
Hepatic Positron Emission Tomography: Applications in Metabolism, Haemodynamics and Cancer




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
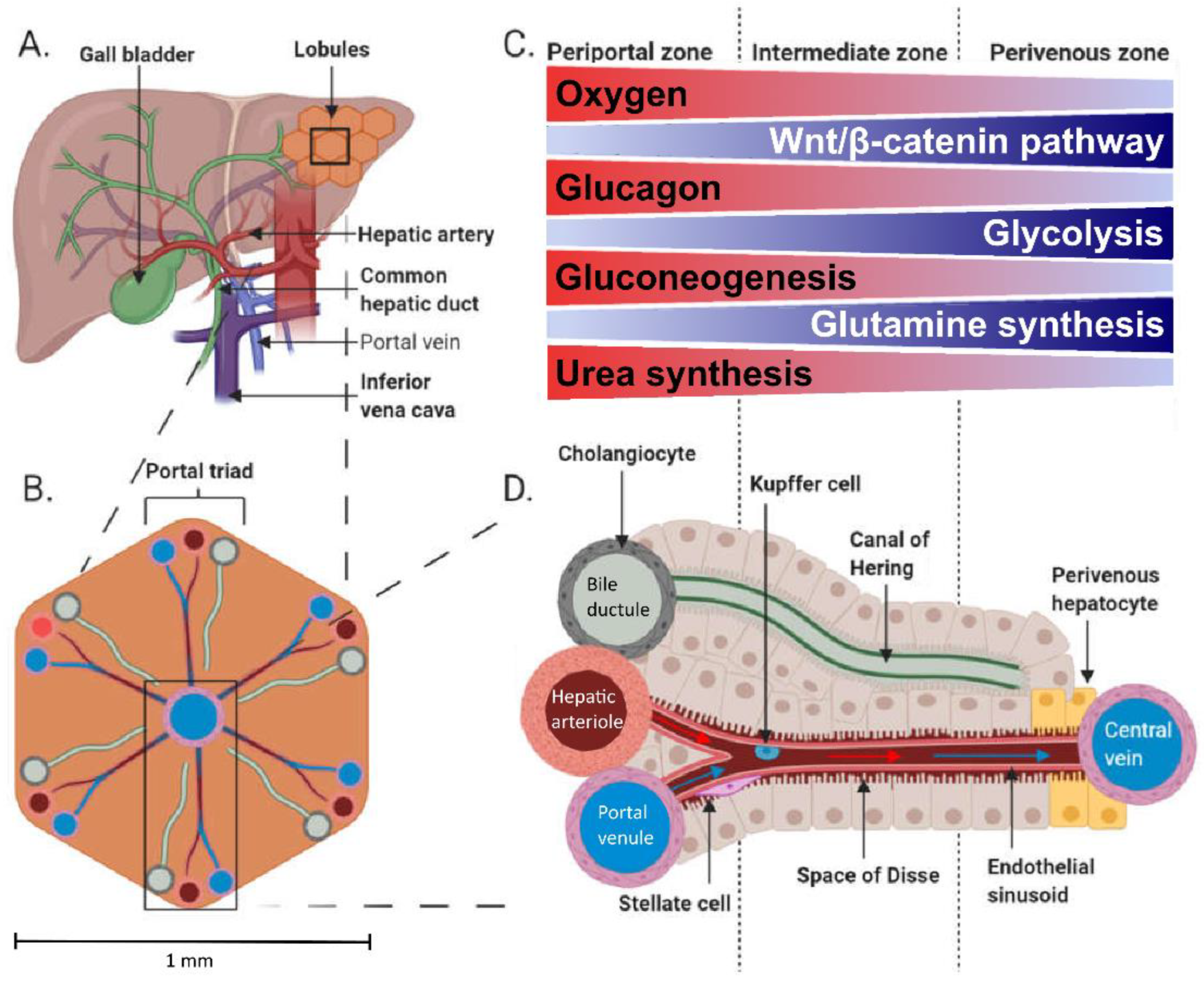
1.1. Hepatic Physiology
1.2. Traditional Nodes of Assessing Tissue Metabolism vs. PET
2. Studying Liver Metabolism with PET
2.1. Principle of PET
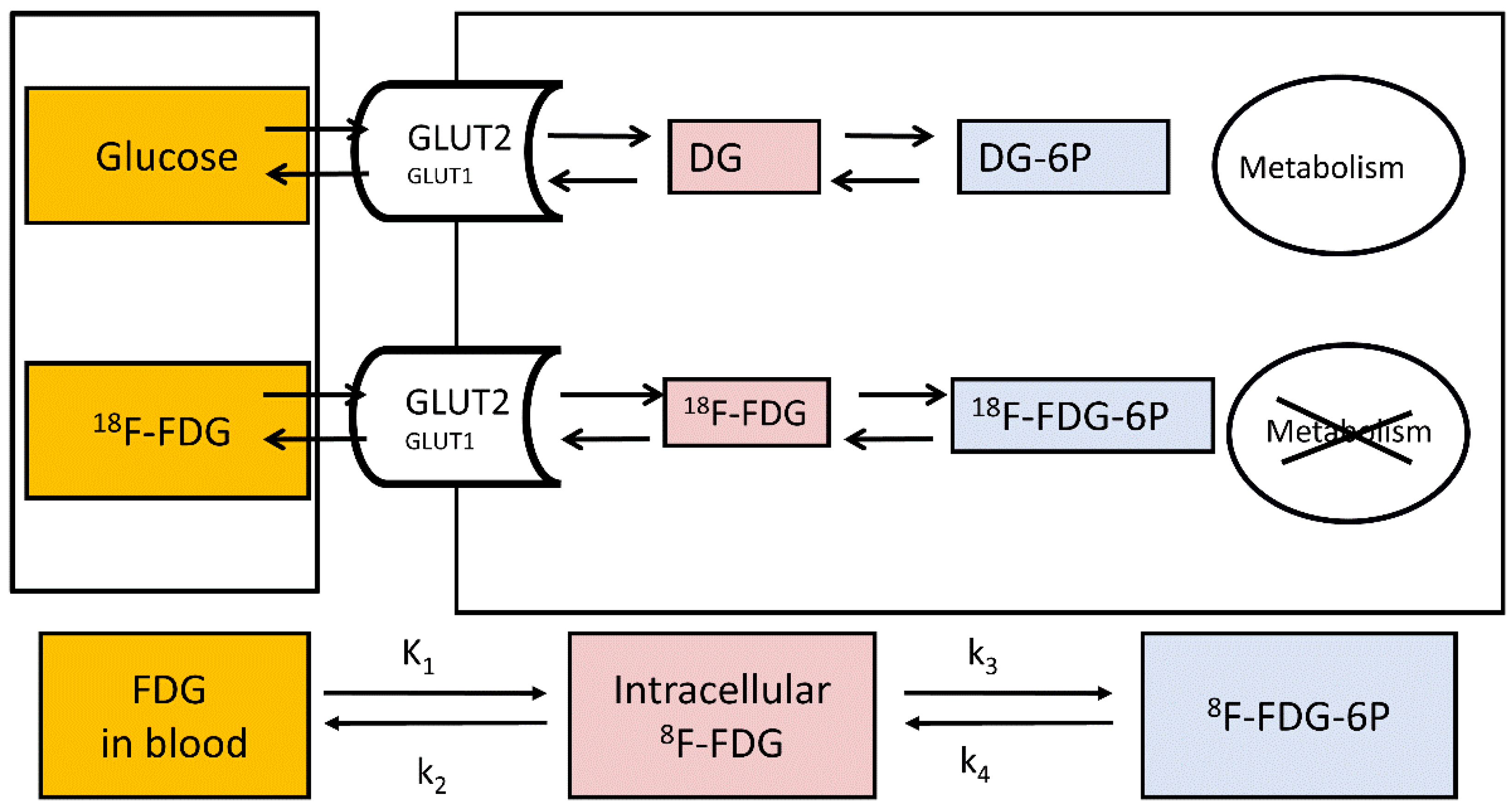
2.2. Study of Hepatic Glucose Uptake Using PET
2.3. Evaluation of Endogenous Glucose Production during 18F-FDG-PET
2.4. Measuring Hepatic GU: Effects of Obesity and Weight Loss
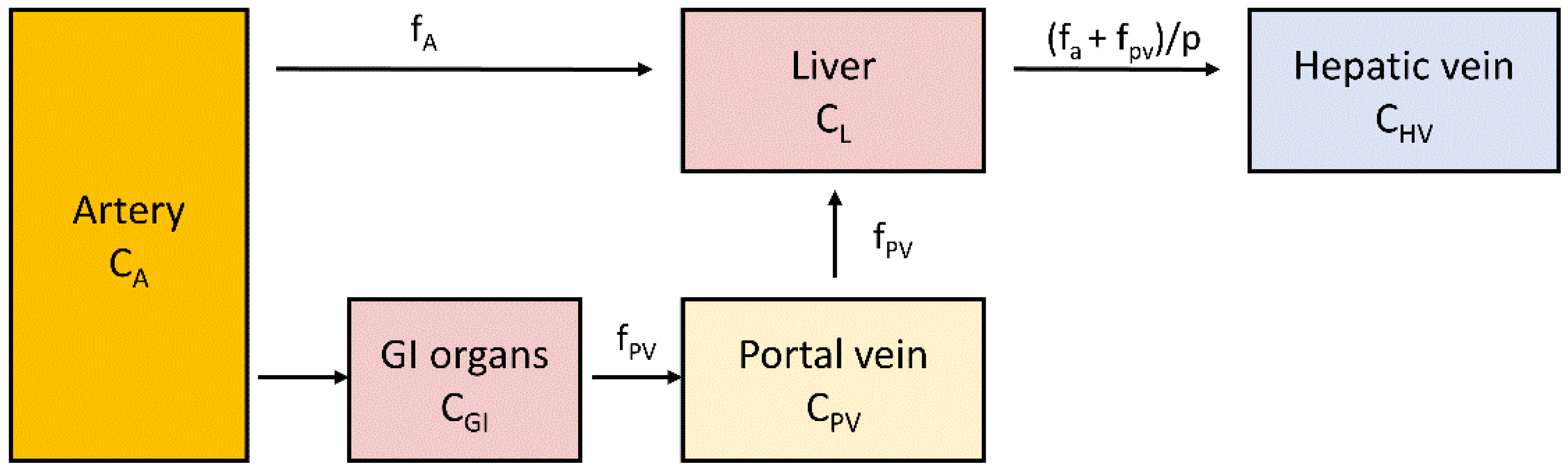
2.5. Measurement of Liver Perfusion
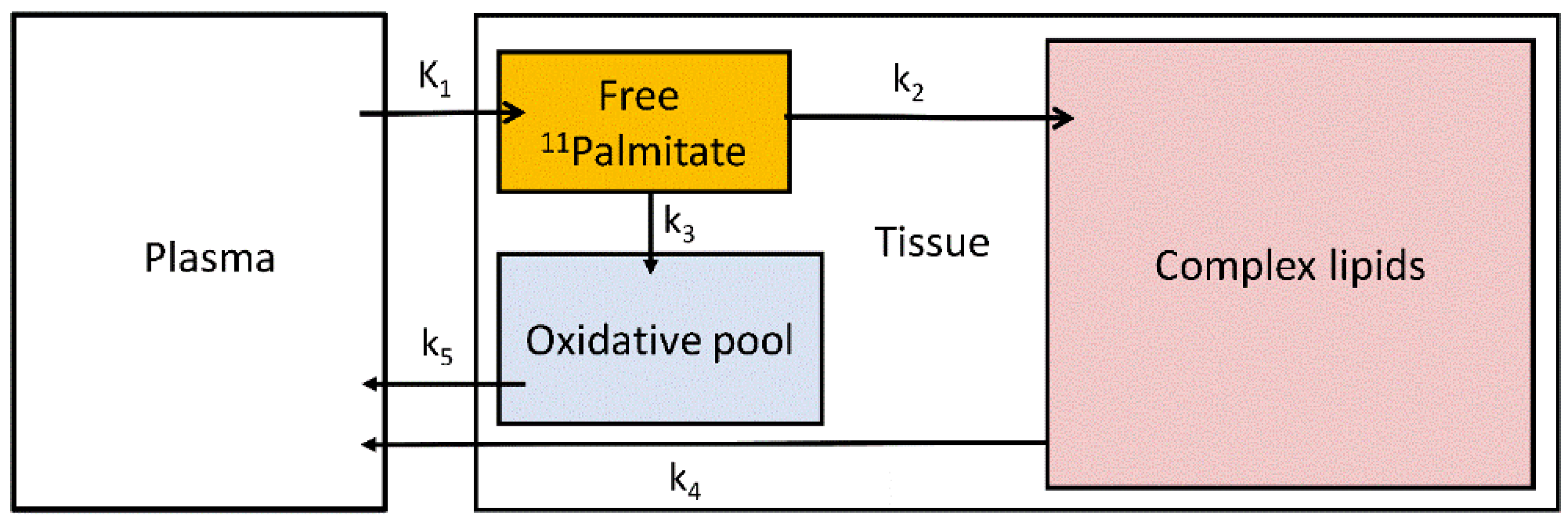
2.6. Measurement of Hepatic Fatty Acid Uptake
2.7. Liver 11C-Acetate: Applications
2.8. 18F-FDG-PET and Nonalcoholic Fatty Liver Disease
2.9. 11C-Acetate-PET and NAFLD
3. PET Metabolic Tracers in Hepatic Malignancies
3.1. 18F-FDG
3.2. 18F-FDG in Hepatocellular Carcinoma
3.3. 18F-FDG in Intrahepatic Cholangiocarcinoma
3.4. 18F-FDG in Liver Metastases
3.5. 11C-Acetate in Liver Malignances
3.6. Limitation of PET Technology in Imaging Liver Malignancies
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schenk, W.G.; McDonald, J.C.; McDonald, K.; Drapanas, T. Direct measurement of hepatic blood flow in surgical patients: With related observations on hepatic flow dynamics in experimental animals. Ann. Surg. 1962, 156, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Pinal-Garcia, D.F.; Nuno-Guzman, C.M.; Gonzalez-Gonzalez, M.E.; Ibarra-Hurtado, T.R. The celiac trunk and its anatomical variations: A cadaveric study. J. Clin. Med. Res. 2018, 10, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Sezai, S.; Sakurabayashi, S.; Yamamoto, Y.; Morita, T.; Hirano, M.; Oka, H. Hepatic arterial and portal venous oxygen content and extraction in liver cirrhosis. Liver 1993, 13, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.L.; Elghajiji, A.; Grimaldos Rodriguez, C.; Weston, S.D.; Burke, Z.D.; Tosh, D. The canonical wnt pathway as a key regulator in liver development, differentiation and homeostatic renewal. Genes 2020, 11, 1163. [Google Scholar] [CrossRef]
- Kang, Y.B.A.; Eo, J.; Mert, S.; Yarmush, M.L.; Usta, O.B. Metabolic patterning on a chip: Towards in vitro liver zonation of primary rat and human hepatocytes. Sci. Rep. 2018, 8, 8951. [Google Scholar] [CrossRef]
- Koffert, J.; Honka, H.; Teuho, J.; Kauhanen, S.; Hurme, S.; Parkkola, R.; Oikonen, V.; Mari, A.; Lindqvist, A.; Wierup, N.; et al. Effects of meal and incretins in the regulation of splanchnic blood flow. Endocr. Connect. 2017, 6, 179–187. [Google Scholar] [CrossRef]
- Someya, N.; Endo, M.Y.; Fukuba, Y.; Hayashi, N. Blood flow responses in celiac and superior mesenteric arteries in the initial phase of digestion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1790–R1796. [Google Scholar] [CrossRef]
- Madsen, J.L.; Søndergaard, S.B.; Møller, S. Meal-induced changes in splanchnic blood flow and oxygen uptake in middle-aged healthy humans. Scand. J. Gastroenterol. 2006, 41, 87–92. [Google Scholar] [CrossRef]
