase mutationally altered in regulatory properties. Biochem. Med. 1973, 7, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Begovich, K.; Yelon, D.; Wilhelm, J.E. PRPS polymerization influences lens fiber organization in zebrafish. Dev. Dyn. 2020, 249, 1018–1031. [Google Scholar] [CrossRef] [PubMed]
- de Brouwer, A.P.M.; Christodoulou, J. Phosphoribosylpyrophosphate Synthetase Superactivity. In GeneReviews((R)); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. [Google Scholar]
- Becker, M.A.; Taylor, W.; Smith, P.R.; Ahmed, M. Overexpression of the normal phosphoribosylpyrophosphate synthetase 1 isoform underlies catalytic superactivity of human phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1996, 271, 19894–19899. [Google Scholar] [CrossRef]
- Ahmed, M.; Taylor, W.; Smith, P.R.; Becker, M.A. Accelerated transcription of PRPS1 in X-linked overactivity of normal human phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1999, 274, 7482–7488. [Google Scholar] [CrossRef]
- Becker, M.A.; Puig, J.G.; Mateos, F.A.; Jimenez, M.L.; Kim, M.; Simmonds, H.A. Inherited superactivity of phosphoribosylpyrophosphate synthetase: Association of uric acid overproduction and sensorineural deafness. Am. J. Med. 1988, 85, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Zikanova, M.; Wahezi, D.; Hay, A.; Stiburkova, B.; Pitts, C., 3rd; Musalkova, D.; Skopova, V.; Baresova, V.; Souckova, O.; Hodanova, K.; et al. Clinical manifestations and molecular aspects of phosphoribosylpyrophosphate synthetase superactivity in females. Rheumatology 2018, 57, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Micheli, V.; Simmonds, H.A.; Ricci, C. Regulation of nicotinamide-adenine dinucleotide synthesis in erythrocytes of patients with hypoxanthine-guanine phosphoribosyltransferase deficiency and a patient with phosphoribosylpyrophosphate synthetase superactivity. Clin. Sci. 1990, 78, 239–245. [Google Scholar] [CrossRef] [PubMed]
- de Brouwer, A.P.; van Bokhoven, H.; Nabuurs, S.B.; Arts, W.F.; Christodoulou, J.; Duley, J. PRPS1 mutations: Four distinct syndromes and potential treatment. Am. J. Hum. Genet. 2010, 86, 506–518. [Google Scholar] [CrossRef]
- Simmonds, H.A.; Fairbanks, L.D.; Morris, G.S.; Webster, D.R.; Harley, E.H. Altered erythrocyte nucleotide patterns are characteristic of inherited disorders of purine or pyrimidine metabolism. Clin. Chim. Acta 1988, 171, 197–210. [Google Scholar] [CrossRef]
- Micheli, V.; Simmonds, H.A.; Bari, M.; Pompucci, G. HPLC determination of oxidized and reduced pyridine coenzymes in human erythrocytes. Clin. Chim. Acta 1993, 220, 1–17. [Google Scholar] [CrossRef]
- Delos Santos, K.; Kwon, E.; Moon, N.S. PRPS-Associated Disorders and the Drosophila Model of Arts Syndrome. Int. J. Mol. Sci. 2020, 21, 4824. [Google Scholar] [CrossRef]
- Almoguera, B.; He, S.; Corton, M.; Fernandez-San Jose, P.; Blanco-Kelly, F.; Lopez-Molina, M.I.; Garcia-Sandoval, B.; Del Val, J.; Guo, Y.; Tian, L.; et al. Expanding the phenotype of PRPS1 syndromes in females: Neuropathy, hearing loss and retinopathy. Orphanet J. Rare Dis. 2014, 9, 190. [Google Scholar] [CrossRef]
- Ugbogu, E.A.; Schweizer, L.M.; Schweizer, M. Contribution of Model Organisms to Investigating the Far-Reaching Consequences of PRPP Metabolism on Human Health and Well-Being. Cells 2022, 11, 1909. [Google Scholar] [CrossRef]
- Kim, H.J.; Sohn, K.M.; Shy, M.E.; Krajewski, K.M.; Hwang, M.; Park, J.H.; Jang, S.Y.; Won, H.H.; Choi, B.O.; Hong, S.H.; et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5). Am. J. Hum. Genet. 2007, 81, 552–558. [Google Scholar] [CrossRef]
- de Brouwer, A.P.; Williams, K.L.; Duley, J.A.; van Kuilenburg, A.B.; Nabuurs, S.B.; Egmont-Petersen, M.; Lugtenberg, D.; Zoetekouw, L.; Banning, M.J.; Roeffen, M.; et al. Arts syndrome is caused by loss-of-function mutations in PRPS1. Am. J. Hum. Genet. 2007, 81, 507–518. [Google Scholar] [CrossRef]
- Lenherr, N.; Christodoulou, J.; Duley, J.; Dobritzsch, D.; Fairbanks, L.; Datta, A.N.; Filges, I.; Gurtler, N.; Roelofsen, J.; van Kuilenburg, A.B.P.; et al. Co-therapy with S-adenosylmethionine and nicotinamide riboside improves t-cell survival and function in Arts Syndrome (PRPS1 deficiency). Mol. Genet. Metab. Rep. 2021, 26, 100709. [Google Scholar] [CrossRef]
- Pei, W.; Xu, L.; Varshney, G.K.; Carrington, B.; Bishop, K.; Jones, M.; Huang, S.C.; Idol, J.; Pretorius, P.R.; Beirl, A.; et al. Additive reductions in zebrafish PRPS1 activity result in a spectrum of deficiencies modeling several human PRPS1-associated diseases. Sci. Rep. 2016, 6, 29946. [Google Scholar] [CrossRef]
- DeSmidt, A.A.; Zou, B.; Grati, M.; Yan, D.; Mittal, R.; Yao, Q.; Richmond, M.T.; Denyer, S.; Liu, X.Z.; Lu, Z. Zebrafish Model for Nonsyndromic X-Linked Sensorineural Deafness, DFNX1. Anat. Rec. 2020, 303, 544–555. [Google Scholar] [CrossRef]
- Jinnah, H.A.; Friedmann, T. Lesch-Nyhan Disease and Its Variants. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Adams, A.; Harkness, R.A. Developmental changes in purine phosphoribosyltransferases in human and rat tissues. Biochem. J. 1976, 160, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.V.; Naviaux, R.K.; Paik, K.K.; Nyhan, W.L. Lesch-Nyhan syndrome: mRNA expression of HPRT in patients with enzyme proven deficiency of HPRT and normal HPRT coding region of the DNA. Mol. Genet. Metab. 2012, 106, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Sutcliffe, D.; Zhao, H.; Huang, X.; Schretlen, D.J.; Benkovic, S.; Jinnah, H.A. Clinical severity in Lesch-Nyhan disease: The role of residual enzyme and compensatory pathways. Mol. Genet. Metab. 2015, 114, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Sebesta, I.; Stiburkova, B.; Dvorakova, L.; Hrebicek, M.; Minks, J.; Stolnaja, L.; Vernerova, Z.; Rychlik, I. Unusual presentation of Kelley-Seegmiller syndrome. Nucleosides Nucleotides Nucleic Acids 2008, 27, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Bozano, A.; Schiaffino, A.; Spessa, A.; Valeriani, F.; Mancinelli, R.; Micheli, V.; Dolcetta, D. Description of the Lesch-Nyhan neurobehavioral disorder and its management through participant observation of three young individuals. JIMD Rep. 2020, 52, 63–71. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ceballos-Picot, I.; Auge, F.; Fu, R.; Olivier-Bandini, A.; Cahu, J.; Chabrol, B.; Aral, B.; de Martinville, B.; Lecain, J.P.; Jinnah, H.A. Phenotypic variation among seven members of one family with deficiency of hypoxanthine-guanine phosphoribosyltransferase. Mol. Genet. Metab. 2013, 110, 268–274. [Google Scholar] [CrossRef][Green Version]
- Cakmakli, H.F.; Torres, R.J.; Menendez, A.; Yalcin-Cakmakli, G.; Porter, C.C.; Puig, J.G.; Jinnah, H.A. Macrocytic anemia in Lesch-Nyhan disease and its variants. Genet. Med. 2019, 21, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.J.; Prior, C.; Puig, J.G. Efficacy and safety of allopurinol in patients with hypoxanthine-guanine phosphoribosyltransferase deficiency. Metabolism 2007, 56, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Ronda, L.; Marchetti, M.; Piano, R.; Liuzzi, A.; Corsini, R.; Percudani, R.; Bettati, S. A Trivalent Enzymatic System for Uricolytic Therapy of HPRT Deficiency and Lesch-Nyhan Disease. Pharm. Res. 2017, 34, 1477–1490. [Google Scholar] [CrossRef] [PubMed]
- Jacomelli, G.; Baldini, E.; Mugnaini, C.; Micheli, V.; Bernardini, G.; Santucci, A. Inhibiting PNP for the therapy of hyperuricemia in Lesch-Nyhan disease: Preliminary in vitro studies with analogues of immucillin-G. J. Inherit. Metab. Dis. 2019, 42, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Visser, J.E.; Schretlen, D.J.; Bloem, B.R.; Jinnah, H.A. Levodopa is not a useful treatment for Lesch-Nyhan disease. Mov. Disord. 2011, 26, 746–749. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, M.; Piccinini, L.; Gallo, M.; Motta, F.; Radice, S.; Clementi, E. Treatment of motor and behavioural symptoms in three Lesch-Nyhan patients with intrathecal baclofen. Orphanet J. Rare Dis. 2014, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Piedimonte, F.; Andreani, J.C.; Piedimonte, L.; Micheli, F.; Graff, P.; Bacaro, V. Remarkable clinical improvement with bilateral globus pallidus internus deep brain stimulation in a case of Lesch-Nyhan disease: Five-year follow-up. Neuromodulation 2015, 18, 118–122. [Google Scholar] [CrossRef]
- Pesi, R.; Micheli, V.; Jacomelli, G.; Peruzzi, L.; Camici, M.; Garcia-Gil, M.; Allegrini, S.; Tozzi, M.G. Cytosolic 5′-nucleotidase hyperactivity in erythrocytes of Lesch-Nyhan syndrome patients. Neuroreport 2000, 11, 1827–1831. [Google Scholar] [CrossRef]
- Del Bene, V.A.; Crawford, J.L.; Gomez-Gastiasoro, A.; Vannorsdall, T.D.; Buchholz, A.; Ojeda, N.; Harris, J.C.; Jinnah, H.A.; Schretlen, D.J. Microstructural white matter abnormalities in Lesch-Nyhan disease. Eur. J. Neurosci. 2022, 55, 264–276. [Google Scholar] [CrossRef]
- Micheli, V.; Jacomelli, G.; Santucci, A.; Bernardini, G. Animal and cell models for Lesch-Nyhan syndrome. Drug Discov. Today Dis. Model. 2020, 31, 45–57. [Google Scholar] [CrossRef]
- Micheli, V.; Gathof, B.S.; Rocchigiani, M.; Jacomelli, G.; Sestini, S.; Peruzzi, L.; Notarantonio, L.; Cerboni, B.; Hayek, G.; Pompucci, G. Biochemical and molecular study of mentally retarded patient with partial deficiency of hypoxanthine-guanine phosphoribosyltransferase. Biochim. Biophys. Acta 2002, 1587, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Sestini, S.; Pescaglini, M.; Magagnoli, C.; Jacomelli, G.; Rocchigiani, M.; Simmonds, H.A. NAD synthesis in human erythrocytes: Study of adenylyl transferase activities in patients bearing purine enzyme disorders. Adv. Exp. Med. Biol. 1991, 309B, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.M.; Outtrim, E.L.; Fu, R.; Sutcliffe, D.J.; Torres, R.J.; Jinnah, H.A. Physiological levels of folic acid reveal purine alterations in Lesch-Nyhan disease. Proc. Natl. Acad. Sci. USA 2020, 117, 12071–12079. [Google Scholar] [CrossRef]
