



Severe Hypertriglyceridaemia and Chylomicronaemia Syndrome—Causes, Clinical Presentation, and Therapeutic Options

 , ,
, , 
Abstract
1. Introduction

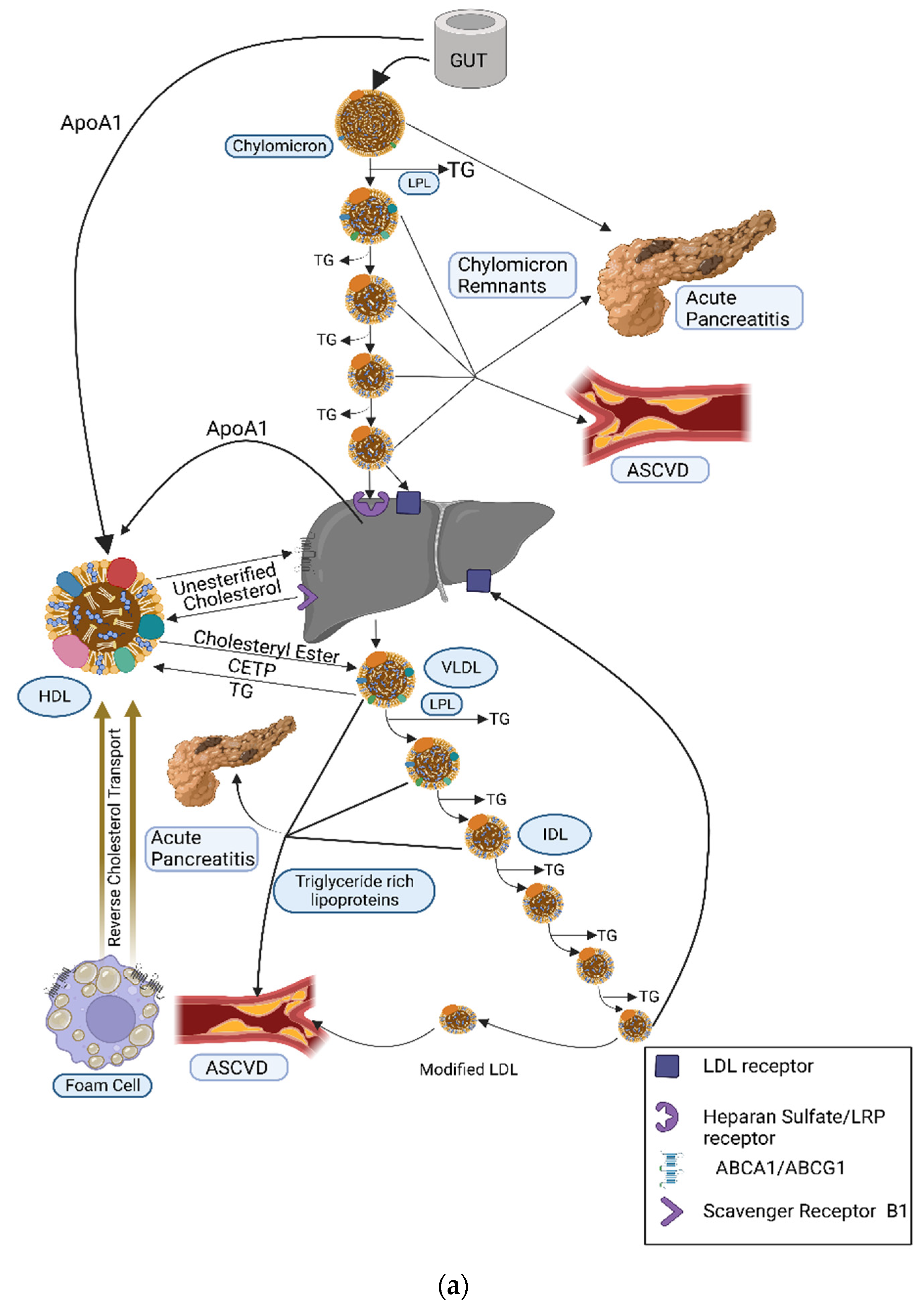{kind=link}
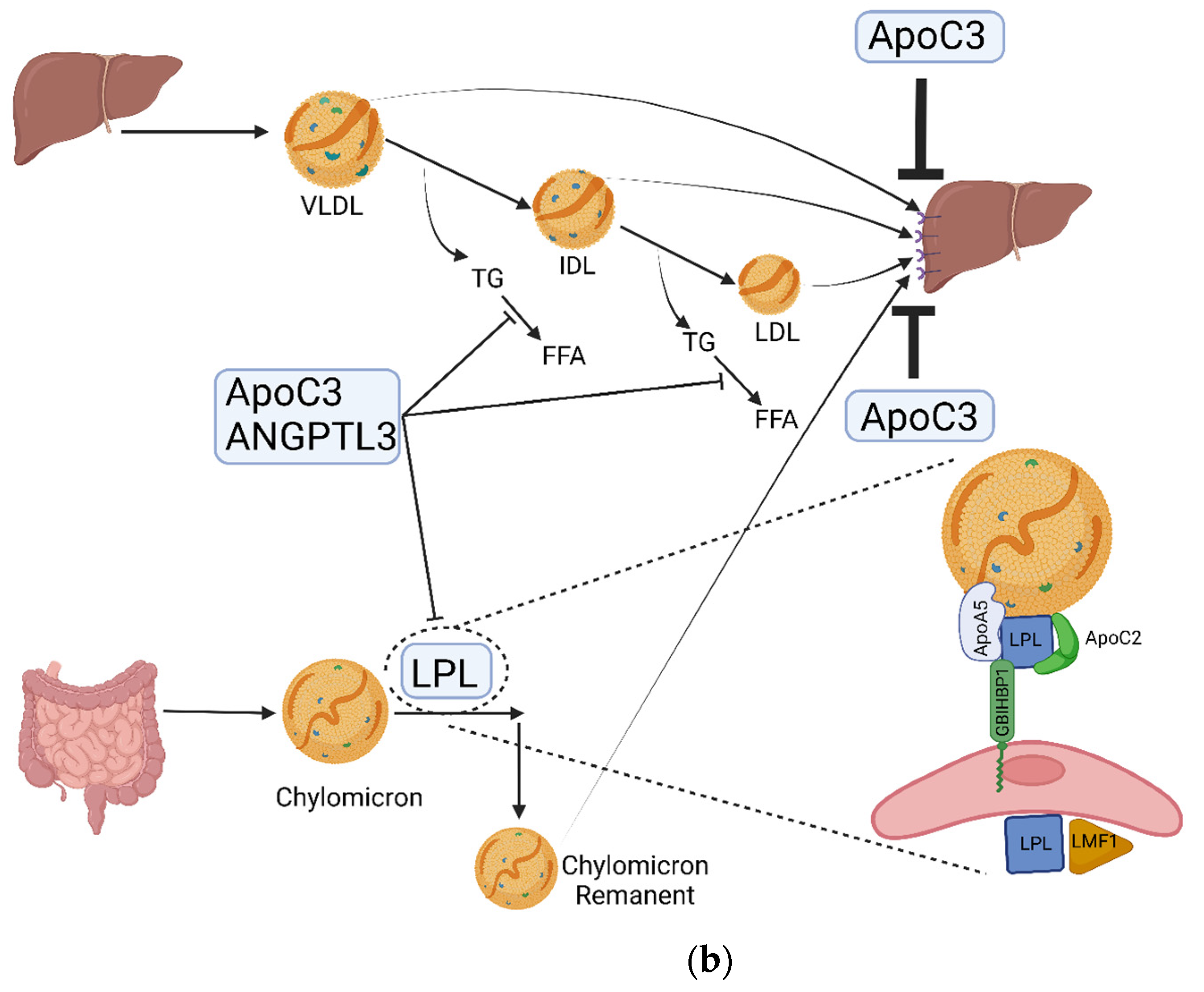{kind=link}
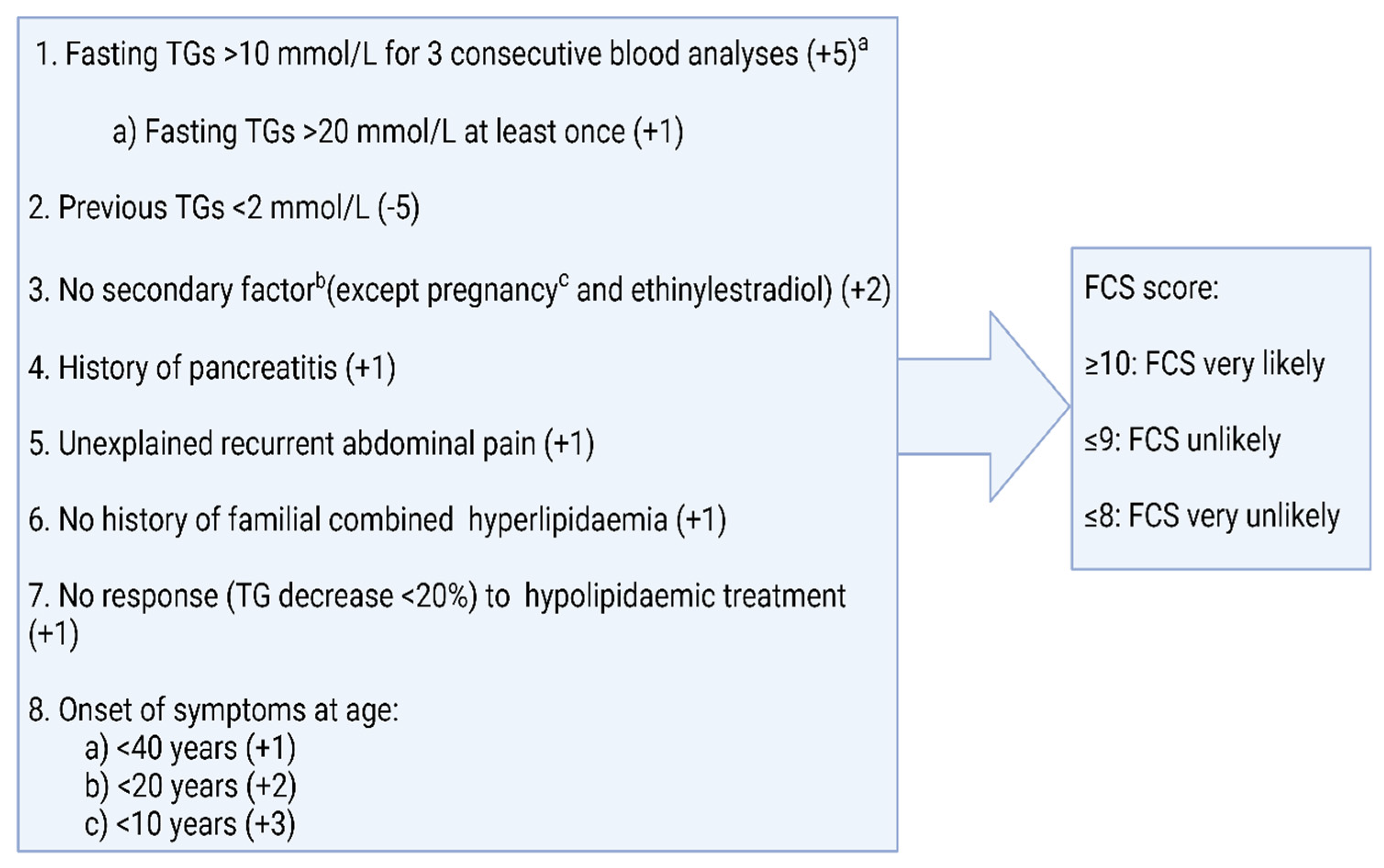{kind=link}
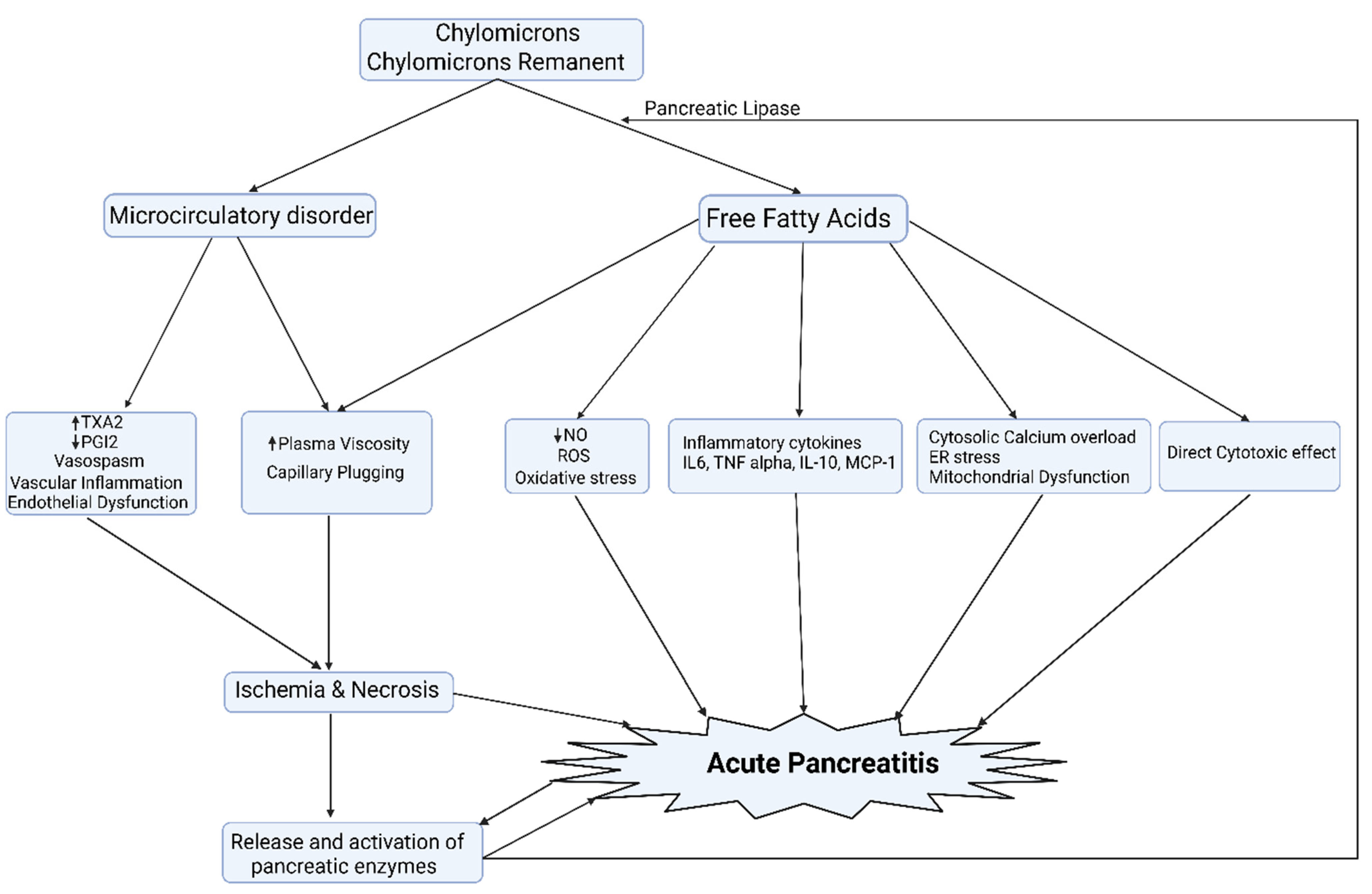{kind=link}
{kind=link}
{kind=link}
| 2018 AHA/ACC Clinical Practice Guidelines * | NCEP ATP III | Endocrine Society | ESC/EAS Guidelines |
|---|---|---|---|
| Normal: <2.0 mmol/L Moderate: 2.0–5.6 mmol/L Severe: >5.6 mmol/L | Normal: <1.7 mmol/L Borderline high: 1.7–2.3 mmol/L High: 2.3–5.6 mmol/L Very High: >5.6 mmol/L | Normal: <1.7 mmol/L Mild: 1.7–2.3 mmol/L Moderate: 2.3–11.2 mmol/L Severe: 11.2–22.4 mmol/L Very severe: >22.4 mmol/L | Normal: <1.7 mmol/L Mild to Moderate: >1.7 mmol/L Severe: >10.0 mmol/L |


2. Epidemiology of Severe Hypertriglyceridaemia
3. Primary Hypertriglyceridaemia
3.1. Chylomicronaemia Syndromes
- Eruptive xanthomata;
- Lipemia retinalis;
- Recurrent abdominal pain;
- Acute/chronic pancreatitis;
- Hepatosplenomegaly;
- Neuropsychiatric and cognitive complications.
3.1.1. Differentiation between FCS and MCS
3.1.2. Genetic Basis of Familial Chylomicronaemia Syndrome (FCS)
3.2. Familial Dysbetalipoproteinaemia
3.3. Lipodystrophies
4. Secondary Hypertriglyceridaemia
5. Complications of Severe Hypertriglyceridaemia
5.1. Acute Pancreatitis
5.2. Atherosclerotic Cardiovascular Disease
5.3. Microvascular Disease
5.3.1. Retinopathy
5.3.2. Neuropathy
5.3.3. Nephropathy
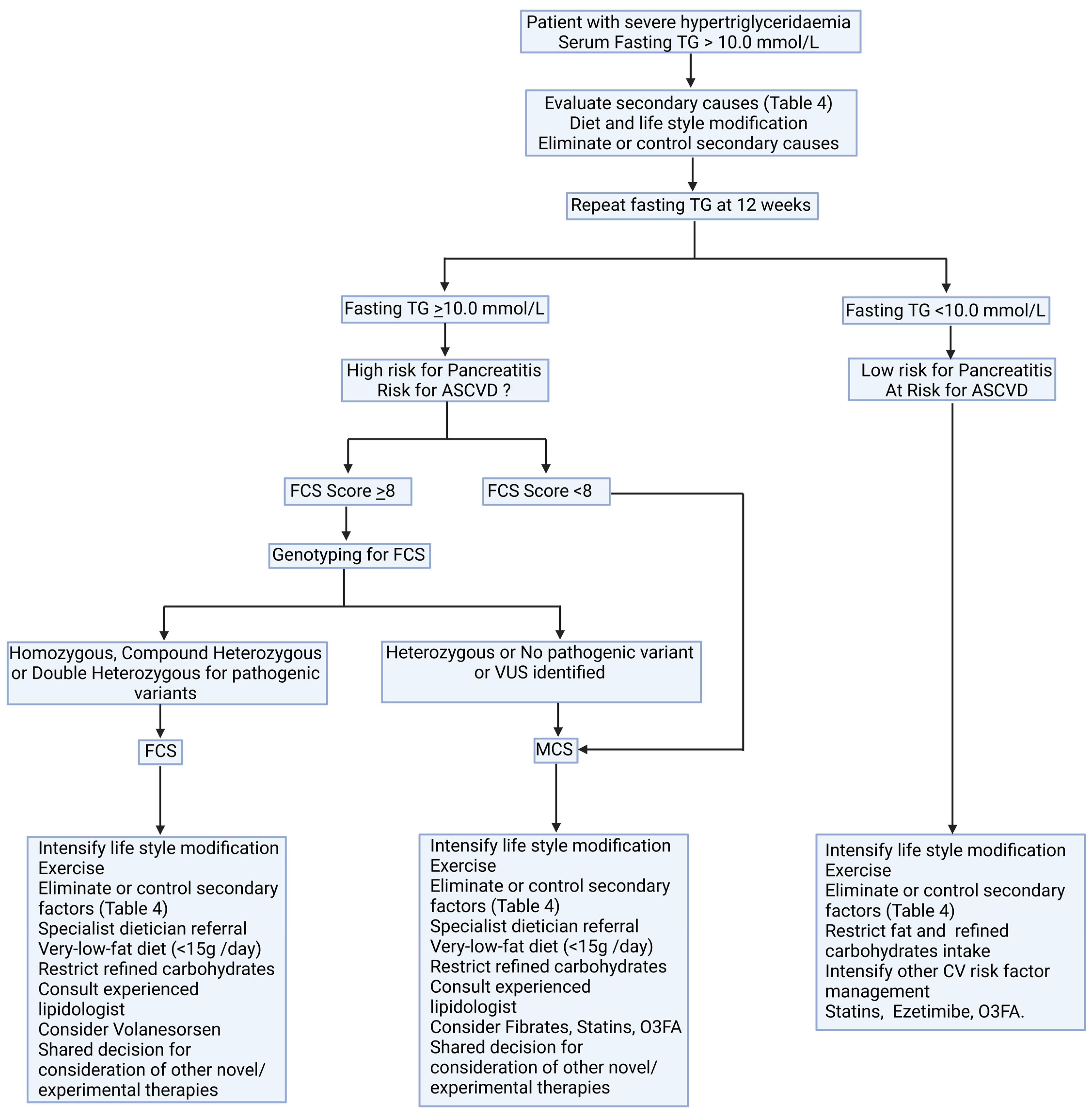
6. Management
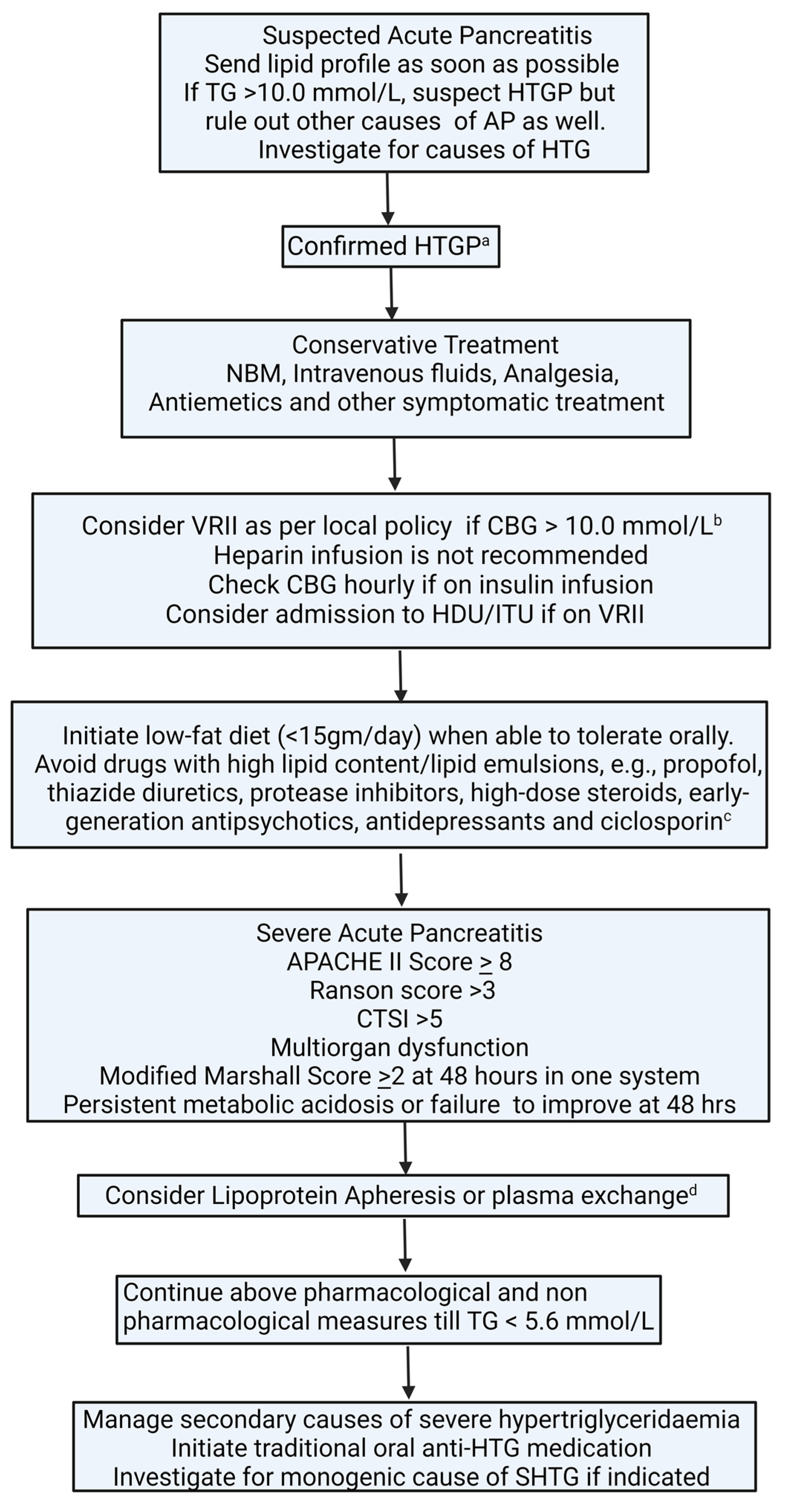
6.1. Acute Management of Hypertriglyceridaemia in Acute Pancreatitis
6.2. Diet and Lifestyle
6.3. Pharmacotherapy
Novel Agents
6.4. Bariatric Surgery
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef]
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, J.M.; Drexel, H.; Hoes, A.V.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur. Heart J. 2016, 37, 2999–3058. [Google Scholar] [CrossRef] [PubMed]
- Berglund, L.; Brunzell, J.D.; Goldberg, A.C.; Goldberg, I.J.; Sacks, F.; Murad, M.H.; Stalenhoef, A.F. Evaluation and treatment of hypertriglyceridemia: An Endo-crine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2012, 97, 2969–2989. [Google Scholar] [CrossRef] [PubMed]
- Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002, 106, 3143–3421. [CrossRef]
- Virani, S.S.; Morris, P.B.; Agarwala, A.; Ballantyne, C.M.; Birtcher, K.K.; Kris-Etherton, P.M.; Stone, N.J. 2021 ACC Expert Consensus Decision Pathway on the Management of ASCVD Risk Reduction in Patients with Persistent Hypertriglyceridemia: A Report of the American College of Car-diology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2021, 78, 960–993. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Yeboah, J. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar]
- Bhatnagar, D.; Soran, H.; Durrington, P.N. Hypercholesterolaemia and its management. BMJ 2008, 337, a993. [Google Scholar] [CrossRef]
- Karpov, Y.; Khomitskaya, Y. PROMETHEUS: An observational, cross-sectional, retrospective study of hypertriglyceridemia in Russia. Cardiovasc. Diabetol. 2015, 14, 115. [Google Scholar] [CrossRef]
- Dron, J.S.; Wang, J.; Cao, H.; McIntyre, A.D.; Iacocca, M.A.; Menard, J.R.; Movsesyan, I.; Malloy, M.J.; Pullinger, C.R.; Kane, J.P.; et al. Severe hypertriglyceridemia is primarily polygenic. J. Clin. Lipidol. 2019, 13, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Chyzhyk, V.; Kozmic, S.; Brown, A.S.; Hudgins, L.C.; Starc, T.J.; Davila, A.D.; Blevins, T.C.; Diffenderfer, M.R.; He, L.; Geller, A.S.; et al. Extreme hypertriglyceridemia: Genetic diversity, pancreatitis, pregnancy, and prevalence. J. Clin. Lipidol. 2018, 13, 89–99. [Google Scholar] [CrossRef]
- Fan, W.; Philip, S.; Granowitz, C.; Toth, P.P.; Wong, N.D. Hypertriglyceridemia in statin-treated US adults: The National Health and Nutri-tion Examination Survey. J. Clin. Lipidol. 2019, 13, 100–108. [Google Scholar] [CrossRef]
- Patel, R.S.; Pasea, L.; Soran, H.; Downie, P.; Jones, R.; Hingorani, A.D.; Neely, D.; Denaxas, S.; Hemingway, H. Elevated plasma triglyceride concentration and risk of adverse clini-cal outcomes in 1.5 million people: A CALIBER linked electronic health record study. Cardiovasc. Diabetol. 2022, 21, 102. [Google Scholar] [CrossRef] [PubMed]
- Ferrieres, J.; Combis, M.S.; Verdier, C.; Genoux, A.L.; Gennero, I.; Hamdi, S.; Perret, B.; Ruidavets, J.B. P5389Big data and severe hypertriglyceridemia: Prevalence in 297 909 individuals. Eur. Heart J. 2018, 39 (Suppl. S1), ehy566.P5389. [Google Scholar] [CrossRef]
- Retterstøl, K.; Narverud, I.; Selmer, R.; Berge, K.E.; Osnes, I.V.; Ulven, S.M.; Halvorsen, B.; Aukrust, P.; Holven, K.B. Severe hypertriglyceridemia in Norway: Prevalence, clinical and genetic characteristics. Lipids Health Dis. 2017, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Hegele, R.A.; Ginsberg, H.N.; Chapman, M.J.; Nordestgaard, B.G.; Kuivenhoven, J.A.; Averna, M.; Wiklund, O. The polygenic nature of hypertriglyceridaemia: Implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014, 2, 655–666. [Google Scholar] [CrossRef]
- Dewey, F.E.; Murray, M.F.; Overton, J.D.; Habegger, L.; Leader, J.B.; Fetterolf, S.N.; O’Dushlaine, C.; Van Hout, C.V.; Staples, J.; Gonzaga-Jauregui, C.; et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016, 354, aaf6814. [Google Scholar] [CrossRef]
