
Urinary Untargeted Metabolic Profile Differentiates Children with Autism from Their Unaffected Siblings

 ,
,  ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Material and Method
2.1. Sample Collection
2.1.1. Subjects Recruitment
2.1.2. Sample Preparation
2.2. Metabolomic Analysis and Data Processing
2.3. Calculation of Concentration Ratios of Selected Urinary Metabolites to Specific Gravity
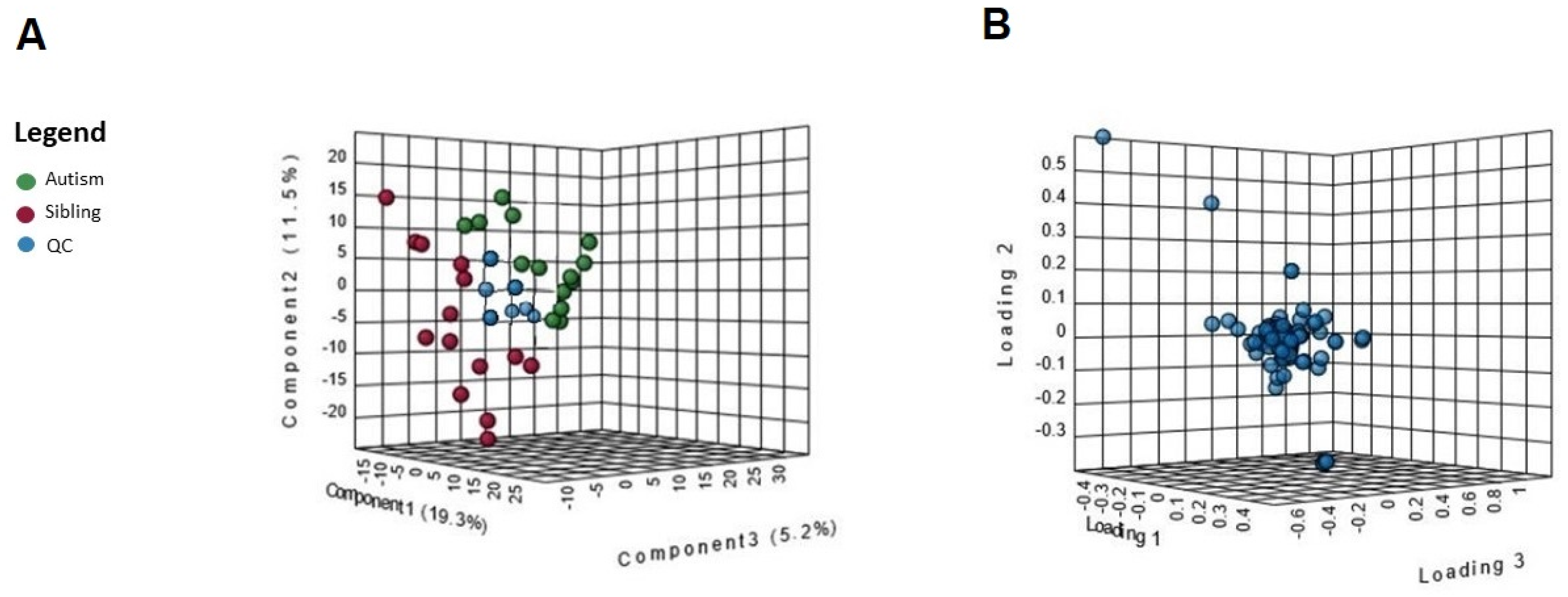
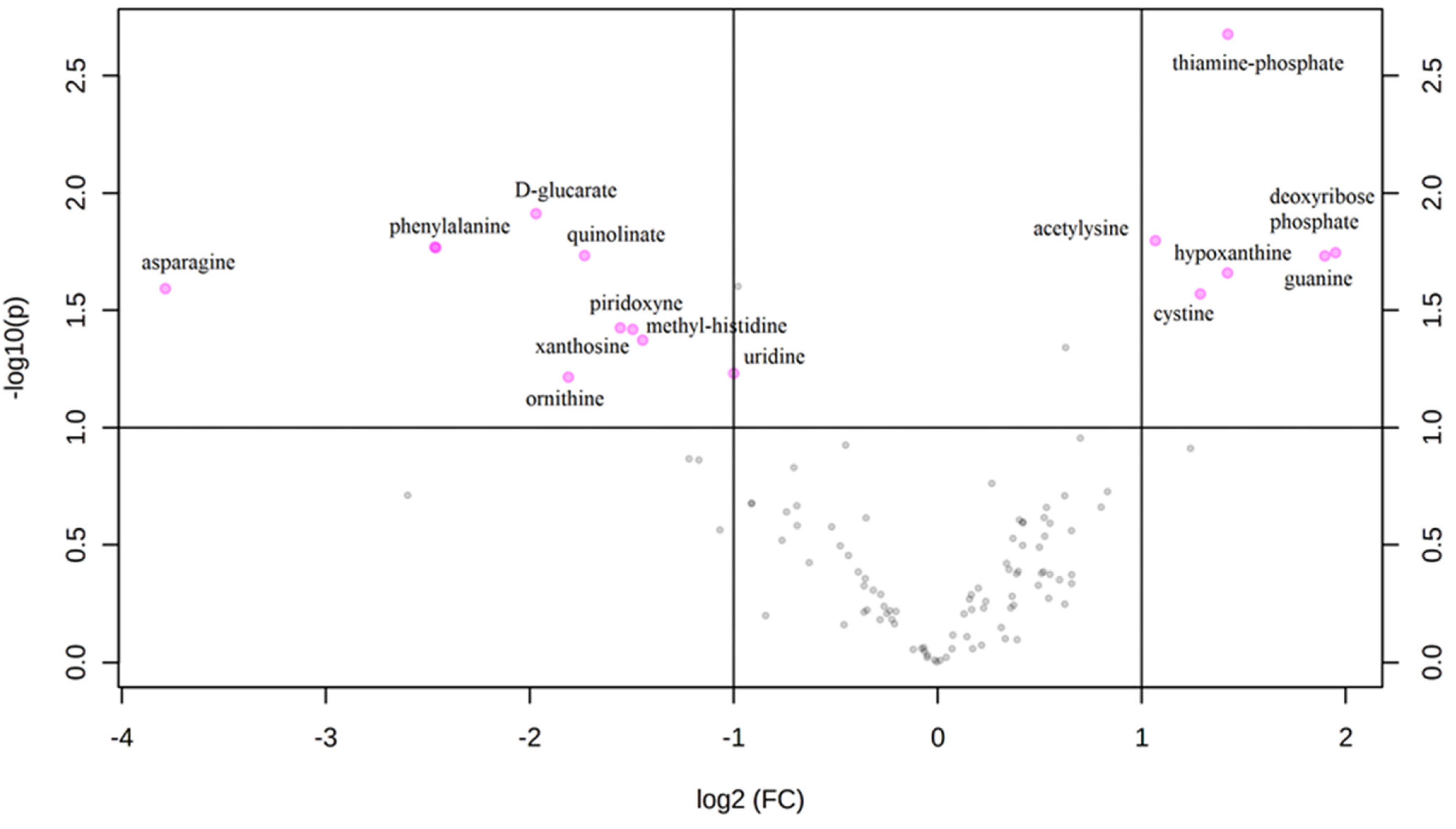
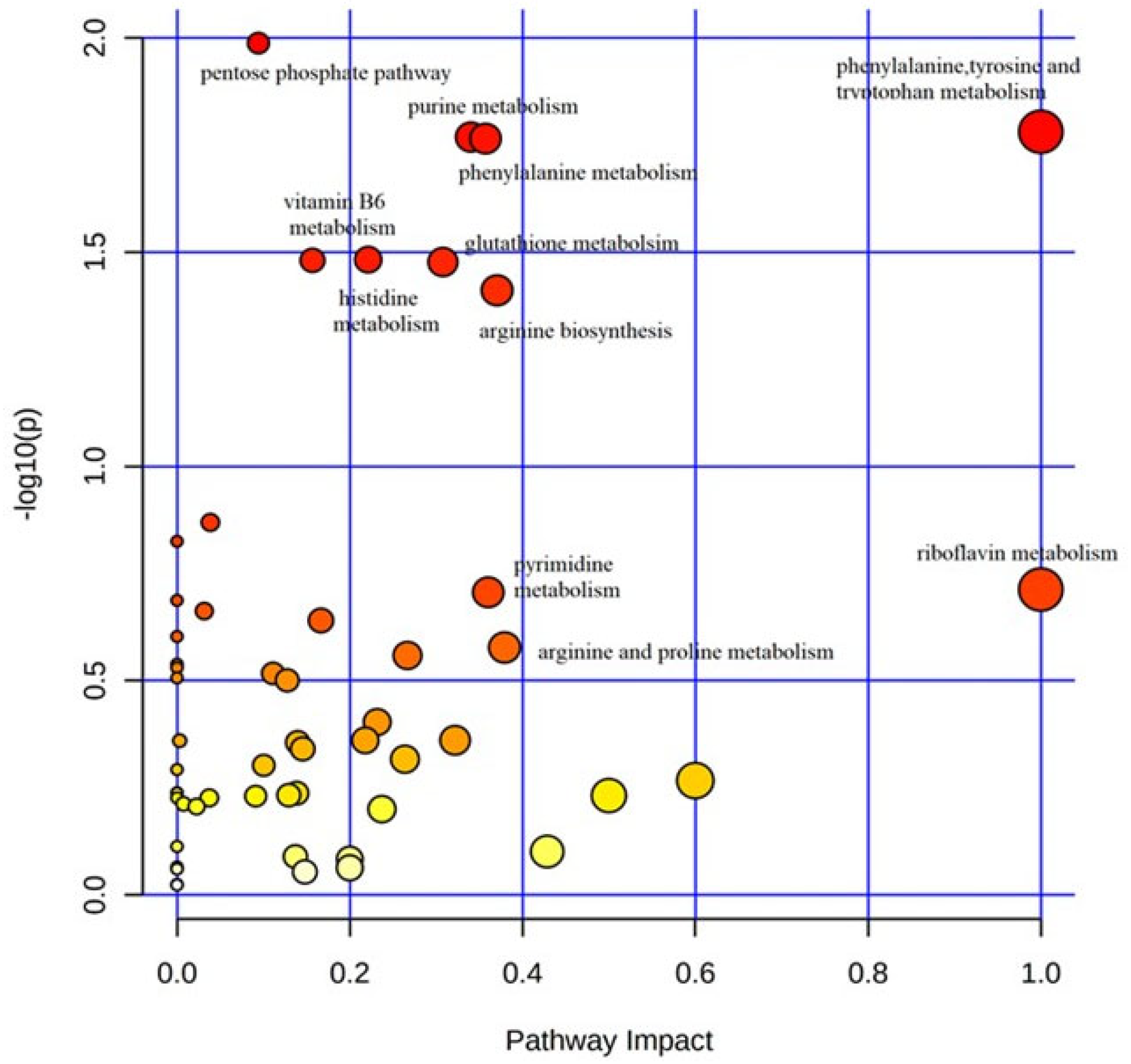
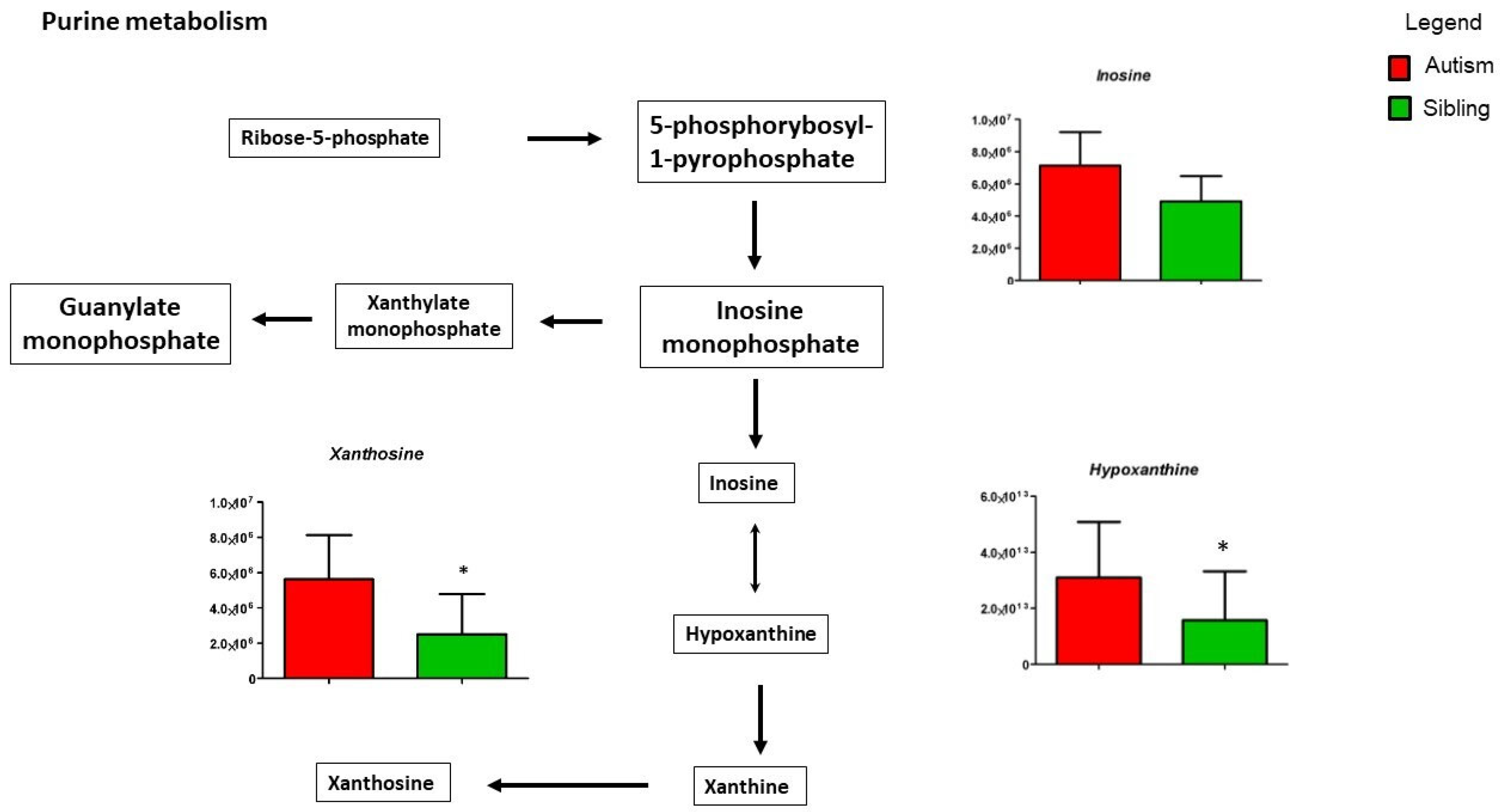
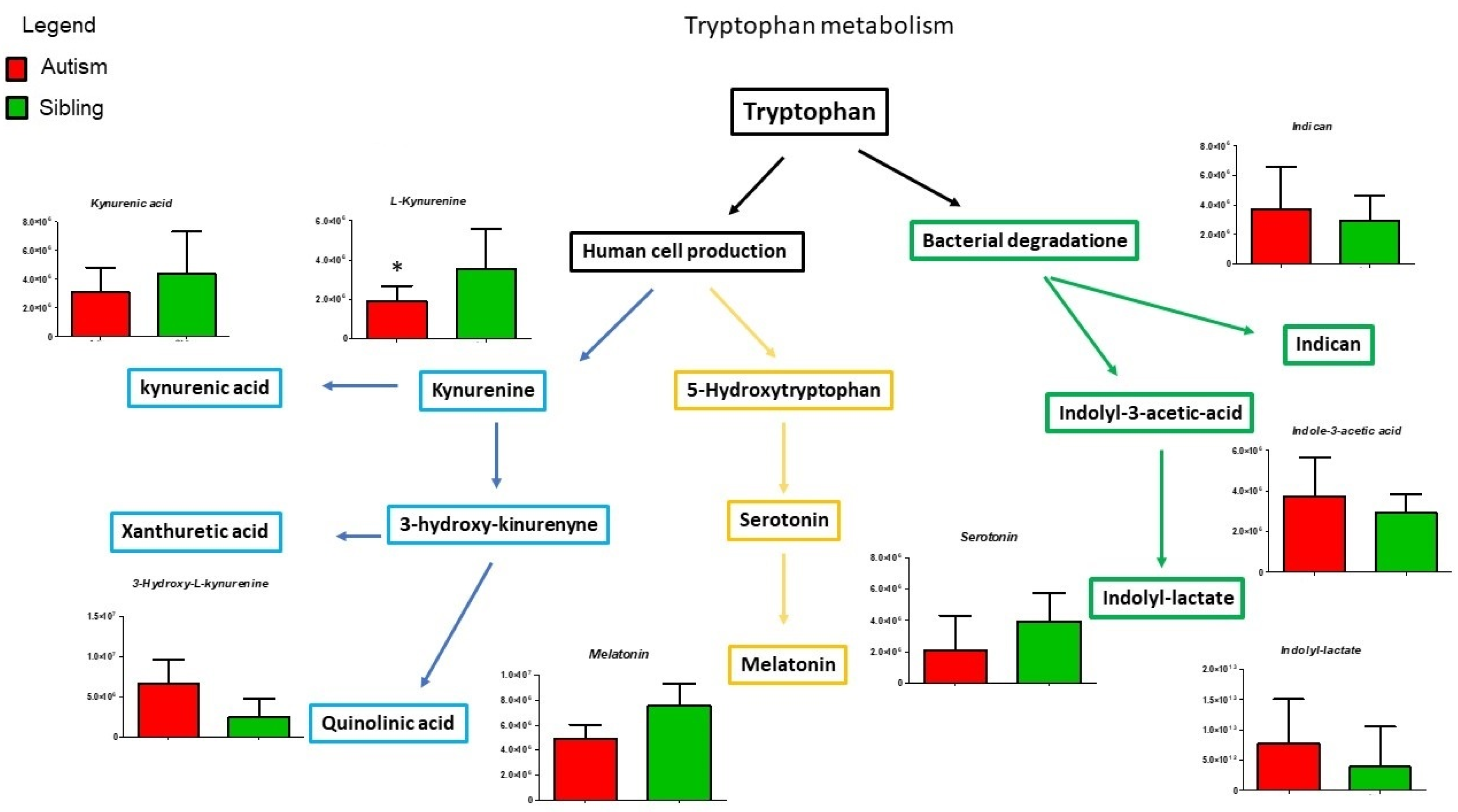
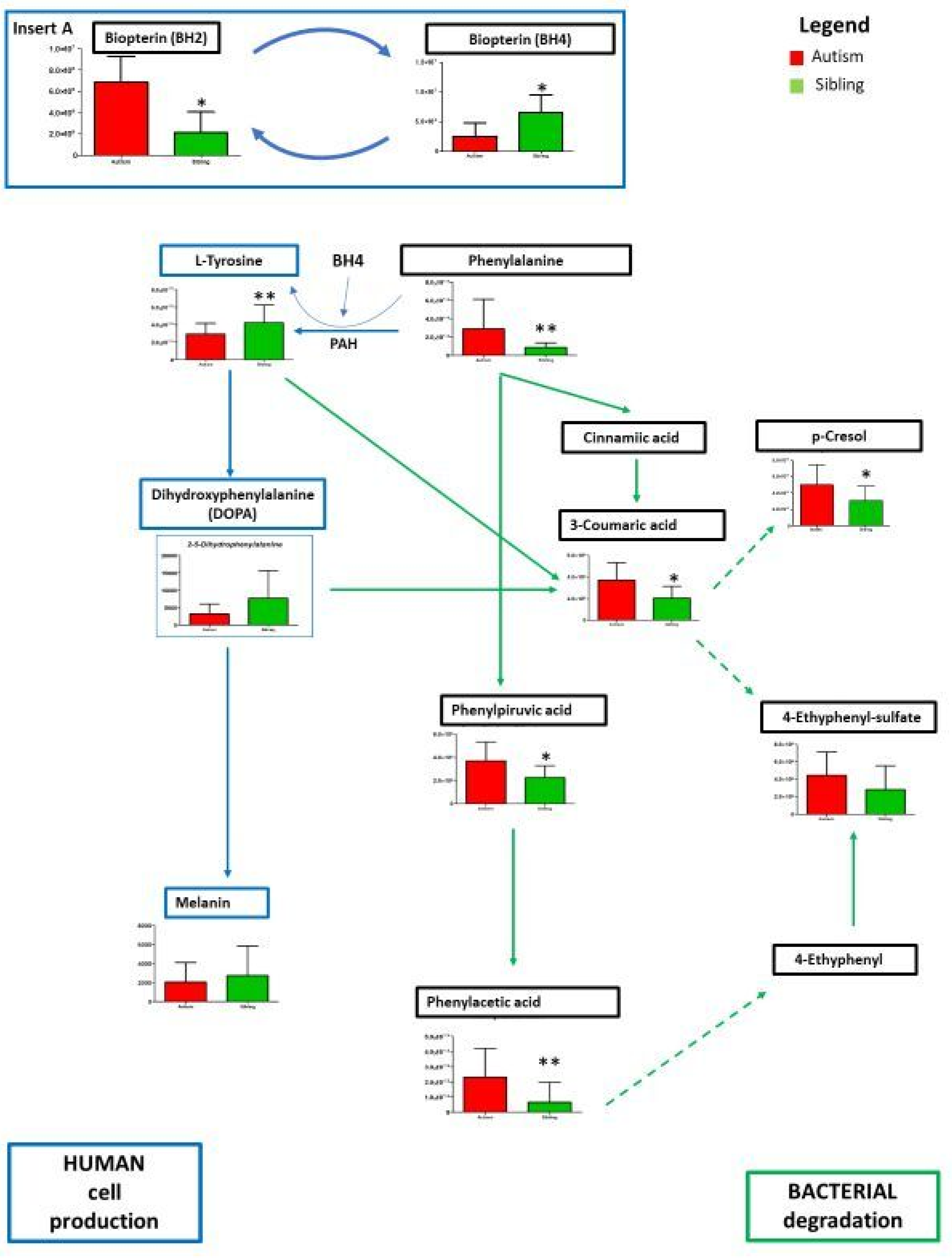
3. Results
Metabolic Profiling Using Untargeted Metabolomics in LC-MS Platform
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of autism spectrum disorder among children aged 8 years—Autism and developmental disabilities monitoring network, 11 Sites, United States, 2016. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2020, 69, 1. [Google Scholar] [CrossRef] [PubMed]
- Dietz, P.M.; Rose, C.E.; McArthur, D.; Maenner, M. National and state estimates of adults with autism spectrum disorder. J. Autism Dev. Disord. 2020, 50, 4258–4266. [Google Scholar] [CrossRef]
- Narzisi, A.; Posada, M.; Barbieri, F.; Chericoni, N.; Ciuffolini, D.; Pinzino, M.; Romano, R.; Scattoni, M.L.; Tancredi, R.; Calderoni, S.; et al. Prevalence of Autism Spectrum Disorder in a large Italian catchment area: A school-based population study within the ASDEU project. Epidemiol. Psychiatr. Sci. 2018, 29, e35. [Google Scholar] [CrossRef] [PubMed]
- Brugha, T.S.; McManus, S.; Bankart, J.; Scott, F.; Purdon, S.; Smith, J.; Bebbington, P.; Jenkins, R.; Meltzer, H. Epidemiology of autism spectrum disorders in adults in the community in England. Arch. Gen. Psychiatry 2011, 68, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Persico, A.M.; Cucinotta, F.; Ricciardello, A.; Turriziani, L. Autisms. Comprehensive Developmental Neuroscience. Neurodevelopmental Disorders, 1st ed.; Rubenstein, J.L.R., Rakic, P., Chen, B., Kwan, K., Wynshaw-Boris, A., Eds.; Academic Press/Elsevier Inc.: New York, NY, USA, 2020; pp. 35–77. ISBN 978-0-12-814409-1. [Google Scholar]
