Abstract
Disrupted fatty acid metabolism is one of the most important metabolic features in heart failure. The heart obtains energy from fatty acids via oxidation. However, heart failure results in markedly decreased fatty acid oxidation and is accompanied by the accumulation of excess lipid moieties that lead to cardiac lipotoxicity. Herein, we summarized and discussed the current understanding of the integrated regulation of fatty acid metabolism (including fatty acid uptake, lipogenesis, lipolysis, and fatty acid oxidation) in the pathogenesis of heart failure. The functions of many enzymes and regulatory factors in fatty acid homeostasis were characterized. We reviewed their contributions to the development of heart failure and highlighted potential targets that may serve as promising new therapeutic strategies.
1. Introduction
Heart failure is a complex clinical syndrome characterized by elevated intracardiac pressure or inadequate cardiac output due to structural or functional cardiac abnormalities [1]. It is a major cause of death in patients with heart disease. Coronary artery disease and hypertension are the predominant factors leading to heart failure [2]. The large improvement in healthcare worldwide has failed to improve the five-year mortality rate of heart failure, which remains at 52.6% [3]. Therefore, there is an urgent need to discover new therapeutic targets for the treatment of heart failure.
A normal heart predominantly relies on fatty acid oxidation (FAO) to produce energy; however, a failing heart is characterized by a decrease in FAO [4,5]. Fatty acids (FAs) are carboxylic acids that contain hydrocarbon chains, ranging from 4–36 carbon atoms. Based on the number of carbon atoms, FAs can be divided into four groups: short-chain FAs (up to 6 carbon atoms), medium-chain FAs (8–12 carbon atoms), long-chain FAs (14–18 carbon atoms), and very long-chain FAs (above 20 carbon atoms). The chains of FAs are either fully saturated (do not contain double bonds) or contain one or more monounsaturated or polyunsaturated double bonds. The most commonly occurring FAs contain an even number of carbon atoms with lengths of 12–24 carbon atoms (Table 1). The heart prefers to use long-chain fatty acids as oxidizing substrates to maintain normal function [6]. The utilization of FAs requires complex metabolic processes. Alterations in the key enzymes involved in this process result in aberrant FA metabolism, which contributes to the severity of heart failure.
This review summarized recent knowledge regarding advances in FA metabolism during heart failure. We discussed the function of each key enzyme involved in the regulation of FA uptake, lipogenesis, lipolysis, and FAO in heart failure. We also examined the transcriptional and phosphorylation regulation of FA homeostasis. Based on these results, this is the first review to explore the potential roles of enzymes and regulatory proteins, which might be considered in the treatment of heart failure.
2. Fatty Acid Uptake
Cells obtain FAs via de novo lipogenesis and exogenous uptake (Figure 1). The entry of exogenous FAs from the surrounding microenvironment into cells is facilitated by specific transporters, including fatty acid translocase (FAT/CD36), fatty acid transport protein family (FATPs), and heart-type fatty acid-binding proteins (H-FABP) [6].
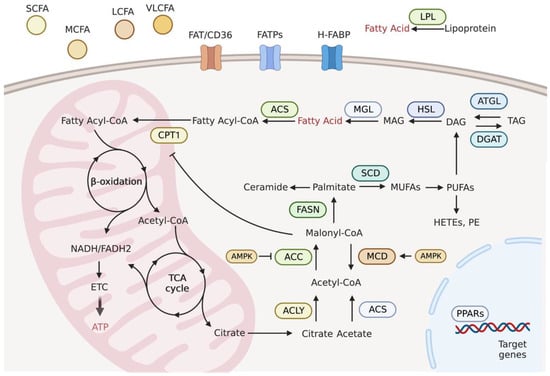
Figure 1.
The cardiomyocytes obtain fatty acids (FAs) from exogenous uptake and de novo lipogenesis. The entry of FAs from microenvironment needs specific transporters, including FAT/CD36, FATPs, and H-FABP. The cellular FAs obtained from extracellular uptake or intracellular lipolysis can be used for ATP production through fatty acid oxidation in the mitochondria. De novo lipogenesis relies on citrate and acetate. With the help of ACLY, ACS, ACC and FASN, palmitate is ultimately generated, which is further desaturated and elongated to form other lipid species. The transcription factor PPARs with the integrated capacity of several enzymes in FA metabolism control the entire system. Abbreviations: SCFA, short-chain FA; MCFA, medium-chain FA; LCFA, long-chain FA; VLCFA, very long-chain FA; LPL, lipoprotein lipase; FAT/CD36, fatty acid translocase/cluster of differentiation 36; FATPs, fatty acid transport proteins; H-FABP, heart-type fatty acid-binding protein; ACS, acetyl-CoA synthetase; CPT1, carnitine acyltransferase I; ETC, electron transport chain; ATP, adenosine triphosphate; ACLY, ATP-citrate lyase; ACC, acetyl-CoA carboxylases; MCD, malonyl-CoA decarboxylase; FASN, fatty acid synthase; SCD, stearoyl-CoA desaturase; ATGL, adipose triglyceride lipase; DGAT, diacylglycerol acyltransferase; HSL, hormone-sensitive lipase; MGL, monoglycerol lipase; TAG, triacylglycerol; DAG, diacylglycerol; MAG, monoacylglycerol; PPARs, peroxisome proliferator-activated receptors; AMPK, AMP-activated protein kinase.
2.1. Fatty Acid Translocase (FAT/CD36)
Fatty acid translocase (FAT) was discovered in rat adipocytes as an 88 kDa integral membrane protein participating in FA transport. The primary protein sequence is 85% homologous to glycoprotein IV (CD36) [7]. FAT/CD36 is abundantly expressed in the heart, intestine, fat, and muscle. Several studies reported a high prevalence of FAT/CD36 deficiency in patients with dilated [8], ischemic [9], or hypertrophic cardiomyopathy with asymmetric septal hypertrophy [10]. Clinical studies in the Japanese population show that point mutations in FAT/CD36 (478C>T) may result in deficiency [11,12]. Similarly, patients with heart failure have decreased FAT/CD36 and overall FA content, whereas FAT/CD36 gene expression is elevated after left ventricular assist device implantation, although the FA content remained unchanged [9]. These studies imply that FAT/CD36 deficiency might be an etiology of heart failure.
Animal studies conducted over the last two decades further clarified the function of FAT/CD36 in heart failure. The myocardial FAT/CD36 expression level correlates with the cardiac ejection fraction in the infarcted rat heart, with a 36% reduction in the protein level compared to sham-operated controls [13]. FAT/CD36 decreases in hypertrophic hearts in response to pressure overload. Tamoxifen-inducible cardiomyocyte-specific CD36 knockout mice exhibit rapid progression from compensated hypertrophy to heart failure [14]. These phenotypes may be caused by sufficient adenosine triphosphate (ATP) production and increased glycolytic flux-mediated structural remodeling [15]. However, FAT/CD36 is highly induced in cardiac lipotoxicity models, including age-induced murine cardiomyopathy and diabetic cardiomyopathy. FAT/CD36 deficient mice show significantly enhanced cardiac function, with lower intramyocardial lipid levels and improved ATP production [16]. Similarly, FAT/CD36 deficient mice show reduced cardiomyocyte triacylglycerol (TAG) accumulation and cardiac dysfunction after high-fat diet feeding [17]. These findings strongly support that moderate levels of FAT/CD36 protect against hemodynamic stress, whereas high expression is associated with cardiac lipotoxicity.
2.2. Fatty Acid Transport Protein Family (FATPs)
Fatty acid transport proteins facilitate the transmembrane transport of FA [18]. These candidate proteins were firstly identified by cloning an adipocyte cDNA library in COS7 cells and exhibiting an uptake of fluorescent FA analogs. This protein was named FATP1. FATP1 is expressed in most mammalian tissues, including the heart, brain, adipocytes, and kidney. Cardiac-specific overexpression of FATP1 using the α-myosin heavy chain gene promoter increases cardiomyocyte FA accumulation and stimulates FAO. However, the heart predominantly exhibits diastolic dysfunction and a prolonged corrected QT interval (QTc) after three months. These results confirm the role of FATP1 in FA import into the heart [19]. Another predominant cardiac FATP is FATP6, which is over 20 times more abundant than FATP1 in mouse heart lysates. The heart preferentially takes up palmitate compared to oleate, and the FATP6-stable cell line has the same uptake pattern. The authors suggest that a significant portion of FA uptake in the heart is mediated by FATP6 [20]. A clinical study revealed that a variation in the 5′-untranslated region of the FATP6 gene (7T>A polymorphism) is associated with features of metabolic syndrome and signs of myocardial alteration or heart failure [21]. The protein levels of FATP6 and FATP1 decrease in infarcted heart failure mice [13]; however, the detailed mechanism of reduced FATPs is less clear in these animal models.
2.3. Heart-Type Fatty Acid-Binding Protein (H-FABP)
Fatty acid-binding proteins are low-molecular-weight proteins (approximately 15 kDa) that facilitate FA uptake. Heart-type FABP (H-FABP) is probably the best-known member of the FABP family. It is encoded by FABP3, which is located in the 1p33-p32 region of chromosome 1 [22]. Tissues with a high demand for FAs (including the heart, brain, kidney, and adrenal gland) express H-FABP. H-FABP is more abundantly expressed in the ventricle and atrium than in other organs [23]. It can be rapidly released from myocytes into circulation, owing to its small molecular weight and free cytoplasmic localization; therefore, it is thought to be a biomarker in the pathogenesis of heart failure [24]. H-FABP levels are significantly increased in patients with chronic heart failure [25], which correlates with the New York Heart Association (NYHA) functional class and inversely correlates with ejection fraction. Furthermore, persistently high H-FABP levels are associated with adverse events during the follow-up of patients with heart failure [26]. Mice with disrupted H-FABP show elevated plasma long-chain FA (LCFA) levels, decreased cardiac deposition of an LCFA analog, and increased cardiac deoxyglucose uptake; therefore, there is a requirement for H-FABP in cardiac LCFA utilization [27]. Furthermore, glucose oxidation increases in H-FABP-knockout mice to compensate for cardiac energy production, as the palmitate uptake and oxidation are reduced [28].
3. Lipogenesis
Lipogenesis is the process of FA synthesis from non-lipid precursors and mainly occurs in the liver and adipose tissues. Fatty acids are synthesized in the heart using several lipogenic enzymes [29]. Lipogenesis encompasses two major processes: the activation of acetyl-coenzyme A (acetyl-CoA) and FA biosynthesis. The main substrate for lipogenesis is acetyl-CoA, which is derived from acetate by acetyl-CoA synthetase (ACS) or citrate by ATP-citrate lyase (ACLY). The carboxylation of acetyl-CoA to malonyl-CoA by acetyl-CoA carboxylase (ACC) is an irreversible rate-limiting step, and the opposite reaction is catalyzed by malonyl-CoA decarboxylase (MCD). Next, seven molecules of malonyl-CoA and one molecule of acetyl-CoA are condensed by fatty acid synthase (FASN), which finally produces saturated palmitate. Palmitate desaturation by stearoyl-CoA desaturase (SCD) generates monounsaturated FA (MUFA). Polyunsaturated FAs (PUFAs) are formed after a series of elongation and desaturation steps (Figure 1) [30].
FA membrane transport proteins are supposed to induce endogenous lipogenesis by increasing FA uptake into the heart. However, muscle-specific overexpression of FAT/CD36 failed to show significant changes in cellular lipids, such as TAG and phospholipids [31]. Consistently, overexpression of FATP1 or H-FABP in the heart did not affect the cardiac TAG content [19,28]. These results suggest that the activation of lipogenesis may require integrated control of particular enzymes.
3.1. Acetyl-CoA-Producing Enzymes ACS and ACLY
Acetyl-CoA synthetase (ACS) produces acetyl-CoA via the ligation of acetate and CoA, and acetyl-CoA is utilized to synthesize FAs in an anabolic pathway or via mitochondrial oxidation via a catabolic pathway. The series of reactions begins with acetyl-CoA; therefore, the acetyl-CoA level is a key element in FA metabolism. The ACS family comprises a large group of enzymes subdivided into five subfamilies: short-chain ACS (ACSS), medium-chain ACS (ACSM), long-chain ACS (ACSL), long-chain synthetase (ACSVL), and bubblegum ACS (ACSBG) [32].