- Fairbanks, L.D.; Jacomelli, G.; Micheli, V.; Slade, T.; Simmonds, H.A. Severe pyridine nucleotide depletion in fibroblasts from Lesch-Nyhan patients. Biochem. J. 2002, 366, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Micheli, V.; Jacomelli, G.; Di Marcello, F.; Notarantonio, L.; Sestini, S.; Cerboni, B.; Bertelli, M.; Pompucci, G.; Jinnah, H.A. NAD metabolism in HPRT-deficient mice. Metab. Brain Dis. 2009, 24, 311–319. [Google Scholar] [CrossRef]
- Guibinga, G.H.; Hsu, S.; Friedmann, T. Deficiency of the housekeeping gene hypoxanthine-guanine phosphoribosyltransferase (HPRT) dysregulates neurogenesis. Mol. Ther. 2010, 18, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Picot, I.; Mockel, L.; Potier, M.C.; Dauphinot, L.; Shirley, T.L.; Torero-Ibad, R.; Fuchs, J.; Jinnah, H.A. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: Implications for Lesch-Nyhan disease pathogenesis. Hum. Mol. Genet. 2009, 18, 2317–2327. [Google Scholar] [CrossRef]
- Cristini, S.; Navone, S.; Canzi, L.; Acerbi, F.; Ciusani, E.; Hladnik, U.; de Gemmis, P.; Alessandri, G.; Colombo, A.; Parati, E.; et al. Human neural stem cells: A model system for the study of Lesch-Nyhan disease neurological aspects. Hum. Mol. Genet. 2010, 19, 1939–1950. [Google Scholar] [CrossRef]
- Gottle, M.; Prudente, C.N.; Fu, R.; Sutcliffe, D.; Pang, H.; Cooper, D.; Veledar, E.; Glass, J.D.; Gearing, M.; Visser, J.E.; et al. Loss of dopamine phenotype among midbrain neurons in Lesch-Nyhan disease. Ann. Neurol. 2014, 76, 95–107. [Google Scholar] [CrossRef]
- Witteveen, J.S.; Loopstok, S.R.; Ballesteros, L.L.; Boonstra, A.; van Bakel, N.H.M.; van Boekel, W.H.P.; Martens, G.J.M.; Visser, J.E.; Kolk, S.M. HGprt deficiency disrupts dopaminergic circuit development in a genetic mouse model of Lesch-Nyhan disease. Cell. Mol. Life Sci. 2022, 79, 341. [Google Scholar] [CrossRef]
- Bertelli, M.; Cecchin, S.; Lapucci, C.; Jacomelli, G.; Jinnah, H.A.; Pandolfo, M.; Micheli, V. Study of the adenosinergic system in the brain of HPRT knockout mouse (Lesch-Nyhan disease). Clin. Chim. Acta 2006, 373, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, M.; Alushi, B.; Veicsteinas, A.; Jinnah, H.A.; Micheli, V. Gene expression and mRNA editing of serotonin receptor 2C in brains of HPRT gene knock-out mice, an animal model of Lesch-Nyhan disease. J. Clin. Neurosci. 2009, 16, 1061–1063. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garcia, M.G.; Puig, J.G.; Torres, R.J. Adenosine, dopamine and serotonin receptors imbalance in lymphocytes of Lesch-Nyhan patients. J. Inherit. Metab. Dis. 2012, 35, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.H.; Stacey, N.C.; Connolly, G.P. Hypoxanthine impairs morphogenesis and enhances proliferation of a neuroblastoma model of Lesch Nyhan syndrome. J. Neurosci. Res. 2001, 63, 500–508. [Google Scholar] [CrossRef]
- Bavaresco, C.S.; Chiarani, F.; Wannmacher, C.M.; Netto, C.A.; Wyse, A.T. Intrastriatal hypoxanthine reduces Na+,K+-ATPase activity and induces oxidative stress in the rats. Metab. Brain Dis. 2007, 22, 1–11. [Google Scholar] [CrossRef]
- Biasibetti-Brendler, H.; Schmitz, F.; Pierozan, P.; Zanotto, B.S.; Prezzi, C.A.; de Andrade, R.B.; Wannmacher, C.M.D.; Wyse, A.T.S. Hypoxanthine Induces Neuroenergetic Impairment and Cell Death in Striatum of Young Adult Wistar Rats. Mol. Neurobiol. 2018, 55, 4098–4106. [Google Scholar] [CrossRef]
- Kinast, L.; von der Ohe, J.; Burhenne, H.; Seifert, R. Impairment of adenylyl cyclase 2 function and expression in hypoxanthine phosphoribosyltransferase-deficient rat B103 neuroblastoma cells as model for Lesch-Nyhan disease: BODIPY-forskolin as pharmacological tool. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 671–683. [Google Scholar] [CrossRef]
- Messina, E.; Micheli, V.; Giacomello, A. Guanine nucleotide depletion induces differentiation and aberrant neurite outgrowth in human dopaminergic neuroblastoma lines: A model for basal ganglia dysfunction in Lesch-Nyhan disease. Neurosci. Lett. 2005, 375, 97–100. [Google Scholar] [CrossRef]
- Hyland, K.; Kasim, S.; Egami, K.; Arnold, L.A.; Jinnah, H.A. Tetrahydrobiopterin deficiency and dopamine loss in a genetic mouse model of Lesch-Nyhan disease. J. Inherit. Metab. Dis. 2004, 27, 165–178. [Google Scholar] [CrossRef]
- Kang, T.H.; Park, Y.; Bader, J.S.; Friedmann, T. The housekeeping gene hypoxanthine guanine phosphoribosyltransferase (HPRT) regulates multiple developmental and metabolic pathways of murine embryonic stem cell neuronal differentiation. PLoS ONE 2013, 8, e74967. [Google Scholar] [CrossRef]
- Dinasarapu, A.R.; Sutcliffe, D.J.; Seifar, F.; Visser, J.E.; Jinnah, H.A. Abnormalities of neural stem cells in Lesch-Nyhan disease. J. Neurogenet. 2022, 36, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Guibinga, G.H.; Jinnah, H.A.; Friedmann, T. HPRT deficiency coordinately dysregulates canonical Wnt and presenilin-1 signaling: A neuro-developmental regulatory role for a housekeeping gene? PLoS ONE 2011, 6, e16572. [Google Scholar] [CrossRef]
- Torres, R.J.; Puig, J.G. Hypoxanthine deregulates genes involved in early neuronal development. Implications in Lesch-Nyhan disease pathogenesis. J. Inherit. Metab. Dis. 2015, 38, 1109–1118. [Google Scholar] [CrossRef]
- Bell, S.; McCarty, V.; Peng, H.; Jefri, M.; Hettige, N.; Antonyan, L.; Crapper, L.; O’Leary, L.A.; Zhang, X.; Zhang, Y.; et al. Lesch-Nyhan disease causes impaired energy metabolism and reduced developmental potential in midbrain dopaminergic cells. Stem Cell Rep. 2021, 16, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Guibinga, G.H.; Murray, F.; Barron, N. HPRT-deficiency dysregulates cAMP-PKA signaling and phosphodiesterase 10A expression: Mechanistic insight and potential target for Lesch-Nyhan Disease? PLoS ONE 2013, 8, e63333. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, L.; Kim, J.E.; Miyanohara, A.; Kang, T.H.; Friedmann, T. Purinergic signaling in human pluripotent stem cells is regulated by the housekeeping gene encoding hypoxanthine guanine phosphoribosyltransferase. Proc. Natl. Acad. Sci. USA 2012, 109, 3377–3382. [Google Scholar] [CrossRef]
- Nguyen, K.V. Potential molecular link between the beta-amyloid precursor protein (APP) and hypoxanthine-guanine phosphoribosyltransferase (HGprt) enzyme in Lesch-Nyhan disease and cancer. AIMS Neurosci. 2021, 8, 548–557. [Google Scholar] [CrossRef]
- Jaeken, J.; Van den Berghe, G. An infantile autistic syndrome characterised by the presence of succinylpurines in body fluids. Lancet 1984, 2, 1058–1061. [Google Scholar]
- Banerjee, A.; Bhatia, V.; Didwal, G.; Singh, A.K.; Saini, A.G. ADSL Deficiency—The Lesser-Known Metabolic Epilepsy in Infancy. Indian J. Pediatr. 2021, 88, 263–265. [Google Scholar] [CrossRef]
- Sitaram, S.; Banka, H.C.; Vassallo, G.; Pavaine, J.; Fairclough, A.; Wright, R.; Fairbanks, L.; Bierau, J.; Bowden, L.; Schwahn, B.; et al. Anticipatory banking of samples enables diagnosis of adenylosuccinase deficiency following molecular autopsy in an infant with vacuolating leukoencephalopathy. Am. J. Med. Genet. A 2023, 191, 234–237. [Google Scholar] [CrossRef]
- Wang, X.C.; Wang, T.; Liu, R.H.; Jiang, Y.; Chen, D.D.; Wang, X.Y.; Kong, Q.X. Child with adenylosuccinate lyase deficiency caused by a novel complex heterozygous mutation in the ADSL gene: A case report. World J. Clin. Cases 2022, 10, 11082–11089. [Google Scholar] [CrossRef]
- Mastrogiorgio, G.; Macchiaiolo, M.; Buonuomo, P.S.; Bellacchio, E.; Bordi, M.; Vecchio, D.; Brown, K.P.; Watson, N.K.; Contardi, B.; Cecconi, F.; et al. Clinical and molecular characterization of patients with adenylosuccinate lyase deficiency. Orphanet J. Rare Dis. 2021, 16, 112. [Google Scholar] [CrossRef]
- Jurecka, A.; Zikanova, M.; Kmoch, S.; Tylki-Szymanska, A. Adenylosuccinate lyase deficiency. J. Inherit. Metab. Dis. 2015, 38, 231–242. [Google Scholar] [CrossRef]
- Salerno, C.; Crifo, C.; Giardini, O. Adenylosuccinase deficiency: A patient with impaired erythrocyte activity and anomalous response to intravenous fructose. J. Inherit. Metab. Dis. 1995, 18, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Mouchegh, K.; Zikanova, M.; Hoffmann, G.F.; Kretzschmar, B.; Kuhn, T.; Mildenberger, E.; Stoltenburg-Didinger, G.; Krijt, J.; Dvorakova, L.; Honzik, T.; et al. Lethal fetal and early neonatal presentation of adenylosuccinate lyase deficiency: Observation of 6 patients in 4 families. J. Pediatr. 2007, 150, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Li, K.; Tang, B.; Luo, Y.; Ding, D.; Zhao, Y.; Wang, C.; Zhou, X.; Liu, Z.; Zhang, Y.; et al. Novel mutations in ADSL for Adenylosuccinate Lyase Deficiency identified by the combination of Trio-WES and constantly updated guidelines. Sci. Rep. 2017, 7, 1625. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Jurkiewicz, E.; Tylki-Szymanska, A. Magnetic resonance imaging of the brain in adenylosuccinate lyase deficiency: A report of seven cases and a review of the literature. Eur. J. Pediatr. 2012, 171, 131–138. [Google Scholar] [CrossRef]
- Zikanova, M.; Skopova, V.; Hnizda, A.; Krijt, J.; Kmoch, S. Biochemical and structural analysis of 14 mutant adsl enzyme complexes and correlation to phenotypic heterogeneity of adenylosuccinate lyase deficiency. Hum. Mutat. 2010, 31, 445–455. [Google Scholar] [CrossRef]