- Geschwind, D.H.; Levitt, P. Autism spectrum disorders: Developmental disconnection syndromes. Curr. Opin. Neurobiol. 2007, 17, 103–111. [Google Scholar] [CrossRef]
- Chaste, P.; Roeder, K.; Devlin, B. The Yin and Yang of autism genetics: How rare de novo and common variations affect liability. Annu. Rev. Genom. Hum. Genet. 2017, 18, 167–187. [Google Scholar] [CrossRef] [PubMed]
- Persico, A.M.; Merelli, S. Environmental factors and Autism Spectrum Disorder. In Autism Spectrum Disorders: Phenotypes, Mechanisms and Treatments; Leboyer, M., Chaste, P., Eds.; Basilea (SU): Basel, Switzerland; Karger: Berlin, Germany, 2015; Volume 180, pp. 113–134. [Google Scholar]
- Bhandari, R.; Paliwal, J.K.; Kuhad, A. Neuropsychopathology of Autism Spectrum Disorder: Complex interplay of genetic, epigenetic, and environmental factors. Adv. Neurobiol. 2020, 24, 97–141. [Google Scholar] [CrossRef]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of genetic and environmental factors with autism in a 5-country cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef]
- Altieri, L.; Neri, C.; Sacco, R.; Curatolo, P.; Benvenuto, A.; Muratori, F.; Santocchi, E.; Militerni, R.; Bravaccio, C.; Lenti, C.; et al. Urinary p-cresol is elevated in small children with Autism Spectrum Disorder. Biomarkers 2011, 16, 252–260. [Google Scholar] [CrossRef]
- Guzmán-Salas, S.; Weber, A.; Malci, A.; Lin, X.; Herrera-Molina, R.; Cerpa, W.; Dorador, C.; Signorelli, J.; Zamorano, P. The metabolite p-cresol impairs dendritic development, synaptogenesis, and synapse function in hippocampal neurons: Implications for autism spectrum disorder. J. Neurochem. 2022, 161, 335–349. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef]