Long-chain ACS is the most intensively studied enzyme in the ACS family, owing to its essential role in cardiac function. It participates in FA thioesterification to produce long-chain fatty acyl-CoA, together with acetyl-CoA production [33]. The five identified ACSL isoforms (ACSL1, ACSL3-6) exhibit specific substrate preferences, and responses to nutritional and hormonal regulation. ACSL1 is the major isoform expressed in highly oxidative tissues, including the heart, skeletal muscle, and brown adipose tissues. Mice lacking cardiac ACSL1 show impaired FAO and cardiac hypertrophy [34]. However, studies using ACSL1 transgenic mice have exhibited opposite results. For example, cardiac overexpression of ACSL1 leads to cardiac lipotoxicity, resulting in mild left ventricular hypertrophy with modest systolic dysfunction, which is associated with increased production of reactive oxygen species (ROS) and FA uptake, followed by mitochondrial remodeling [35]. In contrast, cardiac dysfunction induced by transverse aortic constriction (TAC) is improved in ACSL1 transgenic hearts by reducing cardiac lipotoxicity [36].
ATP citrate lyase (ACLY) is a cytosolic enzyme that generates acetyl-CoA for fatty acid biosynthesis. Citrate was used as the substrate for ACLY, unlike ACS. It is derived from pyruvate oxidation in the tricarboxylic acid (TCA) cycle (which is derived from glucose) [37], or through reductive carboxylation (which is derived from glutamine) [38]. Therefore, ACLY represents a combination of three different metabolic pathways. Homozygous ACLY knockout mice died early in development, and heterozygous mice were healthy, fertile, and normolipidemic on both chow- and high-fat diets. Half-normal amounts of ACLY in heterozygous mice did not perturb triglyceride or cholesterol synthesis or ACS expression [39]. Genetic variants in genes encoding ACLY are associated with the risk of cardiovascular events [40]; however, the role of ACLY in heart failure remains to be explored.
3.2. Fatty Acid Biosynthesis Enzymes
Acetyl-CoA carboxylase (ACC) and malonyl-CoA decarboxylase (MCD) are responsible for malonyl-CoA production. The heart predominantly relies on FAO to fuel ATP production, and the FAO flux is regulated at the level of entry of long-chain fatty acyl-CoA into the mitochondria by carnitine acyltransferase I (CPT1). Malonyl-CoA is a CPT1 inhibitor, which is essential for regulating cardiac function. Cardiac contractile stimulation activates MCD, which decreases the malonyl-CoA content and, hence, increases FAO [41]. MCD levels are highly induced in high-fat diet-fed rats and streptozotocin-treated diabetic model. Peroxisome proliferator-activated receptor-α (PPARα) is activated by the increasing concentrations of non-esterified FAs in the plasma, and thereby stimulates the expression of MCD. In contrast, pressure overload-induced cardiac hypertrophy has reduced MCD activity owing to PPARα inhibition. This results in the suppression of FAO [42]. Notably, pharmacological inhibition or genetic deletion of MCD protects the ischemic heart by inhibiting FAO and stimulating glucose oxidation [43,44,45].
In contrast, acetyl-CoA activates ACC; this results in increased malonyl-CoA levels that inhibit FAO [46]. The two main ACC isoforms in mammals (ACC1 and ACC2) have distinct tissue distributions and functions: ACC1 is predominantly expressed in the liver and adipocytes, whereas ACC2 is enriched in the heart and skeletal muscle [47]. Furthermore, ACC1 is localized in the cytosol, and ACC2 is associated with the mitochondria [48]. Mice with a null mutation in ACC1 show embryonic lethality, while ACC2-null mice are viable [49]. ACC2 knockout mice have a normal lifespan, a higher rate of FAO, and a lower amount of fat compared to wild-type mice [50]. Moreover, ACC2 mutant hearts display normal functional parameters despite a significant decrease in size, with a marked preference for the oxidation of glucose and Fas [51]. Cardiac-specific deletion of ACC2 leads to a reduction in cardiac malonyl-CoA with an increase in FAO and decreased glucose utilization. An ACC2 knockout in the heart did not affect left ventricular function or oxygen consumption. A genetic deletion of ACC2 attenuates cardiac hypertrophy and prevents metabolic remodeling during pressure overload hypertrophy [52]. Furthermore, an ACC2 deletion with pre-existing cardiac pathology improves cardiac function during the transition from pathological hypertrophy to heart failure, but only in female mice. This different response derives from a sex-dependent regulation of the PPARα signaling pathway in heart failure. ACC2 knockout mice resist obesity and retain insulin sensitivity in a high-fat diet-induced diabetes model [49]. This is thought to protect the heart from diabetic cardiomyopathy.
3.3. Fatty Acid Synthase (FASN) and Stearoyl-CoA Desaturase (SCD)
Fatty acid synthase is a multi-enzyme complex that catalyzes the conversion of acetyl-CoA and malonyl-CoA to long-chain saturated FAs. Heterozygous FASN mutants are ostensibly normal; however, the null FASN mutant results in embryonic lethality [53]. FASN also generates an endogenous ligand for PPARα signaling [54,55]. It is highly induced in murine heart failure models, including ACS transgenic mice. Moreover, the mRNA level of FASN increases in cardiac tissue from individuals with sudden cardiac death and in patients with class IV cardiomyopathy requiring left ventricular assist device (LVAD) support. Cardiac specific FASN knockout mice manifested normal resting heart function, cardiac PPARα signaling, and FAO. However, most mice died within one hour of pressure overload induced by transverse aortic constriction, probably due to arrhythmia. Calcium–calmodulin (CaM)-dependent protein kinase II (CaMKII) signaling appears to be pathogenic because inhibition of this signaling rescues mice from early mortality after transverse aortic constriction [56].
Stearoyl-CoA desaturase is the rate-limiting enzyme that catalyzes the synthesis of MUFA, such as palmitoleic acid and oleic acid, which are substrates for the synthesis of phospholipids, cholesterol, and triacylglycerol. Four isoforms (SCD1–4) are identified in mice, and SCD1, SCD2, and SCD4 are expressed in the heart. SCD1 and SCD5 were identified in humans, and each isoform is found in the heart. Compared with wild-type mice, the heart tissues from SCD1 knockout mice have reduced FA transport and oxidation, as well as increased glucose uptake and oxidation. The left ventricular weight is higher in SCD-deficient mice; however, cardiac function is not significantly affected [57]. In addition, there is no significant difference in SCD activity and palmitoleic acid levels in SCD1 knockout mice, as SCD4 compensates for the lack of SCD1 in the heart [58]. Human plasma SCD activity is also associated with the risk of heart failure [59]. SCD1 expression is induced in several heart failure models [60]. The myocardium-specific expression of SCD1 mice triggers cardiac hypertrophy and symptoms of heart failure at an age of eight months with an overload of cardiotoxic saturated lipids [61]. The loss of SCD1 by RNAi still led to cardiac dysfunction and lipotoxicity [62]; however, systemic SCD1 deficiency in ob/ob obese mice improves impaired cardiac function owing to the inhibition of apoptosis [63]. Together, these findings suggest that SCD1 participates in pathological remodeling during heart failure.
3.4. Diacylglycerol Acyltransferase (DGAT)
Diacylglycerol acyltransferase catalyzes the final step in the formation of triglycerides from diacylglycerol (DAG) and fatty acyl-CoA. The mammalian heart has two DGAT isoforms: DGAT1 and DGAT2. Failing human hearts show severely reduced DGAT1 expression with the accumulation of DAG and ceramides [64]. DGAT1-null mice show no cardiac phenotype, with normal TAG and DAG levels in the heart [65]. However, the cardiac-specific deletion of DGAT1 increases DAG levels without altering TAG levels. Half of the cardiac DGAT1 knockout mice died by nine months of age, with an increase in heart failure markers and cardiac dysfunction [66]. In contrast, transgenic overexpression of DGAT1 in the heart results in approximately normal cardiac function at three to four months of age. ACS and DGAT1 double-transgenic mice show improved heart function compared to ACS transgenic mice [64]. However, DGAT1 transgenic mice develop severe cardiomyopathy with moderate systolic dysfunction at 52 weeks of age [67]. DGAT1 knockout mice are resistant to obesity and insulin without changes in TAG levels in a high-fat diet-induced murine model. Co-inhibition of DGAT1/2 in the cardiomyocytes still protects the heart against high-fat diet-induced lipid accumulation and decreases TAG levels [68]. Recent data suggest that DGAT plays an important role in the pathogenesis and development of heart failure.
4. Lipolysis and FAO
Lipolysis is a catabolic process, involving the breakdown of TAGs into FAs and glycerol. Adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL) are key enzymes in lipolysis that generate FAs as energy substrates for FAO and subsequent ATP production. Fatty acid oxidation enzymes are located in the mitochondrial matrix, and FAs in the cytosol must be further activated and transported into the mitochondria. The first reaction of FA activation is catalyzed by ACS via the formation of fatty acyl-CoA, which is transiently attached to the hydroxyl group of carnitine by CPT1 to form fatty acyl-carnitine. Fatty acyl-carnitine ester diffuses into the inner mitochondrial membrane with the help of the acyl-carnitine/carnitine transporter. The final step of the carnitine shuttle pathway involves the regeneration of fatty acyl-CoA by carnitine acyltransferase II (CPT2), and carnitine reenters the intermembrane space via the acyl-carnitine/carnitine transporter. Fatty acyl-CoA then undergoes FAO to produce acetyl-CoA, which enters the TCA cycle and generates nicotinamide adenine dinucleotide (NADH) and FADH2 (flavin adenine dinucleotide) for the electron transport chain to produce ATP (Figure 1).
4.1. Intracellular LIPOLYSIS Enzymes ATGL and HSL
Patients with heart failure show high TAG levels in the left ventricular tissue [69]. Lipid accumulation can result from increased TAG synthesis or defective lipolysis. Adipose triglyceride lipase (ATGL) is a rate-limiting enzyme in intracellular lipolysis that releases FAs from TAG. Most patients carrying the ATGL mutation develop cardiac steatosis and present with cardiomyopathy [70]. In heart failure, most ATGL-deficient patients develop severe cardiac dysfunction that requires heart transplantation [71]. Cardiomyocyte-specific ATGL overexpression protects against cardiac dysfunction in a murine model of pressure overload-induced heart failure. The mice show reduced myocardial TAG content; however, the levels of DAG and ceramides remained unchanged. Notably, the FAO rate decreases, whereas the glucose oxidation rate increases [72]. In contrast, ATGL deficient mice develop excessive lipid accumulation, cardiac insufficiency, and lethal cardiomyopathy [73]. ATGL generates essential mediators that serve as ligands for PPAR activation. ATGL knockout mice treated with PPARα agonist completely restore normal heart function and prevent premature death [74,75]. Cardiac ATGL protein and TAG levels significantly increased in a murine model of diabetic cardiomyopathy, and ATGL deficiency results in lipotoxicity and diastolic dysfunction, whereas ATGL overexpression in cardiomyocytes is resistant to cardiac dysfunction [76]. These studies strongly indicate that ATGL activity affects cardiac function and that ATGL is a promising target for heart failure treatment.