- Ray, S.P.; Duval, N.; Wilkinson, T.G., 2nd; Shaheen, S.E.; Ghosh, K.; Patterson, D. Inherent properties of adenylosuccinate lyase could explain S-Ado/SAICAr ratio due to homozygous R426H and R303C mutations. Biochim. Biophys. Acta 2013, 1834, 1545–1553. [Google Scholar] [CrossRef]
- Kmoch, S.; Hartmannova, H.; Stiburkova, B.; Krijt, J.; Zikanova, M.; Sebesta, I. Human adenylosuccinate lyase (ADSL), cloning and characterization of full-length cDNA and its isoform, gene structure and molecular basis for ADSL deficiency in six patients. Hum. Mol. Genet. 2000, 9, 1501–1513. [Google Scholar] [CrossRef]
- Van den Bergh, F.; Vincent, M.F.; Jaeken, J.; Van den Berghe, G. Residual adenylosuccinase activities in fibroblasts of adenylosuccinase-deficient children: Parallel deficiency with adenylosuccinate and succinyl-AICAR in profoundly retarded patients and non-parallel deficiency in a mildly retarded girl. J. Inherit. Metab. Dis. 1993, 16, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Roberts, L.A.; Morris, B.J.; Jones, P.A.; Ogilvy, H.A.; Behan, W.M.; Duley, J.A.; Simmonds, H.A.; Vincent, M.F.; van den Berghe, G. Succinylpurines induce neuronal damage in the rat brain. Adv. Exp. Med. Biol. 1998, 431, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Souckova, O.; Skopova, V.; Baresova, V.; Sedlak, D.; Bleyer, A.J.; Kmoch, S.; Zikanova, M. Metabolites of De Novo Purine Synthesis: Metabolic Regulators and Cytotoxic Compounds. Metabolites 2022, 12, 1210. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, F.; Aronson, N. Ketogenic diet for the treatment of refractory epilepsy in children: A systematic review of efficacy. Pediatrics 2000, 105, E46. [Google Scholar] [CrossRef] [PubMed]
- Shehata, N.I.; Abdelsamad, M.A.; Amin, H.A.A.; Sadik, N.A.H.; Shaheen, A.A. Ameliorating effect of ketogenic diet on acute status epilepticus: Insights into biochemical and histological changes in rat hippocampus. J. Food Biochem. 2022, 46, e14217. [Google Scholar] [CrossRef]
- Jurecka, A.; Opoka-Winiarska, V.; Rokicki, D.; Tylki-Szymanska, A. Neurologic presentation, diagnostics, and therapeutic insights in a severe case of adenylosuccinate lyase deficiency. J. Child Neurol. 2012, 27, 645–649. [Google Scholar] [CrossRef]
- Salerno, C.; D’Eufemia, P.; Finocchiaro, R.; Celli, M.; Spalice, A.; Iannetti, P.; Crifo, C.; Giardini, O. Effect of D-ribose on purine synthesis and neurological symptoms in a patient with adenylosuccinase deficiency. Biochim. Biophys. Acta 1999, 1453, 135–140. [Google Scholar] [CrossRef]
- Jurecka, A.; Tylki-Szymanska, A.; Zikanova, M.; Krijt, J.; Kmoch, S. D-ribose therapy in four Polish patients with adenylosuccinate lyase deficiency: Absence of positive effect. J. Inherit. Metab. Dis. 2008, 31 (Suppl. S2), S329–S332. [Google Scholar] [CrossRef]
- Van den Bergh, F.; Vincent, M.F.; Jaeken, J.; Van den Berghe, G. Functional studies in fibroblasts of adenylosuccinase-deficient children. J. Inherit. Metab. Dis. 1993, 16, 425–434. [Google Scholar] [CrossRef]
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 2008, 320, 103–106. [Google Scholar] [CrossRef]
- Baresova, V.; Skopova, V.; Sikora, J.; Patterson, D.; Sovova, J.; Zikanova, M.; Kmoch, S. Mutations of ATIC and ADSL affect purinosome assembly in cultured skin fibroblasts from patients with AICA-ribosiduria and ADSL deficiency. Hum. Mol. Genet. 2012, 21, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.E.; Tan, I.S.; Lee, Y.S. SAICAR stimulates pyruvate kinase isoform M2 and promotes cancer cell survival in glucose-limited conditions. Science 2012, 338, 1069–1072. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Dutto, I.; Gerhards, J.; Herrera, A.; Souckova, O.; Skopova, V.; Smak, J.A.; Junza, A.; Yanes, O.; Boeckx, C.; Burkhalter, M.D.; et al. Pathway-specific effects of ADSL deficiency on neurodevelopment. elife 2022, 11, e70518. [Google Scholar] [CrossRef]
- Bruce-Gregorios, J.H.; Agarwal, R.P.; Oracion, A.; Ramirez, A.; Lin, L. Effects of methotrexate on RNA and purine synthesis of astrocytes in primary culture. J. Neuropathol. Exp. Neurol. 1991, 50, 770–778. [Google Scholar] [CrossRef]
- Zimmermann, A.G.; Spychala, J.; Mitchell, B.S. Characterization of the human inosine-5′-monophosphate dehydrogenase type II gene. J. Biol. Chem. 1995, 270, 6808–6814. [Google Scholar] [CrossRef]
- Senda, M.; Natsumeda, Y. Tissue-differential expression of two distinct genes for human IMP dehydrogenase (E.C.1.1.1.205). Life Sci. 1994, 54, 1917–1926. [Google Scholar] [CrossRef]
- Gu, J.J.; Tolin, A.K.; Jain, J.; Huang, H.; Santiago, L.; Mitchell, B.S. Targeted disruption of the inosine 5′-monophosphate dehydrogenase type I gene in mice. Mol. Cell. Biol. 2003, 23, 6702–6712. [Google Scholar] [CrossRef]
- Zimmermann, A.; Gu, J.J.; Spychala, J.; Mitchell, B.S. Inosine monophosphate dehydrogenase expression: Transcriptional regulation of the type I and type II genes. Adv. Enzym. Regul. 1996, 36, 75–84. [Google Scholar] [CrossRef]
- Gunter, J.H.; Thomas, E.C.; Lengefeld, N.; Kruger, S.J.; Worton, L.; Gardiner, E.M.; Jones, A.; Barnett, N.L.; Whitehead, J.P. Characterisation of inosine monophosphate dehydrogenase expression during retinal development: Differences between variants and isoforms. Int. J. Biochem. Cell Biol. 2008, 40, 1716–1728. [Google Scholar] [CrossRef]
- Burrell, A.L.; Nie, C.; Said, M.; Simonet, J.C.; Fernandez-Justel, D.; Johnson, M.C.; Quispe, J.; Buey, R.M.; Peterson, J.R.; Kollman, J.M. IMPDH1 retinal variants control filament architecture to tune allosteric regulation. Nat. Struct. Mol. Biol. 2022, 29, 47–58. [Google Scholar] [CrossRef]
- Fernandez-Justel, D.; Nunez, R.; Martin-Benito, J.; Jimeno, D.; Gonzalez-Lopez, A.; Soriano, E.M.; Revuelta, J.L.; Buey, R.M. A Nucleotide-Dependent Conformational Switch Controls the Polymerization of Human IMP Dehydrogenases to Modulate their Catalytic Activity. J. Mol. Biol. 2019, 431, 956–969. [Google Scholar] [CrossRef]
- Burrell, A.L.; Kollman, J.M. IMPDH dysregulation in disease: A mini review. Biochem. Soc. Trans. 2022, 50, 71–82. [Google Scholar] [CrossRef]
- Kennan, A.; Aherne, A.; Palfi, A.; Humphries, M.; McKee, A.; Stitt, A.; Simpson, D.A.; Demtroder, K.; Orntoft, T.; Ayuso, C.; et al. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho−/− mice. Hum. Mol. Genet. 2002, 11, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Bowne, S.J.; Sullivan, L.S.; Blanton, S.H.; Cepko, C.L.; Blackshaw, S.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Daiger, S.P. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2002, 11, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.A.; Farrar, G.J.; Kenna, P.; Humphries, M.M.; Sheils, D.M.; Kumar-Singh, R.; Sharp, E.M.; Soriano, N.; Ayuso, C.; Benitez, J.; et al. Localization of an autosomal dominant retinitis pigmentosa gene to chromosome 7q. Nat. Genet. 1993, 4, 54–58. [Google Scholar] [CrossRef]
- Coussa, R.G.; Chakarova, C.; Ajlan, R.; Taha, M.; Kavalec, C.; Gomolin, J.; Khan, A.; Lopez, I.; Ren, H.; Waseem, N.; et al. Genotype and Phenotype Studies in Autosomal Dominant Retinitis Pigmentosa (adRP) of the French Canadian Founder Population. Investg. Ophthalmol. Vis. Sci. 2015, 56, 8297–8305. [Google Scholar] [CrossRef] [PubMed]
- Zech, M.; Jech, R.; Boesch, S.; Skorvanek, M.; Weber, S.; Wagner, M.; Zhao, C.; Jochim, A.; Necpal, J.; Dincer, Y.; et al. Monogenic variants in dystonia: An exome-wide sequencing study. Lancet Neurol. 2020, 19, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Kuukasjarvi, A.; Landoni, J.C.; Kaukonen, J.; Juhakoski, M.; Auranen, M.; Torkkeli, T.; Velagapudi, V.; Suomalainen, A. IMPDH2: A new gene associated with dominant juvenile-onset dystonia-tremor disorder. Eur. J. Hum. Genet. 2021, 29, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, Q.; Shen, T.; Xiao, X.; Li, S.; Guan, L.; Zhang, J.; Zhu, Z.; Yin, Y.; Wang, P.; et al. Comprehensive mutation analysis by whole-exome sequencing in 41 Chinese families with Leber congenital amaurosis. Investg. Ophthalmol. Vis. Sci. 2013, 54, 4351–4357. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tam, L.C.; Kiang, A.S.; Kennan, A.; Kenna, P.F.; Chadderton, N.; Ader, M.; Palfi, A.; Aherne, A.; Ayuso, C.; Campbell, M.; et al. Therapeutic benefit derived from RNAi-mediated ablation of IMPDH1 transcripts in a murine model of autosomal dominant retinitis pigmentosa (RP10). Hum. Mol. Genet. 2008, 17, 2084–2100. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Nuzbrokh, Y.; Ragi, S.D.; Tsang, S.H. Gene therapy for inherited retinal diseases. Ann. Transl. Med. 2021, 9, 1278. [Google Scholar] [CrossRef] [PubMed]
- Wong-Riley, M.T. Energy metabolism of the visual system. Eye Brain 2010, 2, 99–116. [Google Scholar] [CrossRef]
- Arshavsky, V.Y.; Burns, M.E. Photoreceptor signaling: Supporting vision across a wide range of light intensities. J. Biol. Chem. 2012, 287, 1620–1626. [Google Scholar] [CrossRef]
- Aherne, A.; Kennan, A.; Kenna, P.F.; McNally, N.; Lloyd, D.G.; Alberts, I.L.; Kiang, A.S.; Humphries, M.M.; Ayuso, C.; Engel, P.C.; et al. On the molecular pathology of neurodegeneration in IMPDH1-based retinitis pigmentosa. Hum. Mol. Genet. 2004, 13, 641–650. [Google Scholar] [CrossRef]
- Bowne, S.J.; Sullivan, L.S.; Mortimer, S.E.; Hedstrom, L.; Zhu, J.; Spellicy, C.J.; Gire, A.I.; Hughbanks-Wheaton, D.; Birch, D.G.; Lewis, R.A.; et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Investg. Ophthalmol. Vis. Sci. 2006, 47, 34–42. [Google Scholar] [CrossRef]