- D’Erasmo, L.; Di Costanzo, A.; Cassandra, F.; Minicocci, I.; Polito, L.; Montali, A.; Ceci, F.; Arca, M. Spectrum of Mutations and Long-Term Clinical Outcomes in Genetic Chylomicronemia Syndromes. Arter. Thromb. Vasc. Biol. 2019, 39, 2531–2541. [Google Scholar] [CrossRef]
- Moulin, P.; Dufour, R.; Averna, M.; Arca, M.; Cefalù, A.B.; Noto, D.; D’Erasmo, L.; Di Costanzo, A.; Marçais, C.; Walther, L.A.A.-S.; et al. Identification and diagnosis of patients with familial chylomicronaemia syndrome (FCS): Expert panel recommendations and proposal of an “FCS score”. Atherosclerosis 2018, 275, 265–272. [Google Scholar] [CrossRef]
- Hegele, R.A. Is Genetic Testing in Hypertriglyceridemia Useful? Arter. Thromb. Vasc. Biol. 2022, 42, 1468–1470. [Google Scholar] [CrossRef]
- Goldberg, R.B.; Chait, A. A Comprehensive Update on the Chylomicronemia Syndrome. Front. Endocrinol. 2020, 11, 593931. [Google Scholar] [CrossRef]
- Sandhu, S.; Al-Sarraf, A.; Taraboanta, C.; Frohlich, J.; Francis, G.A. Incidence of pancreatitis, secondary causes, and treatment of patients re-ferred to a specialty lipid clinic with severe hypertriglyceridemia: A retrospective cohort study. Lipids Health Dis. 2011, 10, 157. [Google Scholar] [CrossRef]
- Scherer, J.; Singh, V.P.; Pitchumoni, C.S.; Yadav, D. Issues in hypertriglyceridemic pancreatitis: An update. J. Clin. Gastroenterol. 2014, 48, 195–203. Available online: https://pubmed.ncbi.nlm.nih.gov/24172179 (accessed on 14 March 2023). [CrossRef] [PubMed]
- Paquette, M.; Bernard, S.; Hegele, R.A.; Baass, A. Chylomicronemia: Differences between familial chylomicronemia syndrome and multi-factorial chylomicronemia. Atherosclerosis 2019, 283, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, Z.; Kwok, S.; Donn, R.; Ho, J.; Jones, A.; Mansfield, M.; Wierzbick, A.; Gupta, P.; Dawson, C.; Miedzybrodzka, Z.; et al. An initial analysis of the United Kingdom registry of familial chylomicronaemia syndrome patients. Atherosclerosis 2020, 315, e44–e45. [Google Scholar] [CrossRef]
- O’Dea, L.S.L.; MacDougall, J.; Alexander, V.J.; Digenio, A.; Hubbard, B.; Arca, M.; Moriarty, P.M.; Kastelein, J.J.P.; Bruckert, E.; Soran, H.; et al. Differentiating Familial Chylomicronemia Syndrome from Multifactorial Severe Hypertriglyceridemia by Clinical Profiles. J. Endocr. Soc. 2019, 3, 2397–2410. [Google Scholar] [CrossRef]
- Rioja, J.; Ariza, M.; García-Casares, N.; Coca-Prieto, I.; Arrobas, T.; Muñiz-Grijalvo, O.; Mangas, A.; Ibarretxe, D.; Sánchez-Chaparro, M.; Valdivielso, P. Evaluation of the chylomicron-TG to VLDL-TG ratio for type I hyperlipoproteinemia diagnostic. Eur. J. Clin. Investig. 2020, 50, e13345. [Google Scholar] [CrossRef]
- Sparkes, R.S.; Zollman, S.; Klisak, I.; Kirchgessner, T.G.; Komaromy, M.C.; Mohandas, T.; Lusis, A.J. Human genes involved in lipolysis of plasma lipo-proteins: Mapping of loci for lipoprotein lipase to 8p22 and hepatic lipase to 15q21. Genomics 1987, 1, 138–144. [Google Scholar] [CrossRef]
- Hegele, R.A.; Berberich, A.J.; Ban, M.R.; Wang, J.; Digenio, A.; Alexander, V.J.; Gaudet, D. Clinical and biochemical features of different molecular etiol-ogies of familial chylomicronemia. J. Clin. Lipidol. 2018, 12, 920–927.e4. [Google Scholar] [CrossRef]
- Brahm, A.J.; Hegele, R.A. Chylomicronaemia—Current diagnosis and future therapies. Nat. Rev. Endocrinol. 2015, 11, 352–362. [Google Scholar] [CrossRef]
- Joshi, M.; Eagan, J.; Desai, N.K.; Newton, S.A.; Towne, M.C.; Marinakis, N.S.; Agrawal, P.B. A compound heterozygous mutation in GPD1 causes hepato-megaly, steatohepatitis, and hypertriglyceridemia. Eur. J. Hum. Genet. 2014, 22, 1229–1232. [Google Scholar] [CrossRef]
- Dron, J.S.; Dilliott, A.A.; Lawson, A.; McIntyre, A.D.; Davis, B.D.; Wang, J.; Cao, H.; Movsesyan, I.; Malloy, M.J.; Pullinger, C.R.; et al. Loss-of-Function CREB3L3 Variants in Patients with Severe Hypertriglyceridemia. Arter. Thromb. Vasc. Biol. 2020, 40, 1935–1941. [Google Scholar] [CrossRef]
- Koopal, C.; Marais, A.D.; Visseren, F.L.J. Familial dysbetalipoproteinemia: An underdiagnosed lipid disorder. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 133–139. [Google Scholar] [CrossRef]
- Blom, D.J.; O’Neill, F.H.; Marais, A.D. Screening for Dysbetalipoproteinemia by Plasma Cholesterol and Apolipoprotein B Concentrations. Clin. Chem. 2005, 51, 904–907. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, M.; Duhon, G.; Riaz, R.; Jetty, V.; Goldenberg, N.; Glueck, C.J.; Wang, P. Pathognomonic Palmar Crease Xanthomas of Apolipoprotein E2 Homozygosity-Familial Dysbetalipoproteinemia. JAMA Dermatol. 2016, 152, 1275–1276. [Google Scholar] [CrossRef]
- Blom, D.; Byrnes, P.; Jones, S.; Marais, A.D. Dysbetalipoproteinaemia--clinical and pathophysiological features. S. Afr. Med. J. 2002, 92, 892–897. [Google Scholar]
- Koopal, C.; Retterstøl, K.; Sjouke, B.; Hovingh, G.; Ros, E.; de Graaf, J.; Dullaart, R.; Bertolini, S.; Visseren, F. Vascular risk factors, vascular disease, lipids and lipid targets in patients with familial dysbetalipoproteinemia: A European cross-sectional study. Atherosclerosis 2015, 240, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D.; de Graaf, J.; Thanassoulis, G.; Tremblay, A.J.; Martin, S.S.; Couture, P. The spectrum of type III hyperlipoproteinemia. J. Clin. Lipidol. 2018, 12, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Bagias, C.; Xiarchou, A.; Bargiota, A.; Tigas, S. Familial Partial Lipodystrophy (FPLD): Recent Insights. Diabetes Metab. Syndr. Obes. Targets Ther. 2020, 13, 1531–1544. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Durrington, P.N.; O’Rahilly, S.; Laing, I.; Humphreys, P.J.; Olukoga, A.O.; Bhatnagar, D.; Mackness, M.I.; Davis, J.R.; Boulton, A.J. Severe insulin resistance, diabetes mellitus, hypertriglyceridemia, and pseudoacromegaly. J. Clin. Endocrinol. Metab. 1996, 81, 3465–3468. [Google Scholar] [CrossRef]