Hormone-sensitive lipase (HSL) is the second lipolytic enzyme next to ATGL that catalyzes the breakdown of DAG into monoacylglycerol (MAG) and FAs. The substrates for HSL are much broader than that for ATGL, which including cholesteryl esters, retinyl esters, and TAG [77]. Humans with HSL deficiency show dyslipidemia, hepatic steatosis, systemic insulin resistance, and diabetes; however, no cardiac phenotype was found [78]. Murine studies have primarily focused on diabetic cardiomyopathy. Cardiac overexpression of HSL inhibits myocardial steatosis and fibrosis in diabetic mice by hydrolyzing toxic lipid metabolites [79]. Moreover, cardiac overexpression of HSL in ATGL-null mice rescues the cardiac dysfunction caused by ATGL knockout. This indicates that HSL compensates for ATGL deficiency [80]. HSL knockout mice have impaired adipose lipolysis and male infertility without severe cardiac defects. However, HSL-deficient mice are still protected from high-fat diet-induced cardiac insulin resistance associated with reduced intramuscular TAG [81]. The role of HSL in lipolysis may be more complex, and the elucidation of a more detailed mechanism requires further exploration.
Triacylglycerols are stored in the heart as lipid droplets. The presence of Perilipin proteins on the surface of lipid droplets protects TAG from lipolysis. It comprises five members: Perilipin 1–5. Perilipin 5 is abundantly expressed in the heart, Perilipins 1 and 4 are mainly expressed in adipose tissue, whereas Perilipin 2 and 3 are found in a variety of tissues. Compared to wild-type mice, Perilipin 5 knockout mice lacked detectable lipid droplets and more actively oxidized FA. Mutant mice show a greater decline in cardiac function with age [82]. However, type 1 diabetic Perilipin 5-null mice exhibit lower levels of lipotoxic molecules and resistance to diabetes-induced cardiac malfunction [83]. Mice that specifically overexpress Perilipin 5 in the heart have increased TAG content, and a chronic excess of lipid droplets causes mild heart dysfunction [84]. In addition, Perilipin phosphorylation by protein kinase A is required for the translocation of HSL from the cytosol to lipid droplets, a key event in triggering lipolysis [85]. However, cardiac overexpression of Perilipin 5 with a mutant phosphorylation site shows a comparable level of cardiac TAG compared to non-mutated Perilipin 5 transgenic mice, with completely normal heart function in these two mice. This finding suggests that Perilipin 5-mediated cardiac lipolysis requires multiple Perilipin 5 phosphorylation sites [86].
4.2. Extracellular Lipolysis Enzyme Lipoprotein Lipase (LPL)
The energy of the heart mainly relies on FA oxidation. Therefore, the supply of FAs to the heart is essential for cardiac function. The hydrolysis of TAG-rich lipoproteins by lipoprotein lipase (LPL) is another source of FAs in the heart in addition to the uptake of albumin-bound FAs derived from adipose tissue. Lipoprotein lipase-null mice exhibit severe hypertriglyceridemia, reduced high-density lipoprotein levels, and neonatal death [87]. LPL is expressed at its highest level in the heart, and its expression in the heart alone is sufficient to maintain normal plasma triglyceride levels and rescue LPL-null mice from neonatal death [88]. LPL overexpression in cardiomyocytes causes dilated cardiomyopathy with lipid accumulation in the heart [89]. In contrast, cardiac-specific LPL knockout mice have significantly elevated plasma triglyceride levels and increased glucose oxidation; however, the cardiac uptake of albumin-bound FAs is unchanged [90]. Cardiac LPL deletion mice died within 48 h of pressure overload, and older mice developed cardiac dysfunction [91]. Thus, LPL may play a central role in maintaining the balance of plasma TAGs during heart failure.
Recently, several LPL regulators were discovered, including lipase maturation factor 1 (LMF1), Sel suppressor of Lin-12-like 1 (SEL1L), glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1), and angiopoietin-like proteins (ANGPTLs) [92]. LMF1 and SEL1L in endoplasmic reticulum functionally ensure the maturation of LPL [93]. The mature LPL will be captured by GPIHBP1 on the surface of endothelial cell, and then it moves to the endothelial lumen through transcytosis [94,95]. ANGPTL3, 4, and 8 have been reported to inhibit LPL activity and increase plasma triglyceride level in a tissue-specific manner [96,97,98]. Additionally, the ANGPTL3–ANGPTL8 complex has been shown to dramatically suppress the activity of LPL compared to either protein alone [99]. As ANGPTL3 is exclusively expressed in the liver, it is likely to be released as a complex with ANGPTL8 to suppress LPL activity in fat and muscle tissues. ANGPTL4 is expressed in numerous tissues and may act as a local LPL inhibitor [100]. Interestingly, transgenic overexpression of ANGPTL4 in the heart developed left-ventricular dysfunction. The mice exhibited hypertriglyceridemia after inhibiting LPL activity [101]. Importantly, patients in a dyslipidemia cohort showed association between LMF1 gene variants and postheparin LPL activity [102]. Mutation or deletion in the GPIHBP1 gene also resulted in severe hypertriglyceridemia in patients [103]. Moreover, people carrying loss-of-function variants in ANGPTL3 or ANGPTL4 have a reduced risk of coronary artery disease with decreased plasma level of triglycerides [104,105,106]. It suggests that the modulation of these LPL regulators could offer a therapeutic treatment for hypertriglyceridemia and reduce the risk for related heart disease.
4.3. Carnitine Acyltransferase I (CPT1)
Carnitine acyltransferase I (CPT1) is responsible for the formation of fatty acyl-carnitines by transferring the acyl group of fatty acyl-CoA to carnitine. It is also known as carnitine palmitoyltransferase I as its main product is palmitoylcarnitine. A decreased rate of FAO is suggested to occur secondary to the reduced carnitine content in the hypertrophied myocardium [107,108,109]. The role of propionyl L-carnitine in cardiac dysfunction was extensively investigated in the last century because it is a naturally occurring carnitine derivative that increases tissue carnitine levels [110]. Multiple studies show that the administration of propionyl-L-carnitine improves the contractile function of hypertrophied hearts [109,111]. However, the drug failed to normalize FAO, and the beneficial effect was due to the increased efficiency of ATP in cardiac function [112]. CPT1 is the rate-limiting enzyme in FAO, and it was actively investigated as a potential therapeutic target. CPT1 activity significantly decreases in hypertrophied hearts under pressure overload [113] and advanced heart failure [9]. CPT1 has three isoforms: CPT1a, CPT1b, and CPT1c. CTP1a is predominant isoform in the liver, and it is also known as a liver isoform. CPT1b is highly expressed in the heart and skeletal muscles, whereas CPT1c is only expressed in neurons. Heterozygous CPT1b knockout mice exhibit exacerbated cardiac hypertrophy and are susceptible to premature death due to congestive heart failure after pressure overload. Moreover, the mice presented with severe mitochondrial abnormalities and myocardial lipotoxicity, leading to cardiomyocyte apoptosis [114]. However, clinical trials show that specific CPT1 inhibitors (such as etomoxir) improve the cardiac function in patients with heart failure [115]. In contrast, overexpression of the CPT1 liver isoform elevates atrial natriuretic peptide levels [116]. This indicates that it induces hypertrophic signaling. CPT1 may be beneficial in the progression of heart failure; however, further studies are required to elucidate its role.
4.4. Fatty Acid Oxidation
Efficient energy production in cardiomyocytes requires three mitochondrial metabolic pathways: FAO, the TCA cycle, and the electron transfer chain. Acetyl-CoA produced from FAO enters the TCA cycle to generate NADH and FADH2 for the electron transport chain to produce ATP in normal hearts. Therefore, the rate of FAO is regulated by FAs supply and uptake, malonyl-CoA content, the ratio of FAD/FADH2 and NAD+/NADH, the mitochondrial acetyl-CoA/CoA ratio, and transcriptional and post-translational modifications of FAO enzymes [117]. Decreased FAO is reported in humans with idiopathic dilated cardiomyopathy [118,119,120,121], post-infarction heart failure [122], heart failure in Dahl salt-sensitive hypertensive rats [123], pressure overload-induced heart failure models [124,125,126], and canine models of cardiac pacing [127]. This alteration may be caused by a reduction in the number of genes and enzymes involved in FAO [9,124,126]. In contrast, FAO is highly induced in type 2 diabetes [128], obesity [129], and heart failure with a preserved ejection fraction [130]. Activated FAO also consistently occurs in diabetic and obese mice lacking leptin [131,132]. The increased FAs and induction of FAO enzymes by lysine acetylation may be the underlying mechanism [133,134].
Nicotinamide adenine dinucleotide (NAD) serves as the major electron carrier coenzyme in all three FAO stages. Initially, FAs undergo β-oxidation and remove successive two-carbon units to form acetyl-CoA. The formation of acetyl-CoA releases two pairs of electrons. NAD+ and FAD serve as electron acceptors and carry electrons in the form of NADH and FADH2. In the second stage, acetyl-CoA enters the TCA cycle to generate NADH and FADH2. Finally, NADH and FADH2 electron carriers produced in the above two stages donate electrons to the mitochondrial respiratory chain for ATP synthesis (Figure 1). Therefore, the NAD+/NADH redox pathway is critical for FA metabolism in patients with heart failure. A reduction in NAD or a decreased NAD+/NADH ratio is observed in patients with heart failure and in a murine model with pathologic hypertrophy [135,136]. Consistently, impaired expression of genes involved in NAD biosynthesis was confirmed in cardiac tissue from patients with ejection fraction preserved heart failure [137]. Mechanistically, a low level of NAD stimulates the hyperacetylation of mitochondrial proteins by inhibiting sirtuin, which impairs the cytosolic redox state and energy deficiency. Notably, the elevation of NAD+ levels by stimulating the nicotinamide phosphoribosyl transferase (Nampt)-dependent NAD+ salvage pathway or supplying NAD precursors improves cardiac function in response to stress (Figure 2) [135,136]. Furthermore, a clinical trial revealed that the administration of NAD precursor in patients with advanced heart failure enhances respiratory capacity through complex I activity in peripheral blood mononuclear cells and reduces proinflammatory cytokine gene expression [138].
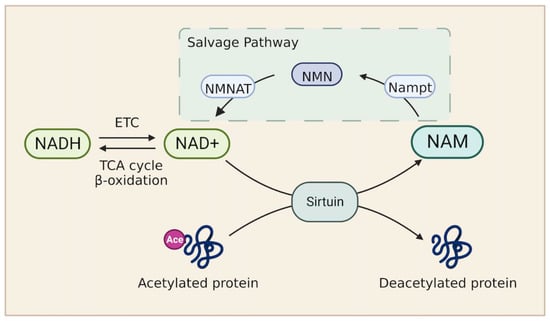
Figure 2.
The nicotinamide adenine dinucleotide (NAD) metabolism. The level of NAD+ is determined by NAD+ synthesis from the salvage pathway or NAD+/NADH ratio. NAD+ is required in the Sirtuin-mediated deacetylase reaction. This reaction also generates NAM. NAM then enters the salvage pathway. Nampt is the rate limiting enzyme in this pathway, catalyzing the conversion from NAM to NMN. NMN is thereby converted to NAD+ by NMNAT. Abbreviations: NAD, nicotinamide adenine dinucleotide; Nampt, nicotinamide phosphoribosyl transferase; NAM, nicotinamide; NMN, nicotinamide mononucleotide; NMNAT, nicotinamide mononucleotide adenyltransferase.
The mitochondrial respiratory chain consists of five members (complexes I–IV) that are the engines producing energy in the heart. In contrast, complex I is considered a potential source of oxygen free radicals in the failing myocardium [139]. Therefore, determining the role of complex I in heart failure is challenging. Rong Tian et al. modeled the impairment of respiratory capacity by knocking out the Ndufs4 gene encoding a critical protein for complex I assembly. Cardiac deletion in Ndufs4 mice maintains normal cardiac function that is consistent with a >40% reduction in respiration. However, the heart develops accelerated dysfunction under pressure overload or repeated pregnancies. The authors thought that this phenotype was derived from a decreased NAD+/NADH ratio owing to complex I deficiency and increased protein acetylation [140]. Moreover, the knockout of Ndufs4 in macrophages worsens cardiac function 30 days after myocardial infarction. Suppression of respiration in macrophages impairs efferocytosis, leading to a low expression of anti-inflammatory cytokines and tissue repair factors [141]. The mitochondrial respiratory chain has multiple functions; these studies confirmed the cell type-dependent functions of these proteins in heart failure.