- Du, J.; An, J.; Linton, J.D.; Wang, Y.; Hurley, J.B. How Excessive cGMP Impacts Metabolic Proteins in Retinas at the Onset of Degeneration. Adv. Exp. Med. Biol. 2018, 1074, 289–295. [Google Scholar] [CrossRef]
- Mortimer, S.E.; Xu, D.; McGrew, D.; Hamaguchi, N.; Lim, H.C.; Bowne, S.J.; Daiger, S.P.; Hedstrom, L. IMP dehydrogenase type 1 associates with polyribosomes translating rhodopsin mRNA. J. Biol. Chem. 2008, 283, 36354–36360. [Google Scholar] [CrossRef]
- Plana-Bonamaiso, A.; Lopez-Begines, S.; Fernandez-Justel, D.; Junza, A.; Soler-Tapia, A.; Andilla, J.; Loza-Alvarez, P.; Rosa, J.L.; Miralles, E.; Casals, I.; et al. Post-translational regulation of retinal IMPDH1 in vivo to adjust GTP synthesis to illumination conditions. elife 2020, 9, e56418. [Google Scholar] [CrossRef]
- Jinnah, H.A.; Sun, Y.V. Dystonia genes and their biological pathways. Neurobiol. Dis. 2019, 129, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, H.; Ohye, T.; Takahashi, E.; Seki, N.; Hori, T.; Segawa, M.; Nomura, Y.; Endo, K.; Tanaka, H.; Tsuji, S.; et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat. Genet. 1994, 8, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Ipata, P.L. Sheep brain 5′-nucleotidase. Some enzymic properties and allosteric inhibition by nucleoside triphosphates. Biochemistry 1968, 7, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Zebisch, M.; Strater, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef]
- Zimmermann, H. History of ectonucleotidases and their role in purinergic signaling. Biochem. Pharmacol. 2021, 187, 114322. [Google Scholar] [CrossRef]
- Sala-Newby, G.B.; Skladanowski, A.C.; Newby, A.C. The mechanism of adenosine formation in cells. Cloning of cytosolic 5′-nucleotidase-I. J. Biol. Chem. 1999, 274, 17789–17793. [Google Scholar] [CrossRef]
- Tozzi, M.G.; Pesi, R.; Allegrini, S. On the physiological role of cytosolic 5′-nucleotidase II (cN-II): Pathological and therapeutical implications. Curr. Med. Chem. 2013, 20, 4285–4291. [Google Scholar] [CrossRef]
- Page, T.; Yu, A.; Fontanesi, J.; Nyhan, W.L. Developmental disorder associated with increased cellular nucleotidase activity. Proc. Natl. Acad. Sci. USA 1997, 94, 11601–11606. [Google Scholar] [CrossRef]
- Page, T. Metabolic approaches to the treatment of autism spectrum disorders. J. Autism Dev. Disord. 2000, 30, 463–469. [Google Scholar] [CrossRef]
- Pesi, R.; Camici, M.; Micheli, V.; Notarantonio, L.; Jacomelli, G.; Tozzi, M.G. Identification of the nucleotidase responsible for the AMP hydrolysing hyperactivity associated with neurological and developmental disorders. Neurochem. Res. 2008, 33, 59–65. [Google Scholar] [CrossRef]
- St Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Price, T.P.; Sundberg, J.P.; Uitto, J. Juxta-articular joint-capsule mineralization in CD73 deficient mice: Similarities to patients with NT5E mutations. Cell Cycle 2014, 13, 2609–2615. [Google Scholar] [CrossRef] [PubMed]
- Moorhead, W.J., 3rd; Chu, C.C.; Cuevas, R.A.; Callahan, J., 4th; Wong, R.; Regan, C.; Boufford, C.K.; Sur, S.; Liu, M.; Gomez, D.; et al. Dysregulation of FOXO1 (Forkhead Box O1 Protein) Drives Calcification in Arterial Calcification due to Deficiency of CD73 and Is Present in Peripheral Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1680–1694. [Google Scholar] [CrossRef] [PubMed]
- Daniels, G.; Ballif, B.A.; Helias, V.; Saison, C.; Grimsley, S.; Mannessier, L.; Hustinx, H.; Lee, E.; Cartron, J.P.; Peyrard, T.; et al. Lack of the nucleoside transporter ENT1 results in the Augustine-null blood type and ectopic mineralization. Blood 2015, 125, 3651–3654. [Google Scholar] [CrossRef] [PubMed]
- Warraich, S.; Bone, D.B.; Quinonez, D.; Ii, H.; Choi, D.S.; Holdsworth, D.W.; Drangova, M.; Dixon, S.J.; Seguin, C.A.; Hammond, J.R. Loss of equilibrative nucleoside transporter 1 in mice leads to progressive ectopic mineralization of spinal tissues resembling diffuse idiopathic skeletal hyperostosis in humans. J. Bone Miner. Res. 2013, 28, 1135–1149. [Google Scholar] [CrossRef]
- Carluccio, M.; Ziberi, S.; Zuccarini, M.; Giuliani, P.; Caciagli, F.; Di Iorio, P.; Ciccarelli, R. Adult mesenchymal stem cells: Is there a role for purine receptors in their osteogenic differentiation? Purinergic Signal. 2020, 16, 263–287. [Google Scholar] [CrossRef]
- Pike, A.F.; Kramer, N.I.; Blaauboer, B.J.; Seinen, W.; Brands, R. An alkaline phosphatase transport mechanism in the pathogenesis of Alzheimer’s disease and neurodegeneration. Chem. Biol. Interact. 2015, 226, 30–39. [Google Scholar] [CrossRef]
- Nagai, K.; Nagasawa, K.; Fujimoto, S. Transport mechanisms for adenosine and uridine in primary-cultured rat cortical neurons and astrocytes. Biochem. Biophys. Res. Commun. 2005, 334, 1343–1350. [Google Scholar] [CrossRef]
- Newby, A.C. The pigeon heart 5′-nucleotidase responsible for ischaemia-induced adenosine formation. Biochem. J. 1988, 253, 123–130. [Google Scholar] [CrossRef]
- Sala-Newby, G.B.; Freeman, N.V.; Skladanowski, A.C.; Newby, A.C. Distinct roles for recombinant cytosolic 5′-nucleotidase-I and -II in AMP and IMP catabolism in COS-7 and H9c2 rat myoblast cell lines. J. Biol. Chem. 2000, 275, 11666–11671. [Google Scholar] [CrossRef]
- Sala-Newby, G.B.; Freeman, N.V.; Curto, M.A.; Newby, A.C. Metabolic and functional consequences of cytosolic 5′-nucleotidase-IA overexpression in neonatal rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H991–H998. [Google Scholar] [CrossRef]
- Hunsucker, S.A.; Mitchell, B.S.; Spychala, J. The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol. Ther. 2005, 107, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Rietveld, A.; van den Hoogen, L.L.; Bizzaro, N.; Blokland, S.L.M.; Dahnrich, C.; Gottenberg, J.E.; Houen, G.; Johannsen, N.; Mandl, T.; Meyer, A.; et al. Autoantibodies to Cytosolic 5′-Nucleotidase 1A in Primary Sjogren’s Syndrome and Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 1200. [Google Scholar] [CrossRef] [PubMed]
- Amlani, A.; Choi, M.Y.; Tarnopolsky, M.; Brady, L.; Clarke, A.E.; Garcia-De La Torre, I.; Mahler, M.; Schmeling, H.; Barber, C.E.; Jung, M.; et al. Anti-NT5c1A Autoantibodies as Biomarkers in Inclusion Body Myositis. Front. Immunol. 2019, 10, 745. [Google Scholar] [CrossRef] [PubMed]
- Damian, L.; Login, C.C.; Solomon, C.; Belizna, C.; Encica, S.; Urian, L.; Jurcut, C.; Stancu, B.; Vulturar, R. Inclusion Body Myositis and Neoplasia: A Narrative Review. Int. J. Mol. Sci. 2022, 23, 7358. [Google Scholar] [CrossRef] [PubMed]
- Dursun, U.; Koroglu, C.; Kocasoy Orhan, E.; Ugur, S.A.; Tolun, A. Autosomal recessive spastic paraplegia (SPG45) with mental retardation maps to 10q24.3–q25.1. Neurogenetics 2009, 10, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P. Expanding the clinical relevance of the 5′-nucleotidase cN-II/NT5C2. Purinergic Signal. 2018, 14, 321–329. [Google Scholar] [CrossRef]
- Fink, J.K. Hereditary spastic paraplegia: Clinical principles and genetic advances. Semin. Neurol. 2014, 34, 293–305. [Google Scholar] [CrossRef]
- Straussberg, R.; Onoufriadis, A.; Konen, O.; Zouabi, Y.; Cohen, L.; Lee, J.Y.W.; Hsu, C.K.; Simpson, M.A.; McGrath, J.A. Novel homozygous missense mutation in NT5C2 underlying hereditary spastic paraplegia SPG45. Am. J. Med. Genet. A 2017, 173, 3109–3113. [Google Scholar] [CrossRef]
- Naseer, M.I.; Abdulkareem, A.A.; Pushparaj, P.N.; Bibi, F.; Chaudhary, A.G. Exome Analysis Identified Novel Homozygous Splice Site Donor Alteration in NT5C2 Gene in a Saudi Family Associated With Spastic Diplegia Cerebral Palsy, Developmental Delay, and Intellectual Disability. Front. Genet. 2020, 11, 14. [Google Scholar] [CrossRef]
- Cividini, F.; Tozzi, M.G.; Galli, A.; Pesi, R.; Camici, M.; Dumontet, C.; Jordheim, L.P.; Allegrini, S. Cytosolic 5′-nucleotidase II interacts with the leucin rich repeat of NLR family member Ipaf. PLoS ONE 2015, 10, e0121525. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Zhang, T.; Li, L.; Fu, D.; Lin, H.; Chen, G.; Chen, T. Two-stage replication of previous genome-wide association studies of AS3MT-CNNM2-NT5C2 gene cluster region in a large schizophrenia case-control sample from Han Chinese population. Schizophr. Res. 2016, 176, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: A genome-wide analysis. Lancet 2013, 381, 1371–1379. [Google Scholar] [CrossRef]
- Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011, 43, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jiang, J.; Long, J.; Ling, W.; Huang, G.; Guo, X.; Su, L. The rs11191580 variant of the NT5C2 gene is associated with schizophrenia and symptom severity in a South Chinese Han population: Evidence from GWAS. Braz. J. Psychiatry 2017, 39, 104–109. [Google Scholar] [CrossRef]
- Gonzalez, S.; Gupta, J.; Villa, E.; Mallawaarachchi, I.; Rodriguez, M.; Ramirez, M.; Zavala, J.; Armas, R.; Dassori, A.; Contreras, J.; et al. Replication of genome-wide association study (GWAS) susceptibility loci in a Latino bipolar disorder cohort. Bipolar Disord. 2016, 18, 520–527. [Google Scholar] [CrossRef]
- Newton-Cheh, C.; Johnson, T.; Gateva, V.; Tobin, M.D.; Bochud, M.; Coin, L.; Najjar, S.S.; Zhao, J.H.; Heath, S.C.; Eyheramendy, S.; et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat. Genet. 2009, 41, 666–676. [Google Scholar] [CrossRef]
- Zhao, X.C.; Yang, S.H.; Yan, Y.Q.; Zhang, X.; Zhang, L.; Jiao, B.; Jiang, S.; Yu, Z.B. Identification of differential gene expression profile from peripheral blood cells of military pilots with hypertension by RNA sequencing analysis. BMC Med. Genom. 2018, 11, 59. [Google Scholar] [CrossRef]