- Patni, N.; Garg, A. Congenital generalized lipodystrophies—New insights into metabolic dysfunction. Nat. Rev. Endocrinol. 2015, 11, 522–534. [Google Scholar] [CrossRef]
- Herbst, K.L.; Tannock, L.R.; Deeb, S.S.; Purnell, J.Q.; Brunzell, J.D.; Chait, A. Köbberling type of familial partial lipodystrophy: An underrecognized syndrome. Diabetes Care 2003, 26, 1819–1824. [Google Scholar] [CrossRef]
- Guillín-Amarelle, C.; Sánchez-Iglesias, S.; Castro-Pais, A.; Rodriguez-Cañete, L.; Ordóñez-Mayán, L.; Pazos, M.; Araújo-Vilar, D. Type 1 familial partial lipo-dystrophy: Understanding the Köbberling syndrome. Endocrine 2016, 54, 411–421. [Google Scholar] [CrossRef]
- Patni, N.; Li, X.; Adams-Huet, B.; Vasandani, C.; Gomez-Diaz, R.A.; Garg, A. Regional Body Fat Changes and Metabolic Complications in Children with Dunnigan Lipodystrophy-Causing LMNA Variants. J. Clin. Endocrinol. Metab. 2018, 104, 1099–1108. [Google Scholar] [CrossRef]
- Lazarte, J.; Wang, J.; McIntyre, A.D.; Hegele, R.A. Prevalence of severe hypertriglyceridemia and pancreatitis in familial partial lipodystrophy type 2. J. Clin. Lipidol. 2021, 15, 653–657. [Google Scholar] [CrossRef]
- Garg, A. Lipodystrophies: Genetic and Acquired Body Fat Disorders. J. Clin. Endocrinol. Metab. 2011, 96, 3313–3325. [Google Scholar] [CrossRef]
- Soran, H.; Adam, S.; Mohammad, J.B.; Ho, J.H.; Schofield, J.D.; Kwok, S.; Siahmansur, T.; Liu, Y.; Syed, A.A.; Dhage, S.S.; et al. Hypercholesterolaemia—Practical information for non-specialists. Arch. Med. Sci. 2018, 1, 1–21. [Google Scholar] [CrossRef]
- Durrington, P.N. Hyperlipidaemia: Diagnosis and Management, 3rd ed.; Hodder Arnold: London, UK, 2007. [Google Scholar]
- Koutroumpakis, E.; Slivka, A.; Furlan, A.; Dasyam, A.K.; Dudekula, A.; Greer, J.B.; Whitcomb, D.C.; Yadav, D.; Papachristou, G.I. Management and outcomes of acute pancreatitis patients over the last decade: A US tertiary-center experience. Pancreatology 2017, 17, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pan, X.; Zeng, H.; He, W.; Xia, L.; Liu, P.; Zhu, Y.; Chen, Y.; Lv, N. A Study on the Etiology, Severity, and Mortality of 3260 Patients with Acute Pancreatitis According to the Revised Atlanta Classification in Jiangxi, China Over an 8-Year Period. Pancreas 2017, 46, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Bálint, E.R.; Fűr, G.; Kiss, L.; Németh, D.I.; Soós, A.; Hegyi, P.; Rakonczay, Z., Jr. Assessment of the course of acute pancreatitis in the light of aetiology: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 17936. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, G.; Qiu, Z.; He, X.; Liu, C. Elevated Serum Triglycerides in the Prognostic Assessment of Acute Pancreatitis: A Systematic Review and Meta-Analysis of Observational Studies. J. Clin. Gastroenterol. 2017, 51, 586–593. [Google Scholar] [CrossRef]
- Belhassen, M.; van Ganse, E.; Nolin, M.; Bérard, M.; Bada, H.; Bruckert, E.; Krempf, M.; Rebours, V.; Valero, R.; Moulin, P. 10-Year Comparative Follow-up of Familial versus Multifactori-al Chylomicronemia Syndromes. J. Clin. Endocrinol. Metab. 2021, 106, e1332–e1342. [Google Scholar] [CrossRef]
- Paquette, M.; Amyot, J.; Fantino, M.; Baass, A.; Bernard, S. Rare Variants in Triglycerides-Related Genes Increase Pancreatitis Risk in Multi-factorial Chylomicronemia Syndrome. J. Clin. Endocrinol Metab. 2021, 106, e3473–e3482. [Google Scholar] [CrossRef]
- Ariza, M.J.; Rioja, J.; Ibarretxe, D.; Camacho, A.; Díaz-Díaz, J.L.; Mangas, A.; Carbayo-Herencia, J.A.; Ruiz-Ocaña, P.; Lamíquiz-Moneo, I.; Mosquera, D.; et al. Molecular basis of the familial chylomicronemia syndrome in patients from the National Dyslipidemia Registry of the Spanish Atherosclerosis Society. J. Clin. Lipidol. 2018, 12, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Blom, D.; Bruckert, E.; Stroes, E.; Kastelein, J.; John, K.; Malloy, M.; Moulin, P.; Retterstøļl, K.; Hughes, S.; et al. Acute Pancreatitis is Highly Prevalent and Complications can be Fatal in Patients with Familial Chylomicronemia: Results from a Survey of Lipidologist. J. Clin. Lipidol. 2016, 10, 680–681. [Google Scholar] [CrossRef]
- Gaudet, D.; Signorovitch, J.; Swallow, E.; Fan, L.; Tremblay, K.; Brisson, D.; Meyers, C.; Gruenberger, J.B. Medical resource use and costs associated with chylomicronemia. J. Med. Econ. 2013, 16, 657–666. [Google Scholar] [CrossRef]
- Davidson, M.; Stevenson, M.; Hsieh, A.; Ahmad, Z.; Roeters van Lennep, J.; Crowson, C.; Witztum, J.L. The burden of familial chylomicronemia syndrome: Results from the global IN-FOCUS study. J. Clin. Lipidol. 2018, 12, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Ewald, N.; Hardt, P.D.; Kloer, H.U. Severe hypertriglyceridemia and pancreatitis: Presentation and management. Curr. Opin. Lipidol. 2009, 20, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Havel, R.J. Pathogenesis, differentiation and management of hypertriglyceridemia. Adv. Intern. Med. 1969, 15, 117–154. [Google Scholar]