- Ekataksin, W. The isolated artery: An intrahepatic arterial pathway that can bypass the lobular parenchyma in mammalian livers. Hepatology 2000, 31, 269–279. [Google Scholar] [CrossRef]
- Ben-Moshe, S.; Itzkovitz, S. Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 395–410. [Google Scholar] [CrossRef]
- Berndt, N.; Kolbe, E.; Gajowski, R.; Eckstein, J.; Ott, F.; Meierhofer, D.; Holzhütter, H.-G.; Matz-Soja, M. Functional consequences of metabolic zonation in murine livers: Insights for an old story. Hepatology 2021, 73, 795–810. [Google Scholar] [CrossRef] [PubMed]
- Droin, C.; Kholtei, J.E.; Bahar Halpern, K.; Hurni, C.; Rozenberg, M.; Muvkadi, S.; Itzkovitz, S.; Naef, F. Space-time logic of liver gene expression at sub-lobular scale. Nat. Metab. 2021, 3, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, R.; Matz-Soja, M. Liver zonation: Novel aspects of its regulation and its impact on homeostasis. World J. Gastroenterol. 2014, 20, 8491–8504. [Google Scholar] [CrossRef] [PubMed]
- Schleicher, J.; Tokarski, C.; Marbach, E.; Matz-Soja, M.; Zellmer, S.; Gebhardt, R.; Schuster, S. Zonation of hepatic fatty acid metabolism—The diversity of its regulation and the benefit of modeling. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2015, 1851, 641–656. [Google Scholar] [CrossRef]
- Guzmán, M.; Bijleveld, C.; Geelen, M.J. Flexibility of zonation of fatty acid oxidation in rat liver. Biochem. J. 1995, 311, 853–860. [Google Scholar] [CrossRef]
- Kietzmann, T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. 2017, 11, 622–630. [Google Scholar] [CrossRef]
- Tygstrup, N.; Winkler, K.; Mellemgaard, K.; Andreassen, M. Determination of the hepatic arterial blood flow and oxygen supply in man by clamping the hepatic artery during surgery. J. Clin. Investig. 1962, 41, 447–454. [Google Scholar] [CrossRef]
- Brooks, A.J.; Eastwood, J.; Beckingham, I.J.; Girling, K.J. Liver tissue partial pressure of oxygen and carbon dioxide during partial hepatectomy. Br. J. Anaesth. 2004, 92, 735–737. [Google Scholar] [CrossRef]
- Brooks, A.J.; Hammond, J.S.; Girling, K.; Beckingham, I.J. The effect of hepatic vascular inflow occlusion on liver tissue PH, carbon dioxide, and oxygen partial pressures: Defining the optimal clamp/release regime for intermittent portal clamping. J. Surg. Res. 2007, 141, 247–251. [Google Scholar] [CrossRef]
- Leary, T.S.; Klinck, J.R.; Hayman, G.; Friend, P.; Jamieson, N.V.; Gupta, A.K. Measurement of liver tissue oxygenation after orthotopic liver transplantation using a multiparameter sensor. Anaesthesia 2002, 57, 1128–1133. [Google Scholar] [CrossRef]
- Kolbe, E.; Aleithe, S.; Rennert, C.; Spormann, L.; Ott, F.; Meierhofer, D.; Gajowski, R.; Stöpel, C.; Hoehme, S.; Kücken, M.; et al. Mutual zonated interactions of Wnt and Hh signaling are orchestrating the metabolism of the adult liver in mice and human. Cell Rep. 2019, 29, 4553–4567.e7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Kim, S.Y.; Okamoto, H.; Xin, Y.; Yancopoulos, G.D.; Murphy, A.J.; Gromada, J. Glucagon contributes to liver zonation. Proc. Natl. Acad. Sci. USA 2018, 115, E4111–E4119. [Google Scholar] [CrossRef] [PubMed]
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [CrossRef]
