
The Glycobiology of Pulmonary Arterial Hypertension

 ,
,  and
and 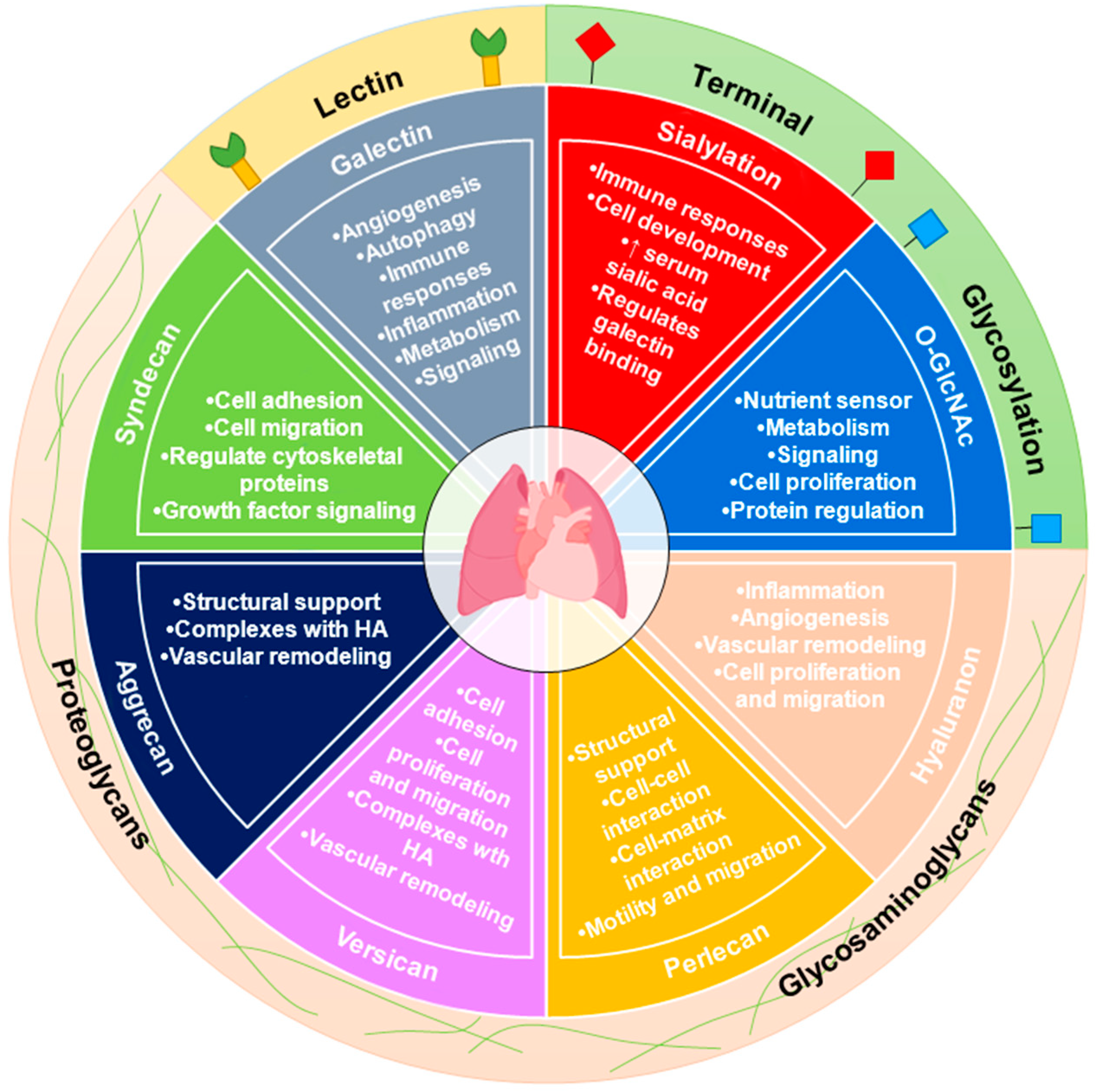{kind=link}
Abstract
1. Introduction
1.1. Metabolism Studies in PAH
1.2. Carbohydrate Metabolism and Glycosylation
1.3. Hexosamine Biosynthetic Pathway
1.4. Intracellular Glycosylation (O-GlcNAc) in PAH
1.5. The Role of Extracellular Matrix (ECM) Glycosaminoglycans (GAGs) in PAH
1.6. Hyaluronan (HA) in PAH
1.7. Perlecan in PAH
1.8. Versican and Aggrecan in PAH
1.9. Other PGs in PAH
1.10. Galectins (Carbohydrate Lectins) in PAH
1.11. Sialylation in PAH
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mélot, C.; Naeije, R. Pulmonary vascular diseases. Compr. Physiol. 2011, 1, 593–619. [Google Scholar] [PubMed]
- Barnes, J.W.; Tonelli, A.R.; Heresi, G.A.; Newman, J.E.; Mellor, N.E.; Grove, D.E.; Dweik, R.A. Novel Methods in Pulmonary Hypertension Phenotyping in the Age of Precision Medicine (2015 Grover Conference Series). Pulm. Circ. 2016, 6, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Badesch, D.B.; Champion, H.C.; Sanchez, M.A.G.; Hoeper, M.M.; Loyd, J.E.; Manes, A.; McGoon, M.; Naeije, R.; Olschewski, H.; Oudiz, R.J.; et al. Diagnosis and Assessment of Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2009, 54, S55–S66. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.L.; Barbera, J.A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J.S.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2009, 30, 2493–2537. [Google Scholar]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Simonneau, G.; Hoeper, M.M. The revised definition of pulmonary hypertension: Exploring the impact on patient management. Eur. Heart J. 2019, 21, K4–K8. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and diagnosis of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the Diagnosis and Treatment of Pulmonary Hypertension. Rev. Esp. Cardiol. (Engl. Ed.) 2016, 69, 177. [Google Scholar]
- Memon, H.A.; Park, M.H. Pulmonary Arterial Hypertension in Women. Methodist Debakey Cardiovasc. J. 2017, 13, 224–237. [Google Scholar] [CrossRef]
- D’Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; He, Y.Y.; Jiang, X.; Wang, Y.; Chen, J.W.; Zhao, J.H.; Ye, J.; Lian, T.Y.; Zhang, X.; Zhang, R.J.; et al. DNA methyltransferase 3B deficiency unveils a new pathological mechanism of pulmonary hypertension. Sci. Adv. 2020, 6, eaba2470. [Google Scholar] [CrossRef]
- Wang, X.-J.; Lian, T.-Y.; Jiang, X.; Liu, S.-F.; Li, S.-Q.; Jiang, R.; Wu, W.-H.; Ye, J.; Cheng, C.-Y.; Du, Y.; et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801609. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.; Girerd, B.; Montani, D.; Wang, X.J.; Galie, N.; Austin, E.D.; Elliott, G.; Asano, K.; Grunig, E.; Yan, Y.; et al. BMPR2 mutations and survival in pulmonary arterial hypertension: An individual participant data meta-analysis. Lancet Respir. Med. 2016, 4, 129–137. [Google Scholar] [CrossRef]