- Wen, W.; Zheng, W.; Okada, Y.; Takeuchi, F.; Tabara, Y.; Hwang, J.Y.; Dorajoo, R.; Li, H.; Tsai, F.J.; Yang, X.; et al. Meta-analysis of genome-wide association studies in East Asian-ancestry populations identifies four new loci for body mass index. Hum. Mol. Genet. 2014, 23, 5492–5504. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Hauberg, M.E.; Holm-Nielsen, M.H.; Mattheisen, M.; Askou, A.L.; Grove, J.; Borglum, A.D.; Corydon, T.J. Schizophrenia risk variants affecting microRNA function and site-specific regulation of NT5C2 by miR-206. Eur. Neuropsychopharmacol. 2016, 26, 1522–1526. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Karlsson, H.K.; Szekeres, F.; Chibalin, A.V.; Krook, A.; Zierath, J.R. Suppression of 5′-nucleotidase enzymes promotes AMP-activated protein kinase (AMPK) phosphorylation and metabolism in human and mouse skeletal muscle. J. Biol. Chem. 2011, 286, 34567–34574. [Google Scholar] [CrossRef] [PubMed]
- Plaideau, C.; Liu, J.; Hartleib-Geschwindner, J.; Bastin-Coyette, L.; Bontemps, F.; Oscarsson, J.; Hue, L.; Rider, M.H. Overexpression of AMP-metabolizing enzymes controls adenine nucleotide levels and AMPK activation in HEK293T cells. FASEB J. 2012, 26, 2685–2694. [Google Scholar] [CrossRef]
- Pesi, R.; Allegrini, S.; Garcia-Gil, M.; Piazza, L.; Moschini, R.; Jordheim, L.P.; Camici, M.; Tozzi, M.G. Cytosolic 5′-Nucleotidase II Silencing in Lung Tumor Cells Regulates Metabolism through Activation of the p53/AMPK Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 7004. [Google Scholar] [CrossRef] [PubMed]
- Sorriento, D.; Iaccarino, G. Commentary: Studies in Zebrafish Demonstrate That CNNM2 and NT5C2 Are Most Likely the Causal Genes at the Blood Pressure-Associated Locus on Human Chromosome 10q24.32. Front. Cardiovasc. Med. 2020, 7, 582101. [Google Scholar] [CrossRef] [PubMed]
- Duarte, R.R.R.; Bachtel, N.D.; Cotel, M.C.; Lee, S.H.; Selvackadunco, S.; Watson, I.A.; Hovsepian, G.A.; Troakes, C.; Breen, G.D.; Nixon, D.F.; et al. The Psychiatric Risk Gene NT5C2 Regulates Adenosine Monophosphate-Activated Protein Kinase Signaling and Protein Translation in Human Neural Progenitor Cells. Biol. Psychiatry 2019, 86, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Kviklyte, S.; Vertommen, D.; Yerna, X.; Andersen, H.; Xu, X.; Gailly, P.; Bohlooly, Y.M.; Oscarsson, J.; Rider, M.H. Effects of genetic deletion of soluble 5′-nucleotidases NT5C1A and NT5C2 on AMPK activation and nucleotide levels in contracting mouse skeletal muscles. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E48–E62. [Google Scholar] [CrossRef]
- Johanns, M.; Kviklyte, S.; Chuang, S.J.; Corbeels, K.; Jacobs, R.; Herinckx, G.; Vertommen, D.; Schakman, O.; Duparc, T.; Cani, P.D.; et al. Genetic deletion of soluble 5′-nucleotidase II reduces body weight gain and insulin resistance induced by a high-fat diet. Mol. Genet. Metab. 2019, 126, 377–387. [Google Scholar] [CrossRef]
- Samaan, Z.; Lee, Y.K.; Gerstein, H.C.; Engert, J.C.; Bosch, J.; Mohan, V.; Diaz, R.; Yusuf, S.; Anand, S.S.; Meyre, D.; et al. Obesity genes and risk of major depressive disorder in a multiethnic population: A cross-sectional study. J. Clin. Psychiatry 2015, 76, e1611–e1618. [Google Scholar] [CrossRef]
- Gross, M. Molecular biology of AMP deaminase deficiency. Pharm. World Sci. 1994, 16, 55–61. [Google Scholar] [CrossRef]
- van Kuppevelt, T.H.; Veerkamp, J.H.; Fishbein, W.N.; Ogasawara, N.; Sabina, R.L. Immunolocalization of AMP-deaminase isozymes in human skeletal muscle and cultured muscle cells: Concentration of isoform M at the neuromuscular junction. J. Histochem. Cytochem. 1994, 42, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, W.N.; Armbrustmacher, V.W.; Griffin, J.L. Myoadenylate deaminase deficiency: A new disease of muscle. Science 1978, 200, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, T.; Gross, M.; Morisaki, H.; Pongratz, D.; Zollner, N.; Holmes, E.W. Molecular basis of AMP deaminase deficiency in skeletal muscle. Proc. Natl. Acad. Sci. USA 1992, 89, 6457–6461. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, H.; Morisaki, T.; Newby, L.K.; Holmes, E.W. Alternative splicing: A mechanism for phenotypic rescue of a common inherited defect. J. Clin. Investg. 1993, 91, 2275–2280. [Google Scholar] [CrossRef]
- Perumal, M.B.; Kovac, S.; Shah, A.; Wood, N.; Houlden, H.; Eriksson, S.; Walker, M. Hypersomnia with dilated pupils in adenosine monophosphate deaminase (AMPD) deficiency. J. Sleep Res. 2014, 23, 118–120. [Google Scholar] [CrossRef]
- Hanisch, F.; Joshi, P.; Zierz, S. AMP deaminase deficiency in skeletal muscle is unlikely to be of clinical relevance. J. Neurol. 2008, 255, 318–322. [Google Scholar] [CrossRef]
- Patten, B.M. Beneficial effect of D-ribose in patient with myoadenylate deaminase deficiency. Lancet 1982, 1, 1071. [Google Scholar] [CrossRef]
- Lecky, B.R. Failure of D-ribose in myoadenylate deaminase deficiency. Lancet 1983, 1, 193. [Google Scholar] [CrossRef]
- Zollner, N.; Reiter, S.; Gross, M.; Pongratz, D.; Reimers, C.D.; Gerbitz, K.; Paetzke, I.; Deufel, T.; Hubner, G. Myoadenylate deaminase deficiency: Successful symptomatic therapy by high dose oral administration of ribose. Klin. Wochenschr. 1986, 64, 1281–1290. [Google Scholar] [CrossRef]
- Wagner, D.R.; Gresser, U.; Zollner, N. Effects of oral ribose on muscle metabolism during bicycle ergometer in AMPD-deficient patients. Ann. Nutr. Metab. 1991, 35, 297–302. [Google Scholar] [CrossRef]
- Sabina, R.L.; Swain, J.L.; Olanow, C.W.; Bradley, W.G.; Fishbein, W.N.; DiMauro, S.; Holmes, E.W. Myoadenylate deaminase deficiency. Functional and metabolic abnormalities associated with disruption of the purine nucleotide cycle. J. Clin. Investg. 1984, 73, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Norman, B.; Sabina, R.L.; Jansson, E. Regulation of skeletal muscle ATP catabolism by AMPD1 genotype during sprint exercise in asymptomatic subjects. J. Appl. Physiol. 2001, 91, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Sinkeler, S.P.; Binkhorst, R.A.; Joosten, E.M.; Wevers, R.A.; Coerwinkei, M.M.; Oei, T.L. AMP deaminase deficiency: Study of the human skeletal muscle purine metabolism during ischaemic isometric exercise. Clin. Sci. 1987, 72, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Plaideau, C.; Lai, Y.C.; Kviklyte, S.; Zanou, N.; Lofgren, L.; Andersen, H.; Vertommen, D.; Gailly, P.; Hue, L.; Bohlooly, Y.M.; et al. Effects of pharmacological AMP deaminase inhibition and Ampd1 deletion on nucleotide levels and AMPK activation in contracting skeletal muscle. Chem. Biol. 2014, 21, 1497–1510. [Google Scholar] [CrossRef]
- Hafen, P.S.; Law, A.S.; Matias, C.; Miller, S.G.; Brault, J.J. Skeletal muscle contraction kinetics and AMPK responses are modulated by the adenine nucleotide degrading enzyme AMPD1. J. Appl. Physiol. 2022, 133, 1055–1066. [Google Scholar] [CrossRef]
- Loh, E.; Rebbeck, T.R.; Mahoney, P.D.; DeNofrio, D.; Swain, J.L.; Holmes, E.W. Common variant in AMPD1 gene predicts improved clinical outcome in patients with heart failure. Circulation 1999, 99, 1422–1425. [Google Scholar] [CrossRef]
- Anderson, J.L.; Habashi, J.; Carlquist, J.F.; Muhlestein, J.B.; Horne, B.D.; Bair, T.L.; Pearson, R.R.; Hart, N. A common variant of the AMPD1 gene predicts improved cardiovascular survival in patients with coronary artery disease. J. Am. Coll. Cardiol. 2000, 36, 1248–1252. [Google Scholar] [CrossRef]
- Feng, A.F.; Liu, Z.H.; Zhou, S.L.; Zhao, S.Y.; Zhu, Y.X.; Wang, H.X. Effects of AMPD1 gene C34T polymorphism on cardiac index, blood pressure and prognosis in patients with cardiovascular diseases: A meta-analysis. BMC Cardiovasc. Disord. 2017, 17, 174. [Google Scholar] [CrossRef]
- Kortum, F.; Jamra, R.A.; Alawi, M.; Berry, S.A.; Borck, G.; Helbig, K.L.; Tang, S.; Huhle, D.; Korenke, G.C.; Hebbar, M.; et al. Clinical and genetic spectrum of AMPD2-related pontocerebellar hypoplasia type 9. Eur. J. Hum. Genet. 2018, 26, 695–708. [Google Scholar] [CrossRef]
- Gilboa, T.; Elefant, N.; Meiner, V.; Hacohen, N. Delineating the phenotype and genetic basis of AMPD2-related pontocerebellar hypoplasia. Neurogenetics 2023, 24, 61–66. [Google Scholar] [CrossRef]
- Abreu, N.J.; Koboldt, D.C.; Gastier-Foster, J.M.; Dave-Wala, A.; Flanigan, K.M.; Waldrop, M.A. Homozygous variants in AMPD2 and COL11A1 lead to a complex phenotype of pontocerebellar hypoplasia type 9 and Stickler syndrome type 2. Am. J. Med. Genet. A 2020, 182, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Scola, E.; Ganau, M.; Robinson, R.; Cleary, M.; De Cocker, L.J.L.; Mankad, K.; Triulzi, F.; D’Arco, F. Neuroradiological findings in three cases of pontocerebellar hypoplasia type 9 due to AMPD2 mutation: Typical MRI appearances and pearls for differential diagnosis. Quant. Imaging Med. Surg. 2019, 9, 1966–1972. [Google Scholar] [CrossRef]
- Marsh, A.P.L.; Novarino, G.; Lockhart, P.J.; Leventer, R.J. CUGC for pontocerebellar hypoplasia type 9 and spastic paraplegia-63. Eur. J. Hum. Genet. 2019, 27, 161–166. [Google Scholar] [CrossRef]
- Akizu, N.; Cantagrel, V.; Schroth, J.; Cai, N.; Vaux, K.; McCloskey, D.; Naviaux, R.K.; Van Vleet, J.; Fenstermaker, A.G.; Silhavy, J.L.; et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell 2013, 154, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Courchet, J.; Viollet, B.; Brenman, J.E.; Polleux, F. AMP-activated protein kinase (AMPK) activity is not required for neuronal development but regulates axogenesis during metabolic stress. Proc. Natl. Acad. Sci. USA 2011, 108, 5849–5854. [Google Scholar] [CrossRef]