- Havel, R.J.; Kane, J.P.; Balasse, E.O.; Segel, N.; Basso, L.V. Splanchnic metabolism of free fatty acids and production of triglycerides of very low density lipoproteins in normotriglyceridemic and hypertriglyceridemic humans. J. Clin. Investig. 1970, 49, 2017–2035. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.J. Hepatic energy and substrate metabolism: A possible metabolic basis for early nutritional support in cirrhotic patients. Nutrition 1998, 14, 30–38. [Google Scholar] [CrossRef]
- Hodson, L.; Bickerton, A.S.T.; McQuaid, S.E.; Roberts, R.; Karpe, F.; Frayn, K.N.; Fielding, B.A. The contribution of splanchnic fat to VLDL triglyceride is greater in insulin-resistant than insulin-sensitive men and women: Studies in the postprandial state. Diabetes 2007, 56, 2433–2441. [Google Scholar] [CrossRef]
- Evans, K.; Burdge, G.C.; Wootton, S.A.; Collins, J.M.; Clark, M.L.; Tan, G.D.; Karpe, F.; Frayn, K.N. Tissue-specific stable isotope measurements of postprandial lipid metabolism in familial combined hyperlipidaemia. Atherosclerosis 2008, 197, 164–170. [Google Scholar] [CrossRef]
- Sidossis, L.S.; Mittendorfer, B.; Chinkes, D.; Walser, E.; Wolfe, R.R. Effect of hyperglycemia-hyperinsulinemia on whole body and regional fatty acid metabolism. Am. J. Physiol. Endocrinol. Metab. 1999, 276, E427–E434. [Google Scholar] [CrossRef]
- Jensen, M.D.; Johnson, C.M.; Cryer, P.E.; Murray, M.J. Thermogenesis after a mixed meal: Role of leg and splanchnic tissues in men and women. Am. J. Physiol. Endocrinol. Metab. 1995, 268, E433–E438. [Google Scholar] [CrossRef]
- Hui, S.; Cowan, A.J.; Zeng, X.; Yang, L.; TeSlaa, T.; Li, X.; Bartman, C.; Zhang, Z.; Jang, C.; Wang, L.; et al. Quantitative fluxomics of circulating metabolites. Cell Metab. 2020, 32, 676–688.e4. [Google Scholar] [CrossRef]
- Moore, M.C.; Coate, K.C.; Winnick, J.J.; An, Z.; Cherrington, A.D. Regulation of hepatic glucose uptake and storage in vivo. Adv. Nutr. 2012, 3, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Cline, G.W.; Gerard, D.P.; Magnusson, I.; Rothman, D.L.; Shulman, G.I. Contribution of net hepatic glycogen synthesis to disposal of an oral glucose load in humans. Metabolism 2001, 50, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Mitrakou, A.; Jones, R.; Okuda, Y.; Pena, J.; Nurjhan, N.; Field, J.B.; Gerich, J.E. Pathway and carbon sources for hepatic glycogen repletion in dogs. Am. J. Physiol. Endocrinol. Metab. 1991, 260, E194–E202. [Google Scholar] [CrossRef]
- Taylor, R.; Magnusson, I.; Rothman, D.L.; Cline, G.W.; Caumo, A.; Cobelli, C.; Shulman, G.I. Direct assessment of liver glycogen storage by 13C nuclear magnetic resonance spectroscopy and regulation of glucose homeostasis after a mixed meal in normal subjects. J. Clin. Investig. 1996, 97, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Perseghin, G.; Rothman, D.L.; Cline, G.W.; Magnusson, I.; Petersen, K.F.; Shulman, G.I. Impaired Net Hepatic Glycogen Synthesis in Insulin-Dependent Diabetic Subjects during Mixed Meal Ingestion. A 13C Nuclear Magnetic Resonance Spectroscopy Study. Available online: https://www.jci.org/articles/view/117727/pdf (accessed on 17 January 2022).
