

Differential Expression Analysis Utilizing Condition-Specific Metabolic Pathways

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
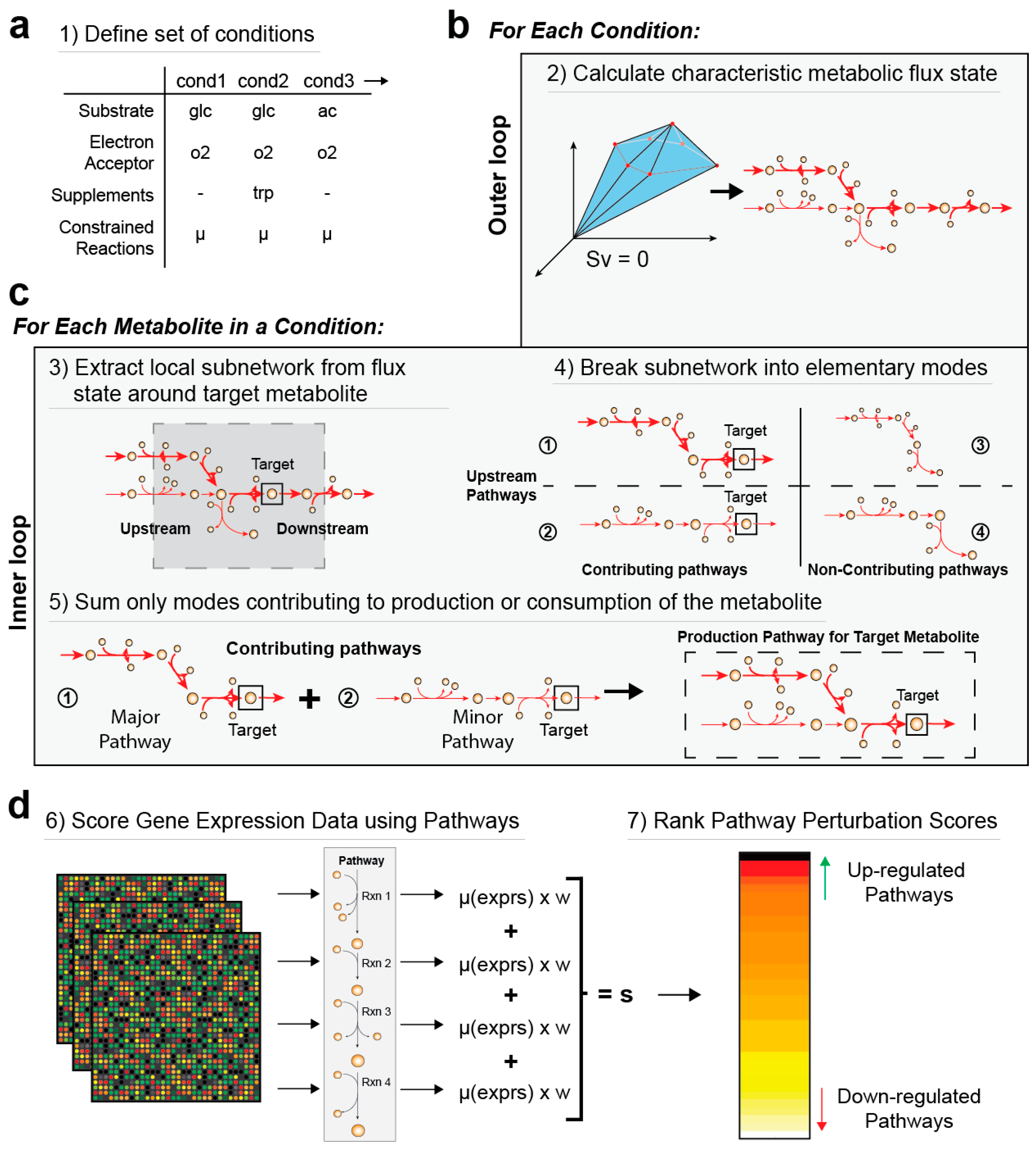
2.1. Overview of MetPath Pathway Calculation and Differential Gene Expression Analysis
2.2. Calculation of a Condition-Specific Flux State
2.3. Calculation of Production and Degradation Pathways for Each Metabolite
2.4. Universal Database Generation
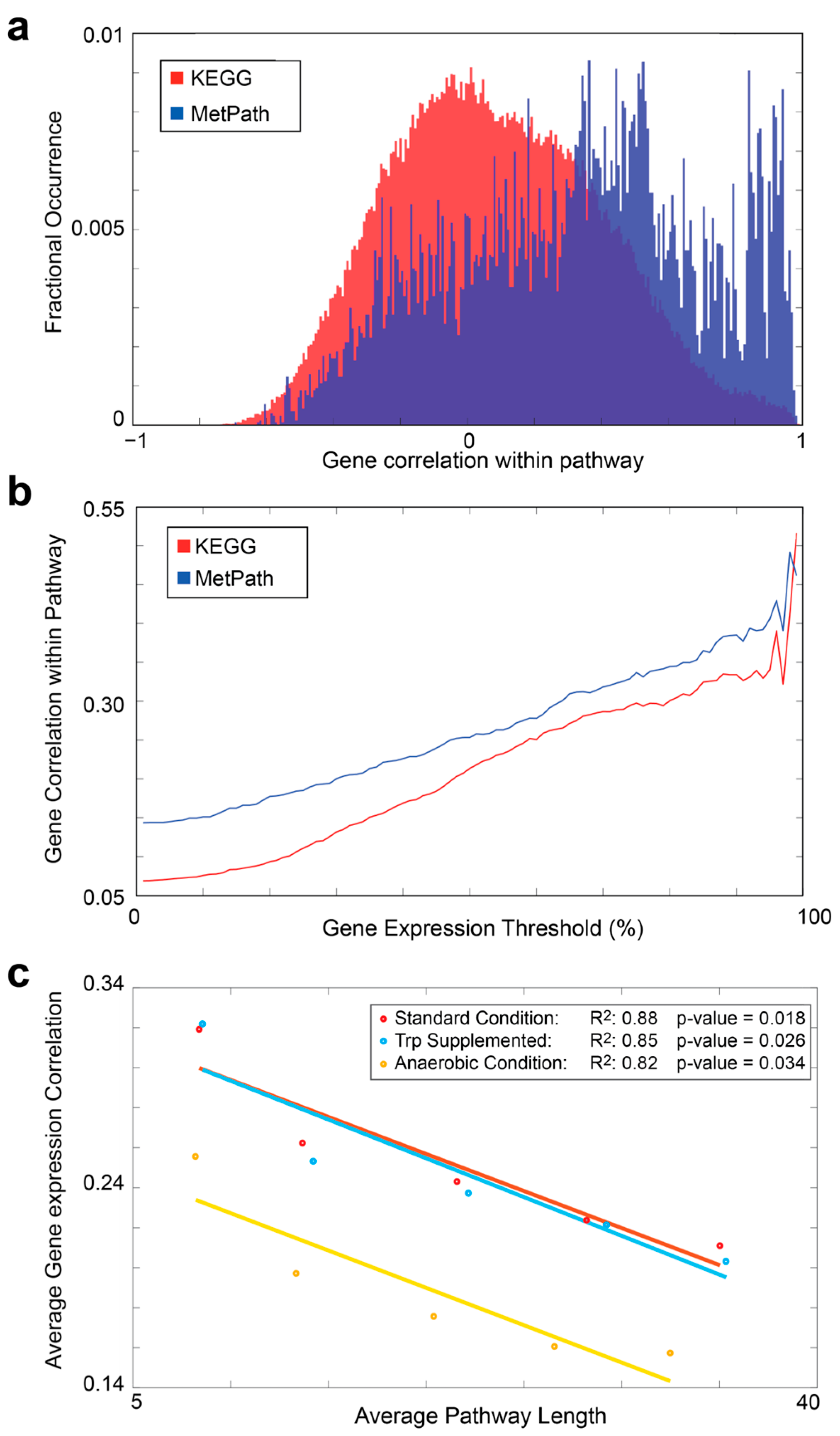
2.5. KEGG Comparison
2.6. Anaerobic Condition Analysis
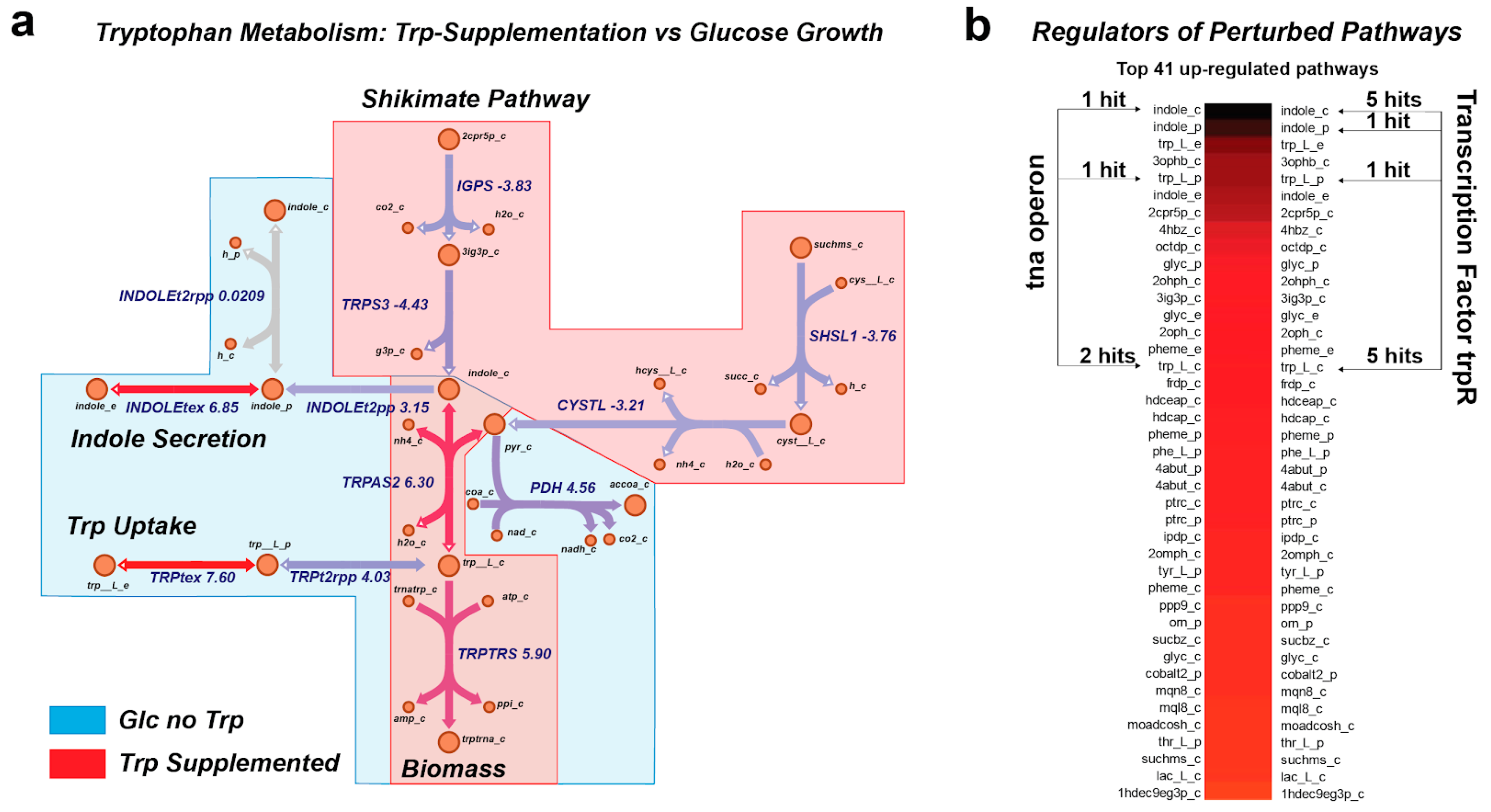
2.7. Tryptophan Supplementation Analysis
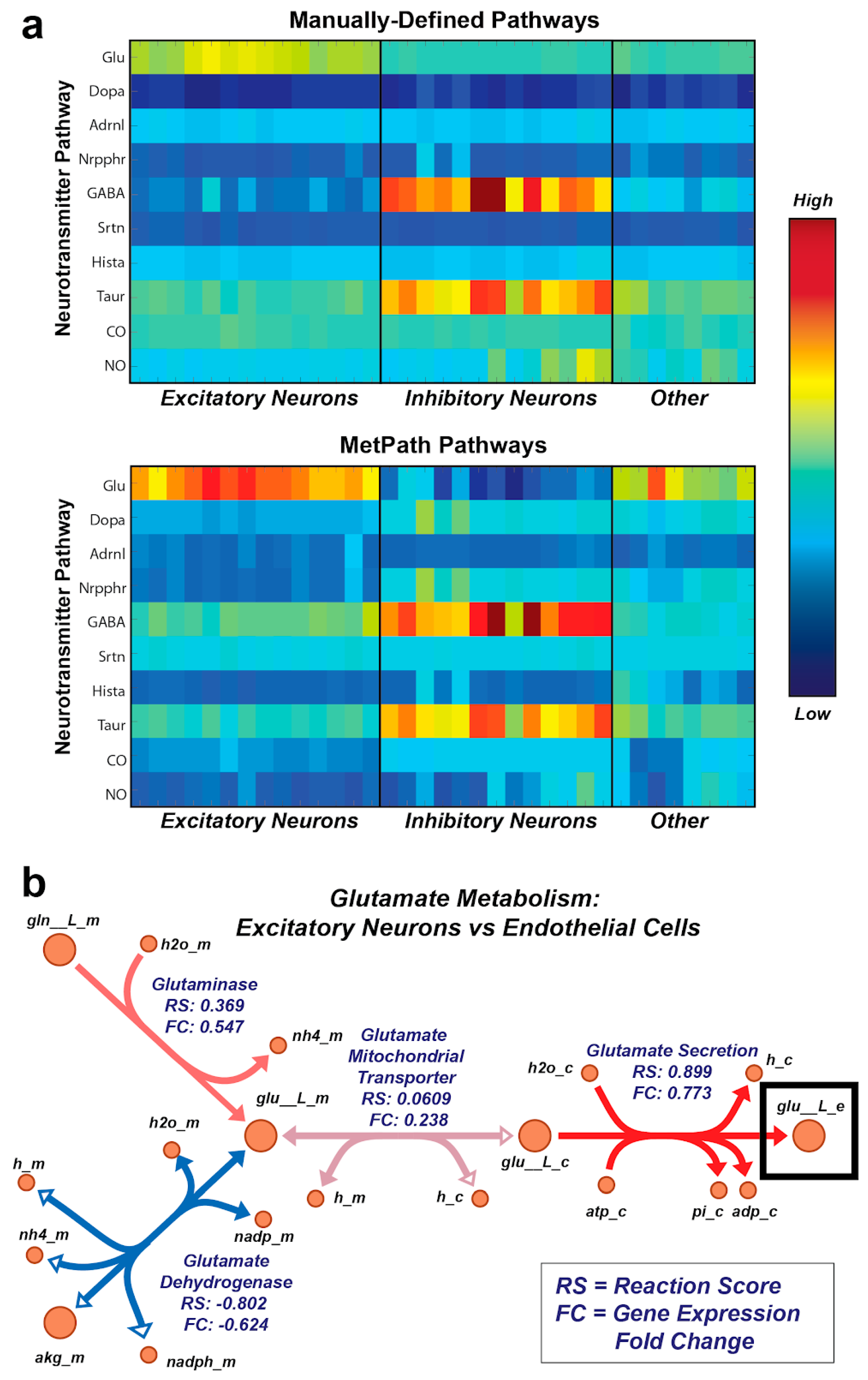
2.8. Neurotransmitter Analysis
3. Results
3.1. Calculation of Condition-Specific Pathways for Production and Consumption of Metabolites
3.2. Definition of a Universal Pathway Database for E. coli Expression Analysis
3.3. Comparison of MetPath Pathways to Manually Curated Pathways
3.4. MetPath Pathways Reveal Coordinated Expression Changes with Shifts in Environment
3.5. MetPath Pathways Recapitulate Canonical Cell Type-Specific Metabolic Functions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joyce, A.R.; Palsson, B.Ø. The Model Organism as a System: Integrating “Omics” Data Sets. Nat. Rev. Mol. Cell Biol. 2006, 7, 198. [Google Scholar] [CrossRef] [PubMed]