- Garcia-Gil, M.; Camici, M.; Allegrini, S.; Pesi, R.; Tozzi, M.G. Metabolic Aspects of Adenosine Functions in the Brain. Front. Pharmacol. 2021, 12, 672182. [Google Scholar] [CrossRef]
- Williams-Karnesky, R.L.; Sandau, U.S.; Lusardi, T.A.; Lytle, N.K.; Farrell, J.M.; Pritchard, E.M.; Kaplan, D.L.; Boison, D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J. Clin. Investg. 2013, 123, 3552–3563. [Google Scholar] [CrossRef]
- Bjursell, M.K.; Blom, H.J.; Cayuela, J.A.; Engvall, M.L.; Lesko, N.; Balasubramaniam, S.; Brandberg, G.; Halldin, M.; Falkenberg, M.; Jakobs, C.; et al. Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am. J. Hum. Genet. 2011, 89, 507–515. [Google Scholar] [CrossRef]
- Almuhsen, N.; Guay, S.P.; Lefrancois, M.; Gauvin, C.; Al Bahlani, A.Q.; Ahmed, N.; Saint-Martin, C.; Gagnon, T.; Waters, P.; Braverman, N.; et al. Clinical utility of methionine restriction in adenosine kinase deficiency. JIMD Rep. 2021, 61, 52–59. [Google Scholar] [CrossRef]
- Lipinski, P.; Ciara, E.; Jurkiewicz, D.; Pronicki, M.; Jurkiewicz, E.; Bogdanska, A.; Ploski, R.; Jankowska, I. Case Report: Adenosine kinase deficiency diagnosed 10 years after liver transplantation: Novel phenotypic insights. Front. Pediatr. 2022, 10, 1061043. [Google Scholar] [CrossRef] [PubMed]
- Staufner, C.; Lindner, M.; Dionisi-Vici, C.; Freisinger, P.; Dobbelaere, D.; Douillard, C.; Makhseed, N.; Straub, B.K.; Kahrizi, K.; Ballhausen, D.; et al. Adenosine kinase deficiency: Expanding the clinical spectrum and evaluating therapeutic options. J. Inherit. Metab. Dis. 2016, 39, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Paz, J.A.; Embirucu, E.K.; Bueno, C.; Ferreira, R.; Oliveira, F.S.; Pereira, A.S.S.; Schwartz, I.V.D.; Paiva, A.R.B.; Lucato, L.T.; Kok, F. Adenosine kinase deficiency presenting with tortuous cervical arteries: A risk factor for recurrent stroke. JIMD Rep. 2021, 62, 49–55. [Google Scholar] [CrossRef]
- Baric, I.; Staufner, C.; Augoustides-Savvopoulou, P.; Chien, Y.H.; Dobbelaere, D.; Grunert, S.C.; Opladen, T.; Petkovic Ramadza, D.; Rakic, B.; Wedell, A.; et al. Consensus recommendations for the diagnosis, treatment and follow-up of inherited methylation disorders. J. Inherit. Metab. Dis. 2017, 40, 5–20. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Caudill, M.A. Elevation in S-adenosylhomocysteine and DNA hypomethylation: Potential epigenetic mechanism for homocysteine-related pathology. J. Nutr. 2002, 132, 2361S–2366S. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.; Caro, P.; Sanchez, I.; Naudi, A.; Jove, M.; Portero-Otin, M.; Lopez-Torres, M.; Pamplona, R.; Barja, G. Effect of methionine dietary supplementation on mitochondrial oxygen radical generation and oxidative DNA damage in rat liver and heart. J. Bioenerg. Biomembr. 2009, 41, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Caro, P.; Gomez, J.; Lopez-Torres, M.; Sanchez, I.; Naudi, A.; Jove, M.; Pamplona, R.; Barja, G. Forty percent and eighty percent methionine restriction decrease mitochondrial ROS generation and oxidative stress in rat liver. Biogerontology 2008, 9, 183–196. [Google Scholar] [CrossRef]
- Boison, D.; Scheurer, L.; Zumsteg, V.; Rulicke, T.; Litynski, P.; Fowler, B.; Brandner, S.; Mohler, H. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc. Natl. Acad. Sci. USA 2002, 99, 6985–6990. [Google Scholar] [CrossRef]
- Sandau, U.S.; Colino-Oliveira, M.; Jones, A.; Saleumvong, B.; Coffman, S.Q.; Liu, L.; Miranda-Lourenco, C.; Palminha, C.; Batalha, V.L.; Xu, Y.; et al. Adenosine Kinase Deficiency in the Brain Results in Maladaptive Synaptic Plasticity. J. Neurosci. 2016, 36, 12117–12128. [Google Scholar] [CrossRef]
- Boison, D. Adenosine dysfunction and adenosine kinase in epileptogenesis. Open Neurosci. J. 2010, 4, 93–101. [Google Scholar] [CrossRef]
- Hall, B.; George, J.G.; Allen, S.P. Adenosine deaminase, not immune to a mechanistic rethink in central nervous system disorders? Histol. Histopathol. 2022, 37, 189–212. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Canet, J.; Gracia, E.; Lluis, C.; Mallol, J.; Canela, E.I.; Cortes, A.; Casado, V. Molecular Evidence of Adenosine Deaminase Linking Adenosine A(2A) Receptor and CD26 Proteins. Front. Pharmacol. 2018, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Homma, K.J.; Tanaka, Y.; Matsushita, T.; Yokoyama, K.; Matsui, H.; Natori, S. Adenosine deaminase activity of insect-derived growth factor is essential for its growth factor activity. J. Biol. Chem. 2001, 276, 43761–43766. [Google Scholar] [CrossRef] [PubMed]
- Aksentijevich, I.; Sampaio Moura, N.; Barron, K. Adenosine Deaminase 2 Deficiency. In GeneReviews((R)); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2023. [Google Scholar]
- Signa, S.; Bertoni, A.; Penco, F.; Caorsi, R.; Cafaro, A.; Cangemi, G.; Volpi, S.; Gattorno, M.; Schena, F. Adenosine Deaminase 2 Deficiency (DADA2): A Crosstalk between Innate and Adaptive Immunity. Front. Immunol. 2022, 13, 935957. [Google Scholar] [CrossRef] [PubMed]
- Zavialov, A.V.; Gracia, E.; Glaichenhaus, N.; Franco, R.; Zavialov, A.V.; Lauvau, G. Human adenosine deaminase 2 induces differentiation of monocytes into macrophages and stimulates proliferation of T helper cells and macrophages. J. Leukoc. Biol. 2010, 88, 279–290. [Google Scholar] [CrossRef]
- Zavialov, A.V.; Yu, X.; Spillmann, D.; Lauvau, G.; Zavialov, A.V. Structural basis for the growth factor activity of human adenosine deaminase ADA2. J. Biol. Chem. 2010, 285, 12367–12377. [Google Scholar] [CrossRef]
- Iwaki-Egawa, S.; Yamamoto, T.; Watanabe, Y. Human plasma adenosine deaminase 2 is secreted by activated monocytes. Biol. Chem. 2006, 387, 319–321. [Google Scholar] [CrossRef]
- Flinn, A.M.; Gennery, A.R. Adenosine deaminase deficiency: A review. Orphanet J. Rare Dis. 2018, 13, 65. [Google Scholar] [CrossRef]
- Kohn, D.B.; Hershfield, M.S.; Puck, J.M.; Aiuti, A.; Blincoe, A.; Gaspar, H.B.; Notarangelo, L.D.; Grunebaum, E. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2019, 143, 852–863. [Google Scholar] [CrossRef]
- Cuvelier, G.D.E.; Logan, B.R.; Prockop, S.E.; Buckley, R.H.; Kuo, C.Y.; Griffith, L.M.; Liu, X.; Yip, A.; Hershfield, M.S.; Ayoub, P.G.; et al. Outcomes following treatment for ADA-deficient severe combined immunodeficiency: A report from the PIDTC. Blood 2022, 140, 685–705. [Google Scholar] [CrossRef]
- Cohen, A.; Hirschhorn, R.; Horowitz, S.D.; Rubinstein, A.; Polmar, S.H.; Hong, R.; Martin, D.W., Jr. Deoxyadenosine triphosphate as a potentially toxic metabolite in adenosine deaminase deficiency. Proc. Natl. Acad. Sci. USA 1978, 75, 472–476. [Google Scholar] [CrossRef]
- Titman, P.; Pink, E.; Skucek, E.; O’Hanlon, K.; Cole, T.J.; Gaspar, J.; Xu-Bayford, J.; Jones, A.; Thrasher, A.J.; Davies, E.G.; et al. Cognitive and behavioral abnormalities in children after hematopoietic stem cell transplantation for severe congenital immunodeficiencies. Blood 2008, 112, 3907–3913. [Google Scholar] [CrossRef]
- Cicalese, M.P.; Ferrua, F.; Castagnaro, L.; Pajno, R.; Barzaghi, F.; Giannelli, S.; Dionisio, F.; Brigida, I.; Bonopane, M.; Casiraghi, M.; et al. Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood 2016, 128, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Sauer, A.V.; Hernandez, R.J.; Fumagalli, F.; Bianchi, V.; Poliani, P.L.; Dallatomasina, C.; Riboni, E.; Politi, L.S.; Tabucchi, A.; Carlucci, F.; et al. Alterations in the brain adenosine metabolism cause behavioral and neurological impairment in ADA-deficient mice and patients. Sci. Rep. 2017, 7, 40136. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Negandhi, J.; Min, W.; Tsui, M.; Post, M.; Harrison, R.V.; Grunebaum, E. Early Enzyme Replacement Therapy Improves Hearing and Immune Defects in Adenosine Deaminase Deficient-Mice. Front. Immunol. 2019, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Manalo, J.M.; Liu, H.; Ding, D.; Hicks, J.; Sun, H.; Salvi, R.; Kellems, R.E.; Pereira, F.A.; Xia, Y. Adenosine A2B receptor: A pathogenic factor and a therapeutic target for sensorineural hearing loss. FASEB J. 2020, 34, 15771–15787. [Google Scholar] [CrossRef] [PubMed]
- Navon Elkan, P.; Pierce, S.B.; Segel, R.; Walsh, T.; Barash, J.; Padeh, S.; Zlotogorski, A.; Berkun, Y.; Press, J.J.; Mukamel, M.; et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N. Engl. J. Med. 2014, 370, 921–931. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, D.; Ombrello, A.K.; Zavialov, A.V.; Toro, C.; Zavialov, A.V.; Stone, D.L.; Chae, J.J.; Rosenzweig, S.D.; Bishop, K.; et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N. Engl. J. Med. 2014, 370, 911–920. [Google Scholar] [CrossRef]
- Pilania, R.K.; Banday, A.Z.; Sharma, S.; Kumrah, R.; Joshi, V.; Loganathan, S.; Dhaliwal, M.; Jindal, A.K.; Vignesh, P.; Suri, D.; et al. Deficiency of Human Adenosine Deaminase Type 2—A Diagnostic Conundrum for the Hematologist. Front. Immunol. 2022, 13, 869570. [Google Scholar] [CrossRef]
- Bulut, E.; Erden, A.; Karadag, O.; Oguz, K.K.; Ozen, S. Deficiency of adenosine deaminase 2; special focus on central nervous system imaging. J. Neuroradiol. 2019, 46, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Belot, A.; Wassmer, E.; Twilt, M.; Lega, J.C.; Zeef, L.A.; Oojageer, A.; Kasher, P.R.; Mathieu, A.L.; Malcus, C.; Demaret, J.; et al. Mutations in CECR1 associated with a neutrophil signature in peripheral blood. Pediatr. Rheumatol. Online J. 2014, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Kasapcopur, O.; Foster, J., 2nd; Barut, K.; Tekin, A.; Kizilkilic, O.; Tekin, M. Novel adenosine deaminase 2 mutations in a child with a fatal vasculopathy. Eur. J. Pediatr. 2014, 173, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.Y.; Kellner, E.S.; Huang, Y.; Furutani, E.; Huang, Z.; Bainter, W.; Alosaimi, M.F.; Stafstrom, K.; Platt, C.D.; Stauber, T.; et al. Genotype and functional correlates of disease phenotype in deficiency of adenosine deaminase 2 (DADA2). J. Allergy Clin. Immunol. 2020, 145, 1664–1672. [Google Scholar] [CrossRef]