5. The Regulatory Factors of Fatty Acid Homeostasis
Transcriptional factors, such as PPARs and phosphorylation kinase (such as AMP-activated protein kinase (AMPK)), are central mechanisms that control complex metabolic networks in the heart during hemodynamic stress, as they can tune energy utilization of the heart by FA or glucose [4]. Transcription control requires three components: upstream events that activate signaling, molecular mechanisms that cooperate with transcription factors, and downstream actions that transcribe target genes [142]. Additionally, protein kinase also needs molecular triggering, as well as spatial and temporal factor cross-talking. Modification of these components has emerged as a potential target for therapeutic intervention.
5.1. Peroxisome Proliferator-Activated Receptors (PPARs)
Peroxisome proliferator-activated receptors are nuclear receptors sharing a common structure, including a conserved DNA-binding domain and a ligand binding domain [143]. They comprise three subtypes (PPARα, PPARδ, and PPARγ), and each is expressed in the heart. PPARα and PPARδ are relatively abundant [144]. PPARα expression is relatively decreased in heart failure patients with hypertensive heart disease compared to patients with normal cardiac function [145,146]. Consistently, PPARα is deactivated in ventricular pressure overload studies in mice, which is accompanied by the downregulation of FAO enzymes [147]. The activation of PPARα increases several downstream targets, including FAT/CD36, CPT1, and MCD [148]. PPARα deficiency in the heart is sufficient to maintain normal energy homeostasis and cardiac function. However, knockout mice show accelerated cardiac remodeling and contractile dysfunction during hemodynamic overload [149,150]. Transcriptome analysis reveals the activation of glucose metabolism genes and a switch in substrate utilization from FA to glucose. Notably, overexpression of glucose transporter GLUT1 in PPARα-deficient mice improves the metabolic and functional defects. This suggests that the upregulation of glucose metabolism from intrinsic responses to PPARα deficiency or from glucose transporter overexpression has different effects on heart failure. In contrast, PPARα activation by inducible transgenic mice maintains myocardial function with enhanced FAO in the early stages of heart failure [151]. PPARα knockout mice are protected from the severe cardiomyopathic phenotype in diabetes-induced cardiac hypertrophy. However, PPARα overexpression in diabetic hearts exacerbated the hypertrophy with myocardial triglyceride accumulation [152,153]. These results suggest that PPARα modulation could be a promising therapeutic strategy for heart failure.
PPARγ/δ is differentially regulated in the heart. Unlike the expression of PPARα, PPARγ is highly induced in heart failure. As PPARγ is a downstream target of hypoxia-inducible factor 1 (HIF1α), the activation of PPARγ induced by HIF1α stimulated the FA uptake and lipid accumulation. Ventricular HIF1a deficiency inhibits PPARγ expression and attenuates stress-induced pathological hypertrophy [154]. Besides, cardiac PPARγ overexpression develops a dilated cardiomyopathy associated with increased lipid stores [155]. The detailed mechanism is unclear; however, PPARγ-induced cardiolipotoxicity is ameliorated by deleting PPARα [156], and PPARγ activation seems to be protective in sepsis-related cardiac dysfunction [157]. PPARδ deletion downregulates the expression of FAO genes and decreases the basal myocardial FAO rate. These mice exhibit cardiac dysfunction and progressive lipid accumulation [158].
PPARγ coactivator 1 (PGC1) is defined as a protein that interacts with transcription factors and increases the probability of target gene transcription. PPARα, PPARδ, and PPARγ are all subject to transcriptional coactivation by PGC1. There are three PGC1 members: PGC1α, PGC1β, and PGC1-related coactivator [159]. PGC1α repression is a signature of the failing human heart [160]. PGC1 activity induces the expression of nuclear and mitochondrial genes in cultured cardiomyocytes, in addition to its role in cooperation with PPARs. However, cardiac-specific PGC1 overexpression develops dilated cardiomyopathy with uncontrolled mitochondrial proliferation [161]. Consistently, serious mitochondrial structural derangements were observed in the hearts of PGC1α/β-deficient mice during postnatal growth associated with the development of lethal cardiomyopathy. However, PGC1α/β-deficient adult mice did not result in heart failure and mitochondrial abnormalities [162]. PGC1α knockout mice develop marked dilated cardiomyopathy with the downregulation of several target genes in FA metabolism and electron transport chain in the pressure overload heart failure model [163].
5.2. AMP-Activated Protein Kinase (AMPK)
AMP-activated protein kinase serves as a cellular fuel gauge and metabolic regulator in the heart. It is a heterotrimeric complex, comprising a catalytic α subunit and two regulatory β and γ subunits. The α subunit contains a serine–threonine kinase domain that includes an activating Thr172 residue. Phosphorylation of this site is critical for AMPK activation [164]. AMPK regulates FA synthesis by phosphorylating ACC and HMG-CoA reductase [165]. It induces the expression and translocation of FAT/CD36 to the cell membrane and improves FA transport [166,167,168]. Furthermore, it regulates genes, such as PGC1, MCD, CPT1, and GLUT4 [169,170,171]. However, AMPK activation during pressure overload stress mainly increases the rates of glucose transport and glycolysis [172].
AMPKα2 knockout mice exacerbate ventricular hypertrophy and cardiac dysfunction in the pressure overload model [173] or high-fat diet-induced heart failure model [174]. In contrast, long-term activation of AMPK by the specific chemical activator AICAR attenuates pressure overload-induced cardiac hypertrophy [175]. In addition, AMPK deficiency in the ischemic heart increases cardiac injury and impairs contractile function with exacerbated ATP depletion [176,177]. Thus, AMPK exerts protective effects against heart failure. Several studies confirm that the pharmacological AMPK activator (metformin) improves ventricular function and survival in heart failure [178,179].

Table 1.
The classification and function of fatty acids.
Table 1.
The classification and function of fatty acids.
| Classification | Typical FAs | Carbon Skeleton | Common Name | Function |
|---|---|---|---|---|
| Saturated FAs | n-Dodecanoic acid | 12:0 | Lauric acid | Increased risk of cardiovascular disease by aggravating dyslipidemia [180] |
| n-Tetradecanoic acid | 14:0 | Myristic acid | Increased risk of coronary heart disease [180] | |
| n-Hexadecanoic acid | 16:0 | Palmitic acid | Increased risk of coronary heart disease [180] | |
| n-Octadecanoic acid | 18:0 | Stearic acid | Increased risk of coronary heart disease [180] | |
| n-Eicosanoic acid | 20:0 | Arachidic acid | Decreased risk of incident heart failure [181] | |
| n-Teracosanoic acid | 24:0 | Lignoceric acid | Decreased risk of incident heart failure [181] | |
| Monounsaturated FAs | cis-9- Hexadecanoic acid | 16:1 (Δ9) | Palmitoleic acid | Increased risk of heart failure [59] |
| cis-9- Octadecanoic acid | 18:1 (Δ9) | Oleic acid | Not associated with heart failure risk [59] | |
| Polyunsaturated FAs | cis, cis-9,12-Octadecadienoic acid | 18:2 (Δ9,12) | Linoleic acid | Decreased risk of coronary heart disease [182] |
| cis, cis, cis-9,12,15-Octadecatrienoic acid | 18:3 (Δ9,12,15) | α-Linolenic acid | Increased risk of cardiovascular disease [183] | |
| cis, cis, cis, cis-5,8,11,14-Icosatetraenoic acid | 20:4 (Δ5,8,11,14) | Arachidonic acid | Increased risk of cardiovascular disease [184] | |
| cis, cis, cis, cis, cis-5,8,11,14,17-Eicosapentaenoic acid | 20:5(Δ5,8,11,14,17) | Eicosapentaenoic acid | Decreased risk of cardiovascular disease and incident heart failure [185,186] | |
| cis, cis, cis, cis, cis, cis-4,7,10,13,16,19-Docosahexaenoic acid | 24:6 (Δ4,7,10,13,16,19) | Docosahexaenoic acid | Decreased risk of cardiovascular disease [185] |
6. Conclusions and Perspectives
Emerging evidence supports the notion that unbalanced FA metabolism occurs in heart failure, which is characterized by the repression of FAO and the accumulation of toxic lipids. Almost all enzymes are downregulated in heart failure, including FAT/CD36, FATPs, ACS, MCD, and CPT1. However, FASN and SCD lipogenic enzymes are highly induced, thereby causing excess DAG or ceramide storage (Figure 3). Consequently, the heart suffers from a double burden of insufficient energy supply and lipotoxicity. ACC and ATGL may serve as potential targets to treat hemodynamic stress-induced heart failure. In contrast, FAT/CD36, DGAT, ATGL, HSL, and Perilipin may be candidates to treat cardiac dysfunction related to metabolic syndrome. Fatty acid metabolism involves a tight and cooperative network; therefore, transcriptional factors and protein kinase with the integrated capacity of several enzymes provide another potential therapeutic strategy in failing hearts. Importantly, clinical trials in patients with heart failure show that supplementation with NAD precursors has beneficial effects. The limitation of this review is we only focus on some key enzymes and important regulatory factors, and other proteins, such as cholesterol biosynthesis enzymes, are not involved here. Overall, significant progress has been made in broadening our understanding of the pathophysiology of cardiac dysfunction. However, further comprehensive studies are warranted to identify therapeutic targets to treat heart failure.
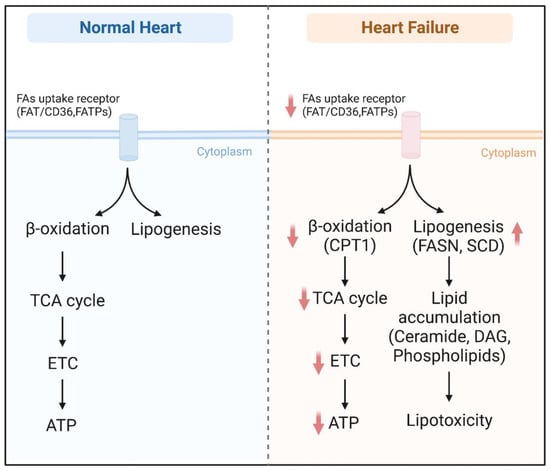
Figure 3.
Overview of fatty acid metabolism in the normal heart and failing heart. Abbreviations: FAT/CD36, fatty acid translocase/cluster of differentiation 36; FATPs, fatty acid transport proteins; TCA cycle, tricarboxylic acid cycle; ETC, electron transport chain; ATP, adenosine triphosphate; FASN, fatty acid synthase; SCD, stearoyl-CoA desaturase; DAG, diacylglycerol.
Author Contributions
Original draft preparation, X.L.; writing review and editing, X.B. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by research grants from the National Natural Science Foundation of China (81900232 to X.B. and 81800706 to X.L.), the Natural Science Foundation of Zhejiang Province (LQ19H020011 to X.B.), and the Chinese Postdoctoral Science Foundation (2019M652118 to X.B.).