- Alsahli, M.; Shrayyef, M.Z.; Gerich, J.E. Normal glucose homeostasis. In Principles of Diabetes Mellitus; Poretsky, L., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 23–42. ISBN 978-3-319-18741-9. [Google Scholar]
- Katz, J.; Tayek, J.A. Gluconeogenesis and the Cori cycle in 12-, 20-, and 40-h-fasted humans. Am. J. Physiol. Endocrinol. Metab. 1998, 275, E537–E542. [Google Scholar] [CrossRef]
- Landau, B.R.; Wahren, J.; Chandramouli, V.; Schumann, W.C.; Ekberg, K.; Kalhan, S.C. Contributions of gluconeogenesis to glucose production in the fasted state. J. Clin. Investig. 1996, 98, 378–385. [Google Scholar] [CrossRef]
- Magnusson, I.; Rothman, D.L.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J. Clin. Investig. 1992, 90, 1323–1327. [Google Scholar] [CrossRef]
- Rothman, D.L.; Magnusson, I.; Katz, L.D.; Shulman, R.G.; Shulman, G.I. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science 1991, 254, 573–576. [Google Scholar] [CrossRef]
- Zierler, K.L. Theory of the use of arteriovenous concentration differences for measuring metabolism in steady and non-steady states. J. Clin. Investig. 1961, 40, 2111–2125. [Google Scholar] [CrossRef]
- Defronzo, R.A. 4 use of the splanchnic/hepatic balance technique in the study of glucose metabolism. Baillière’s Clin. Endocrinol. Metab. 1987, 1, 837–862. [Google Scholar] [CrossRef]
- Honka, M.-J.; Latva-Rasku, A.; Bucci, M.; Virtanen, K.A.; Hannukainen, J.C.; Kalliokoski, K.K.; Nuutila, P. Insulin-stimulated glucose uptake in skeletal muscle, adipose tissue and liver: A positron emission tomography study. Eur. J. Endocrinol. 2018, 178, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Immonen, H.; Hannukainen, J.C.; Iozzo, P.; Soinio, M.; Salminen, P.; Saunavaara, V.; Borra, R.; Parkkola, R.; Mari, A.; Lehtimäki, T.; et al. Effect of bariatric surgery on liver glucose metabolism in morbidly obese diabetic and non-diabetic patients. J. Hepatol. 2014, 60, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Sala-Rabanal, M.; Hirayama, B.A.; Ghezzi, C.; Liu, J.; Huang, S.-C.; Kepe, V.; Koepsell, H.; Yu, A.; Powell, D.R.; Thorens, B.; et al. Revisiting the physiological roles of SGLTs and GLUT.Ts using positron emission tomography in mice. J. Physiol. 2016, 594, 4425–4438. [Google Scholar] [CrossRef] [PubMed]
- Barrio, J.R.; Huang, S.-C.; Satyamurthy, N.; Scafoglio, C.S.; Yu, A.S.; Alavi, A.; Krohn, K.A. Does 2-FDG PET accurately reflect quantitative in vivo glucose utilization? J. Nucl. Med. 2020, 61, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.Y.; Jun, H.S.; Mansfield, B.C. The SLC37 family of phosphate-linked sugar phosphate antiporters. Mol. Asp. Med. 2013, 34, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, Y.; Umayahara, K.; Shimmura, T.; Yamashita, T. 19F NMR of 2-deoxy-2-fluoro-D-glucose for tumor diagnosis in mice. An NDP-bound hexose analog as a new NMR target for imaging. NMR Biomed. 1997, 10, 35–41. [Google Scholar] [CrossRef]
- Bender, D.; Munk, O.L.; Feng, H.-Q.; Keiding, S. Metabolites of 18F-FDG and 3-O-11C-methylglucose in pig liver. J. Nucl. Med. 2001, 42, 1673–1678. [Google Scholar]
- Iozzo, P.; Jarvisalo, M.J.; Kiss, J.; Borra, R.; Naum, G.A.; Viljanen, A.; Viljanen, T.; Gastaldelli, A.; Buzzigoli, E.; Guiducci, L.; et al. Quantification of liver glucose metabolism by positron emission tomography: Validation study in pigs. Gastroenterology 2007, 132, 531–542. [Google Scholar] [CrossRef]
- Southworth, R.; Parry, C.R.; Parkes, H.G.; Medina, R.A.; Garlick, P.B. Tissue-specific differences in 2-fluoro-2-deoxyglucose metabolism beyond FDG-6-P: A 19F NMR spectroscopy study in the rat. NMR Biomed. 2003, 16, 494–502. [Google Scholar] [CrossRef]
- Rokka, J.; Grönroos, T.J.; Viljanen, T.; Solin, O.; Haaparanta-Solin, M. HPLC and TLC methods for analysis of [18F] FDG and its metabolites from biological samples. J. Chromatogr. B 2017, 1048, 140–149. [Google Scholar] [CrossRef]
- Iozzo, P.; Geisler, F.; Oikonen, V.; Mäki, M.; Takala, T.; Solin, O.; Ferrannini, E.; Knuuti, J.; Nuutila, P. Insulin stimulates liver glucose uptake in humans: An 18F-FDG PET study. J. Nucl. Med. 2003, 44, 682–689. [Google Scholar] [PubMed]
- Rijzewijk, L.J.; van der Meer, R.W.; Lubberink, M.; Lamb, H.J.; Romijn, J.A.; de Roos, A.; Twisk, J.W.; Heine, R.J.; Lammertsma, A.A.; Smit, J.W.A.; et al. Liver fat content in type 2 diabetes: Relationship with hepatic perfusion and substrate metabolism. Diabetes 2010, 59, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Van der Weerdt, A.P.; Klein, L.J.; Visser, C.A.; Visser, F.C.; Lammertsma, A.A. Use of arterialised venous instead of arterial blood for measurement of myocardial glucose metabolism during euglycaemic-hyperinsulinaemic clamping. Eur. J. Nucl. Med. 2002, 29, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Phelps, M.E.; Huang, S.C.; Hoffman, E.J.; Selin, C.; Sokoloff, L.; Kuhl, D.E. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: Validation of method. Ann. Neurol. 1979, 6, 371–388. [Google Scholar] [CrossRef] [PubMed]
- Keiding, S. Bringing physiology into PET of the liver. J. Nucl. Med. 2012, 53, 425–433. [Google Scholar] [CrossRef]
- Munk, O.L.; Bass, L.; Roelsgaard, K.; Bender, D.; Hansen, S.B.; Keiding, S. Liver kinetics of glucose analogs measured in pigs by PET: Importance of dual-input blood sampling. J. Nucl. Med. 2001, 42, 795–801. [Google Scholar]
- Winterdahl, M.; Munk, O.L.; Sørensen, M.; Mortensen, F.V.; Keiding, S. Hepatic blood perfusion measured by 3-min dynamic 18F-FDG PET in pigs. J. Nucl. Med. 2011, 52, 1119–1124. [Google Scholar] [CrossRef]
- Winterdahl, M.; Keiding, S.; Sørensen, M.; Mortensen, F.V.; Alstrup, A.K.O.; Munk, O.L. Tracer input for kinetic modelling of liver physiology determined without sampling portal venous blood in pigs. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 263–270. [Google Scholar] [CrossRef]
- Ackerman, N.B. Experimental studies on the circulatory dynamics of intrahepatic tumor blood supply. Cancer 1972, 29, 435–439. [Google Scholar] [CrossRef]
- Fukuda, K.; Taniguchi, H.; Koh, T.; Kunishima, S.; Yamagishi, H. Relationships between oxygen and glucose metabolism in human liver tumours: Positron emission tomography using 15O and 18F-deoxyglucose. Nucl. Med. Commun. 2004, 25, 577–583. [Google Scholar] [CrossRef]
- Breedis, C.; Young, G. The blood supply of neoplasms in the liver. Am. J. Pathol. 1954, 30, 969–985. [Google Scholar] [PubMed]