- Geraldo, A.F.; Caorsi, R.; Tortora, D.; Gandolfo, C.; Ammendola, R.; Alessio, M.; Conti, G.; Insalaco, A.; Pastore, S.; Martino, S.; et al. Widening the Neuroimaging Features of Adenosine Deaminase 2 Deficiency. AJNR Am. J. Neuroradiol. 2021, 42, 975–979. [Google Scholar] [CrossRef] [PubMed]
- Hashem, H.; Bucciol, G.; Ozen, S.; Unal, S.; Bozkaya, I.O.; Akarsu, N.; Taskinen, M.; Koskenvuo, M.; Saarela, J.; Dimitrova, D.; et al. Hematopoietic Cell Transplantation Cures Adenosine Deaminase 2 Deficiency: Report on 30 Patients. J. Clin. Immunol. 2021, 41, 1633–1647. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Khaznadar, S.S.; Shwin, K.W.; Irizarry-Caro, J.A.; O’Neil, L.J.; Liu, Y.; Jacobson, K.A.; Ombrello, A.K.; Stone, D.L.; Tsai, W.L.; et al. Deficiency of adenosine deaminase 2 triggers adenosine-mediated NETosis and TNF production in patients with DADA2. Blood 2019, 134, 395–406. [Google Scholar] [CrossRef]
- Schnappauf, O.; Ombrello, A.K.; Kastner, D.L. Deficiency of adenosine deaminase 2: Is it an elephant after all? J. Allergy Clin. Immunol. 2020, 145, 1560–1561. [Google Scholar] [CrossRef]
- Schoen, J.; Euler, M.; Schauer, C.; Schett, G.; Herrmann, M.; Knopf, J.; Yaykasli, K.O. Neutrophils’ Extracellular Trap Mechanisms: From Physiology to Pathology. Int. J. Mol. Sci. 2022, 23, 2855. [Google Scholar] [CrossRef]
- Watanabe, N.; Gao, S.; Wu, Z.; Batchu, S.; Kajigaya, S.; Diamond, C.; Alemu, L.; Raffo, D.Q.; Hoffmann, P.; Stone, D.; et al. Analysis of deficiency of adenosine deaminase 2 pathogenesis based on single-cell RNA sequencing of monocytes. J. Leukoc. Biol. 2021, 110, 409–424. [Google Scholar] [CrossRef]
- Dhanwani, R.; Takahashi, M.; Mathews, I.T.; Lenzi, C.; Romanov, A.; Watrous, J.D.; Pieters, B.; Hedrick, C.C.; Benedict, C.A.; Linden, J.; et al. Cellular sensing of extracellular purine nucleosides triggers an innate IFN-beta response. Sci. Adv. 2020, 6, eaba3688. [Google Scholar] [CrossRef]
- Nihira, H.; Izawa, K.; Ito, M.; Umebayashi, H.; Okano, T.; Kajikawa, S.; Nanishi, E.; Keino, D.; Murakami, K.; Isa-Nishitani, M.; et al. Detailed analysis of Japanese patients with adenosine deaminase 2 deficiency reveals characteristic elevation of type II interferon signature and STAT1 hyperactivation. J. Allergy Clin. Immunol. 2021, 148, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Blausen Medical. Medical gallery of Blausen Medical 2014. WikiJournal Med. 2014, 1, 1–79. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Castelli, L.M.; Francis, L.; Woof, R.; Siskos, A.P.; Kouloura, E.; Gray, E.; Thompson, A.G.; Talbot, K.; et al. Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 2019, 142, 586–605. [Google Scholar] [CrossRef] [PubMed]
- Kutryb-Zajac, B.; Kawecka, A.; Caratis, F.; Urbanowicz, K.; Braczko, A.; Furihata, T.; Karaszewski, B.; Smolenski, R.T.; Rutkowska, A. The impaired distribution of adenosine deaminase isoenzymes in multiple sclerosis plasma and cerebrospinal fluid. Front. Mol. Neurosci. 2022, 15, 998023. [Google Scholar] [CrossRef]
- Beluzic, L.; Grbesa, I.; Beluzic, R.; Park, J.H.; Kong, H.K.; Kopjar, N.; Espadas, G.; Sabido, E.; Lepur, A.; Rokic, F.; et al. Knock-down of AHCY and depletion of adenosine induces DNA damage and cell cycle arrest. Sci. Rep. 2018, 8, 14012. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kong, H.K.; Kim, Y.S.; Lee, Y.S.; Park, J.H. Inhibition of S-adenosylhomocysteine hydrolase decreases cell mobility and cell proliferation through cell cycle arrest. Am. J. Cancer Res. 2015, 5, 2127–2138. [Google Scholar]
- Aranda, S.; Alcaine-Colet, A.; Blanco, E.; Borras, E.; Caillot, C.; Sabido, E.; Di Croce, L. Chromatin capture links the metabolic enzyme AHCY to stem cell proliferation. Sci. Adv. 2019, 5, eaav2448. [Google Scholar] [CrossRef]
- Greco, C.M.; Cervantes, M.; Fustin, J.M.; Ito, K.; Ceglia, N.; Samad, M.; Shi, J.; Koronowski, K.B.; Forne, I.; Ranjit, S.; et al. S-adenosyl-l-homocysteine hydrolase links methionine metabolism to the circadian clock and chromatin remodeling. Sci. Adv. 2020, 6, eabc5629. [Google Scholar] [CrossRef]
- Gaull, G.E.; Bender, A.N.; Vulovic, D.; Tallan, H.H.; Schaffner, F. Methioninemia and myopathy: A new disorder. Ann. Neurol. 1981, 9, 423–432. [Google Scholar] [CrossRef]
- Labrune, P.; Perignon, J.L.; Rault, M.; Brunet, C.; Lutun, H.; Charpentier, C.; Saudubray, J.M.; Odievre, M. Familial hypermethioninemia partially responsive to dietary restriction. J. Pediatr. 1990, 117, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Baric, I.; Fumic, K.; Glenn, B.; Cuk, M.; Schulze, A.; Finkelstein, J.D.; James, S.J.; Mejaski-Bosnjak, V.; Pazanin, L.; Pogribny, I.P.; et al. S-adenosylhomocysteine hydrolase deficiency in a human: A genetic disorder of methionine metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 4234–4239. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Chang, R.; Abdenur, J.E. The biochemical profile and dietary management in S-adenosylhomocysteine hydrolase deficiency. Mol. Genet. Metab. Rep. 2022, 32, 100885. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, R.; Vugrek, O.; Deisch, J.; Wagner, C.; Stabler, S.; Allen, R.; Baric, I.; Rados, M.; Mudd, S.H. S-adenosylhomocysteine hydrolase deficiency: Two siblings with fetal hydrops and fatal outcomes. J. Inherit. Metab. Dis. 2010, 33, 705–713. [Google Scholar] [CrossRef]
- Stender, S.; Chakrabarti, R.S.; Xing, C.; Gotway, G.; Cohen, J.C.; Hobbs, H.H. Adult-onset liver disease and hepatocellular carcinoma in S-adenosylhomocysteine hydrolase deficiency. Mol. Genet. Metab. 2015, 116, 269–274. [Google Scholar] [CrossRef]
- Baric, I.; Cuk, M.; Fumic, K.; Vugrek, O.; Allen, R.H.; Glenn, B.; Maradin, M.; Pazanin, L.; Pogribny, I.; Rados, M.; et al. S-Adenosylhomocysteine hydrolase deficiency: A second patient, the younger brother of the index patient, and outcomes during therapy. J. Inherit. Metab. Dis. 2005, 28, 885–902. [Google Scholar] [CrossRef]
- Strauss, K.A.; Ferreira, C.; Bottiglieri, T.; Zhao, X.; Arning, E.; Zhang, S.; Zeisel, S.H.; Escolar, M.L.; Presnick, N.; Puffenberger, E.G.; et al. Liver transplantation for treatment of severe S-adenosylhomocysteine hydrolase deficiency. Mol. Genet. Metab. 2015, 116, 44–52. [Google Scholar] [CrossRef]
- Petkovic Ramadza, D.; Kuhtic, I.; Zarkovic, K.; Lochmuller, H.; Cavka, M.; Kovac, I.; Baric, I.; Prutki, M. Case Report: Advanced Skeletal Muscle Imaging in S-Adenosylhomocysteine Hydrolase Deficiency and Further Insight Into Muscle Pathology. Front. Pediatr. 2022, 10, 847445. [Google Scholar] [CrossRef]
- Michel, V.; Singh, R.K.; Bakovic, M. The impact of choline availability on muscle lipid metabolism. Food Funct. 2011, 2, 53–62. [Google Scholar] [CrossRef]
- Buist, N.R.; Glenn, B.; Vugrek, O.; Wagner, C.; Stabler, S.; Allen, R.H.; Pogribny, I.; Schulze, A.; Zeisel, S.H.; Baric, I.; et al. S-adenosylhomocysteine hydrolase deficiency in a 26-year-old man. J. Inherit. Metab. Dis. 2006, 29, 538–545. [Google Scholar] [CrossRef]
- Miller, M.W.; Duhl, D.M.; Winkes, B.M.; Arredondo-Vega, F.; Saxon, P.J.; Wolff, G.L.; Epstein, C.J.; Hershfield, M.S.; Barsh, G.S. The mouse lethal nonagouti (a(x)) mutation deletes the S-adenosylhomocysteine hydrolase (Ahcy) gene. EMBO J. 1994, 13, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Erion, M.D.; Takabayashi, K.; Smith, H.B.; Kessi, J.; Wagner, S.; Honger, S.; Shames, S.L.; Ealick, S.E. Purine nucleoside phosphorylase. 1. Structure-function studies. Biochemistry 1997, 36, 11725–11734. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.M.; Li, C.; Allan, P.W.; Parker, W.B.; Ealick, S.E. Structural basis for substrate specificity of Escherichia coli purine nucleoside phosphorylase. J. Biol. Chem. 2003, 278, 47110–47118. [Google Scholar] [CrossRef]
- Bzowska, A.; Kulikowska, E.; Shugar, D. Purine nucleoside phosphorylases: Properties, functions, and clinical aspects. Pharmacol. Ther. 2000, 88, 349–425. [Google Scholar] [CrossRef] [PubMed]
- Giblett, E.R.; Ammann, A.J.; Wara, D.W.; Sandman, R.; Diamond, L.K. Nucleoside-phosphorylase deficiency in a child with severely defective T-cell immunity and normal B-cell immunity. Lancet 1975, 1, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Markert, M.L. Purine nucleoside phosphorylase deficiency. Immunodefic. Rev. 1991, 3, 45–81. [Google Scholar] [PubMed]
- Ozkinay, F.; Pehlivan, S.; Onay, H.; van den Berg, P.; Vardar, F.; Koturoglu, G.; Aksu, G.; Unal, D.; Tekgul, H.; Can, S.; et al. Purine nucleoside phosphorylase deficiency in a patient with spastic paraplegia and recurrent infections. J. Child Neurol. 2007, 22, 741–743. [Google Scholar] [CrossRef]
- Tabarki, B.; Yacoub, M.; Tlili, K.; Trabelsi, A.; Dogui, M.; Essoussi, A.S. Familial spastic paraplegia as the presenting manifestation in patients with purine nucleoside phosphorylase deficiency. J. Child Neurol. 2003, 18, 140–141. [Google Scholar] [CrossRef]
- Arduini, A.; Marasco, E.; Marucci, G.; Pardeo, M.; Insalaco, A.; Caiello, I.; Moneta, G.M.; Prencipe, G.; De Benedetti, F.; Bracaglia, C. An unusual presentation of purine nucleoside phosphorylase deficiency mimicking systemic juvenile idiopathic arthritis complicated by macrophage activation syndrome. Pediatr. Rheumatol. Online J. 2019, 17, 25. [Google Scholar] [CrossRef]