- Feinberg, J.I.; Bakulski, K.M.; Jaffe, A.E.; Tryggvadottir, R.; Brown, S.C.; Goldman, L.R.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Fallin, M.D.; et al. Paternal sperm DNA methylation associated with early signs of autism risk in an autism-enriched cohort. Int. J. Epidemiol. 2015, 44, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.N.; Zhang, Y.I.; Frazier, T.; Abbacchi, A.M.; Law, P. Sibling recurrence and the genetic epidemiology of autism. Am. J. Psychiatry 2010, 167, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.; Beam, A.; Agniel, D.; Eran, A.; Manrai, A.; Spettell, C.; Steinberg, G.; Mandl, K.; Fox, K.; Nelson, S.F.; et al. Association of sex with recurrence of autism spectrum disorder among siblings. JAMA Pediatrics 2017, 171, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Constantino, J.N.; Lajonchere, C.; Lutz, M.; Gray, T.; Abbacchi, A.; McKenna, K.; Singh, D.; Todd, R.D. Autistic social impairment in the siblings of children with pervasive developmental disorders. Am. J. Psychiatry 2006, 163, 294–296. [Google Scholar] [CrossRef]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Hegarty, J.P.; Lazzeroni, L.C.; Raman, M.M.; Pegoraro, L.F.L.; Monterrey, J.C.; Cleveland, S.C.; Hallmayer, J.F.; Wolke, O.N.; Phillips, J.M.; Reiss, A.L.; et al. Genetic and environmental influences on lobar brain structures in twins with autism. Cereb. Cortex 2020, 30, 1946–1956. [Google Scholar] [CrossRef]
- D’Abate, L.; Walker, S.; Yuen, R.K.C.; Tammimies, K.; Buchanan, J.A.; Davies, R.W.; Thiruvahindrapuram, B.; Wei, J.; Brian, J.; Bryson, S.E.; et al. Predictive impact of rare genomic copy number variations in siblings of individuals with autism spectrum disorders. Nat. Commun. 2019, 10, 5519. [Google Scholar] [CrossRef]
- Kang, D.W.; Ilhan, Z.E.; Isern, N.G.; Hoyt, D.W.; Howsmon, D.P.; Shaffer, M.; Lozupone, C.A.; Hahn, J.; Adams, J.B.; Krajmalnik-Brown, R. Differences in fecal microbial metabolites and microbiota of children with autism spectrum disorders. Anaerobe 2018, 49, 121–131. [Google Scholar] [CrossRef]