- Nordestgaard, B.G.; Varbo, A. Triglycerides and cardiovascular disease. Lancet 2014, 384, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Arca, M.; Veronesi, C.; D’Erasmo, L.; Borghi, C.; Colivicchi, F.; De Ferrari, G.M.; Desideri, G.; Pontremoli, R.; Temporelli, P.L.; Perrone, V.; et al. Association of Hypertriglyceridemia with All-Cause Mortality and Atherosclerotic Cardiovascular Events in a Low-Risk Italian Population: The TG-REAL Retrospective Cohort Analysis. J. Am. Heart. Assoc. 2020, 9, e015801. [Google Scholar] [CrossRef]
- Mitropoulos, K.; Miller, G.; Watts, G.; Durrington, P. Lipolysis of triglyceride-rich lipoproteins activates coagulant factor XII: A study in familial lipoprotein-lipase deficiency. Atherosclerosis 1992, 95, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Junker, R.; Heinrich, J.; Schulte, H.; van de Loo, J.; Assmann, G. Coagulation Factor VII and the Risk of Coronary Heart Disease in Healthy Men. Arter. Thromb. Vasc. Biol. 1997, 17, 1539–1544. [Google Scholar] [CrossRef]
- Woodmansey, C.; McGovern, A.P.; McCullough, K.A.; Whyte, M.B.; Munro, N.M.; Correa, A.C.; Gatenby, P.A.; Jones, S.A.; de Lusignan, S. Incidence, Demographics, and Clinical Characteristics of Diabetes of the Exocrine Pancreas (Type 3c): A Retrospective Cohort Study. Diabetes Care 2017, 40, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, I.J.; Mohrschladt, M.F.; Westendorp, R.G.; van der Laarse, A.; Smelt, A.H. Severe hypertriglyceridemia with insulin resistance is associated with systemic inflammation: Reversal with bezafibrate therapy in a randomized controlled trial. Am. J. Med. 2002, 112, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.D.; Fisher, M.R.; Gangnon, R.E.; Barton, F.; Aiello, L.M.; Chew, E.Y.; Ferris, F.L.; Knatterud, G.L. Risk factors for high-risk proliferative diabetic retinopathy and severe visual loss: Early Treatment Diabetic Retinopathy Study Report #18. Investig. Opthalmology Vis. Sci. 1998, 39, 233–252. [Google Scholar]
- Diabetes Control and Complications Trial Research Group. Progression of retinopathy with intensive versus conventional treatment in the Diabetes Control and Complications Trial. Ophthalmology 1995, 102, 647–661. [Google Scholar] [CrossRef]
- Miljanovic, B.; Glynn, R.J.; Nathan, D.M.; Manson, J.E.; Schaumberg, D.A. A Prospective Study of Serum Lipids and Risk of Diabetic Macular Edema in Type 1 Diabetes. Diabetes 2004, 53, 2883–2892. [Google Scholar] [CrossRef]
- Chew, E.Y.; Klein, M.L.; Ferris, F.L., 3rd; Remaley, N.A.; Murphy, R.P.; Chantry, K.; Hoogwerf, B.J.; Miller, D. Association of elevated serum lipid levels with retinal hard exudate in diabetic retinopathy. Early Treatment Diabetic Retinopathy Study (ETDRS) Report 22. Arch. Ophthalmol. 1996, 114, 1079–1084. [Google Scholar] [CrossRef]
- van Leiden, H.A.; Dekker, J.M.; Moll, A.C.; Nijpels, G.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D.; Polak, B.C. Blood Pressure, Lipids, and Obesity Are Associated with Retinopathy. Diabetes Care 2002, 25, 1320–1325. [Google Scholar] [CrossRef]
- Jende, J.M.E.; Groener, J.B.; Oikonomou, D.; Heiland, S.; Kopf, S.; Pham, M.; Nawroth, P.; Bendszus, M.; Kurz, F.T. Diabetic neuropathy differs between type 1 and type 2 diabetes: Insights from magnetic resonance neurography. Ann. Neurol. 2018, 83, 588–598. [Google Scholar] [CrossRef]
- Braffett, B.H.; Gubitosi-Klug, R.A.; Albers, J.W.; Feldman, E.L.; Martin, C.L.; White, N.H.; Orchard, T.J.; Lopes-Virella, M.; Lachin, J.M.; Pop-Busui, R.; et al. Risk Factors for Diabetic Peripheral Neuropathy and Cardiovascular Autonomic Neuropathy in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study. Diabetes 2020, 69, 1000–1010. [Google Scholar] [CrossRef]
- Tesfaye, S.; Chaturvedi, N.; Eaton, S.E.; Ward, J.D.; Manes, C.; Ionescu-Tirgoviste, C.; Witte, D.R.; Fuller, J.H. Vascular Risk Factors and Diabetic Neuropathy. N. Engl. J. Med. 2005, 352, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Maser, R.E.; Pfeifer, M.A.; Dorman, J.S.; Kuller, L.H.; Becker, D.J.; Orchard, T.J. Diabetic autonomic neuropathy and cardiovascular risk. Pittsburgh Epidemiology of Diabetes Complications Study III. Arch. Intern. Med. 1990, 150, 1218–1222. [Google Scholar] [CrossRef]
- D’Onofrio, L.; Kalteniece, A.; Iqbal, Z.; Adam, S.; Ho, J.; Ferdousi, M.; Liu, Y.; Donn, R.; Petropoulos, I.; Ponirakis, G.; et al. Patients with severe hypertriglyceridaemia have neuropathy and small nerve fibre damage. Atherosclerosis 2020, 315, e162–e163. [Google Scholar] [CrossRef]
- D’Onofrio, L.; Ferdousi, M.; Kalteniece, A.; Iqbal, Z.; Petropoulos, I.N.; Ponirakis, G.; Buzzetti, R.; Malik, R.A.; Soran, H. Corneal confocal microscopy identifies small nerve fibre damage in patients with hypertriglyceridemia. J. Clin. Lipidol. 2022, 16, 463–471. [Google Scholar] [CrossRef]
- Adam, S.; Azmi, S.; Ho, J.H.; Liu, Y.; Ferdousi, M.; Siahmansur, T.; Kalteniece, A.; Marshall, A.; Dhage, S.S.; Iqbal, Z.; et al. Improvements in Diabetic Neuropathy and Nephropathy After Bariatric Surgery: A Prospective Cohort Study. Obes. Surg. 2020, 31, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Azmi, S.; Ferdousi, M.; Liu, Y.; Adam, S.; Iqbal, Z.; Dhage, S.; Ponirakis, G.; Siahmansur, T.; Marshall, A.; Petropoulos, I.; et al. Bariatric surgery leads to an improvement in small nerve fibre damage in subjects with obesity. Int. J. Obes. 2021, 45, 631–638. [Google Scholar] [CrossRef]
- Cai, Z.; Yang, Y.; Zhang, J. A systematic review and meta-analysis of the serum lipid profile in prediction of diabetic neuropathy. Sci. Rep. 2021, 11, 499. [Google Scholar] [CrossRef]