- Berger, B.; Peng, J.; Singh, M. Computational Solutions for Omics Data. Nat. Rev. Genet. 2013, 14, 333. [Google Scholar] [CrossRef] [PubMed]
- Hyduke, D.R.; Lewis, N.E.; Palsson, B.Ø. Analysis of Omics Data with Genome-Scale Models of Metabolism. Mol. Biosyst. 2013, 9, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Bordbar, A.; Nagarajan, H.; Lewis, N.E.; Latif, H.; Ebrahim, A.; Federowicz, S.; Schellenberger, J.; Palsson, B.O. Minimal Metabolic Pathway Structure Is Consistent with Associated Biomolecular Interactions. Mol. Syst. Biol. 2014, 10, 737. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Monk, J.M.; Charusanti, P.; Aziz, R.K.; Lerman, J.A.; Premyodhin, N.; Orth, J.D.; Feist, A.M.; Palsson, B.Ø. Genome-Scale Metabolic Reconstructions of Multiple Escherichia coli Strains Highlight Strain-Specific Adaptations to Nutritional Environments. Proc. Natl. Acad. Sci. USA 2013, 110, 20338–20343. [Google Scholar] [CrossRef]
- Wiback, S.J.; Mahadevan, R.; Palsson, B.Ø. Reconstructing Metabolic Flux Vectors from Extreme Pathways: Defining the Alpha-Spectrum. J. Theor. Biol. 2003, 224, 313–324. [Google Scholar] [CrossRef]