- Trägårdh, M.; Møller, N.; Sørensen, M. Methodologic considerations for quantitative 18F-FDG PET/CT studies of hepatic glucose metabolism in healthy subjects. J. Nucl. Med. 2015, 56, 1366–1371. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reivich, M.; Alavi, A.; Wolf, A.; Fowler, J.; Russell, J.; Arnett, C.; MacGregor, R.R.; Shiue, C.Y.; Atkins, H.; Anand, A.; et al. Glucose metabolic rate kinetic model parameter determination in humans: The lumped constants and rate constants for [18F] fluorodeoxyglucose and [11C] deoxyglucose. J. Cereb. Blood Flow Metab. 1985, 5, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-M.; Bergsneider, M.; Glenn, T.C.; Yeh, E.; Hovda, D.A.; Phelps, M.E.; Huang, S.-C. Measurement of the global lumped constant for 2-deoxy-2-[18F] fluoro-D-glucose in normal human brain using [15O] water and 2-deoxy-2-[18F] fluoro-D-glucose positron emission tomography imaging: A method with validation based on multiple methodologies. Mol. Imaging Biol. 2003, 5, 32–41. [Google Scholar] [CrossRef]
- Keiding, S. How should lumped constant be estimated for hepatic 18F-FDG glucose in humans? J. Nucl. Med. 2015, 56, 1302–1303. [Google Scholar] [CrossRef]
- Keiding, S.; Sørensen, M.; Frisch, K.; Gormsen, L.C.; Munk, O.L. Quantitative PET of liver functions. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 73–85. [Google Scholar]
- Patlak, C.S.; Blasberg, R.G. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J. Cereb. Blood Flow Metab. 1985, 5, 584–590. [Google Scholar] [CrossRef]
- Patlak, C.S.; Blasberg, R.G.; Fenstermacher, J.D. Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. J. Cereb. Blood Flow Metab. 1983, 3, 1–7. [Google Scholar] [CrossRef]
- Lammertsma, A.A.; Brooks, D.J.; Frackowiak, R.S.J.; Beaney, R.P.; Herold, S.; Heather, J.D.; Palmer, A.J.; Jones, T. Measurement of glucose utilisation with [18F] 2-fluoro-2-deoxy-d-glucose: A comparison of different analytical methods. J. Cereb. Blood Flow Metab. 1987, 7, 161–172. [Google Scholar] [CrossRef]
- Keramida, G.; Hunter, J.; Peters, A.M. Hepatic glucose utilization in hepatic steatosis and obesity. Biosci. Rep. 2016, 36, e00402. [Google Scholar] [CrossRef]
- Thie, J.A. Clarification of a fractional uptake concept. J. Nucl. Med. 1995, 36, 711–712. [Google Scholar] [PubMed]
- Oikonen, V. Fractional Uptake Rate (FUR). Available online: http://www.turkupetcentre.net/petanalysis/model_fur.html (accessed on 5 February 2022).
- Keyes, J.W. SUV: Standard uptake or silly useless value? J. Nucl. Med. 1995, 36, 1836–1839. [Google Scholar] [PubMed]
- Bach-Gansmo, T.; Dybvik, J.; Adamsen, T.; Naum, A. Variation in urinary excretion of FDG, yet another uncertainty in quantitative PET. Acta Radiol. Short Rep. 2012, 1, 120038. [Google Scholar] [CrossRef] [PubMed]
- Cysouw, M.C.F.; Jansen, B.H.E.; Yaqub, M.; Voortman, J.; Vis, A.N.; van Moorselaar, R.J.A.; Hoekstra, O.S.; Boellaard, R.; Oprea-Lager, D.E. Letter to the editor Re: Semiquantitative parameters in PSMA-targeted PET imaging with [18F] DCFPyL: Impact of tumor burden on normal organ uptake. Mol. Imaging Biol. 2020, 22, 15–17. [Google Scholar] [CrossRef]
- Bertoldo, A.; Rizzo, G.; Veronese, M. Deriving physiological information from PET images: From SUV to Compartmental modelling. Clin. Transl. Imaging 2014, 2, 239–251. [Google Scholar] [CrossRef]
- Van den Hoff, J.; Oehme, L.; Schramm, G.; Maus, J.; Lougovski, A.; Petr, J.; Beuthien-Baumann, B.; Hofheinz, F. The PET-derived tumor-to-blood standard uptake ratio (SUR) is superior to tumor SUV as a surrogate parameter of the metabolic rate of FDG. EJNMMI Res. 2013, 3, 77. [Google Scholar] [CrossRef]
- Iozzo, P.; Gastaldelli, A.; Järvisalo, M.J.; Kiss, J.; Borra, R.; Buzzigoli, E.; Viljanen, A.; Naum, G.; Viljanen, T.; Oikonen, V.; et al. 18F-FDG assessment of glucose disposal and production rates during fasting and insulin stimulation: A validation study. J. Nucl. Med. 2006, 47, 1016–1022. [Google Scholar]
- Rebelos, E.; Immonen, H.; Bucci, M.; Hannukainen, J.C.; Nummenmaa, L.; Honka, M.-J.; Soinio, M.; Salminen, P.; Ferrannini, E.; Iozzo, P.; et al. Brain glucose uptake is associated with endogenous glucose production in obese patients before and after bariatric surgery and predicts metabolic outcome at follow-up. Diabetes Obes. Metab. 2019, 21, 218–226. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tobin, J.D.; Andres, R. Glucose clamp technique: A method for quantifying insulin secretion and resistance. Am. J. Physiol. 1979, 237, E214–E223. [Google Scholar] [CrossRef]
- Iozzo, P.; Hallsten, K.; Oikonen, V.; Virtanen, K.A.; Kemppainen, J.; Solin, O.; Ferrannini, E.; Knuuti, J.; Nuutila, P. Insulin-mediated hepatic glucose uptake is impaired in type 2 diabetes: Evidence for a relationship with glycemic control. J. Clin. Endocrinol. Metab. 2003, 88, 2055–2060. [Google Scholar] [CrossRef]
- Basu, A.; Basu, R.; Shah, P.; Vella, A.; Johnson, C.M.; Nair, K.S.; Jensen, M.D.; Schwenk, W.F.; Rizza, R.A. Effects of type 2 diabetes on the ability of insulin and glucose to regulate splanchnic and muscle glucose metabolism: Evidence for a defect in hepatic glucokinase activity. Diabetes 2000, 49, 272–283. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Basu, A.; Basu, R.; Shah, P.; Vella, A.; Johnson, C.M.; Jensen, M.; Nair, K.S.; Schwenk, W.F.; Rizza, R.A. Type 2 diabetes impairs splanchnic uptake of glucose but does not alter intestinal glucose absorption during enteral glucose feeding: Additional evidence for a defect in hepatic glucokinase activity. Diabetes 2001, 50, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Krssak, M.; Brehm, A.; Bernroider, E.; Anderwald, C.; Nowotny, P.; Man, C.D.; Cobelli, C.; Cline, G.W.; Shulman, G.I.; Waldhäusl, W.; et al. Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 2004, 53, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Petersen, M.C.; Zhang, D.; Vatner, D.F.; Perry, R.J.; Abulizi, A.; Haedersdal, S.; Zhang, X.-M.; Butrico, G.M.; Samuel, V.T.; et al. Metabolic control analysis of hepatic glycogen synthesis in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 8166–8176. [Google Scholar] [CrossRef] [PubMed]
- Ruotsalainen, U.; Raitakari, M.; Nuutila, P.; Oikonen, V.; Sipilä, H.; Teräs, M.; Knuuti, M.J.; Bloomfield, P.M.; Iida, H. Quantitative blood flow measurement of skeletal muscle using oxygen-15-water and PET. J. Nucl. Med. 1997, 38, 314–319. [Google Scholar]