- Soran, H.; Charlton-Menys, V.; Hegele, R.; Wang, J.; Benbow, E.W.; Roberts, I.; Wood, G.; Durrington, P. Proteinuria and severe mixed dyslipidemia associated with a novel APOAV gene mutation. J. Clin. Lipidol. 2010, 4, 310–313. [Google Scholar] [CrossRef]
- Liberopoulos, E.; Siamopoulos, K.; Elisaf, M. Apolipoprotein E and renal disease. Am. J. Kidney Dis. 2004, 43, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Matsunaga, A.; Oikawa, S. Impact of Lipoprotein Glomerulopathy on the Relationship Between Lipids and Renal Diseases. Am. J. Kidney Dis. 2006, 47, 199–211. [Google Scholar] [CrossRef]
- Asghari, G.; Momenan, M.; Yuzbashian, E.; Mirmiran, P.; Azizi, F. Dietary pattern and incidence of chronic kidney disease among adults: A pop-ulation-based study. Nutr. Metab. 2018, 15, 88. [Google Scholar] [CrossRef]
- Liang, X.; Ye, M.; Tao, M.; Zheng, D.; Cai, R.; Zhu, Y.; Jin, J.; He, Q. The association between dyslipidemia and the incidence of chronic kidney disease in the general Zhejiang population: A retrospective study. BMC Nephrol. 2020, 21, 1–9. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for Insulin Resistance: Common Threads and Missing Links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Adeosun, S.O.; Gordon, D.M.; Weeks, M.F.; Moore, K.H.; Hall, J.E.; Hinds, T.D.J.; Stec, D.E. Loss of biliverdin reductase-A promotes lipid accumulation and lipotoxicity in mouse proximal tubule cells. Am. J. Physiol. Renal. Physiol. 2018, 315, F323–F331. [Google Scholar] [CrossRef]
- McMahon, G.M.; Preis, S.R.; Hwang, S.J.; Fox, C.S. Mid-adulthood risk factor profiles for CKD. J. Am. Soc. Nephrol. 2014, 25, 2633–2641. [Google Scholar] [CrossRef] [PubMed]
- Cases, A.; Coll, E. Dyslipidemia and the progression of renal disease in chronic renal failure patients. Kidney Int. 2005, 68, S87–S93. [Google Scholar] [CrossRef]
- Mahley, R.W. Atherogenic hyperlipoproteinemia. The cellular and molecular biology of plasma lipoproteins altered by dietary fat and cholesterol. Med. Clin. N. Am. 1982, 66, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Gyebi, L.; Soltani, Z.; Reisin, E. Lipid Nephrotoxicity: New Concept for an Old Disease. Curr. Hypertens. Rep. 2012, 14, 177–181. [Google Scholar] [CrossRef]
- Stefanutti, C.; Labbadia, G.; Morozzi, C. Severe Hypertriglyceridemia-Related Acute Pancreatitis. Ther. Apher. Dial. 2013, 17, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Rustagi, T. Management of Hypertriglyceridemia Induced Acute Pancreatitis. BioMed Res. Int. 2018, 2018, 4721357. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Li, M.; Guo, F.; Zhang, G.; Song, S.; Liu, N.; Wang, D. Timely Reduction of Triglyceride Levels Is Associated with Decreased Persistent Organ Failure in Hypertriglyceridemic Pancreatitis. Pancreas 2020, 49, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Huang, X.; Zhang, M.; Han, N.; Ning, Y. Rapid reduction in triglyceride levels by therapeutic plasma exchange in patients with hypertriglyceridemic pancreatitis. J. Clin. Apher. 2021, 37, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Dhindsa, S.; Sharma, A.; Al-Khazaali, A.; Sitaula, S.; Nadella, S.; McKee, A.; Albert, S.; Bourey, R.; Dandona, P. Intravenous Insulin Versus Conservative Management in Hypertriglyceridemia-Associated Acute Pancreatitis. J. Endocr. Soc. 2019, 4, bvz019. [Google Scholar] [CrossRef]
- Berberich, A.J.; Ziada, A.; Zou, G.Y.; Hegele, R.A. Conservative management in hypertriglyceridemia-associated pancreatitis. J. Intern. Med. 2019, 286, 644–650. [Google Scholar] [CrossRef]
- Gubensek, J.; Andonova, M.; Jerman, A.; Persic, V.; Vajdic-Trampuz, B.; Zupunski-Cede, A.; Sever, N.; Plut, S. Comparable Triglyceride Reduction with Plasma Exchange and Insulin in Acute Pancreatitis—A Randomized Trial. Front. Med. 2022, 12, 9. [Google Scholar] [CrossRef]
- He, W.H.; Yu, M.; Zhu, Y.; Xia, L.; Liu, P.; Zeng, H.; Lv, N.H. Emergent Triglyceride-lowering Therapy with Early High-volume Hemofiltration Against Low–Molecular-Weight Heparin Combined with Insulin in Hypertriglyceridemic Pancreatitis: A Prospective Randomized Con-trolled Trial. J. Clin. Gastroenterol. 2016, 50, 772–778. Available online: https://journals.lww.com/jcge/Fulltext/2016/10000/Emergent_Triglyceride_lowering_Therapy_With_Early.15.aspx (accessed on 5 December 2022). [CrossRef]
- Zhang, Y.; Lin, J.; Wu, L.; Lin, J.; Liang, Y. Blood Purification for Hypertriglyceridemia-Induced Acute Pancreatitis: A Meta-analysis. Pancreas 2022, 51, 531–539. Available online: https://journals.lww.com/pancreasjournal/Fulltext/2022/05000/Blood_Purification_for.18.aspx (accessed on 5 December 2022). [CrossRef]
- He, W.; Cai, W.; Yang, X.; Camilleri, G.; Zheng, X.; Wang, Q.; Li, Y.; Mukherjee, R.; Huang, W.; Sutton, R. Insulin or blood purification treatment for hypertriglyceridaemia-associated acute pancreatitis: A systematic review and meta-analysis. Pancreatology 2022, 22, 846–857. [Google Scholar] [CrossRef]
- Gubensek, J.; Buturovic-Ponikvar, J.; Romozi, K.; Ponikvar, R. Factors Affecting Outcome in Acute Hypertriglyceridemic Pancreatitis Treated with Plasma Exchange: An Observational Cohort Study. PLoS ONE 2014, 9, e102748. [Google Scholar] [CrossRef]