- Chan, S.H.J.; Ji, P. Decomposing Flux Distributions into Elementary Flux Modes in Genome-Scale Metabolic Networks. Bioinformatics 2011, 27, 2256–2262. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.B.; von Meyenburg, K. Are Growth Rates of Escherichia coli in Batch Cultures Limited by Respiration? J. Bacteriol. 1980, 144, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Covert, M.W.; Knight, E.M.; Reed, J.L.; Herrgard, M.J.; Palsson, B.O. Integrating High-Throughput and Computational Data Elucidates Bacterial Networks. Nature 2004, 429, 92–96. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A Platform for Integrating, Standardizing and Sharing Genome-Scale Models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Thiele, I.; Palsson, B.Ø. What Is Flux Balance Analysis? Nat. Biotechnol. 2010, 28, 245–248. [Google Scholar] [CrossRef]
- Orth, J.D.; Conrad, T.M.; Na, J.; Lerman, J.A.; Nam, H.; Feist, A.M.; Palsson, B.Ø. A Comprehensive Genome-Scale Reconstruction of Escherichia coli Metabolism—2011. Mol. Syst. Biol. 2011, 7, 535. [Google Scholar] [CrossRef]
- Kharchenko, P.; Church, G.M.; Vitkup, D. Expression Dynamics of a Cellular Metabolic Network. Mol. Syst. Biol. 2005, 1, 2005.0016. [Google Scholar] [CrossRef]
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative Single-Cell Analysis of Transcriptional and Epigenetic States in the Human Adult Brain. Nat. Biotechnol. 2018, 36, 70–80. [Google Scholar] [CrossRef]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.Ø. Global Reconstruction of the Human Metabolic Network Based on Genomic and Bibliomic Data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar] [CrossRef]
- Chubukov, V.; Uhr, M.; Le Chat, L.; Kleijn, R.J.; Jules, M.; Link, H.; Aymerich, S.; Stelling, J.; Sauer, U. Transcriptional Regulation Is Insufficient to Explain Substrate-Induced Flux Changes in Bacillus Subtilis. Mol. Syst. Biol. 2013, 9, 709. [Google Scholar] [CrossRef]
- Fendt, S.-M.; Buescher, J.M.; Rudroff, F.; Picotti, P.; Zamboni, N.; Sauer, U. Tradeoff between Enzyme and Metabolite Efficiency Maintains Metabolic Homeostasis upon Perturbations in Enzyme Capacity. Mol. Syst. Biol. 2010, 6, 356. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.D.; Kimball, E.H.; Gao, M.; Osterhout, R.; Van Dien, S.J.; Rabinowitz, J.D. Absolute Metabolite Concentrations and Implied Enzyme Active Site Occupancy in Escherichia coli. Nat. Chem. Biol. 2009, 5, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing Protein Abundance and mRNA Expression Levels on a Genomic Scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mattei, G.; Gan, Z.; Ramazzotti, M.; Palsson, B.O.; Zielinski, D.C. Differential Expression Analysis Utilizing Condition-Specific Metabolic Pathways. Metabolites 2023, 13, 1127. https://doi.org/10.3390/metabo13111127
Mattei G, Gan Z, Ramazzotti M, Palsson BO, Zielinski DC. Differential Expression Analysis Utilizing Condition-Specific Metabolic Pathways. Metabolites. 2023; 13(11):1127. https://doi.org/10.3390/metabo13111127
Chicago/Turabian StyleMattei, Gianluca, Zhuohui Gan, Matteo Ramazzotti, Bernhard O. Palsson, and Daniel C. Zielinski. 2023. "Differential Expression Analysis Utilizing Condition-Specific Metabolic Pathways" Metabolites 13, no. 11: 1127. https://doi.org/10.3390/metabo13111127
APA StyleMattei, G., Gan, Z., Ramazzotti, M., Palsson, B. O., & Zielinski, D. C. (2023). Differential Expression Analysis Utilizing Condition-Specific Metabolic Pathways. Metabolites, 13(11), 1127. https://doi.org/10.3390/metabo13111127