- Wang, X.-J.; Xu, X.-Q.; Sun, K.; Liu, K.-Q.; Li, S.-Q.; Jiang, X.; Zhao, Q.-H.; Wang, L.; Peng, F.-H.; Ye, J.; et al. Association of Rare PTGIS Variants With Susceptibility and Pulmonary Vascular Response in Patients With Idiopathic Pulmonary Arterial Hypertension. JAMA Cardiol. 2020, 5, 677–684. [Google Scholar] [CrossRef]
- Klouda, T.; Yuan, K. Inflammation in Pulmonary Arterial Hypertension. Adv. Exp. Med. Biol. 2021, 1303, 351–372. [Google Scholar]
- Voelkel, N.F.; Gomez-Arroyo, J.; Abbate, A.; Bogaard, H.J.; Nicolls, M.R. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur. Respir. J. 2012, 40, 1555–1565. [Google Scholar] [CrossRef]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef]
- Meloche, J.; Renard, S.; Provencher, S.; Bonnet, S. Anti-inflammatory and immunosuppressive agents in PAH. Handb. Exp. Pharmacol. 2013, 218, 437–476. [Google Scholar]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmuller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and pathobiology of pulmonary hypertension: State of the art and research perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, T.K.; Morrell, N.W. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog. Cardiovasc. Dis. 2002, 45, 173–202. [Google Scholar] [CrossRef]
- Koress, C.; Swan, K.; Kadowitz, P. Soluble Guanylate Cyclase Stimulators and Activators: Novel Therapies for Pulmonary Vascular Disease or a Different Method of Increasing cGMP? Curr. Hypertens. Rep. 2016, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Kaneko, F.T.; Zheng, S.; Comhair, S.A.; Janocha, A.J.; Goggans, T.; Thunnissen, F.B.; Farver, C.; Hazen, S.L.; Jennings, C.; et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004, 18, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, F.T.; Arroliga, A.C.; Dweik, R.A.; Comhair, S.A.; Laskowski, D.; Oppedisano, R.; Thomassen, M.J.; Erzurum, S.C. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1998, 158, 917–923. [Google Scholar] [CrossRef]
- Giaid, A.; Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef]
- Rubin, L.J. Primary pulmonary hypertension. N. Engl. J. Med. 1997, 336, 111–117. [Google Scholar] [CrossRef]
- Brunner, N.W.; Skhiri, M.; Fortenko, O.; Hsi, A.; Haddad, F.; Khazeni, N.; Zamanian, R.T. Impact of insulin resistance on ventricular function in pulmonary arterial hypertension. J. Heart Lung Transplant. 2014, 33, 721–726. [Google Scholar] [CrossRef]