- Näsström, B.; Olivecrona, G.; Stegmayr, B.G. Lipoprotein lipase during heparin infusion: Lower activity in hemodialysis patients. Scand. J. Clin. Lab. Investig. 2003, 63, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.F.; Cameron, J.; Henderson, A.; Richmond, W. Lipoprotein lipase deficiency due to long-term heparinization presenting as severe hypertriglyceridaemia in pregnancy. Postgrad. Med. J. 1991, 67, 1062. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, M.; Rassin, T.; Eisenberg, S.; Ringel, Y.; Grosskopf, I.; Iaina, A.; Charach, G.; Liron, M.; Rubinstein, A. Continuous intravenous heparin administration in humans causes a decrease in serum lipolytic activity and accumulation of chylomicrons in circulation. J. Lipid Res. 1994, 35, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, J.; Li, G.; Tong, Z.; Dong, J.; Pan, Y.; Ke, L.; Li, W.; Li, J. Early Spontaneous Abdominal Bleeding is associated with Poor Outcome in Moderate to Severe Acute Pancreatitis Patients: A Propensity Matched Study. Sci. Rep. 2017, 7, 42607. [Google Scholar] [CrossRef] [PubMed]
- Tümer, A.R.; Dener, C. Diagnostic Dilemma of Sudden Deaths Due to Acute Hemorrhagic Pancreatitis. J. Forensic Sci. 2007, 52, 180–182. [Google Scholar] [CrossRef]
- Stoppacher, R. Sudden Death Due to Acute Pancreatitis. Acad. Forensic Pathol. 2018, 8, 239–255. [Google Scholar] [CrossRef]
- Padmanabhan, A.; Connelly-Smith, L.; Aqui, N.; Balogun, R.A.; Klingel, R.; Meyer, E.; Pham, H.P.; Schneiderman, J.; Witt, V.; Wu, Y.; et al. Guidelines on the Use of Therapeutic Apheresis in Clinical Practice—Evidence-Based Approach from the Writing Committee of the American Society for Apheresis: The Eighth Special Issue. J. Clin. Apher. 2019, 34, 171–354. [Google Scholar] [CrossRef]
- Zádori, N.; Gede, N.; Antal, J.; Szentesi, A.; Alizadeh, H.; Vincze, Á.; Hegyi, P. EarLy Elimination of Fatty Acids iN hypertriglyceridemia-induced acuTe pancreatitis (ELEFANT trial): Protocol of an open-label, multicenter, adaptive randomized clinical trial. Pancreatology 2019, 20, 369–376. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Insulin and Standard Management in Hypertriglyceridemic Acute Pancreatitis [Internet]. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05487833 (accessed on 3 December 2022).
- McCloy, R. Chronic Pancreatitis at Manchester, UK. Digestion 1998, 59, 36–48. [Google Scholar] [CrossRef]
- Gooshe, M.; Abdolghaffari, A.H.; Nikfar, S.; Mahdaviani, P.; Abdollahi, M. Antioxidant therapy in acute, chronic and post-endoscopic retrograde cholangiopancreatography pancreatitis: An updated systematic review and meta-analysis. World J. Gastroenterol. 2015, 21, 9189–9208. [Google Scholar] [CrossRef]
- Heaney, A.P.; Sharer, N.; Rameh, B.; Braganza, J.M.; Durrington, P.N. Prevention of Recurrent Pancreatitis in Familial Lipoprotein Lipase Defi-ciency with High-Dose Antioxidant Therapy. J. Clin. Endocrinol. Metab. 1999, 84, 1203–1205. [Google Scholar] [CrossRef] [PubMed]
- Ellerton, C.; Firman, S.; Khan, F.; Robertson, L.; Curran, O.; Telford, C.; Donald, S.; Dunlop, C.; Flanagan, C.; Gaff, F.; et al. Current Dietary Practice in the Management of Adults with Familial Chylomicronaemia Syndrome—An Expert Panel Opinion Piece. 2021. Available online: https://www.heartuk.org.uk/downloads/health-professionals/nutrition-acacdemy/ellerton-et-al-2021.-fcs-dietary-management-uk-expert-panel-opinion.pdf (accessed on 5 December 2022).
- Asakura, L.; Lottenberg, A.M.; Neves, M.Q.; Nunes, V.S.; Rocha, J.C.; Passarelli, M.; Nakandakare, E.R.; Quintão, E.C. Dietary medium-chain triacylglycerol prevents the postprandial rise of plasma triacylglycerols but induces hypercholesterolemia in primary hypertriglyceridemic subjects. Am. J. Clin. Nutr. 2000, 71, 701–705. [Google Scholar] [CrossRef]
- Hauenschild, A.; Bretzel, R.G.; Schnell-Kretschmer, H.; Kloer, H.-U.; Hardt, P.D.; Ewald, N. Successful Treatment of Severe Hypertriglyceridemia with a Formula Diet Rich in Omega–3 Fatty Acids and Medium-Chain Triglycerides. Ann. Nutr. Metab. 2010, 56, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.; Rhodes, K.S.; Karmally, W.; Welstead, L.A.; Alexander, L.; Sutton, L. Familial chylomicronemia syndrome: Bringing to life dietary recommendations throughout the life span. J. Clin. Lipidol. 2018, 12, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Dekker, M.J.; Graham, T.E.; Ooi, T.C.; Robinson, L.E. Exercise prior to fat ingestion lowers fasting and postprandial VLDL and decreases adipose tissue IL-6 and GIP receptor mRNA in hypertriacylglycerolemic men. J. Nutr. Biochem. 2010, 21, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Klop, B.; Elte, J.; Cabezas, M. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218–1240. [Google Scholar] [CrossRef]
- Neelamekam, S.; Kwok, S.; Malone, R.; Wierzbicki, A.S.; Soran, H. The impact of lipoprotein lipase deficiency on health-related quality of life: A detailed, structured, qualitative study. Orphanet J. Rare Dis. 2017, 12, 156. [Google Scholar] [CrossRef]
- Frick, M.H.; Elo, O.; Haapa, K.; Heinonen, O.P.; Heinsalmi, P.; Helo, P.; Huttunen, J.K.; Kaitaniemi, P.; Koskinen, P.; Manninen, V.; et al. Helsinki Heart Study: Primary-Prevention Trial with Gemfibrozil in Middle-Aged Men with Dyslipidemia. N. Engl. J. Med. 1987, 317, 1237–1245. [Google Scholar] [CrossRef]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Ehnholm, C.; Keech, A.; Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study Investigators. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2018, 380, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Orlistat for the Treatment of Type I Hyperlipoproteinemia (T1HLP) [Internet]. Available online: https://clinicaltrials.gov/ct2/show/results/NCT02767531 (accessed on 5 December 2022).