- Smith, A.M.; King, J.J.; West, P.R.; Ludwig, M.A.; Donley, E.L.R.; Burrier, R.E.; Amaral, D.G. Amino acid dysregulation metabotypes: Potential biomarkers for diagnosis and individualized treatment for subtypes of autism spectrum disorder. Biol. Psychiatry 2019, 85, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Gevi, F.; Zolla, L.; Gabriele, S.; Persico, A.M. Urinary metabolomics of young Italian autistic children supports abnormal tryptophan and purine metabolism. Mol. Autism. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, X.; Zhang, H.; Lin, J.; Feng, C.; Iqbal, J. Biomarkers in autism spectrum disorders: Current progress. Clin. Chim. Acta 2020, 502, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Likhitweerawong, N.; Thonusin, C.; Boonchooduang, N.; Louthrenoo, O.; Nookaew, I.; Chattipakorn, N.; Chattipakorn, S.C. Profiles of urine and blood metabolomics in autism spectrum disorders. Metab. Brain Dis. 2021, 36, 1641–1671. [Google Scholar] [CrossRef]
- Garcia-Gutierrez, E.; Narbad, A.; Rodríguez, J.M. Autism spectrum disorder associated with gut microbiota at immune, metabolomic, and neuroactive level. Front. Neurosci. 2020, 14, 578666. [Google Scholar] [CrossRef] [PubMed]
- Belardo, A.; Gevi, F.; Zolla, L. The concomitant lower concentrations of vitamins B6, B9 and B12 may caus e methylation deficiency in autistic children. J. Nutr. Biochem. 2017, 70, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Gevi, F.; Belardo, A.; Zolla, L. A metabolomics approach to investigate urine levels of neurotransmitters and related metabolites in autistic children. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165859. [Google Scholar] [CrossRef]
- Lord, C.; Rutter, M.; DiLavore, P.C.; Risi, S.; Gotham, K.; Bishop, S. Autism Diagnostic Observation Scales, 2nd ed.; Western Psychological Services: Torrance, CA, USA, 2012. [Google Scholar]
- Rutter, M.; Le Couter, A.; Lord, C. ADI-R, Autism Diagnostic Interview—Revised; Faggioli, R., Saccani, M., Persico, A.M., Tancredi, R., Parrini, B., Igliozzi, R., Eds.; Western Psychological Services: Los Angeles, CA, USA, 2003. [Google Scholar]