- Hansmann, G.; Wagner, R.A.; Schellong, S.; Perez, V.A.; Urashima, T.; Wang, L.; Sheikh, A.Y.; Suen, R.S.; Stewart, D.J.; Rabinovitch, M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 2007, 115, 1275–1284. [Google Scholar] [CrossRef]
- Pugh, M.E.; Robbins, I.M.; Rice, T.W.; West, J.; Newman, J.H.; Hemnes, A.R. Unrecognized glucose intolerance is common in pulmonary arterial hypertension. J. Heart Lung Transplant. 2011, 30, 904–911. [Google Scholar] [CrossRef]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 2009, 33, 318–324. [Google Scholar] [CrossRef]
- Aytekin, M.; Tonelli, A.R.; Farver, C.F.; Feldstein, A.E.; Dweik, R.A. Leptin deficiency recapitulates the histological features of pulmonary arterial hypertension in mice. Int. J. Clin. Exp. Pathol. 2014, 7, 1935–1946. [Google Scholar]
- Santos, M.; Reis, A.; Goncalves, F.; Ferreira-Pinto, M.J.; Cabral, S.; Torres, S.; Leite-Moreira, A.F.; Henriques-Coelho, T. Adiponectin levels are elevated in patients with pulmonary arterial hypertension. Clin. Cardiol. 2014, 37, 21–25. [Google Scholar] [CrossRef]
- Tonelli, A.R.; Aytekin, M.; Feldstein, A.E.; Dweik, R.A. Leptin Levels Predict Survival in Pulmonary Arterial Hypertension. Pulm. Circ. 2012, 2, 214–219. [Google Scholar] [CrossRef]
- Talati, M.; Hemnes, A. Fatty acid metabolism in pulmonary arterial hypertension: Role in right ventricular dysfunction and hypertrophy. Pulm. Circ. 2015, 5, 269–278. [Google Scholar] [CrossRef]
- Cracowski, J.L.; Cracowski, C.; Bessard, G.; Pepin, J.L.; Bessard, J.; Schwebel, C.; Stanke-Labesque, F.; Pison, C. Increased lipid peroxidation in patients with pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2001, 164, 1038–1042. [Google Scholar] [CrossRef]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef]
- Cathey, S.S. Breath analysis in pulmonary arterial hypertension. Eur. J. Hum. Genet. EJHG 2014, 145, 551–558. [Google Scholar]
- Chen, J.; Tang, H.; Sysol, J.R.; Moreno-Vinasco, L.; Shioura, K.M.; Chen, T.; Gorshkova, I.; Wang, L.; Huang, L.S.; Usatyuk, P.V.; et al. The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2014, 190, 1032–1043. [Google Scholar] [CrossRef]
- Ross, D.J.; Hough, G.; Hama, S.; Aboulhosn, J.; Belperio, J.A.; Saggar, R.; Van Lenten, B.J.; Ardehali, A.; Eghbali, M.; Reddy, S.; et al. Proinflammatory high-density lipoprotein results from oxidized lipid mediators in the pathogenesis of both idiopathic and associated types of pulmonary arterial hypertension. Pulm. Circ. 2015, 5, 640–648. [Google Scholar] [CrossRef]
- Sutendra, G.; Bonnet, S.; Rochefort, G.; Haromy, A.; Folmes, K.D.; Lopaschuk, G.D.; Dyck, J.R.; Michelakis, E.D. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci. Transl. Med. 2010, 2, 44ra58. [Google Scholar] [CrossRef]