- Al-Saud, B.; Al Alawi, Z.; Hussain, F.B.; Hershfield, M.; Alkuraya, F.S.; Al-Mayouf, S.M. A case with purine nucleoside phosphorylase deficiency suffering from late-onset systemic lupus erythematosus and lymphoma. J. Clin. Immunol. 2020, 40, 833–839. [Google Scholar] [CrossRef]
- Grunebaum, E.; Campbell, N.; Leon-Ponte, M.; Xu, X.; Chapdelaine, H. Partial Purine Nucleoside Phosphorylase Deficiency Helps Determine Minimal Activity Required for Immune and Neurological Development. Front. Immunol. 2020, 11, 1257. [Google Scholar] [CrossRef] [PubMed]
- Parvaneh, N.; Teimourian, S.; Jacomelli, G.; Badalzadeh, M.; Bertelli, M.; Zakharova, E.; Tabatabaei, P.; Parvaneh, L.; Pourakbari, B.; Yeganeh, M.; et al. Novel mutations of NP in two patients with purine nucleoside phosphorylase deficiency. Clin. Biochem. 2008, 41, 350–352. [Google Scholar] [CrossRef] [PubMed]
- la Marca, G.; Canessa, C.; Giocaliere, E.; Romano, F.; Malvagia, S.; Funghini, S.; Moriondo, M.; Valleriani, C.; Lippi, F.; Ombrone, D.; et al. Diagnosis of immunodeficiency caused by a purine nucleoside phosphorylase defect by using tandem mass spectrometry on dried blood spots. J. Allergy Clin. Immunol. 2014, 134, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Schejter, Y.D.; Even-Or, E.; Shadur, B.; NaserEddin, A.; Stepensky, P.; Zaidman, I. The Broad Clinical Spectrum and Transplant Results of PNP Deficiency. J. Clin. Immunol. 2020, 40, 123–130. [Google Scholar] [CrossRef]
- Torun, B.; Bilgin, A.; Orhan, D.; Gocmen, R.; Kilic, S.S.; Kuskonmaz, B.; Cetinkaya, D.; Tezcan, I.; Cagdas, D. Combined immunodeficiency due to purine nucleoside phosphorylase deficiency: Outcome of three patients. Eur. J. Med. Genet. 2022, 65, 104428. [Google Scholar] [CrossRef]
- Baguette, C.; Vermylen, C.; Brichard, B.; Louis, J.; Dahan, K.; Vincent, M.F.; Cornu, G. Persistent developmental delay despite successful bone marrow transplantation for purine nucleoside phosphorylase deficiency. J. Pediatr. Hematol. Oncol. 2002, 24, 69–71. [Google Scholar] [CrossRef]
- Abt, E.R.; Rashid, K.; Le, T.M.; Li, S.; Lee, H.R.; Lok, V.; Li, L.; Creech, A.L.; Labora, A.N.; Mandl, H.K.; et al. Purine nucleoside phosphorylase enables dual metabolic checkpoints that prevent T cell immunodeficiency and TLR7-associated autoimmunity. J. Clin. Investg. 2022, 132, e160852. [Google Scholar] [CrossRef]
- Papinazath, T.; Min, W.; Sujiththa, S.; Cohen, A.; Ackerley, C.; Roifman, C.M.; Grunebaum, E. Effects of purine nucleoside phosphorylase deficiency on thymocyte development. J. Allergy Clin. Immunol. 2011, 128, 854–863. [Google Scholar] [CrossRef]
- Seet, C.S.; He, C.; Bethune, M.T.; Li, S.; Chick, B.; Gschweng, E.H.; Zhu, Y.; Kim, K.; Kohn, D.B.; Baltimore, D.; et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 2017, 14, 521–530. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Arkatkar, T.; Du, S.W.; Jacobs, H.M.; Dam, E.M.; Hou, B.; Buckner, J.H.; Rawlings, D.J.; Jackson, S.W. B cell-derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J. Exp. Med. 2017, 214, 3207–3217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Dror, Y.; Grunebaum, E.; Hitzler, J.; Narendran, A.; Ye, C.; Tellier, R.; Edwards, V.; Freedman, M.H.; Roifman, C.M. Purine nucleoside phosphorylase deficiency associated with a dysplastic marrow morphology. Pediatr. Res. 2004, 55, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Min, W.; Cole, C.J.; Josselyn, S.A.; Henderson, J.T.; van Eede, M.; Henkelman, R.M.; Ackerley, C.; Grunebaum, E.; Roifman, C.M. Cerebellar abnormalities in purine nucleoside phosphorylase deficient mice. Neurobiol. Dis. 2012, 47, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Toro, A.; Grunebaum, E. TAT-mediated intracellular delivery of purine nucleoside phosphorylase corrects its deficiency in mice. J. Clin. Investg. 2006, 116, 2717–2726. [Google Scholar] [CrossRef]
- Tsui, M.; Biro, J.; Chan, J.; Min, W.; Dobbs, K.; Notarangelo, L.D.; Grunebaum, E. Purine nucleoside phosphorylase deficiency induces p53-mediated intrinsic apoptosis in human induced pluripotent stem cell-derived neurons. Sci. Rep. 2022, 12, 9084. [Google Scholar] [CrossRef]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef]
- Schuler, M.; Bossy-Wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J. Biol. Chem. 2000, 275, 7337–7342. [Google Scholar] [CrossRef]
- Pasquini, S.; Contri, C.; Merighi, S.; Gessi, S.; Borea, P.A.; Varani, K.; Vincenzi, F. Adenosine Receptors in Neuropsychiatric Disorders: Fine Regulators of Neurotransmission and Potential Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 1219. [Google Scholar] [CrossRef]
- Dai, S.; Lin, J.; Hou, Y.; Luo, X.; Shen, Y.; Ou, J. Purine signaling pathway dysfunction in autism spectrum disorders: Evidence from multiple omics data. Front. Mol. Neurosci. 2023, 16, 1089871. [Google Scholar] [CrossRef]
- Gevi, F.; Zolla, L.; Gabriele, S.; Persico, A.M. Urinary metabolomics of young Italian autistic children supports abnormal tryptophan and purine metabolism. Mol. Autism 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Argudo-Ramirez, A.; Martin-Nalda, A.; Gonzalez de Aledo-Castillo, J.M.; Lopez-Galera, R.; Marin-Soria, J.L.; Pajares-Garcia, S.; Martinez-Gallo, M.; Garcia-Prat, M.; Colobran, R.; Riviere, J.G.; et al. Newborn Screening for SCID. Experience in Spain (Catalonia). Int. J. Neonatal Screen. 2021, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.W.; Grossman, W.J.; Laessig, R.H.; Hoffman, G.L.; Brokopp, C.D.; Kurtycz, D.F.; Cogley, M.F.; Litsheim, T.J.; Katcher, M.L.; Routes, J.M. Development of a routine newborn screening protocol for severe combined immunodeficiency. J. Allergy Clin. Immunol. 2009, 124, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Biggs, C.M.; Haddad, E.; Issekutz, T.B.; Roifman, C.M.; Turvey, S.E. Newborn screening for severe combined immunodeficiency: A primer for clinicians. CMAJ 2017, 189, E1551–E1557. [Google Scholar] [CrossRef]
- Kwan, A.; Church, J.A.; Cowan, M.J.; Agarwal, R.; Kapoor, N.; Kohn, D.B.; Lewis, D.B.; McGhee, S.A.; Moore, T.B.; Stiehm, E.R.; et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: Results of the first 2 years. J. Allergy Clin. Immunol. 2013, 132, 140–150. [Google Scholar] [CrossRef]
- Torres, R.J.; Puig, J.G. Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet J. Rare Dis. 2007, 2, 48. [Google Scholar] [CrossRef]
- Puck, J.M. Neonatal screening for severe combined immunodeficiency. Curr. Opin. Pediatr. 2011, 23, 667–673. [Google Scholar] [CrossRef]







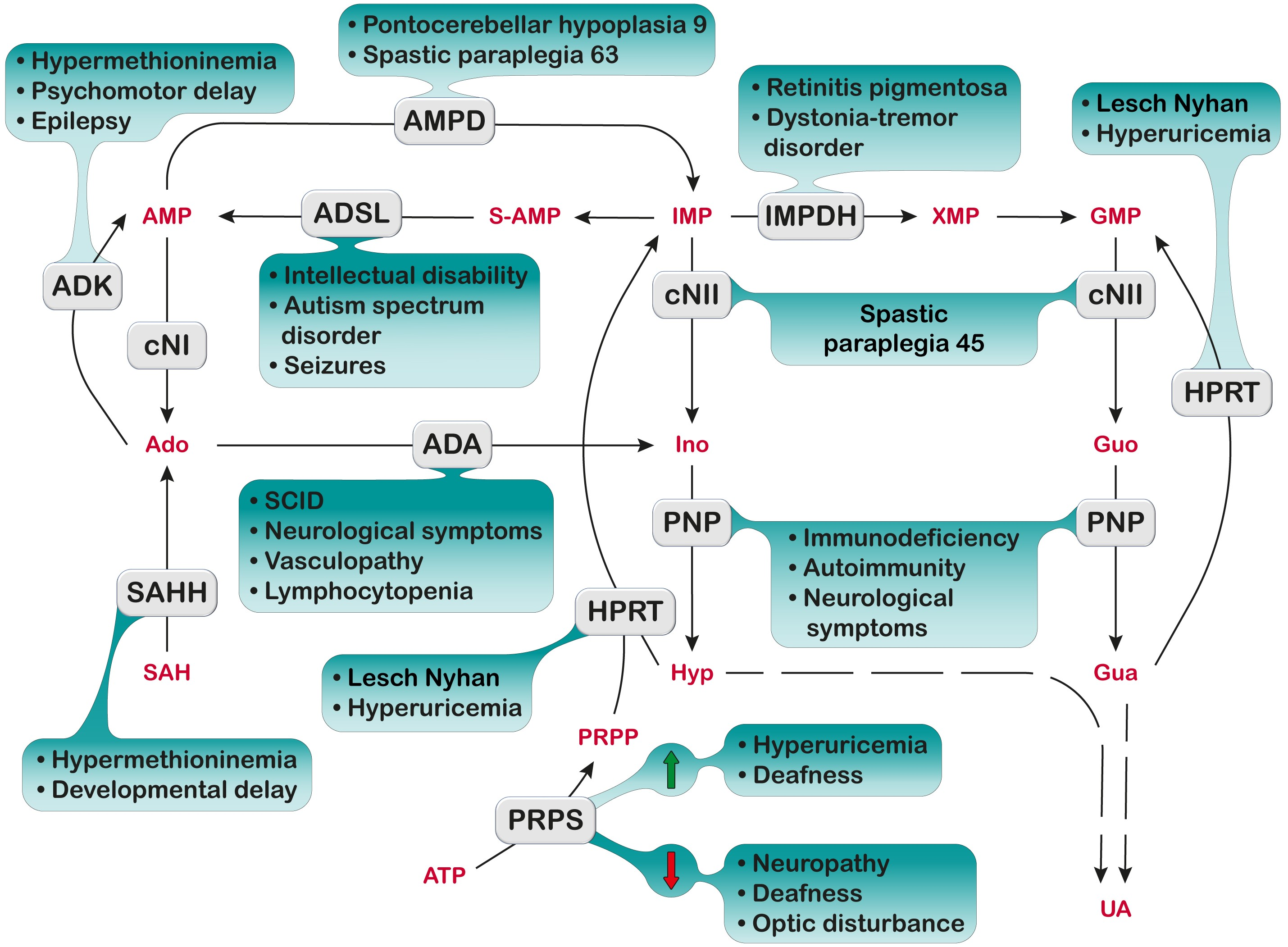{kind=link}
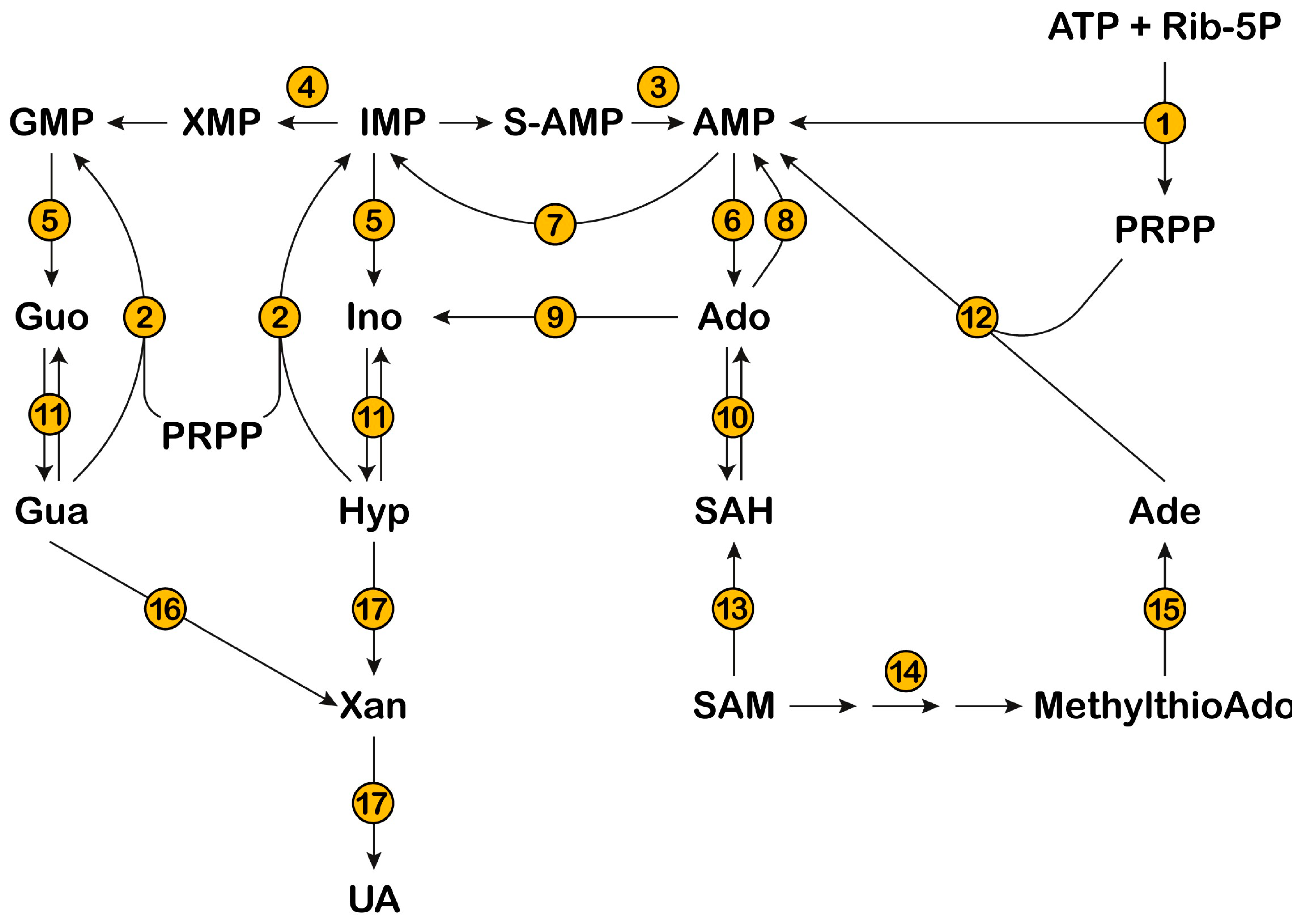{kind=link}
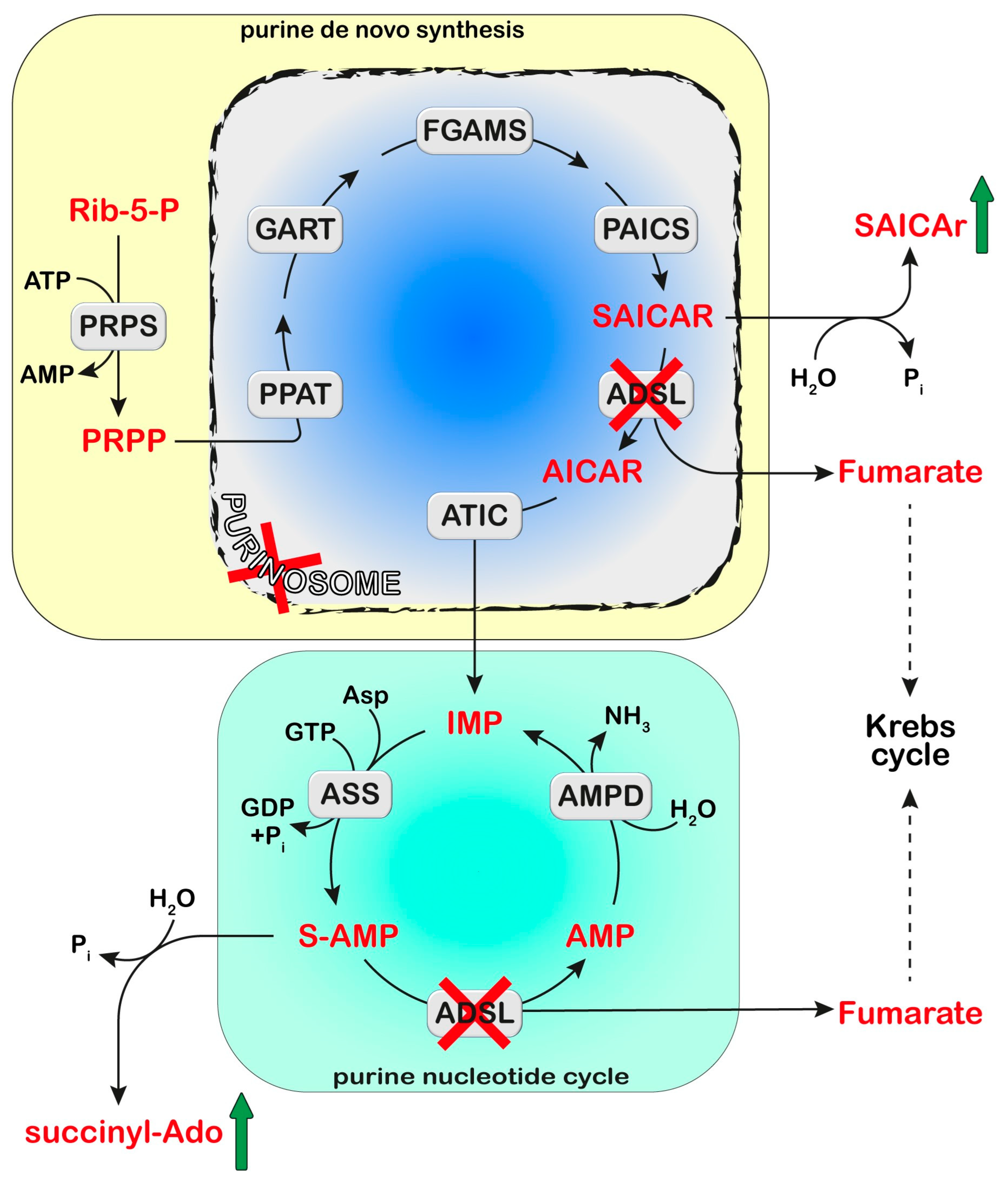{kind=link}
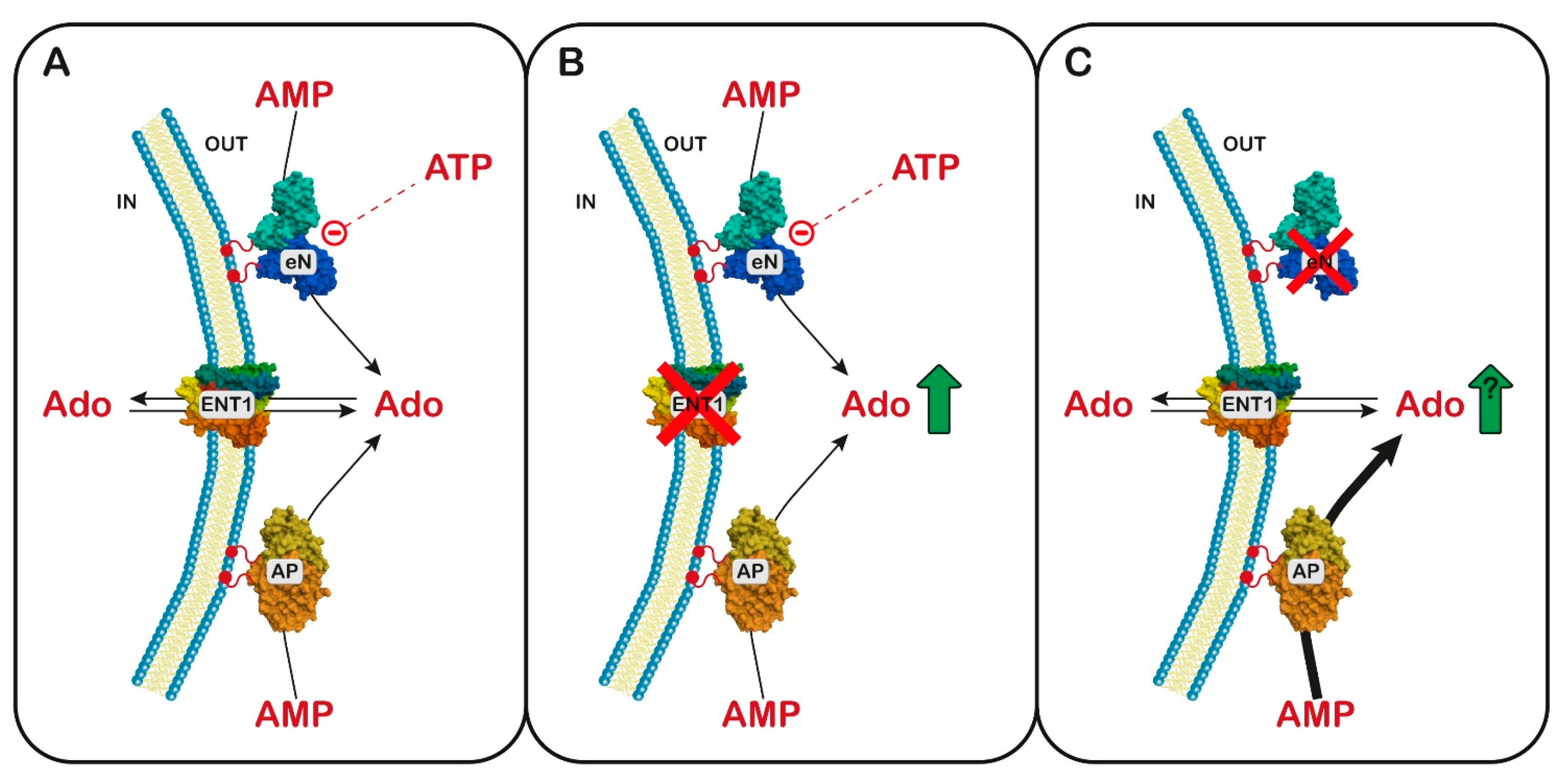{kind=link}
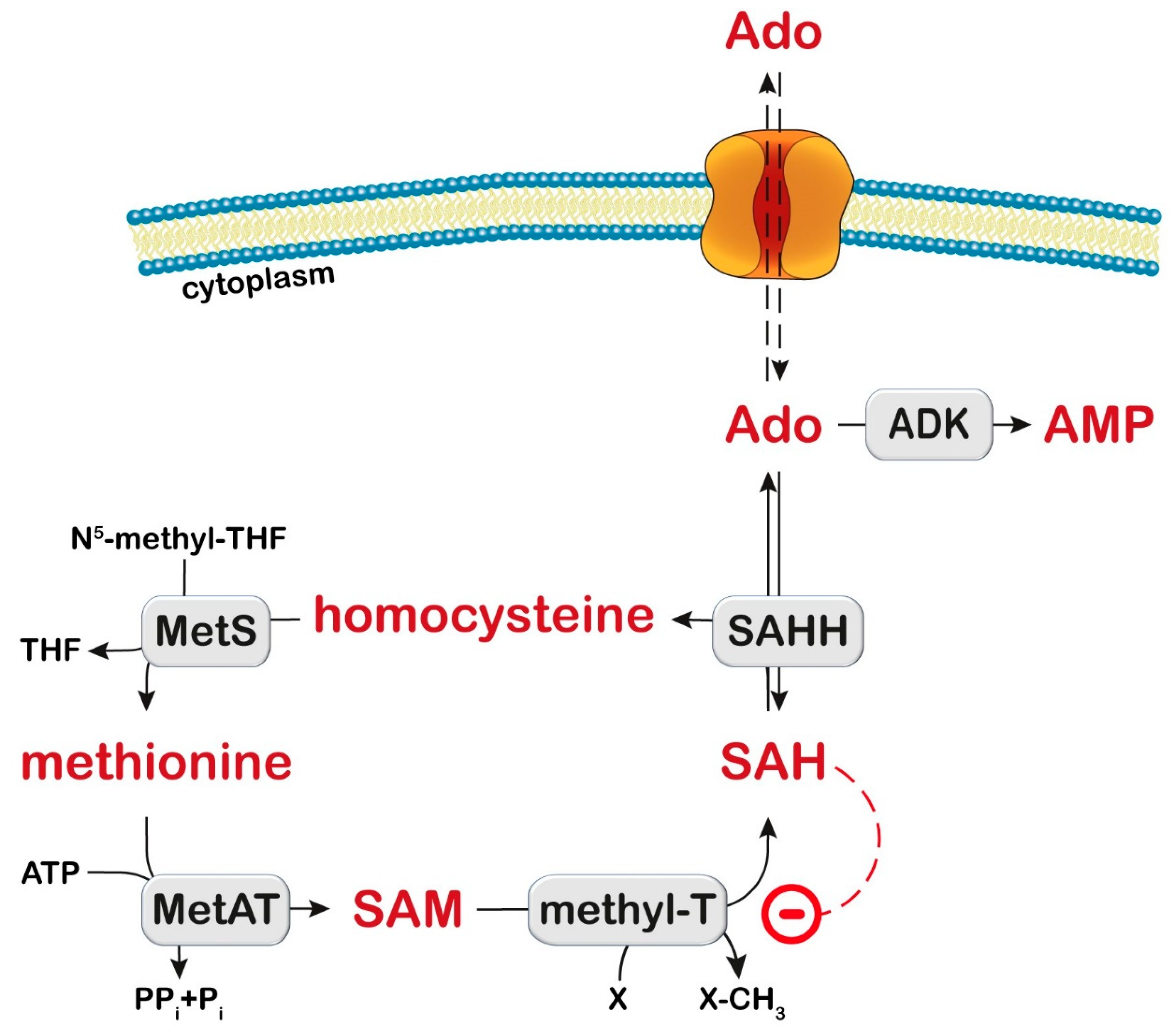{kind=link}
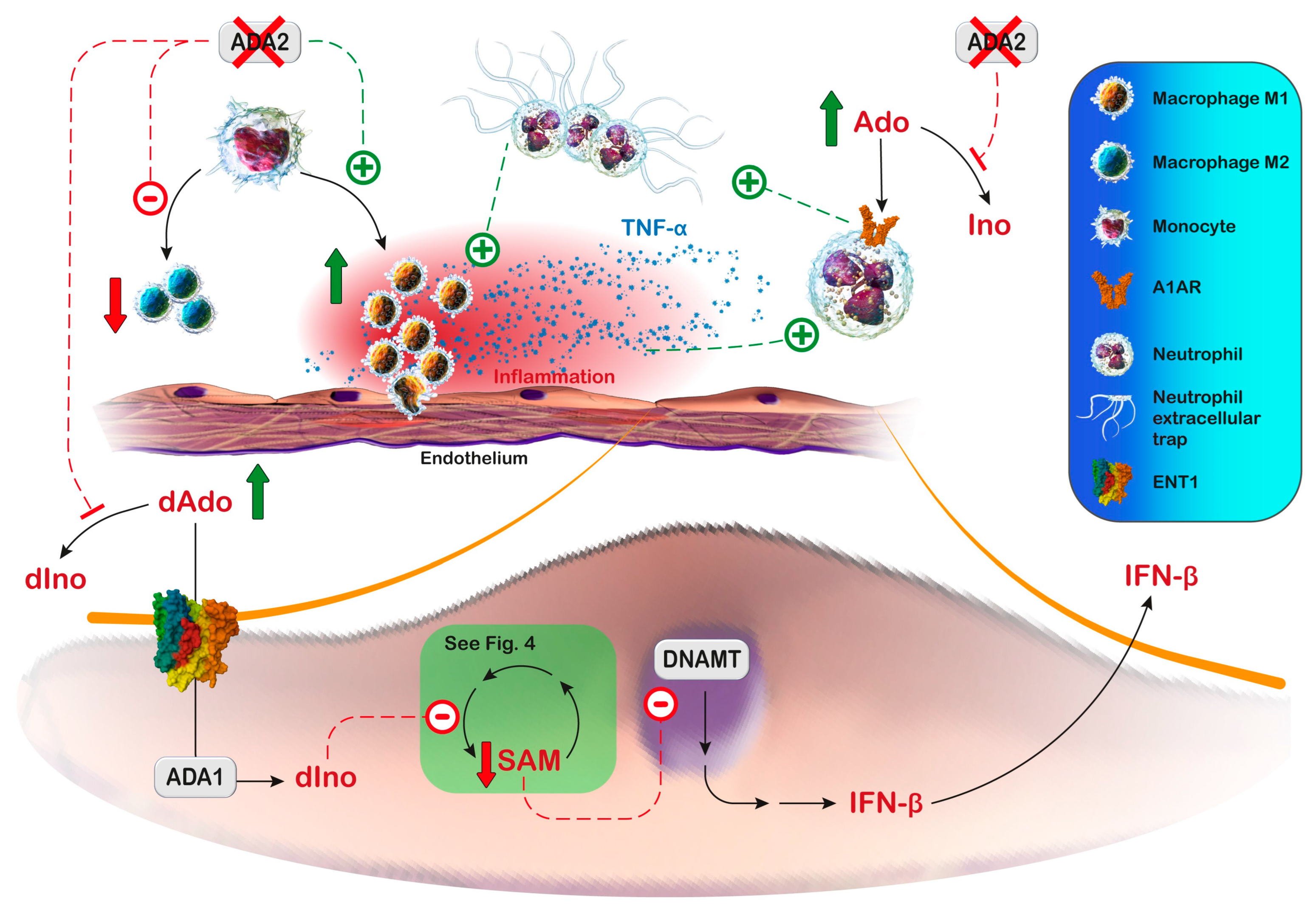{kind=link}
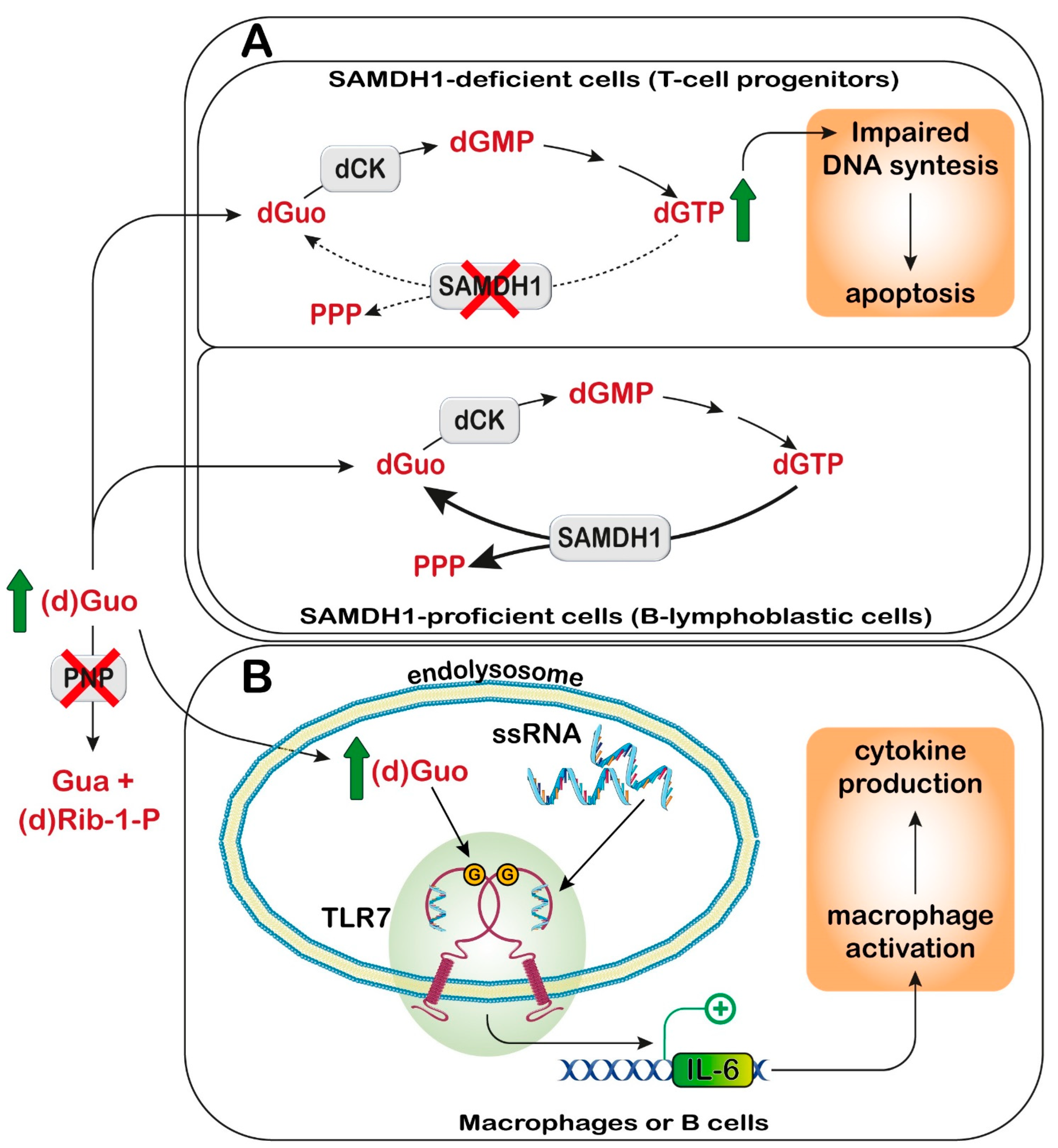{kind=link}
| Enzyme (Name; Symbol; EC) | Gene (Symbol; Location; Omim Code) | Pathology (and Variants) | Clinical Features | Diagnostics |
|---|---|---|---|---|
| Phosphoribosylpyrophosphate Synthetase; PRPS1; EC 2.7.6.1 | PRPS1; Xq22.3; OMIM 311850 | PRPS superactivity |
|
|
| PRPS deficiency(DFNX1; CMTX5; Arts syndrome) |
|
| ||
| Hypoxanthine-Guanine Phosphoribosyltyransferase; HPRT; EC 2.4.2.8 | HPRT; Xq26.2-q26.3; OMIM 308000 | Lesch–Nyhan Disease (total deficiency)Lesch–Nyhan variants (partial deficiency) |
|
|
| Adenylosuccinate lyase; ADSL; EC 4.3.2.2 | ADSL; 22q13.1; OMIM 608222 | ADSL deficiency
|
|
|
| Inosine-5′-monophosphate dehydrogenase IMPDH; EC 1.1.1.205 | IMPDH1; 7q32.1; OMIM 146690 | IMPDH1 malfunction:
|
|
|
| IMPDH2; 3p21.31; OMIM 146691 | IMPDH2 deficiency:
|
|
| |
| Ectosolic 5′-nucleotidase; eN; EC 3.1.3.5 | NT5E; 6q14.3; OMIM 129190 | eN hyperactivity:
|
|
|
eN deficiency:
|
|
| ||
| Cytosolic 5′-nucleotidase; cN-II; EC 3.1.3.5 | NT5C2; 10q24.32-q24.33; OMIM 600417 | Hereditary spastic paraplegia type 45 (HSP-cN-II deficiency) |
|
|
| Adenosine monophosphate deaminase; AMPD; EC 3.5.4.6 | AMPD1; 1p13.2; OMIM 102770 | Myoadenylate deaminase deficiency (AMPD1) |
|
|
| AMPD2; 1p13.3; OMIM 102771 | Pontocerebellar hypoplasia type 9 (AMPD2 deficiency) |
|
| |
| Adenosine kinase; ADK; EC 2.7.1.20 | ADK; 10q22.2; OMIM 102750 | ADK deficiency |
|
|
| Adenosine deaminase; ADA; EC 3.5.4.4 | ADA1; 20q13.12; OMIM 608958 | Severe combined immunodeficiency (SCID-ADA1 deficiency) |
|
|
| ADA2; 22q11.1; OMIM 607575 | ADA2 deficiency |
|
| |
| S-Adenosylhomocysteine hydrolase; SAHH; EC 3.3.1.1 | AHCY; 20q11.22; OMIM 180960 | SAHH deficiency |
|
|
| Purine nucleoside phosphorylase; PNP; EC 2.4.2.1 | PNP; 14q11.2; OMIM 164050 | PNP deficiency (SCID) |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camici, M.; Garcia-Gil, M.; Allegrini, S.; Pesi, R.; Bernardini, G.; Micheli, V.; Tozzi, M.G. Inborn Errors of Purine Salvage and Catabolism. Metabolites 2023, 13, 787. https://doi.org/10.3390/metabo13070787
Camici M, Garcia-Gil M, Allegrini S, Pesi R, Bernardini G, Micheli V, Tozzi MG. Inborn Errors of Purine Salvage and Catabolism. Metabolites. 2023; 13(7):787. https://doi.org/10.3390/metabo13070787
Chicago/Turabian StyleCamici, Marcella, Mercedes Garcia-Gil, Simone Allegrini, Rossana Pesi, Giulia Bernardini, Vanna Micheli, and Maria Grazia Tozzi. 2023. "Inborn Errors of Purine Salvage and Catabolism" Metabolites 13, no. 7: 787. https://doi.org/10.3390/metabo13070787
APA StyleCamici, M., Garcia-Gil, M., Allegrini, S., Pesi, R., Bernardini, G., Micheli, V., & Tozzi, M. G. (2023). Inborn Errors of Purine Salvage and Catabolism. Metabolites, 13(7), 787. https://doi.org/10.3390/metabo13070787