- Sacco, R.; Curatolo, P.; Manzi, B.; Militerni, R.; Bravaccio, C.; Frolli, A.; Lenti, C.; Saccani, M.; Elia, M.; Reichelt, K.L.; et al. Principal pathogenetic components and biological endophenotypes in autism spectrum disorders. Autism. Res. 2010, 3, 237–252. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Rose, C.R.; Felix, L.; Zeug, A.; Dietrich, D.; Reiner, A.; Henneberger, C. Astroglial glutamate signaling and uptake in the hippocampus. Front. Mol. Neurosci. 2018, 10, 451. [Google Scholar] [CrossRef]
- Naviaux, R.K. Metabolic features of the cell danger response. Mitochondrion 2014, 16, 7–17. [Google Scholar] [CrossRef]
- Persico, A.M.; Napolioni, V. Urinary p-cresol in autism spectrum disorder. Neurotoxicol. Teratol. 2013, 36, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Martin, P.; Becker, J.A.J.; Caramello, N.; Fernandez, S.P.; Costa-Campos, R.; Canaguier, J.; Barbosa, S.; Martinez-Gili, L.; Myridakis, A.; Dumas, M.E.; et al. The microbial metabolite p-Cresol induces autistic-like behaviors in mice by remodeling the gut microbiota. Microbiome 2021, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Needham, B.D.; Funabashi, M.; Adame, M.D.; Wang, Z.; Boktor, J.C.; Haney, J.; Wu, W.L.; Rabut, C.; Ladinsky, M.S.; Hwang, S.J.; et al. A gut-derived metabolite alters brain activity and anxiety behaviour in mice. Nature 2022, 602, 647–653. [Google Scholar] [CrossRef]
- Pascucci, T.; Colamartino, M.; Fiori, E.; Sacco, R.; Coviello, A.; Ventura, R.; Puglisi-Allegra, S.; Turriziani, L.; Persico, A.M. P-cresol alters brain dopamine metabolism and exacerbates autism-like behaviors in the BTBR mouse. Brain Sci. 2020, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Persico, A.M.; Sacco, R. Endophenotypes in autism spectrum disorders. In The Comprehensive Guide to Autism; Patel, V.B., Preedy, V.R., Martin, C.R., Eds.; Springer Science + Business Media: Berlin/Heidelberg, Germany, 2014; pp. 77–96. [Google Scholar]
- Dodd, D.; Spitzer, M.H.; Van Treuren, W.; Merrill, B.D.; Hryckowian, A.J.; Higginbottom, S.K.; Le, A.; Cowan, T.M.; Nolan, G.P.; Fischbach, M.A.; et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 2017, 551, 648–652. [Google Scholar] [CrossRef]