- Kudomi, N.; Slimani, L.; Järvisalo, M.J.; Kiss, J.; Lautamäki, R.; Naum, G.A.; Savunen, T.; Knuuti, J.; Iida, H.; Nuutila, P.; et al. Non-invasive estimation of hepatic blood perfusion from H215O-PET images using tissue-derived arterial and portal input functions. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1899–1911. [Google Scholar] [CrossRef][Green Version]
- Ziegler, S.I.; Haberkorn, U.; Byrne, H.; Tong, C.; Schosser, R.; Krieter, H.; Kaja, S.; Richolt, J.A.; Lammertsma, A.A.; Price, P. Measurement of liver blood flow using oxygen-15 labelled water and dynamic positron emission tomography: Limitations of model description. Eur. J. Nucl. Med. 1996, 23, 169–177. [Google Scholar] [CrossRef]
- Slimani, L.; Kudomi, N.; Oikonen, V.; Jarvisalo, M.; Kiss, J.; Naum, A.; Borra, R.; Viljanen, A.; Sipila, H.; Ferrannini, E.; et al. Quantification of liver perfusion with [15O] H2O-PET and its relationship with glucose metabolism and substrate levels. J. Hepatol. 2008, 48, 974–982. [Google Scholar] [CrossRef]
- Immonen, H.; Hannukainen, J.C.; Kudomi, N.; Pihlajamäki, J.; Saunavaara, V.; Laine, J.; Salminen, P.; Lehtimäki, T.; Pham, T.; Iozzo, P.; et al. Increased liver fatty acid uptake is partly reversed and liver fat content normalized after bariatric surgery. Diabetes Care 2018, 41, 368–371. [Google Scholar] [CrossRef]
- Honka, H.; Koffert, J.; Kauhanen, S.; Kudomi, N.; Hurme, S.; Mari, A.; Lindqvist, A.; Wierup, N.; Parkkola, R.; Groop, L.; et al. Liver blood dynamics after bariatric surgery: The effects of mixed-meal test and incretin infusions. Endocr. Connect. 2018, 7, 888–896. [Google Scholar] [CrossRef]
- Kiss, J.; Naum, A.; Kudomi, N.; Knuuti, J.; Iozzo, P.; Savunen, T.; Nuutila, P. Non-invasive diagnosis of acute mesenteric ischaemia using PET. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1338. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L.; Skeaff, C.M.; Fielding, B.A. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog. Lipid Res. 2008, 47, 348–380. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, P.; Bucci, M.; Roivainen, A.; Någren, K.; Järvisalo, M.J.; Kiss, J.; Guiducci, L.; Fielding, B.; Naum, A.G.; Borra, R.; et al. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology 2010, 139, 846–856.e6. [Google Scholar] [CrossRef] [PubMed]
- DeGrado, T.R.; Coenen, H.H.; Stöcklin, G. 14(R,S)-[18F] fluoro-6-thia-heptadecanoic acid (FTHA): Evaluation in mouse of a new probe of myocardial utilization of long chain fatty acids. J. Nucl. Med. 1991, 32, 1888–1896. [Google Scholar]
- Guiducci, L.; Grönroos, T.; Järvisalo, M.J.; Kiss, J.; Viljanen, A.; Naum, A.G.; Viljanen, T.; Savunen, T.; Knuuti, J.; Ferrannini, E.; et al. Biodistribution of the fatty acid analogue 18F-FTHA: Plasma and tissue partitioning between lipid pools during fasting and hyperinsulinemia. J. Nucl. Med. 2007, 48, 455–462. [Google Scholar]
- Ci, X.; Frisch, F.; Lavoie, F.; Germain, P.; Lecomte, R.; van Lier, J.E.; Bénard, F.; Carpentier, A.C. The effect of insulin on the intracellular distribution of 14(R,S)-[18F] fluoro-6-thia-heptadecanoic acid in rats. Mol. Imaging Biol. 2006, 8, 237–244. [Google Scholar] [CrossRef]
- Iozzo, P.; Turpeinen, A.K.; Takala, T.; Oikonen, V.; Solin, O.; Ferrannini, E.; Nuutila, P.; Knuuti, J. Liver uptake of free fatty acids in vivo in humans as determined with 14(R,S)-[18F] fluoro-6-thia-heptadecanoic acid and PET. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1160–1164. [Google Scholar] [CrossRef]
- Viljanen, A.P.M.; Iozzo, P.; Borra, R.; Kankaanpää, M.; Karmi, A.; Lautamäki, R.; Järvisalo, M.; Parkkola, R.; Rönnemaa, T.; Guiducci, L.; et al. Effect of weight loss on liver free fatty acid uptake and hepatic insulin resistance. J. Clin. Endocrinol. Metab. 2009, 94, 50–55. [Google Scholar] [CrossRef]
- Oldgren, J.; Laurila, S.; Åkerblom, A.; Latva-Rasku, A.; Rebelos, E.; Isackson, H.; Saarenhovi, M.; Eriksson, O.; Heurling, K.; Johansson, E.; et al. Effects of 6 weeks of treatment with dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on myocardial function and metabolism in patients with type 2 diabetes: A randomized, placebo-controlled, exploratory study. Diabetes Obes. Metab. 2021, 23, 1505–1517. [Google Scholar] [CrossRef]
- Klein, L.J.; Visser, F.C.; Knaapen, P.; Peters, J.H.; Teule, G.J.J.; Visser, C.A.; Lammertsma, A.A. Carbon-11 acetate as a tracer of myocardial oxygen consumption. Eur. J. Nucl. Med. 2001, 28, 651–668. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic steatohepatitis is the fastest growing cause of hepatocellular carcinoma in liver transplant candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef] [PubMed]
- Rebelos, E.; Iozzo, P.; Guzzardi, M.A.; Brunetto, M.R.; Bonino, F. Brain-gut-liver interactions across the spectrum of insulin resistance in metabolic fatty liver disease. World J. Gastroenterol. 2021, 27, 4999–5018. [Google Scholar] [CrossRef] [PubMed]
- Keramida, G.; Potts, J.; Bush, J.; Dizdarevic, S.; Peters, A.M. Hepatic steatosis is associated with increased hepatic FDG uptake. Eur. J. Radiol. 2014, 83, 751–755. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Lin, W.-Y.; Lin, C.-C.; Shih, C.-M.; Jeng, L.-B.; Kao, C.-H. The negative impact of fatty liver on maximum standard uptake value of liver on FDG PET. Clin. Imaging 2011, 35, 437–441. [Google Scholar] [CrossRef]
- Abikhzer, G.; Alabed, Y.Z.; Azoulay, L.; Assayag, J.; Rush, C. Altered hepatic metabolic activity in patients with hepatic steatosis on FDG PET/CT. Am. J. Roentgenol. 2011, 196, 176–180. [Google Scholar] [CrossRef]
- Goceri, E.; Shah, Z.K.; Layman, R.; Jiang, X.; Gurcan, M.N. Quantification of liver fat: A comprehensive review. Comput. Biol. Med. 2016, 71, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Corwin, M.T.; Olson, K.A.; Badawi, R.D.; Sarkar, S. Dynamic PET of human liver inflammation: Impact of kinetic modeling with optimization-derived dual-blood input function. Phys. Med. Biol. 2018, 63, 155004. [Google Scholar] [CrossRef]
- Sarkar, S.; Corwin, M.T.; Olson, K.A.; Stewart, S.L.; Liu, C.-H.; Badawi, R.D.; Wang, G. Pilot study to diagnose nonalcoholic steatohepatitis with dynamic 18F-FDG PET. Am. J. Roentgenol. 2019, 212, 529–537. [Google Scholar] [CrossRef]
- Guzzardi, M.A.; Guiducci, L.; Campani, D.; la Rosa, F.; Cacciato Insilla, A.; Bartoli, A.; Cabiati, M.; de Sena, V.; del Ry, S.; Burchielli, S.; et al. Leptin resistance before and after obesity: Evidence that tissue glucose uptake underlies adipocyte enlargement and liver steatosis/steatohepatitis in Zucker rats from early-life stages. Int. J. Obes. 2022, 46, 50–58. [Google Scholar] [CrossRef]