Acknowledgments
The authors thank the members of Guosheng Fu‘s laboratory (Zhejiang University School of Medicine) for helpful discussion.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACC | acetyl-CoA carboxylases |
| ACLY | ATP-citrate lyase |
| ACS | acetyl-CoA synthetase |
| AMPK | AMP-activated protein kinase |
| ATGL | adipose triglyceride lipase |
| ATP | adenosine triphosphate |
| CPT1 | carnitine acyltransferase I |
| DAG | diacylglycerol |
| DGAT | diacylglycerol acyltransferase |
| ETC | electron transport chain |
| FA | fatty acid |
| FASN | fatty acid synthase |
| FAT/CD36 | fatty acid translocase/cluster of differentiation 36 |
| FATPs | fatty acid transport proteins |
| FAO | fatty acid oxidation |
| H-FABP | heart-type fatty acid-binding protein |
| HSL | hormone-sensitive lipase |
| LCFA | long-chain FA |
| LPL | lipoprotein lipase |
| MAG | monoacylglycerol |
| MCD | malonyl-CoA decarboxylase |
| MGL | monoglycerol lipase |
| MUFA | monounsaturated FA |
| PPARs | peroxisome proliferator-activated receptors |
| PUFA | polyunsaturated FA |
| SCD | stearoyl-CoA desaturase |
| TAG | triacylglycerol |
| TCA cycle | tricarboxylic acid cycle |
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Disease, G.B.D.; Injury, I.; Prevalence, C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Tsao, C.W.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Beaton, A.Z.; Boehme, A.K.; Buxton, A.E.; et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 2023, 147, e93–e621. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Van der Vusse, G.J.; van Bilsen, M.; Glatz, J.F. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc. Res. 2000, 45, 279–293. [Google Scholar] [CrossRef]
- Abumrad, N.A.; el-Maghrabi, M.R.; Amri, E.Z.; Lopez, E.; Grimaldi, P.A. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J. Biol. Chem. 1993, 268, 17665–17668. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Suematsu, Y.; Kato, S.; Miura, S.I. Type 1 cluster of differentiation 36 deficiency-related cardiomyopathy accelerates heart failure with co-existing mitral valve prolapse: A case report. Eur. Heart J. Case Rep. 2019, 3, ytz116. [Google Scholar] [CrossRef]
- Chokshi, A.; Drosatos, K.; Cheema, F.H.; Ji, R.; Khawaja, T.; Yu, S.; Kato, T.; Khan, R.; Takayama, H.; Knoll, R.; et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 2012, 125, 2844–2853. [Google Scholar] [CrossRef]
- Tanaka, T.; Sohmiya, K.; Kawamura, K. Is CD36 deficiency an etiology of hereditary hypertrophic cardiomyopathy? J. Mol. Cell. Cardiol. 1997, 29, 121–127. [Google Scholar] [CrossRef]
- Tanaka, T.; Okamoto, F.; Sohmiya, K.; Kawamura, K. Lack of myocardial iodine-123 15-(p-iodiphenyl)-3-R,S-methylpentadecanoic acid (BMIPP) uptake and CD36 abnormality--CD36 deficiency and hypertrophic cardiomyopathy. Jpn. Circ. J. 1997, 61, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nakata, T.; Oka, T.; Ogawa, T.; Okamoto, F.; Kusaka, Y.; Sohmiya, K.; Shimamoto, K.; Itakura, K. Defect in human myocardial long-chain fatty acid uptake is caused by FAT/CD36 mutations. J. Lipid Res. 2001, 42, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Heather, L.C.; Cole, M.A.; Lygate, C.A.; Evans, R.D.; Stuckey, D.J.; Murray, A.J.; Neubauer, S.; Clarke, K. Fatty acid transporter levels and palmitate oxidation rate correlate with ejection fraction in the infarcted rat heart. Cardiovasc. Res. 2006, 72, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.M.; Byrne, N.J.; Kim, T.T.; Levasseur, J.; Masson, G.; Boisvenue, J.J.; Febbraio, M.; Dyck, J.R. Cardiomyocyte-specific ablation of CD36 accelerates the progression from compensated cardiac hypertrophy to heart failure. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H552–H560. [Google Scholar] [CrossRef]
- Umbarawan, Y.; Syamsunarno, M.; Koitabashi, N.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Sano, M.; Sunaga, H.; et al. Myocardial fatty acid uptake through CD36 is indispensable for sufficient bioenergetic metabolism to prevent progression of pressure overload-induced heart failure. Sci. Rep. 2018, 8, 12035. [Google Scholar] [CrossRef]
- Koonen, D.P.; Febbraio, M.; Bonnet, S.; Nagendran, J.; Young, M.E.; Michelakis, E.D.; Dyck, J.R. CD36 expression contributes to age-induced cardiomyopathy in mice. Circulation 2007, 116, 2139–2147. [Google Scholar] [CrossRef]
- Yang, J.; Sambandam, N.; Han, X.; Gross, R.W.; Courtois, M.; Kovacs, A.; Febbraio, M.; Finck, B.N.; Kelly, D.P. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ. Res. 2007, 100, 1208–1217. [Google Scholar] [CrossRef]
- Schaffer, J.E.; Lodish, H.F. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994, 79, 427–436. [Google Scholar] [CrossRef]
- Chiu, H.C.; Kovacs, A.; Blanton, R.M.; Han, X.; Courtois, M.; Weinheimer, C.J.; Yamada, K.A.; Brunet, S.; Xu, H.; Nerbonne, J.M.; et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ. Res. 2005, 96, 225–233. [Google Scholar] [CrossRef]
- Gimeno, R.E.; Ortegon, A.M.; Patel, S.; Punreddy, S.; Ge, P.; Sun, Y.; Lodish, H.F.; Stahl, A. Characterization of a heart-specific fatty acid transport protein. J. Biol. Chem. 2003, 278, 16039–16044. [Google Scholar] [CrossRef]
- Auinger, A.; Helwig, U.; Pfeuffer, M.; Rubin, D.; Luedde, M.; Rausche, T.; Eddine El Mokhtari, N.; Folsch, U.R.; Schreiber, S.; Frey, N.; et al. A variant in the heart-specific fatty acid transport protein 6 is associated with lower fasting and postprandial TAG, blood pressure and left ventricular hypertrophy. Br. J. Nutr. 2012, 107, 1422–1428. [Google Scholar] [CrossRef]
- Rezar, R.; Jirak, P.; Gschwandtner, M.; Derler, R.; Felder, T.K.; Haslinger, M.; Kopp, K.; Seelmaier, C.; Granitz, C.; Hoppe, U.C.; et al. Heart-Type Fatty Acid-Binding Protein (H-FABP) and its Role as a Biomarker in Heart Failure: What Do We Know So Far? J. Clin. Med. 2020, 9, 164. [Google Scholar] [CrossRef]
- Yoshimoto, K.; Tanaka, T.; Somiya, K.; Tsuji, R.; Okamoto, F.; Kawamura, K.; Ohkaru, Y.; Asayama, K.; Ishii, H. Human heart-type cytoplasmic fatty acid-binding protein as an indicator of acute myocardial infarction. Heart Vessels 1995, 10, 304–309. [Google Scholar] [CrossRef]
- Fischer, T.A.; McNeil, P.L.; Khakee, R.; Finn, P.; Kelly, R.A.; Pfeffer, M.A.; Pfeffer, J.M. Cardiac myocyte membrane wounding in the abruptly pressure-overloaded rat heart under high wall stress. Hypertension 1997, 30, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Lichtenauer, M.; Jirak, P.; Wernly, B.; Paar, V.; Rohm, I.; Jung, C.; Schernthaner, C.; Kraus, J.; Motloch, L.J.; Yilmaz, A.; et al. A comparative analysis of novel cardiovascular biomarkers in patients with chronic heart failure. Eur. J. Intern. Med. 2017, 44, 31–38. [Google Scholar] [CrossRef]
- Niizeki, T.; Takeishi, Y.; Arimoto, T.; Nozaki, N.; Hirono, O.; Watanabe, T.; Nitobe, J.; Miyashita, T.; Miyamoto, T.; Koyama, Y.; et al. Persistently increased serum concentration of heart-type fatty acid-binding protein predicts adverse clinical outcomes in patients with chronic heart failure. Circ. J. 2008, 72, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Binas, B.; Danneberg, H.; McWhir, J.; Mullins, L.; Clark, A.J. Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization. FASEB J. 1999, 13, 805–812. [Google Scholar] [CrossRef]
- Schaap, F.G.; Binas, B.; Danneberg, H.; van der Vusse, G.J.; Glatz, J.F. Impaired long-chain fatty acid utilization by cardiac myocytes isolated from mice lacking the heart-type fatty acid binding protein gene. Circ. Res. 1999, 85, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Bian, F.; Kasumov, T.; Jobbins, K.A.; Minkler, P.E.; Anderson, V.E.; Kerner, J.; Hoppel, C.L.; Brunengraber, H. Competition between acetate and oleate for the formation of malonyl-CoA and mitochondrial acetyl-CoA in the perfused rat heart. J. Mol. Cell. Cardiol. 2006, 41, 868–875. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimi, A.; Bonen, A.; Blinn, W.D.; Hajri, T.; Li, X.; Zhong, K.; Cameron, R.; Abumrad, N.A. Muscle-specific overexpression of FAT/CD36 enhances fatty acid oxidation by contracting muscle, reduces plasma triglycerides and fatty acids, and increases plasma glucose and insulin. J. Biol. Chem. 1999, 274, 26761–26766. [Google Scholar] [CrossRef]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Rock, C.O.; Jackowski, S. Pathways for the incorporation of exogenous fatty acids into phosphatidylethanolamine in Escherichia coli. J. Biol. Chem. 1985, 260, 12720–12724. [Google Scholar] [CrossRef]
- Ellis, J.M.; Mentock, S.M.; Depetrillo, M.A.; Koves, T.R.; Sen, S.; Watkins, S.M.; Muoio, D.M.; Cline, G.W.; Taegtmeyer, H.; Shulman, G.I.; et al. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs Fatty Acid oxidation and induces cardiac hypertrophy. Mol. Cell. Biol. 2011, 31, 1252–1262. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, J.R.; Carley, A.N.; Ji, R.; Zhang, X.; Fasano, M.; Schulze, P.C.; Lewandowski, E.D. Preservation of Acyl Coenzyme A Attenuates Pathological and Metabolic Cardiac Remodeling Through Selective Lipid Trafficking. Circulation 2019, 139, 2765–2777. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef]
- Beigneux, A.P.; Kosinski, C.; Gavino, B.; Horton, J.D.; Skarnes, W.C.; Young, S.G. ATP-citrate lyase deficiency in the mouse. J. Biol. Chem. 2004, 279, 9557–9564. [Google Scholar] [CrossRef]
- Ference, B.A.; Ray, K.K.; Catapano, A.L.; Ference, T.B.; Burgess, S.; Neff, D.R.; Oliver-Williams, C.; Wood, A.M.; Butterworth, A.S.; Di Angelantonio, E.; et al. Mendelian Randomization Study of ACLY and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 1033–1042. [Google Scholar] [CrossRef]
- Goodwin, G.W.; Taegtmeyer, H. Regulation of fatty acid oxidation of the heart by MCD and ACC during contractile stimulation. Am. J. Physiol. 1999, 277, E772–E777. [Google Scholar] [CrossRef]
- Young, M.E.; Goodwin, G.W.; Ying, J.; Guthrie, P.; Wilson, C.R.; Laws, F.A.; Taegtmeyer, H. Regulation of cardiac and skeletal muscle malonyl-CoA decarboxylase by fatty acids. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E471–E479. [Google Scholar] [CrossRef]
- Dyck, J.R.; Cheng, J.F.; Stanley, W.C.; Barr, R.; Chandler, M.P.; Brown, S.; Wallace, D.; Arrhenius, T.; Harmon, C.; Yang, G.; et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ. Res. 2004, 94, e78–e84. [Google Scholar] [CrossRef]
- Dyck, J.R.; Hopkins, T.A.; Bonnet, S.; Michelakis, E.D.; Young, M.E.; Watanabe, M.; Kawase, Y.; Jishage, K.; Lopaschuk, G.D. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 2006, 114, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, L.; Battiprolu, P.K.; Fukushima, A.; Nguyen, K.; Milner, K.; Gupta, A.; Altamimi, T.; Byrne, N.; Mori, J.; et al. Malonyl CoA Decarboxylase Inhibition Improves Cardiac Function Post-Myocardial Infarction. JACC Basic. Transl. Sci. 2019, 4, 385–400. [Google Scholar] [CrossRef]