- Gabriele, S.; Sacco, R.; Cerullo, S.; Neri, C.; Urbani, A.; Tripi, G.; Malvy, J.; Barthelemy, C.; Bonnet-Brihault, F.; Persico, A.M. Urinary p-cresol is elevated in young French children with autism spectrum disorder: A replication study. Biomarkers 2014, 19, 463–470. [Google Scholar] [CrossRef]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef]
- Kang, D.W.; Adams, J.B.; Vargason, T.; Santiago, M.; Hahn, J.; Krajmalnik-Brown, R. Distinct fecal and plasma metabolites in children with autism spectrum disorders and their modulation after microbiota transfer therapy. Msphere 2020, 5, e00314–e00320. [Google Scholar] [CrossRef]
- Zheng, Y.; Bek, M.K.; Prince, N.Z.; Peralta Marzal, L.N.; Garssen, J.; Perez Pardo, P.; Kraneveld, A.D. The Role of Bacterial-Derived Aromatic Amino Acids Metabolites Relevant in Autism Spectrum Disorders: A Comprehensive Review. Front. Neurosci. 2021, 15, 738220. [Google Scholar] [CrossRef]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 2002, 35, S6–S16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Li, S.; Gan, R.Y.; Zhou, T.; Xu, D.P.; Li, H.B. Impacts of gut bacteria on human health and diseases. Int. J. Mol. Sci. 2015, 16, 7493–7519. [Google Scholar] [CrossRef] [PubMed]
- Parracho, H.M.; Bingham, M.O.; Gibson, G.R.; McCartney, A.L. Differences between the gut microflora of children with autistic spectrum disorders and that of healthy children. J. Med. Microbiol. 2005, 54, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism–comparisons to typical children and correlation with autism severity. BMC Gastroenterol 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Berding, K.; Cryan, J.F. Microbiota-targeted interventions for mental health. Curr. Opin. Psychiatry 2022, 35, 3–9. [Google Scholar] [CrossRef]
- Ligabue, G.; Damiano, F.; Cuoghi, A.; De Biasi, S.; Bellei, E.; Granito, M. P-Cresol and cardiovascular risk in kidney transplant recipients. Transplant. Proc. 2015, 47, 2121–2125. [Google Scholar] [CrossRef]