- Nejabat, M.; Leisser, A.; Karanikas, G.; Wadsak, W.; Mitterhauser, M.; Mayerhöfer, M.; Kienbacher, C.; Trauner, M.; Hacker, M.; Haug, A.R. [11C] acetate PET as a tool for diagnosis of liver steatosis. Abdom. Radiol. 2018, 43, 2963–2969. [Google Scholar] [CrossRef] [PubMed]
- Ong, L.-C.; Jin, Y.; Song, I.-C.; Yu, S.; Zhang, K. 2-[18F]-2-deoxy-d-glucose (FDG) uptake in human tumor cells is related to the expression of glut-1 and hexokinase II. Acta Radiol. 2008, 49, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Kuge, Y.; Mochizuki, T.; Takahashi, T.; Nakada, K.; Sato, M.; Takei, T.; Tamaki, N. Biologic correlates of intratumoral heterogeneity in 18F-FDG distribution with regional expression of glucose transporters and hexokinase-II in experimental tumor. J. Nucl. Med. 2005, 46, 675–682. [Google Scholar] [PubMed]
- Pauwels, E.K.; Ribeiro, M.J.; Stoot, J.H.; McCready, V.R.; Bourguignon, M.; Mazière, B. FDG accumulation and tumor biology. Nucl. Med. Biol. 1998, 25, 317–322. [Google Scholar] [CrossRef]
- Lee, J.D.; Yang, W.I.; Park, Y.N.; Kim, K.S.; Choi, J.S.; Yun, M.; Ko, D.; Kim, T.-S.; Cho, A.E.H.; Kim, H.M.; et al. Different glucose uptake and glycolytic mechanisms between hepatocellular carcinoma and intrahepatic mass-forming cholangiocarcinoma with increased 18F-FDG uptake. J. Nucl. Med. 2005, 46, 1753–1759. [Google Scholar]
- Torizuka, T.; Tamaki, N.; Inokuma, T.; Magata, Y.; Sasayama, S.; Yonekura, Y.; Tanaka, A.; Yamaoka, Y.; Yamamoto, K.; Konishi, J. In vivo assessment of glucose metabolism in hepatocellular carcinoma with FDG-PET. J. Nucl. Med. 1995, 36, 1811–1817. [Google Scholar]
- Khan, M.A.; Combs, C.S.; Brunt, E.M.; Lowe, V.J.; Wolverson, M.K.; Solomon, H.; Collins, B.T.; Bisceglie, A.M.D. Positron emission tomography scanning in the evaluation of hepatocellular carcinoma. J. Hepatol. 2000, 32, 792–797. [Google Scholar] [CrossRef]
- Wudel, L.J.; Delbeke, D.; Morris, D.; Rice, M.; Washington, M.K.; Shyr, Y.; Pinson, C.W.; Chapman, W.C. The role of [18F] fluorodeoxyglucose positron emission tomography imaging in the evaluation of hepatocellular carcinoma. Am. Surg. 2003, 69, 117–124. [Google Scholar]
- Kawamura, E.; Shiomi, S.; Kotani, K.; Kawabe, J.; Hagihara, A.; Fujii, H.; Uchida-Kobayashi, S.; Iwai, S.; Morikawa, H.; Enomoto, M.; et al. Positioning of 18F-fluorodeoxyglucose-positron emission tomography imaging in the management algorithm of hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2014, 29, 1722–1727. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, H.S.; Lee, S.; Lee, J.W. Emerging role of 18F-fluorodeoxyglucose positron emission tomography for guiding management of hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 1289–1306. [Google Scholar] [CrossRef]
- Lee, Y.; Yoo, I.R.; Boo, S.H.; Kim, H.; Park, H.L.; Hyun O, J. The role of F-18 FDG PET/CT in intrahepatic cholangiocarcinoma. Nucl. Med. Mol. Imaging 2017, 51, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Yun, M.; Lee, W.J.; Kim, K.S.; Lee, J.D. Usefulness of 18F-FDG PET in intrahepatic cholangiocarcinoma. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, S.; Caldarella, C.; Pizzuto, D.A.; Galiandro, F.; Sadeghi, R.; Giovanella, L.; Treglia, G. Diagnostic accuracy of fluorine-18-fluorodeoxyglucose positron emission tomography in the evaluation of the primary tumor in patients with cholangiocarcinoma: A meta-analysis. Biomed. Res. Int. 2014, 2014, 247693. [Google Scholar] [CrossRef] [PubMed]
- Sacks, A.; Peller, P.J.; Surasi, D.S.; Chatburn, L.; Mercier, G.; Subramaniam, R.M. Value of PET/CT in the management of liver metastases, part 1. Am. J. Roentgenol. 2011, 197, W256–W259. [Google Scholar] [CrossRef]
- Chua, S.C.; Groves, A.M.; Kayani, I.; Menezes, L.; Gacinovic, S.; Du, Y.; Bomanji, J.B.; Ell, P.J. The impact of 18F-FDG PET/CT in patients with liver metastases. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1906–1914. [Google Scholar] [CrossRef]
- Cornelis, F.; Sotirchos, V.; Violari, E.; Sofocleous, C.T.; Schoder, H.; Durack, J.C.; Siegelbaum, R.H.; Maybody, M.; Humm, J.; Solomon, S.B. 18F-FDG PET/CT is an immediate imaging biomarker of treatment success after liver metastasis ablation. J. Nucl. Med. 2016, 57, 1052–1057. [Google Scholar] [CrossRef]
- Maffione, A.M.; Lopci, E.; Bluemel, C.; Giammarile, F.; Herrmann, K.; Rubello, D. Diagnostic accuracy and impact on management of 18F-FDG PET and PET/CT in colorectal liver metastasis: A meta-analysis and systematic review. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 152–163. [Google Scholar] [CrossRef]
- Bipat, S.; van Leeuwen, M.S.; Comans, E.F.I.; Pijl, M.E.J.; Bossuyt, P.M.M.; Zwinderman, A.H.; Stoker, J. Colorectal liver metastases: CT, MR imaging, and PET for diagnosis—Meta-analysis. Radiology 2005, 237, 123–131. [Google Scholar] [CrossRef]
- Iwano, S.; Ito, S.; Tsuchiya, K.; Kato, K.; Naganawa, S. What causes false-negative PET findings for solid-type lung cancer? Lung Cancer 2013, 79, 132–136. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.-Q.; Liu, J.-L.; Qin, X.-G.; Huang, Y. FDG-PET in diagnosis, staging and prognosis of pancreatic carcinoma: A meta-analysis. World J. Gastroenterol. 2013, 19, 4808–4817. [Google Scholar] [CrossRef]
- Berger, K.L.; Nicholson, S.A.; Dehdashti, F.; Siegel, B.A. FDG PET evaluation of mucinous neoplasms. Am. J. Roentgenol. 2000, 174, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Waki, A.; Yonekura, Y.; Sadato, N.; Murata, T.; Omata, N.; Takahashi, N.; Welch, M.J.; Fujibayashi, Y. Characterization of acetate metabolism in tumor cells in relation to cell proliferation: Acetate metabolism in tumor cells. Nucl. Med. Biol. 2001, 28, 117–122. [Google Scholar] [CrossRef]
- Ho, C.-L.; Yu, S.C.H.; Yeung, D.W.C. 11C-acetate PET imaging in hepatocellular carcinoma and other liver masses. J. Nucl. Med. 2003, 44, 213–221. [Google Scholar] [PubMed]
- Hwang, K.H.; Choi, D.-J.; Lee, S.-Y.; Lee, M.K.; Choe, W. Evaluation of patients with hepatocellular carcinomas using [11C] acetate and [18F] FDG PET/CT: A preliminary study. Appl. Radiat. Isot. 2009, 67, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.T.; Ho, C.L.; Lo, C.M.; Chen, S.; Chan, S.C.; Chok, K.S.H.; Fung, J.Y.; Chan, A.C.Y.; Sharr, W.; Yau, T.; et al. 11C-acetate and 18F-FDG PET/CT for clinical staging and selection of patients with hepatocellular carcinoma for liver transplantation on the basis of milan criteria: Surgeon’s perspective. J. Nucl. Med. 2013, 54, 192–200. [Google Scholar] [CrossRef]