- Saddik, M.; Gamble, J.; Witters, L.A.; Lopaschuk, G.D. Acetyl-CoA carboxylase regulation of fatty acid oxidation in the heart. J. Biol. Chem. 1993, 268, 25836–25845. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elheiga, L.; Jayakumar, A.; Baldini, A.; Chirala, S.S.; Wakil, S.J. Human acetyl-CoA carboxylase: Characterization, molecular cloning, and evidence for two isoforms. Proc. Natl. Acad. Sci. USA 1995, 92, 4011–4015. [Google Scholar] [CrossRef]
- Abu-Elheiga, L.; Brinkley, W.R.; Zhong, L.; Chirala, S.S.; Woldegiorgis, G.; Wakil, S.J. The subcellular localization of acetyl-CoA carboxylase 2. Proc. Natl. Acad. Sci. USA 2000, 97, 1444–1449. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elheiga, L.; Matzuk, M.M.; Kordari, P.; Oh, W.; Shaikenov, T.; Gu, Z.; Wakil, S.J. Mutant mice lacking acetyl-CoA carboxylase 1 are embryonically lethal. Proc. Natl. Acad. Sci. USA 2005, 102, 12011–12016. [Google Scholar] [CrossRef]
- Abu-Elheiga, L.; Matzuk, M.M.; Abo-Hashema, K.A.; Wakil, S.J. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001, 291, 2613–2616. [Google Scholar] [CrossRef]
- Essop, M.F.; Camp, H.S.; Choi, C.S.; Sharma, S.; Fryer, R.M.; Reinhart, G.A.; Guthrie, P.H.; Bentebibel, A.; Gu, Z.; Shulman, G.I.; et al. Reduced heart size and increased myocardial fuel substrate oxidation in ACC2 mutant mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H256–H265. [Google Scholar] [CrossRef]
- Kolwicz, S.C., Jr.; Olson, D.P.; Marney, L.C.; Garcia-Menendez, L.; Synovec, R.E.; Tian, R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 2012, 111, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Chirala, S.S.; Chang, H.; Matzuk, M.; Abu-Elheiga, L.; Mao, J.; Mahon, K.; Finegold, M.; Wakil, S.J. Fatty acid synthesis is essential in embryonic development: Fatty acid synthase null mutants and most of the heterozygotes die in utero. Proc. Natl. Acad. Sci. USA 2003, 100, 6358–6363. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Lodhi, I.J.; Yin, L.; Malapaka, R.R.; Xu, H.E.; Turk, J.; Semenkovich, C.F. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell 2009, 138, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, M.V.; Zhu, Y.; Lopez, M.; Yin, L.; Wozniak, D.F.; Coleman, T.; Hu, Z.; Wolfgang, M.; Vidal-Puig, A.; Lane, M.D.; et al. Brain fatty acid synthase activates PPARalpha to maintain energy homeostasis. J. Clin. Investig. 2007, 117, 2539–2552. [Google Scholar] [CrossRef]
- Razani, B.; Zhang, H.; Schulze, P.C.; Schilling, J.D.; Verbsky, J.; Lodhi, I.J.; Topkara, V.K.; Feng, C.; Coleman, T.; Kovacs, A.; et al. Fatty acid synthase modulates homeostatic responses to myocardial stress. J. Biol. Chem. 2011, 286, 30949–30961. [Google Scholar] [CrossRef]
- Dobrzyn, P.; Sampath, H.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 inhibits fatty acid oxidation and increases glucose utilization in the heart. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E357–E364. [Google Scholar] [CrossRef]
- Miyazaki, M.; Jacobson, M.J.; Man, W.C.; Cohen, P.; Asilmaz, E.; Friedman, J.M.; Ntambi, J.M. Identification and characterization of murine SCD4, a novel heart-specific stearoyl-CoA desaturase isoform regulated by leptin and dietary factors. J. Biol. Chem. 2003, 278, 33904–33911. [Google Scholar] [CrossRef]
- Djousse, L.; Weir, N.L.; Hanson, N.Q.; Tsai, M.Y.; Gaziano, J.M. Plasma phospholipid concentration of cis-palmitoleic acid and risk of heart failure. Circ. Heart Fail. 2012, 5, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, P.; Bednarski, T.; Dobrzyn, A. Metabolic reprogramming of the heart through stearoyl-CoA desaturase. Prog. Lipid Res. 2015, 57, 1–12. [Google Scholar] [CrossRef]
- Abd Alla, J.; Jamous, Y.F.; Quitterer, U. Stearoyl-CoA Desaturase (SCD) Induces Cardiac Dysfunction with Cardiac Lipid Overload and Angiotensin II AT1 Receptor Protein Up-Regulation. Int. J. Mol. Sci. 2021, 22, 9883. [Google Scholar] [CrossRef]
- Tuthill Ii, B.F.; Quaglia, C.J.; O’Hara, E.; Musselman, L.P. Loss of Stearoyl-CoA desaturase 1 leads to cardiac dysfunction and lipotoxicity. J. Exp. Biol. 2021, 224, jeb240432. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, P.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 rescues cardiac function in obese leptin-deficient mice. J. Lipid Res. 2010, 51, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Shi, X.; Bharadwaj, K.G.; Ikeda, S.; Yamashita, H.; Yagyu, H.; Schaffer, J.E.; Yu, Y.H.; Goldberg, I.J. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J. Biol. Chem. 2009, 284, 36312–36323. [Google Scholar] [CrossRef]
- Liu, L.; Yu, S.; Khan, R.S.; Ables, G.P.; Bharadwaj, K.G.; Hu, Y.; Huggins, L.A.; Eriksson, J.W.; Buckett, L.K.; Turnbull, A.V.; et al. DGAT1 deficiency decreases PPAR expression and does not lead to lipotoxicity in cardiac and skeletal muscle. J. Lipid Res. 2011, 52, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Trent, C.M.; Fang, X.; Son, N.H.; Jiang, H.; Blaner, W.S.; Hu, Y.; Yin, Y.X.; Farese, R.V., Jr.; Homma, S.; et al. Cardiomyocyte-specific loss of diacylglycerol acyltransferase 1 (DGAT1) reproduces the abnormalities in lipids found in severe heart failure. J. Biol. Chem. 2014, 289, 29881–29891. [Google Scholar] [CrossRef] [PubMed]
- Glenn, D.J.; Wang, F.; Nishimoto, M.; Cruz, M.C.; Uchida, Y.; Holleran, W.M.; Zhang, Y.; Yeghiazarians, Y.; Gardner, D.G. A murine model of isolated cardiac steatosis leads to cardiomyopathy. Hypertension 2011, 57, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Roe, N.D.; Handzlik, M.K.; Li, T.; Tian, R. The Role of Diacylglycerol Acyltransferase (DGAT) 1 and 2 in Cardiac Metabolism and Function. Sci. Rep. 2018, 8, 4983. [Google Scholar] [CrossRef]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef]
- Schreiber, R.; Xie, H.; Schweiger, M. Of mice and men: The physiological role of adipose triglyceride lipase (ATGL). Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2019, 1864, 880–899. [Google Scholar] [CrossRef]
- Hirano, K.; Tanaka, T.; Ikeda, Y.; Yamaguchi, S.; Zaima, N.; Kobayashi, K.; Suzuki, A.; Sakata, Y.; Sakata, Y.; Kobayashi, K.; et al. Genetic mutations in adipose triglyceride lipase and myocardial up-regulation of peroxisome proliferated activated receptor-gamma in patients with triglyceride deposit cardiomyovasculopathy. Biochem. Biophys. Res. Commun. 2014, 443, 574–579. [Google Scholar] [CrossRef]
- Kienesberger, P.C.; Pulinilkunnil, T.; Sung, M.M.; Nagendran, J.; Haemmerle, G.; Kershaw, E.E.; Young, M.E.; Light, P.E.; Oudit, G.Y.; Zechner, R.; et al. Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol. Cell. Biol. 2012, 32, 740–750. [Google Scholar] [CrossRef]
- Haemmerle, G.; Lass, A.; Zimmermann, R.; Gorkiewicz, G.; Meyer, C.; Rozman, J.; Heldmaier, G.; Maier, R.; Theussl, C.; Eder, S.; et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006, 312, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Buttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef]
- Kienesberger, P.C.; Pulinilkunnil, T.; Nagendran, J.; Young, M.E.; Bogner-Strauss, J.G.; Hackl, H.; Khadour, R.; Heydari, E.; Haemmerle, G.; Zechner, R.; et al. Early structural and metabolic cardiac remodelling in response to inducible adipose triglyceride lipase ablation. Cardiovasc. Res. 2013, 99, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Pulinilkunnil, T.; Kienesberger, P.C.; Nagendran, J.; Waller, T.J.; Young, M.E.; Kershaw, E.E.; Korbutt, G.; Haemmerle, G.; Zechner, R.; Dyck, J.R. Myocardial adipose triglyceride lipase overexpression protects diabetic mice from the development of lipotoxic cardiomyopathy. Diabetes 2013, 62, 1464–1477. [Google Scholar] [CrossRef]
- Kintscher, U.; Foryst-Ludwig, A.; Haemmerle, G.; Zechner, R. The Role of Adipose Triglyceride Lipase and Cytosolic Lipolysis in Cardiac Function and Heart Failure. Cell. Rep. Med. 2020, 1, 100001. [Google Scholar] [CrossRef]
- Albert, J.S.; Yerges-Armstrong, L.M.; Horenstein, R.B.; Pollin, T.I.; Sreenivasan, U.T.; Chai, S.; Blaner, W.S.; Snitker, S.; O’Connell, J.R.; Gong, D.W.; et al. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N. Engl. J. Med. 2014, 370, 2307–2315. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Suzuki, J.; Zenimaru, Y.; Takahashi, S.; Koizumi, T.; Noriki, S.; Yamaguchi, O.; Otsu, K.; Shen, W.J.; Kraemer, F.B.; et al. Cardiac overexpression of hormone-sensitive lipase inhibits myocardial steatosis and fibrosis in streptozotocin diabetic mice. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E1109–E1118. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Suzuki, J.; Sato, S.; Zenimaru, Y.; Saito, R.; Konoshita, T.; Kraemer, F.B.; Ishizuka, T. Hormone-sensitive lipase protects adipose triglyceride lipase-deficient mice from lethal lipotoxic cardiomyopathy. J. Lipid Res. 2022, 63, 100194. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, H.J.; Wang, S.; Higashimori, T.; Dong, J.; Kim, Y.J.; Cline, G.; Li, H.; Prentki, M.; Shulman, G.I.; et al. Hormone-sensitive lipase knockout mice have increased hepatic insulin sensitivity and are protected from short-term diet-induced insulin resistance in skeletal muscle and heart. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E30–E39. [Google Scholar] [CrossRef]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef]
- Kuramoto, K.; Sakai, F.; Yoshinori, N.; Nakamura, T.Y.; Wakabayashi, S.; Kojidani, T.; Haraguchi, T.; Hirose, F.; Osumi, T. Deficiency of a lipid droplet protein, perilipin 5, suppresses myocardial lipid accumulation, thereby preventing type 1 diabetes-induced heart malfunction. Mol. Cell. Biol. 2014, 34, 2721–2731. [Google Scholar] [CrossRef]
- Wang, H.; Sreenivasan, U.; Gong, D.W.; O’Connell, K.A.; Dabkowski, E.R.; Hecker, P.A.; Ionica, N.; Konig, M.; Mahurkar, A.; Sun, Y.; et al. Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J. Lipid Res. 2013, 54, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Souza, S.C.; Zhang, H.H.; Strissel, K.J.; Christoffolete, M.A.; Kovsan, J.; Rudich, A.; Kraemer, F.B.; Bianco, A.C.; Obin, M.S.; et al. Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J. Biol. Chem. 2006, 281, 15837–15844. [Google Scholar] [CrossRef] [PubMed]
- Kolleritsch, S.; Kien, B.; Schoiswohl, G.; Diwoky, C.; Schreiber, R.; Heier, C.; Maresch, L.K.; Schweiger, M.; Eichmann, T.O.; Stryeck, S.; et al. Low cardiac lipolysis reduces mitochondrial fission and prevents lipotoxic heart dysfunction in Perilipin 5 mutant mice. Cardiovasc. Res. 2020, 116, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, P.H.; Bisgaier, C.L.; Aalto-Setala, K.; Radner, H.; Ramakrishnan, R.; Levak-Frank, S.; Essenburg, A.D.; Zechner, R.; Breslow, J.L. Severe hypertriglyceridemia, reduced high density lipoprotein, and neonatal death in lipoprotein lipase knockout mice. Mild hypertriglyceridemia with impaired very low density lipoprotein clearance in heterozygotes. J. Clin. Investig. 1995, 96, 2555–2568. [Google Scholar] [CrossRef]