- Shiba, T.; Makino, I.; Sasaki, T.; Fukuhara, Y.; Kawakami, K.; Kato, I. P-Cresyl sulfate decreases peripheral B cells in mice with adenine-induced renal dysfunction. Toxicol. Appl. Pharmacol. 2018, 342, 50–59. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, Y.M.; Lee, S.H. TUDCA-treated mesenchymal stem cells protect against ER stress in the hippocampus of a murine chronic kidney disease model. Int. J. Mol. Sci. 2019, 20, 613. [Google Scholar] [CrossRef]
- Carmassi, C.; Palagini, L.; Caruso, D.; Masci, I.; Nobili, L.; Vita, A.; Dell’Osso, L. Systematic review of sleep disturbances and circadian sleep desynchronization in Autism Spectrum Disorder: Toward an integrative model of a self-reinforcing loop. Front. Psychiatry 2019, 10, 366. [Google Scholar] [CrossRef]
- McElhanon, B.O.; McCracken, C.; Karpen, S.; Sharp, W.G. Gastrointestinal symptoms in autism spectrum disorder: A meta-analysis. Pediatrics 2014, 133, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Naushad, S.M.; Jain, J.M.N.; Prasad, C.K.; Naik, U.; Akella, R.R.D. Autistic children exhibit distinct plasma amino acid profile. Indian J. Biochem. Biophys. 2013, 50, 474–478. [Google Scholar] [PubMed]
- Bugajska, J.; Berska, J.; Wojtyto, T.; Bik-Multanowski, M.; Sztefko, K. The amino acid profile in blood plasma of young boys with autism. Psychiatry Pol. 2017, 51, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Lv, Q.-Q.; Cong, Y.; Zou, X.-B.; Deng, H.-Z. Reduction of essential amino acid levels and sex-specific alterations in serum amino acid concentration profiles in children with autism spectrum disorder. Psychiatry Res. 2021, 297, 113675. [Google Scholar] [CrossRef] [PubMed]
- Aldred, S.; Moore, K.M.; Fitzgerald, M.; Waring, R.H. Plasma amino acid levels in children with autism and their families. J. Autism Dev. Disord. 2003, 33, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Bala, K.A.; Dogan, M.; Mutluer, T.; Kaba, S.; Aslan, O.; Balahoroglu, R.; Cokluk, E.; Ustyol, L.; Kocaman, S. Plasma amino acid profile in autism spectrum disorder (ASD). Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 923–929. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Mean/Median (Range) or % | ||