- Yadav, R.; Liu, Y.; Kwok, S.; Hama, S.; France, M.; Eatough, R.; Pemberton, P.; Schofield, J.; Siahmansur, T.J.; Malik, R.; et al. Effect of Extended-Release Niacin on High-Density Lipoprotein (HDL) Functionality, Lipoprotein Metabolism, and Mediators of Vascular Inflammation in Statin-Treated Patients. J. Am. Heart. Assoc. 2015, 4, e001508. [Google Scholar] [CrossRef]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.-C. Mechanism of Action of Fibrates on Lipid and Lipoprotein Metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Skulas-Ray, A.C.; Wilson, P.W.; Harris, W.S.; Brinton, E.A.; Kris-Etherton, P.M.; Richter, C.K.; Jacobson, T.A.; Engler, M.B.; Miller, M.; Robinson, J.G.; et al. Omega-3 Fatty Acids for the Management of Hypertriglyceridemia: A Science Advisory From the American Heart Association. Circulation 2019, 140, e673–e691. [Google Scholar] [CrossRef]
- Kamanna, V.S.; Kashyap, M.L. Mechanism of Action of Niacin. Am. J. Cardiol. 2008, 101 (Suppl. S8), pp. S20–S26. Available online: https://www.sciencedirect.com/science/article/pii/S0002914908002531 (accessed on 5 December 2022).
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Gouni-Berthold, I.; Alexander, V.J.; Yang, Q.; Hurh, E.; Steinhagen-Thiessen, E.; Moriarty, P.M.; Hughes, S.G.; Gaudet, D.; Hegele, R.A.; O’Dea, L.S.L.; et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021, 9, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Volanesorsen, Familial Chylomicronemia Syndrome, and Thrombocytopenia. N. Engl. J. Med. 2019, 381, 2582–2584. [CrossRef] [PubMed]
- Durrington, P.N.; Maciver, J.E.; Holdsworth, G.; Galton, D.J. Severe Hypertriglyceridemia Associated with Pancytopenia and Lipoprotein Lipase Deficiency. Ann. Intern. Med. 1981, 94, 211–212. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Identifier: NCT05355402. A Study of Olezarsen (Formerly Known as AKCEA-APOCIII-LRx) in Adults with Hypertriglyceridemia and Atherosclerotic Cardiovascular Disease (Established or at Increased Risk for), and/or with Severe Hypertriglyceridemia [Internet]. Available online: https://clinicaltrials.gov/ct2/show/NCT05355402 (accessed on 5 December 2022).
- Scott, L.J. Alipogene Tiparvovec: A Review of Its Use in Adults with Familial Lipoprotein Lipase Deficiency. Drugs 2015, 75, 175–182. [Google Scholar] [CrossRef]
- Meyers, C.D.; Tremblay, K.; Amer, A.; Chen, J.; Jiang, L.; Gaudet, D. Effect of the DGAT1 inhibitor pradigastat on triglyceride and apoB48 levels in patients with familial chylomicronemia syndrome. Lipids Health. Dis. 2015, 14, 8. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Identifier: NCT01474434. Efficacy of LCQ908 on Cardiovascular Risk [Internet]. Available online: https://clinicaltrials.gov/ct2/show/NCT01474434 (accessed on 5 December 2022).
- Sacks, F.M.; Stanesa, M.; Hegele, R.A. Severe Hypertriglyceridemia with Pancreatitis. JAMA Intern. Med. 2014, 174, 443–447. [Google Scholar] [CrossRef]
- Cefalù, A.B.; D’Erasmo, L.; Iannuzzo, G.; Noto, D.; Giammanco, A.; Montali, A.; Averna, M. Efficacy and safety of lomitapide in familial chylomicro-naemia syndrome. Atherosclerosis 2022, 359, 13–19. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Identifier: NCT04419688. A First in Human Study of STT-5058, an Antibody That Binds ApoC3 [Internet]. Available online: https://clinicaltrials.gov/ct2/show/NCT04419688 (accessed on 5 December 2022).
- ClinicalTrials.gov. Identifier: NCT04832971. Study of ARO-ANG3 in Adults with Mixed Dyslipidemia (ARCHES-2) [Internet]. Available online: https://clinicaltrials.gov/ct2/show/NCT04832971 (accessed on 5 December 2022).
- ClinicalTrials.gov. Identifier: NCT04720534. Study to Evaluate ARO-APOC3 in Adults with Severe Hypertriglyceridemia (SHASTA-2) [In-ternet]. Available online: https://clinicaltrials.gov/ct2/show/NCT04720534 (accessed on 5 December 2022).
- ClinicalTrials.gov. Identifier: NCT04863014. Efficacy and Safety of Evinacumab in Adult Patients with Severe Hypertriglyceridemia for the Prevention of Recurrent Acute Pancreatitis [Internet]. Available online: https://clinicaltrials.gov/ct2/show/NCT04863014 (accessed on 5 December 2022).
- Pfizer Ionis Pharmaceuticals, I. Pfizer and Ionis Announce Discontinuation of Vupanorsen Clinical Development Program [Internet]. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-and-ionis-announce-discontinuation-vupanorsen (accessed on 5 December 2022).
- Gaudet, D.; Karwatowska-Prokopczuk, E.; Baum, S.J.; Hurh, E.; Kingsbury, J.; Bartlett, V.J.; Figueroa, A.L.; Piscitelli, P.; Singleton, W.; Witztum, J.L.; et al. Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur. Heart J. 2020, 41, 3936–3945. [Google Scholar] [CrossRef] [PubMed]
- Padilla, N.; Maraninchi, M.; Béliard, S.; Berthet, B.; Nogueira, J.P.; Wolff, E.; Nicolay, A.; Bégu, A.; Dubois, N.; Grangeot, R.; et al. Effects of bariatric surgery on hepatic and intestinal lipopro-tein particle metabolism in obese, nondiabetic humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2330–2337. [Google Scholar] [CrossRef] [PubMed]
- Castagneto, M.; De Gaetano, A.; Mingrone, G.; Capristo, E.; Benedetti, G.; Tacchino, R.M.; Greco, A.V.; Gasbarrini, G. A Surgical Option for Familial Chylomicronemia Associated with Insulin-Resistant Diabetes Mellitus. Obes. Surg. 1998, 8, 191–198. [Google Scholar] [CrossRef]
- Marco-Benedí, V.; Lamiquiz-Moneo, I.; Álvarez-Sala, L.A.; Civeira, F. Disappearance of recurrent pancreatitis after splenectomy in familial chylomicronemia syndrome. Atherosclerosis 2018, 275, 342–345. [Google Scholar] [CrossRef]




| Features | FCS | MCS |
|---|---|---|
| Age of onset | Paediatric or Early Adolescence | Adulthood |
| Prevalence | 1:100,000–1,000,000 | 1:600 |
| BMI | Normal BMI | Overweight or obese |
| Contribution of secondary factors | Minor | Major |
| Peak TG | Higher | Relatively lower |
| Lowest TG | Higher | Relatively Lower |
| Acute pancreatitis (prevalence) | Significantly higher | High |
| Multiple acute pancreatitis (prevalence) | Significantly higher | At the risk of recurrent pancreatitis |
| Hospital admissions | More frequent | Less frequent |
| Lipoprotein disturbance | Increased number of chylomicron particles. Reduced LDL and HDL. | Increase in the number of TRL, i.e., VLDL, IDL, VLDL, and chylomicron remnant particles. |
| ASCVD | Increased? | Significantly higher than FCS |
| Adverse metabolic phenotype * | Less prevalent | More prevalent |
| Post-heparin LPL activity | Severely reduced | Normal or mildly impaired |
| Inheritance pattern | Autosomal recessive | No discrete pattern |
| Genetic Causes | Presence of two pathogenic variants in LPL, ApoC2, ApoA5, GPIHBP1, LMF1, GPD 1, or CREB3L3 | More than 300 SNVs identified |
| Response to traditional LLT ** | No effect | Mild to moderate effect |
| Treatment | Very-low-fat diet Volanesorsen | Low-fat diet Addressing secondary factors Variable efficacy with pharmacotherapy |
| Genes | Inheritance Pattern | Gene Product Function |
|---|---|---|
| LPL | AR | Hydrolysis of TG, reduction in the size of chylomicrons and VLDL via depleting TG-rich core |
| GPIHBPI1 | AR | Binds LPL from subendothelial interstitial space; transports and anchors it to the luminal surface of endothelial cells |
| ApoA5 | AR | Activation of LPL |
| ApoC2 | AR | Activation of LPL |
| LMF 1 | AR | Maturation, stabilisation, and transport of LPL to the capillary endothelial surface |
| GPD 1 | AR | The exact mechanism is unclear; overexpression of mutated genes leads to overproduction and secretion of TG. |
| CREB3L3 | AR | Functions as a transcription factor for canonical gene expression |
| Causes | Mechanism |
|---|---|
| Obesity, IR, and metabolic syndrome phenotype | Increased production of VLDL due to increased flux of FFA from the expanded adipose tissue mass. |
| Suboptimal diabetes control | Increased VLDL production and reduced chylomicron and VLDL clearance |
| Alcohol | Increased chylomicron and VLDL production, increased lipolysis-free fatty acid fluxes from adipose tissue to the liver |
| Pregnancy | Increased chylomicron and VLDL synthesis, reduced HL and LPL activity, relative IR |
| Chronic renal failure | Downregulation of LPL and LDLR activity |
| Hypothyroidism | Reduced LPL and LDLR activity |
| High-Fat and High-GI food | Increased production of chylomicron and VLDL particles |
| Multiple myeloma | Reduced clearance of TRL particles and reduced function of LPL secondary to paraproteins binding with them |
| SLE | Reduced LPL activity due to endothelial damage and antibodies against LPL |
| Drugs | |
| Thiazide diuretics and beta blockers | Reduced LPL activity |
| Oral Oestrogen, Tamoxifen, Clomiphene | Increased VLDL production |
| Corticosteroids | Increased VLDL production due to IR |
| Protease Inhibitors | Increased VLDL production and reduced LPL activity |
| First- and second-generation antipsychotics and tetracyclic antidepressants | Increased IR and VLDL production and reduced LPL activity |
| Cyclosporin, sirolimus, and everolimus | Increased ApoC3 levels and inhibited LPL |
| Isotretinoin | Increased ApoC3 levels |
| Propofol | Formulated in a 10% oil-in-water lipid emulsion rich in TG and PL and, hence, increased fat delivery |
| Author | Pancreatitis | Recurrent Pancreatitis | CVD | Comments | |||
|---|---|---|---|---|---|---|---|
| FCS % (n) | MCS % (n) | FCS % (n) | MCS % (n) | FCS % (n) | MCS % (n) | ||