- Levak-Frank, S.; Hofmann, W.; Weinstock, P.H.; Radner, H.; Sattler, W.; Breslow, J.L.; Zechner, R. Induced mutant mouse lines that express lipoprotein lipase in cardiac muscle, but not in skeletal muscle and adipose tissue, have normal plasma triglyceride and high-density lipoprotein-cholesterol levels. Proc. Natl. Acad. Sci. USA 1999, 96, 3165–3170. [Google Scholar] [CrossRef] [PubMed]
- Yagyu, H.; Chen, G.; Yokoyama, M.; Hirata, K.; Augustus, A.; Kako, Y.; Seo, T.; Hu, Y.; Lutz, E.P.; Merkel, M.; et al. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J. Clin. Investig. 2003, 111, 419–426. [Google Scholar] [CrossRef]
- Augustus, A.; Yagyu, H.; Haemmerle, G.; Bensadoun, A.; Vikramadithyan, R.K.; Park, S.Y.; Kim, J.K.; Zechner, R.; Goldberg, I.J. Cardiac-specific knock-out of lipoprotein lipase alters plasma lipoprotein triglyceride metabolism and cardiac gene expression. J. Biol. Chem. 2004, 279, 25050–25057. [Google Scholar] [CrossRef]
- Augustus, A.S.; Buchanan, J.; Park, T.S.; Hirata, K.; Noh, H.L.; Sun, J.; Homma, S.; D’Armiento, J.; Abel, E.D.; Goldberg, I.J. Loss of lipoprotein lipase-derived fatty acids leads to increased cardiac glucose metabolism and heart dysfunction. J. Biol. Chem. 2006, 281, 8716–8723. [Google Scholar] [CrossRef]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Peterfy, M.; Ben-Zeev, O.; Mao, H.Z.; Weissglas-Volkov, D.; Aouizerat, B.E.; Pullinger, C.R.; Frost, P.H.; Kane, J.P.; Malloy, M.J.; Reue, K.; et al. Mutations in LMF1 cause combined lipase deficiency and severe hypertriglyceridemia. Nat. Genet. 2007, 39, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Davies, B.S.; Goulbourne, C.N.; Barnes, R.H., 2nd; Turlo, K.A.; Gin, P.; Vaughan, S.; Vaux, D.J.; Bensadoun, A.; Beigneux, A.P.; Fong, L.G.; et al. Assessing mechanisms of GPIHBP1 and lipoprotein lipase movement across endothelial cells. J. Lipid Res. 2012, 53, 2690–2697. [Google Scholar] [CrossRef]
- Sha, H.; Sun, S.; Francisco, A.B.; Ehrhardt, N.; Xue, Z.; Liu, L.; Lawrence, P.; Mattijssen, F.; Guber, R.D.; Panhwar, M.S.; et al. The ER-associated degradation adaptor protein Sel1L regulates LPL secretion and lipid metabolism. Cell. Metab. 2014, 20, 458–470. [Google Scholar] [CrossRef]
- Mandard, S.; Zandbergen, F.; van Straten, E.; Wahli, W.; Kuipers, F.; Muller, M.; Kersten, S. The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J. Biol. Chem. 2006, 281, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Koster, A.; Chao, Y.B.; Mosior, M.; Ford, A.; Gonzalez-DeWhitt, P.A.; Hale, J.E.; Li, D.; Qiu, Y.; Fraser, C.C.; Yang, D.D.; et al. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: Regulation of triglyceride metabolism. Endocrinology 2005, 146, 4943–4950. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.F.; Mintah, I.J.; Shihanian, L.M.; Stevis, P.; Buckler, D.; Alexa-Braun, C.A.; Kleiner, S.; Banfi, S.; Cohen, J.C.; Hobbs, H.H.; et al. ANGPTL8 requires ANGPTL3 to inhibit lipoprotein lipase and plasma triglyceride clearance. J. Lipid Res. 2017, 58, 1166–1173. [Google Scholar] [CrossRef]
- Chi, X.; Britt, E.C.; Shows, H.W.; Hjelmaas, A.J.; Shetty, S.K.; Cushing, E.M.; Li, W.; Dou, A.; Zhang, R.; Davies, B.S.J. ANGPTL8 promotes the ability of ANGPTL3 to bind and inhibit lipoprotein lipase. Mol. Metab. 2017, 6, 1137–1149. [Google Scholar] [CrossRef]
- Kersten, S. New insights into angiopoietin-like proteins in lipid metabolism and cardiovascular disease risk. Curr. Opin. Lipidol. 2019, 30, 205–211. [Google Scholar] [CrossRef]
- Yu, X.; Burgess, S.C.; Ge, H.; Wong, K.K.; Nassem, R.H.; Garry, D.J.; Sherry, A.D.; Malloy, C.R.; Berger, J.P.; Li, C. Inhibition of cardiac lipoprotein utilization by transgenic overexpression of Angptl4 in the heart. Proc. Natl. Acad. Sci. USA 2005, 102, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Ehrhardt, N.; Weissglas-Volkov, D.; Lai, C.M.; Mao, H.Z.; Liao, J.L.; Nikkola, E.; Bensadoun, A.; Taskinen, M.R.; Doolittle, M.H.; et al. Transgenic expression and genetic variation of Lmf1 affect LPL activity in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1204–1210. [Google Scholar] [CrossRef]
- Vallerie, S.N.; Bornfeldt, K.E. GPIHBP1: Two get tangled. Circ. Res. 2015, 116, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’Dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Dewey, F.E.; Gusarova, V.; O’Dushlaine, C.; Gottesman, O.; Trejos, J.; Hunt, C.; Van Hout, C.V.; Habegger, L.; Buckler, D.; Lai, K.M.; et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 1123–1133. [Google Scholar] [CrossRef]
- El Alaoui-Talibi, Z.; Landormy, S.; Loireau, A.; Moravec, J. Fatty acid oxidation and mechanical performance of volume-overloaded rat hearts. Am. J. Physiol. 1992, 262, H1068–H1074. [Google Scholar] [CrossRef]
- Reibel, D.K.; Uboh, C.E.; Kent, R.L. Altered coenzyme A and carnitine metabolism in pressure-overload hypertrophied hearts. Am. J. Physiol. 1983, 244, H839–H843. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Samaja, M.; Tarantola, M.; Micheletti, R.; Bianchi, G. Functional and metabolic effects of propionyl-L-carnitine in the isolated perfused hypertrophied rat heart. Mol. Cell. Biochem. 1992, 116, 139–145. [Google Scholar] [CrossRef]
- Schonekess, B.O.; Allard, M.F.; Lopaschuk, G.D. Propionyl L-carnitine improvement of hypertrophied heart function is accompanied by an increase in carbohydrate oxidation. Circ. Res. 1995, 77, 726–734. [Google Scholar] [CrossRef]
- Bartels, G.L.; Remme, W.J.; Pillay, M.; Schonfeld, D.H.; Cox, P.H.; Kruijssen, H.A.; Knufman, N.M. Acute improvement of cardiac function with intravenous L-propionylcarnitine in humans. J. Cardiovasc. Pharmacol. 1992, 20, 157–164. [Google Scholar]
- Schonekess, B.O.; Allard, M.F.; Lopaschuk, G.D. Propionyl L-carnitine improvement of hypertrophied rat heart function is associated with an increase in cardiac efficiency. Eur. J. Pharmacol. 1995, 286, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Sorokina, N.; O’Donnell, J.M.; McKinney, R.D.; Pound, K.M.; Woldegiorgis, G.; LaNoue, K.F.; Ballal, K.; Taegtmeyer, H.; Buttrick, P.M.; Lewandowski, E.D. Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation 2007, 115, 2033–2041. [Google Scholar] [CrossRef]
- He, L.; Kim, T.; Long, Q.; Liu, J.; Wang, P.; Zhou, Y.; Ding, Y.; Prasain, J.; Wood, P.A.; Yang, Q. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 2012, 126, 1705–1716. [Google Scholar] [CrossRef]
- Schmidt-Schweda, S.; Holubarsch, C. First clinical trial with etomoxir in patients with chronic congestive heart failure. Clin. Sci. 2000, 99, 27–35. [Google Scholar] [CrossRef]
- Lewandowski, E.D.; Fischer, S.K.; Fasano, M.; Banke, N.H.; Walker, L.A.; Huqi, A.; Wang, X.; Lopaschuk, G.D.; O’Donnell, J.M. Acute liver carnitine palmitoyltransferase I overexpression recapitulates reduced palmitate oxidation of cardiac hypertrophy. Circ. Res. 2013, 112, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Davila-Roman, V.G.; Vedala, G.; Herrero, P.; de las Fuentes, L.; Rogers, J.G.; Kelly, D.P.; Gropler, R.J. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2002, 40, 271–277. [Google Scholar] [CrossRef]
- Tuunanen, H.; Engblom, E.; Naum, A.; Nagren, K.; Hesse, B.; Airaksinen, K.E.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; Opie, L.H.; et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation 2006, 114, 2130–2137. [Google Scholar] [CrossRef]
- Neglia, D.; De Caterina, A.; Marraccini, P.; Natali, A.; Ciardetti, M.; Vecoli, C.; Gastaldelli, A.; Ciociaro, D.; Pellegrini, P.; Testa, R.; et al. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3270–H3278. [Google Scholar] [CrossRef]
- Tuunanen, H.; Engblom, E.; Naum, A.; Scheinin, M.; Nagren, K.; Airaksinen, J.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; Knuuti, J. Decreased myocardial free fatty acid uptake in patients with idiopathic dilated cardiomyopathy: Evidence of relationship with insulin resistance and left ventricular dysfunction. J. Card. Fail. 2006, 12, 644–652. [Google Scholar] [CrossRef]
- Rosenblatt-Velin, N.; Montessuit, C.; Papageorgiou, I.; Terrand, J.; Lerch, R. Postinfarction heart failure in rats is associated with upregulation of GLUT-1 and downregulation of genes of fatty acid metabolism. Cardiovasc. Res. 2001, 52, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Tamaki, Y.; Iwanaga, Y.; Narazaki, M.; Matsuda, T.; Soga, T.; et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ. Heart Fail. 2010, 3, 420–430. [Google Scholar] [CrossRef]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 1996, 94, 2837–2842. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.M.; Fields, A.D.; Sorokina, N.; Lewandowski, E.D. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J. Mol. Cell. Cardiol. 2008, 44, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Farber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef]
- Osorio, J.C.; Stanley, W.C.; Linke, A.; Castellari, M.; Diep, Q.N.; Panchal, A.R.; Hintze, T.H.; Lopaschuk, G.D.; Recchia, F.A. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation 2002, 106, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Rijzewijk, L.J.; van der Meer, R.W.; Lamb, H.J.; de Jong, H.W.; Lubberink, M.; Romijn, J.A.; Bax, J.J.; de Roos, A.; Twisk, J.W.; Heine, R.J.; et al. Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: Studies with cardiac positron emission tomography and magnetic resonance imaging. J. Am. Coll. Cardiol. 2009, 54, 1524–1532. [Google Scholar] [CrossRef]
- Peterson, L.R.; Herrero, P.; Schechtman, K.B.; Racette, S.B.; Waggoner, A.D.; Kisrieva-Ware, Z.; Dence, C.; Klein, S.; Marsala, J.; Meyer, T.; et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 2004, 109, 2191–2196. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by beta-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef]
- Barouch, L.A.; Berkowitz, D.E.; Harrison, R.W.; O’Donnell, C.P.; Hare, J.M. Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 2003, 108, 754–759. [Google Scholar] [CrossRef]
- Christoffersen, C.; Bollano, E.; Lindegaard, M.L.; Bartels, E.D.; Goetze, J.P.; Andersen, C.B.; Nielsen, L.B. Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology 2003, 144, 3483–3490. [Google Scholar] [CrossRef]
- Fukushima, A.; Lopaschuk, G.D. Acetylation control of cardiac fatty acid beta-oxidation and energy metabolism in obesity, diabetes, and heart failure. Biochim. Biophys. Acta 2016, 1862, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, A.; Lopaschuk, G.D. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta 2016, 1861, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; Altamirano, F.; Szweda, P.A.; Elnwasany, A.; Lee, D.I.; Yoo, H.; Kass, D.A.; Szweda, L.I.; et al. NAD(+) Repletion Reverses Heart Failure With Preserved Ejection Fraction. Circ. Res. 2021, 128, 1629–1641. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, D.D.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Utsumi, H.; Kang, D.; Hattori, N.; Uchida, K.; Arimura, K.; Egashira, K.; Takeshita, A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ. Res. 1999, 85, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell. Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Zhao, M.; Zhou, B.; Yoshii, A.; Bugg, D.; Villet, O.; Sahu, A.; Olson, G.S.; Davis, J.; Tian, R. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J. Clin. Investig. 2023, 133, e159498. [Google Scholar] [CrossRef]