- Soret, M.; Bacharach, S.L.; Buvat, I. Partial-volume effect in PET tumor imaging. J. Nucl. Med. 2007, 48, 932–945. [Google Scholar] [CrossRef]
- Park, J.-W.; Kim, J.H.; Kim, S.K.; Kang, K.W.; Park, K.W.; Choi, J.-I.; Lee, W.J.; Kim, C.-M.; Nam, B.H. A prospective evaluation of 18F-FDG and 11C-acetate PET/CT for detection of primary and metastatic hepatocellular carcinoma. J. Nucl. Med. 2008, 49, 1912–1921. [Google Scholar] [CrossRef]
- Fong, Y.; Saldinger, P.F.; Akhurst, T.; Macapinlac, H.; Yeung, H.; Finn, R.D.; Cohen, A.; Kemeny, N.; Blumgart, L.H.; Larson, S.M. Utility of 18F-FDG positron emission tomography scanning on selection of patients for resection of hepatic colorectal metastases. Am. J. Surg. 1999, 178, 282–287. [Google Scholar] [CrossRef]
- Delbeke, D.; Vitola, J.V.; Sandler, M.P.; Arildsen, R.C.; Powers, T.A.; Wright, J.K.; Chapman, W.C.; Pinson, C.W. Staging recurrent metastatic colorectal carcinoma with PET. J. Nucl. Med. 1997, 38, 1196–1201. [Google Scholar]
- McDermott, S.; Skehan, S.J. Whole body imaging in the abdominal cancer patient: Pitfalls of PET-CT. Abdom. Imaging 2010, 35, 55–69. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Hendler, R.; Felig, P.; Wahren, J. Regulation of splanchnic and peripheral glucose uptake by insulin and hyperglycemia in man. Diabetes 1983, 32, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Honka, H.; Hannukainen, J.C.; Tarkia, M.; Karlsson, H.; Saunavaara, V.; Salminen, P.; Soinio, M.; Mikkola, K.; Kudomi, N.; Oikonen, V.; et al. Pancreatic metabolism, blood flow, and β-cell function in obese humans. J. Clin. Endocrinol. Metab. 2014, 99, E981–E990. [Google Scholar] [CrossRef] [PubMed]
- Koffert, J.P.; Mikkola, K.; Virtanen, K.A.; Andersson, A.-M.D.; Faxius, L.; Hällsten, K.; Heglind, M.; Guiducci, L.; Pham, T.; Silvola, J.M.U.; et al. Metformin treatment significantly enhances intestinal glucose uptake in patients with type 2 diabetes: Results from a randomized clinical trial. Diabetes Res. Clin. Pract. 2017, 131, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, J.; Hannukainen, J.C.; Karmi, A.; Immonen, H.M.; Soinio, M.; Nelimarkka, L.; Savisto, N.; Helmiö, M.; Ovaska, J.; Salminen, P.; et al. Obesity-associated intestinal insulin resistance is ameliorated after bariatric surgery. Diabetologia 2015, 58, 1055–1062. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Shao, Y.; Liu, B.; Wang, X.; Geist, B.K.; Li, X.; Li, F.; Zhao, H.; Hacker, M.; Ding, H.; et al. Dynamic 18F-FDG PET imaging of liver lesions: Evaluation of a two-tissue compartment model with dual blood input function. BMC Med. Imaging 2021, 21, 90. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Søndergaard, E.; Christensen, N.L.; Jakobsen, S.; Nielsen, E.H.T.; Munk, O.L.; Tolbod, L.P.; Jessen, N.; Nielsen, S. Metformin does not affect postabsorptive hepatic free fatty acid uptake, oxidation or resecretion in humans: A 3-month placebo-controlled clinical trial in patients with type 2 diabetes and healthy controls. Diabetes Obes. Metab. 2018, 20, 1435–1444. [Google Scholar] [CrossRef]
- Gill, A.B.; Hilliard, N.J.; Hilliard, S.T.; Graves, M.J.; Lomas, D.J.; Shaw, A. A semi-automatic method for the extraction of the portal venous input function in quantitative dynamic contrast-enhanced CT of the liver. BJR 2017, 90, 20160875. [Google Scholar] [CrossRef]
- Walker, M.D.; Morgan, A.J.; Bradley, K.M.; McGowan, D.R. Data-driven respiratory gating outperforms device-based gating for clinical 18F-FDG PET/CT. J. Nucl. Med. 2020, 61, 1678–1683. [Google Scholar] [CrossRef]
- Merlin, T.; Visvikis, D.; Fernandez, P.; Lamare, F. Dynamic PET image reconstruction integrating temporal regularization associated with respiratory motion correction for applications in oncology. Phys. Med. Biol. 2018, 63, 045012. [Google Scholar] [CrossRef]
- Kyme, A.Z.; Fulton, R.R. Motion Estimation and Correction in SPECT, PET and CT. Phys. Med. Biol. 2021, 66, 18TR02. [Google Scholar] [CrossRef]
- Munk, O.L.; Keiding, S.; Baker, C.; Bass, L. A microvascular compartment model validated using 11C-methylglucose liver PET in pigs. Phys. Med. Biol. 2017, 63, 015032. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Harada, K.; Terayama, N.; Kosaka, N.; Kimura, H.; Gabata, T. FDG-PET/CT imaging findings of hepatic tumors and tumor-like lesions based on molecular background. Jpn. J. Radiol. 2020, 38, 697–718. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Lee, S.M. Radiomics in oncological PET/CT: Clinical applications. Nucl. Med. Mol. Imaging 2018, 52, 170–189. [Google Scholar] [CrossRef] [PubMed]
- Cherry, S.R.; Jones, T.; Karp, J.S.; Qi, J.; Moses, W.W.; Badawi, R.D. Total-body PET: Maximizing sensitivity to create new opportunities for clinical research and patient care. J. Nucl. Med. 2018, 59, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Alberts, I.; Hünermund, J.-N.; Prenosil, G.; Mingels, C.; Bohn, K.P.; Viscione, M.; Sari, H.; Vollnberg, B.; Shi, K.; Afshar-Oromieh, A.; et al. Clinical performance of long axial field of view PET/CT: A head-to-head intra-individual comparison of the biograph vision quadra with the biograph vision PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2395–2404. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honka, M.-J.; Rebelos, E.; Malaspina, S.; Nuutila, P. Hepatic Positron Emission Tomography: Applications in Metabolism, Haemodynamics and Cancer. Metabolites 2022, 12, 321. https://doi.org/10.3390/metabo12040321
Honka M-J, Rebelos E, Malaspina S, Nuutila P. Hepatic Positron Emission Tomography: Applications in Metabolism, Haemodynamics and Cancer. Metabolites. 2022; 12(4):321. https://doi.org/10.3390/metabo12040321
Chicago/Turabian StyleHonka, Miikka-Juhani, Eleni Rebelos, Simona Malaspina, and Pirjo Nuutila. 2022. "Hepatic Positron Emission Tomography: Applications in Metabolism, Haemodynamics and Cancer" Metabolites 12, no. 4: 321. https://doi.org/10.3390/metabo12040321
APA StyleHonka, M.-J., Rebelos, E., Malaspina, S., & Nuutila, P. (2022). Hepatic Positron Emission Tomography: Applications in Metabolism, Haemodynamics and Cancer. Metabolites, 12(4), 321. https://doi.org/10.3390/metabo12040321