| Paquette et al. 2019 [23] (FCS: 25, MCS: 36) | 60 (15) | 6 (2) | 48 (12) | 3 (1) | 0 (0) | 17 (6) | Acute pancreatitis was the presenting feature that led to the ascertainment of CS in 12% of FCS patients and 3% of MCS patients. The FCS cohort was mostly free of metabolic features (55% vs. 6%) while the majority of MCS patients had a combination of two or more adverse metabolic elements (67% vs. 10%) |
| Iqbal et al. 2020 [24] (FCS: 38, MCS: 40) | 76.3 (29) | 27.5 (11) | 50 (19) | 12.5 (5) | 15.8 (6) | 30 (12) | Peak and trough triglyceride levels were higher in FCS (47.4 (19.8) and 10.2 (7.37)) as compared to MCS (35.7 (22.4) and 5.2 (6.3)). The phenotype of LPL FCS was comparable with non-LPL FCS. |
| Ariza et al. 2018 [54] (FCS: 26, MCS: 212) | 88 (23) | 26 (54) | NR | NR | NR | NR | The median number of AP episodes in FCS was 5 (2–12) vs. 1 (1–2) in MCS. Low-fat and/or low-calorie diet led to a significant reduction in TG levels in MCS. However, TG levels remained unchanged in the FCS cohort regardless of the use of conventional LLT and a low-fat diet. |
| O’Dea et al. 2019 [25] (FCS: 50, MCS: 106) | 86 (43) | 21.7 (23) | NR | NR | 2(1) | 9.4 (10) | Baseline data from two phase III trials for volanesorsen (APPROACH and COMPASS). Patients with FCS in the COMPASS trial were excluded from the analysis. BMI and history of pancreatitis along with ApoB100 or ApoA1 had a sensitivity of >90% for the diagnosis of FCS. |
| Gaudet et al. 2016 * [55] (FCS: 251, MCS: 1981) | 67 (168) | 14 (277) | 50 (125) | NR | NR | NR | 33.3% of AP secondary to FCS required ICU care as compared to 3.4% in non-FCS. Pancreatitis-related mortality was higher in FCS as compared to non-FCS AP (6% vs. 0.55%). |
| Paquette et al. 2021 [53] (FCS: 28 MCS: 75 **) | 61 (17) | 18.7 (14) | 46 (13) | 10.7 (8) | 0 | 16 (12) | Prevalence of acute pancreatitis and recurrent pancreatitis was higher in variant-positive MCS (41% and 23%) as compared to variant-negative MCS (9% and 6%). The prevalence of CVD in FCS (0%) was lower but comparable between variant-positive and variant-negative MCS (18% vs. 15%). |
| D’Erasmo et al. 2019 [17] (FCS: 12, MCS: 19) | 75 (9) | 37 (7) | 42 (5) | 16 (3) | 9.1(1) | 15.8 (3) | The estimated overall incidence rate of AP was 42 per 1000 person-years in FCS and 13 per 1000 person-years in MCS. No difference in the phenotype of LPL FCS and non-LPL FCS. |
| Belhassen et al. 2021 [52] (FCS: 29, MCS: 124) | 58.6 (17) | 19.4 (24) | 55.1 (16) | 12.1 (15) | 10.3 (3) | 25 (31) | Longitudinal observational study with a median follow-up of 10 years. Ischemic CVD events in FCS were lower in FCS as compared to MCS but were comparable with controls. |
| Pooled Results | 321/459 (70%) | 412/2593 (16%) | 65/132 (49%) | 32/294 (11%) | 11/182 (6%) | 74/400 (18%) | - |
| Intervention | Mechanism of Action | Comments |
|---|---|---|
| Bowel rest, NBM, IVF, Analgesia (Standard of Care) | Pancreatic rest maintains blood flow to the pancreas, reduces chylomicrons and VLDL production, and reduces HTG burden. | Severe pancreatitis may require a prolonged period of fasting; consider post-ligament of Treitz, enteral feeding or parenteral feeding. Consider fat-free/low-fat enteral parenteral feed. Avoid the use of oil-based medication, e.g., Propofol. |
| Insulin Infusion | Activates LPL activity to accelerate chylomicron degradation and lower TG levels. | Continuous insulin infusion. The risk of hypoglycaemia may outweigh any potential benefits in patients without diabetes. Consider if CBG is persistently >10.0 mmol/L. |
| Heparin Infusion | The initial increase in lipoprotein lipase activity converts TG to FFA. | Risk of rebound hypertriglyceridaemia, worsening of lipotoxicity from FFA, and risk of bleeding in pancreatic bed. Not recommended. |
| Lipoprotein apheresis/ Plasma exchange | Removes TG and inflammatory cytokines. Provides functional LPL (plasma exchange). | May be considered in SHTG with organ failure, worsening systemic inflammation, or acidosis. However, there is no convincing evidence to support including TPE as one of the standard therapies. |
| Class | Agent | Mechanism of Action | Dose | TG Reduction | Comments |
|---|---|---|---|---|---|
| Fibrates | Fenofibrate | Increases TRL catabolism via activation of PPARα and LPL and inhibition of ApoC3 and hepatic synthesis of VLDL | 160–267 mg OD | Up to 50% | No effect on gut-derived chylomicrons. Reduces VLDL particles. Minimal or no effect in FCS patients. |
| Gemfibrozil * | 600 mg BD | ||||
| Bezafibrate | 200 mg TDS | ||||
| Pemafibrate ** | 0.4 mg OD | ||||
| Vitamin B3 | Niacin | Inhibits HSL, reduces FFA delivery to the liver, inhibits DAGAT II, reduces VLDL production | 2 g daily | 15–30% | Less effective when TG is very high. Improves HDL function and reduces Lp(a). May worsen diabetes. Not available in Europe for clinical use. |
| Omega-3 Fatty acids | O3AEE, EPA+DHA (Omacor/Lovaza) | Inhibits VLDL production, Inhibits ApoC 3, increases chylomicron clearance by activating LPL | 4 g daily | 20–30% | Less effective in chylomicronaemia of monogenic origin. Epanova is not commercially available. Vazkepa is the only purified EPA derivative without DHA. |
| O3CA, EPA+DHA (Epanova) | |||||
| IPE (Vazkepa) | |||||
| Gut lipase inhibitor | Orlistat | Gastric and pancreatic lipase inhibitors. Reduces the absorption of fat and chylomicron production | 120 mg TDS | 30–50% | Phase II clinical trial for FCS is ongoing. |
| Leptin Analogue | Metreleptin | Leptin receptor activator | Weight- and Gender-dependent | 30–35% | Approved by NICE for lipodystrophies. |
| Drug | Mechanism of Action | Effect on TG | Phase of Development | Comments |
|---|---|---|---|---|
| Approved Pharmacotherapies | ||||
| Volanesorsen (ISIS-ApoCIIIRx) | ASO against hepatic ApoC3 | 50–70% reduction | Approved by EMA and NICE for use in FCS in 2019. | Thrombocytopenia remains a predominant side effect requiring close monitoring. Not advisable to start if the platelet count is <140 × 109/L. |
| Lomitapide * | MTP inhibitor | Up to 70% reduction | Approved by EMA in 2013 for HoFH. | Individual case reports of the progression of steatohepatitis to fibrosis occurred after 10 years of treatment. |
| Pharmacotherapies in development | ||||
| Olezarcen (AKCEA ApoCIII-LRx) | GalNAc3 conjugated ASO against hepatic ApoC3 | 70% reduction | First Phase III trial is expected to be completed in 2023. | Targets the ASGPR in hepatocytes with similar efficacy as compared to native ASO with 20–30-fold lower dose, therefore minimizing side effects including thrombocytopenia. |
| Evinacumab | Monoclonal antibody against ANGPTL3 | 55% reduction | Phase II trial for SHTG and AP expected to be completed in 2023. | Reduces LDL cholesterol by 47% and was approved by EMA for use in HoFH in 2019. |
| ARO ApoCIII | siRNA against ApoC3 | 40–70% reduction | Phase II trial is expected to be completed in 2023. | In phase I, along with TG reduction, a dose-dependent increase in HDL (40–80%) was also observed. |
| ARO-ANG3 | siRNA against ANGPTL3 | Up to 66% reduction | Phase II trial is expected to be completed in 2024. - | |
| STT-5058 | Monoclonal antibody against ApoC3 | - | Phase I trial was expected to be completed in December 2022—no updates at the time of writing. | |
| Others, Suspended therapies | ||||
| Alipogene Tiparvovec (Glybera) | Gene replacement | 40–60% reduction initially. | Approved by EMA in 2012 for clinical use but withdrawn from the market owing to poor commercial prospects in 2017. | Sustained gene expression and reduced risk of pancreatitis despite the transient effect on hypertriglyceridaemia. |
| Vupanorsen (AKCEA-ANGPTL3-LRx) | ASO against ANGPTL3 | 50–60% reduction | Development halted in 2022 after a review of the Phase 2b (TRANSLATE-TIMI) study. | Data from the Phase 2b trial did not support the clinical development of the drug for CV risk reduction or SHTG. It was also associated with dose-dependent hepatotoxicity. |
| Pradigastat | DAGT inhibitor | 40% reduction | No updates since 2015. | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bashir, B.; Ho, J.H.; Downie, P.; Hamilton, P.; Ferns, G.; Datta, D.; Cegla, J.; Wierzbicki, A.S.; Dawson, C.; Jenkinson, F.; et al. Severe Hypertriglyceridaemia and Chylomicronaemia Syndrome—Causes, Clinical Presentation, and Therapeutic Options. Metabolites 2023, 13, 621. https://doi.org/10.3390/metabo13050621
Bashir B, Ho JH, Downie P, Hamilton P, Ferns G, Datta D, Cegla J, Wierzbicki AS, Dawson C, Jenkinson F, et al. Severe Hypertriglyceridaemia and Chylomicronaemia Syndrome—Causes, Clinical Presentation, and Therapeutic Options. Metabolites. 2023; 13(5):621. https://doi.org/10.3390/metabo13050621
Chicago/Turabian StyleBashir, Bilal, Jan H. Ho, Paul Downie, Paul Hamilton, Gordon Ferns, Dev Datta, Jaimini Cegla, Anthony S. Wierzbicki, Charlotte Dawson, Fiona Jenkinson, and et al. 2023. "Severe Hypertriglyceridaemia and Chylomicronaemia Syndrome—Causes, Clinical Presentation, and Therapeutic Options" Metabolites 13, no. 5: 621. https://doi.org/10.3390/metabo13050621
APA StyleBashir, B., Ho, J. H., Downie, P., Hamilton, P., Ferns, G., Datta, D., Cegla, J., Wierzbicki, A. S., Dawson, C., Jenkinson, F., Delaney, H., Mansfield, M., Teoh, Y., Miedzybrodzka, Z., Haso, H., Durrington, P. N., & Soran, H. (2023). Severe Hypertriglyceridaemia and Chylomicronaemia Syndrome—Causes, Clinical Presentation, and Therapeutic Options. Metabolites, 13(5), 621. https://doi.org/10.3390/metabo13050621