- Desvergne, B.; Michalik, L.; Wahli, W. Transcriptional regulation of metabolism. Physiol. Rev. 2006, 86, 465–514. [Google Scholar] [CrossRef]
- Scholtes, C.; Giguere, V. Transcriptional control of energy metabolism by nuclear receptors. Nat. Rev. Mol. Cell. Biol. 2022, 23, 750–770. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Ding, G.; Qin, Q.; Xiao, Y.; Woods, D.; Chen, Y.E.; Yang, Q. Peroxisome proliferator-activated receptor delta activates fatty acid oxidation in cultured neonatal and adult cardiomyocytes. Biochem. Biophys. Res. Commun. 2004, 313, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Goikoetxea, M.J.; Beaumont, J.; Gonzalez, A.; Lopez, B.; Querejeta, R.; Larman, M.; Diez, J. Altered cardiac expression of peroxisome proliferator-activated receptor-isoforms in patients with hypertensive heart disease. Cardiovasc. Res. 2006, 69, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Karbowska, J.; Kochan, Z.; Smolenski, R.T. Peroxisome proliferator-activated receptor alpha is downregulated in the failing human heart. Cell. Mol. Biol. Lett. 2003, 8, 49–53. [Google Scholar] [PubMed]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.S.; Oka, S.I.; Zablocki, D.; Sadoshima, J. Metabolic reprogramming via PPARalpha signaling in cardiac hypertrophy and failure: From metabolomics to epigenetics. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H584–H596. [Google Scholar] [CrossRef]
- Luptak, I.; Balschi, J.A.; Xing, Y.; Leone, T.C.; Kelly, D.P.; Tian, R. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation 2005, 112, 2339–2346. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, X.X.; Jiao, S.Y.; Qi, D.; Yu, B.Q.; Xie, G.M.; Liu, Y.; Song, Y.T.; Xu, Q.; Xu, Q.B.; et al. Cardiomyocyte peroxisome proliferator-activated receptor alpha is essential for energy metabolism and extracellular matrix homeostasis during pressure overload-induced cardiac remodeling. Acta Pharmacol. Sin. 2022, 43, 1231–1242. [Google Scholar] [CrossRef]
- Kaimoto, S.; Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Tateishi, S.; Ono, K.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Matoba, S. Activation of PPAR-alpha in the early stage of heart failure maintained myocardial function and energetics in pressure-overload heart failure. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H305–H313. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Han, X.; Courtois, M.; Aimond, F.; Nerbonne, J.M.; Kovacs, A.; Gross, R.W.; Kelly, D.P. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc. Natl. Acad. Sci. USA 2003, 100, 1226–1231. [Google Scholar] [CrossRef]
- Finck, B.N.; Lehman, J.J.; Leone, T.C.; Welch, M.J.; Bennett, M.J.; Kovacs, A.; Han, X.; Gross, R.W.; Kozak, R.; Lopaschuk, G.D.; et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J. Clin. Investig. 2002, 109, 121–130. [Google Scholar] [CrossRef]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell. Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef]
- Son, N.H.; Park, T.S.; Yamashita, H.; Yokoyama, M.; Huggins, L.A.; Okajima, K.; Homma, S.; Szabolcs, M.J.; Huang, L.S.; Goldberg, I.J. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J. Clin. Investig. 2007, 117, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Son, N.H.; Yu, S.; Tuinei, J.; Arai, K.; Hamai, H.; Homma, S.; Shulman, G.I.; Abel, E.D.; Goldberg, I.J. PPARgamma-induced cardiolipotoxicity in mice is ameliorated by PPARalpha deficiency despite increases in fatty acid oxidation. J. Clin. Investig. 2010, 120, 3443–3454. [Google Scholar] [CrossRef] [PubMed]
- Drosatos, K.; Khan, R.S.; Trent, C.M.; Jiang, H.; Son, N.H.; Blaner, W.S.; Homma, S.; Schulze, P.C.; Goldberg, I.J. Peroxisome proliferator-activated receptor-gamma activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ. Heart Fail. 2013, 6, 550–562. [Google Scholar] [CrossRef]
- Cheng, L.; Ding, G.; Qin, Q.; Huang, Y.; Lewis, W.; He, N.; Evans, R.M.; Schneider, M.D.; Brako, F.A.; Xiao, Y.; et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat. Med. 2004, 10, 1245–1250. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1alpha: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Sihag, S.; Cresci, S.; Li, A.Y.; Sucharov, C.C.; Lehman, J.J. PGC-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J. Mol. Cell. Cardiol. 2009, 46, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Investig. 2000, 106, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Martin, O.J.; Lai, L.; Soundarapandian, M.M.; Leone, T.C.; Zorzano, A.; Keller, M.P.; Attie, A.D.; Muoio, D.M.; Kelly, D.P. A role for peroxisome proliferator-activated receptor gamma coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ. Res. 2014, 114, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef] [PubMed]
- Zaha, V.G.; Young, L.H. AMP-activated protein kinase regulation and biological actions in the heart. Circ. Res. 2012, 111, 800–814. [Google Scholar] [CrossRef]
- Yeh, L.A.; Lee, K.H.; Kim, K.H. Regulation of rat liver acetyl-CoA carboxylase. Regulation of phosphorylation and inactivation of acetyl-CoA carboxylase by the adenylate energy charge. J. Biol. Chem. 1980, 255, 2308–2314. [Google Scholar] [CrossRef]
- Luiken, J.J.; Koonen, D.P.; Willems, J.; Zorzano, A.; Becker, C.; Fischer, Y.; Tandon, N.N.; Van Der Vusse, G.J.; Bonen, A.; Glatz, J.F. Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes 2002, 51, 3113–3119. [Google Scholar] [CrossRef]
- Luiken, J.J.; Coort, S.L.; Willems, J.; Coumans, W.A.; Bonen, A.; van der Vusse, G.J.; Glatz, J.F. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes 2003, 52, 1627–1634. [Google Scholar] [CrossRef]
- Chabowski, A.; Momken, I.; Coort, S.L.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Bonen, A. Prolonged AMPK activation increases the expression of fatty acid transporters in cardiac myocytes and perfused hearts. Mol. Cell. Biochem. 2006, 288, 201–212. [Google Scholar] [CrossRef]
- Hu, X.; Xu, X.; Lu, Z.; Zhang, P.; Fassett, J.; Zhang, Y.; Xin, Y.; Hall, J.L.; Viollet, B.; Bache, R.J.; et al. AMP activated protein kinase-alpha2 regulates expression of estrogen-related receptor-alpha, a metabolic transcription factor related to heart failure development. Hypertension 2011, 58, 696–703. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, Q.; Zhang, L.; Fang, Z.; Zhao, F.; Lv, Z.; Gu, Z.; Zhang, J.; Wang, J.; Zen, K.; et al. Hypoxia induces PGC-1alpha expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell. Res. 2010, 20, 676–687. [Google Scholar] [CrossRef]
- McGee, S.L.; van Denderen, B.J.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008, 57, 860–867. [Google Scholar] [CrossRef]
- Allard, M.F.; Parsons, H.L.; Saeedi, R.; Wambolt, R.B.; Brownsey, R. AMPK and metabolic adaptation by the heart to pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H140–H148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hu, X.; Xu, X.; Fassett, J.; Zhu, G.; Viollet, B.; Xu, W.; Wiczer, B.; Bernlohr, D.A.; Bache, R.J.; et al. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension 2008, 52, 918–924. [Google Scholar] [CrossRef]
- Turdi, S.; Kandadi, M.R.; Zhao, J.; Huff, A.F.; Du, M.; Ren, J. Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. J. Mol. Cell. Cardiol. 2011, 50, 712–722. [Google Scholar] [CrossRef]
- Li, H.L.; Yin, R.; Chen, D.; Liu, D.; Wang, D.; Yang, Q.; Dong, Y.G. Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J. Cell. Biochem. 2007, 100, 1086–1099. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.R., 3rd; Li, J.; Coven, D.L.; Pypaert, M.; Zechner, C.; Palmeri, M.; Giordano, F.J.; Mu, J.; Birnbaum, M.J.; Young, L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Investig. 2004, 114, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Musi, N.; Fujii, N.; Zou, L.; Luptak, I.; Hirshman, M.F.; Goodyear, L.J.; Tian, R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J. Biol. Chem. 2003, 278, 28372–28377. [Google Scholar] [CrossRef] [PubMed]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 2009, 104, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Asanuma, H.; Fujita, M.; Takahama, H.; Wakeno, M.; Ito, S.; Ogai, A.; Asakura, M.; Kim, J.; Minamino, T.; et al. Metformin prevents progression of heart failure in dogs: Role of AMP-activated protein kinase. Circulation 2009, 119, 2568–2577. [Google Scholar] [CrossRef]
- Zong, G.; Li, Y.; Wanders, A.J.; Alssema, M.; Zock, P.L.; Willett, W.C.; Hu, F.B.; Sun, Q. Intake of individual saturated fatty acids and risk of coronary heart disease in US men and women: Two prospective longitudinal cohort studies. BMJ 2016, 355, i5796. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, R.N.; McKnight, B.; Sotoodehnia, N.; Fretts, A.M.; Qureshi, W.T.; Song, X.; King, I.B.; Sitlani, C.M.; Siscovick, D.S.; Psaty, B.M.; et al. Circulating Very Long-Chain Saturated Fatty Acids and Heart Failure: The Cardiovascular Health Study. J. Am. Heart Assoc. 2018, 7, e010019. [Google Scholar] [CrossRef]
- Farvid, M.S.; Ding, M.; Pan, A.; Sun, Q.; Chiuve, S.E.; Steffen, L.M.; Willett, W.C.; Hu, F.B. Dietary linoleic acid and risk of coronary heart disease: A systematic review and meta-analysis of prospective cohort studies. Circulation 2014, 130, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Naghshi, S.; Aune, D.; Beyene, J.; Mobarak, S.; Asadi, M.; Sadeghi, O. Dietary intake and biomarkers of alpha linolenic acid and risk of all cause, cardiovascular, and cancer mortality: Systematic review and dose-response meta-analysis of cohort studies. BMJ 2021, 375, n2213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, J.V.; Schooling, C.M. The associations of plasma phospholipid arachidonic acid with cardiovascular diseases: A Mendelian randomization study. EBioMedicine 2021, 63, 103189. [Google Scholar] [CrossRef] [PubMed]
- Mason, R.P.; Libby, P.; Bhatt, D.L. Emerging Mechanisms of Cardiovascular Protection for the Omega-3 Fatty Acid Eicosapentaenoic Acid. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1135–1147. [Google Scholar] [CrossRef]
- Block, R.C.; Liu, L.; Herrington, D.M.; Huang, S.; Tsai, M.Y.; O’Connell, T.D.; Shearer, G.C. Predicting Risk for Incident Heart Failure With Omega-3 Fatty Acids: From MESA. JACC Heart Fail. 2019, 7, 651–661. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).