|---|---|---|---|
| Age in yrs (mean± SEM) | -ASD -Unaffected siblings | 14 14 | 7.06 ± 0.96 (3.2–15.6) 6.68 ± 1.28 (1.0–14.0) |
| Gender: | Male | 11 pairs | 78.6% |
| Female | 3 pairs | 21.4% | |
| M/F ratio | 3.7:1 | ||
| I.Q. | Mean ± SEM | 13 * | 64.7 ± 6.78 (30–104) |
| >70 | 7 | 50.0% | |
| ≤70 | 7 | 50.0% | |
| DSM-IV Diagnosis: | Autistic Disorder | 10 | 71.5% |
| Asperger Syndrome | 1 | 7.1% | |
| PDD-NOS | 3 | 21.4% | |
| Level of Expressive Language | Sentences | 3 | 21.4% |
| Words | 7 | 50.0% | |
| Non-verbal | 4 | 28.6% | |
| Median VABS Scores: | |||
| Communication | 12 | 73.5 (31–115) | |
| Daily Living Skills | 12 | 78.5 (48–113) | |
| Socialization | 12 | 73.0 (55–116) | |
| Motor Skills | 8 | 87.5 (56–111) | |
| Composite | 12 | 73.5 (47–115) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timperio, A.M.; Gevi, F.; Cucinotta, F.; Ricciardello, A.; Turriziani, L.; Scattoni, M.L.; Persico, A.M. Urinary Untargeted Metabolic Profile Differentiates Children with Autism from Their Unaffected Siblings. Metabolites 2022, 12, 797. https://doi.org/10.3390/metabo12090797
Timperio AM, Gevi F, Cucinotta F, Ricciardello A, Turriziani L, Scattoni ML, Persico AM. Urinary Untargeted Metabolic Profile Differentiates Children with Autism from Their Unaffected Siblings. Metabolites. 2022; 12(9):797. https://doi.org/10.3390/metabo12090797
Chicago/Turabian StyleTimperio, Anna Maria, Federica Gevi, Francesca Cucinotta, Arianna Ricciardello, Laura Turriziani, Maria Luisa Scattoni, and Antonio M. Persico. 2022. "Urinary Untargeted Metabolic Profile Differentiates Children with Autism from Their Unaffected Siblings" Metabolites 12, no. 9: 797. https://doi.org/10.3390/metabo12090797
APA StyleTimperio, A. M., Gevi, F., Cucinotta, F., Ricciardello, A., Turriziani, L., Scattoni, M. L., & Persico, A. M. (2022). Urinary Untargeted Metabolic Profile Differentiates Children with Autism from Their Unaffected Siblings. Metabolites, 12(9), 797. https://doi.org/10.3390/metabo12090797