- Heresi, G.A.; Aytekin, M.; Newman, J.; DiDonato, J.; Dweik, R.A. Plasma levels of high-density lipoprotein cholesterol and outcomes in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 661–668. [Google Scholar] [CrossRef]
- Hu, J.; Xu, Q.; McTiernan, C.; Lai, Y.C.; Osei-Hwedieh, D.; Gladwin, M. Novel Targets of Drug Treatment for Pulmonary Hypertension. Am. J. Cardiovasc. Drugs 2015, 15, 225–234. [Google Scholar] [CrossRef]
- Ozkan, M.; Dweik, R.A.; Laskowski, D.; Arroliga, A.C.; Erzurum, S.C. High levels of nitric oxide in individuals with pulmonary hypertension receiving epoprostenol therapy. Lung 2001, 179, 233–243. [Google Scholar] [CrossRef]
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of Endothelin-1 in the Lungs of Patients with Pulmonary Hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef]
- Heresi, G.A.; Malin, S.K.; Barnes, J.W.; Tian, L.; Kirwan, J.P.; Dweik, R.A. Abnormal Glucose Metabolism and High-Energy Expenditure in Idiopathic Pulmonary Arterial Hypertension. Ann. Am. Thorac. Soc. 2016, 14, 190–199. [Google Scholar]
- Lundgrin, E.L.; Park, M.M.; Sharp, J.; Tang, W.H.; Thomas, J.D.; Asosingh, K.; Comhair, S.A.; DiFilippo, F.P.; Neumann, D.R.; Davis, L.; et al. Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann. Am. Thorac. Soc. 2013, 10, 1–9. [Google Scholar] [CrossRef]
- Marsboom, G.; Wietholt, C.; Haney, C.R.; Toth, P.T.; Ryan, J.J.; Morrow, E.; Thenappan, T.; Bache-Wiig, P.; Piao, L.; Paul, J.; et al. Lung (1)(8)F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 670–679. [Google Scholar] [CrossRef]
- Barnes, J.W.; Kucera, E.T.; Tian, L.; Mellor, N.E.; Dvorina, N.; Baldwin, W.W., III; Aldred, M.A.; Farver, C.F.; Comhair, S.A.; Aytekin, M.; et al. BMPR2 Mutation-independent Mechanisms of Disrupted BMP Signaling in IPAH. Am. J. Respir. Cell Mol. Biol. 2016, 55, 564–575. [Google Scholar] [CrossRef]
- Cikach, F.S., Jr.; Tonelli, A.R.; Barnes, J.; Paschke, K.; Newman, J.; Grove, D.; Dababneh, L.; Wang, S.; Dweik, R.A. Breath Analysis in Pulmonary Arterial Hypertension. Chest 2013, 145, 551–558. [Google Scholar] [CrossRef]
- Kao, C.C.; Wedes, S.H.; Hsu, J.W.; Bohren, K.M.; Comhair, S.A.A.; Jahoor, F.; Erzurum, S.C. Arginine Metabolic Endotypes in Pulmonary Arterial Hypertension. Pulm. Circ. 2015, 5, 124–134. [Google Scholar] [CrossRef]
- Demoncheaux, E.A.; Higenbottam, T.W.; Kiely, D.G.; Wong, J.M.; Wharton, S.; Varcoe, R.; Siddons, T.; Spivey, A.C.; Hall, K.; Gize, A.P. Decreased whole body endogenous nitric oxide production in patients with primary pulmonary hypertension. J. Vasc. Res. 2005, 42, 133–136. [Google Scholar] [CrossRef]
- Izquierdo-Garcia, J.L.; Arias, T.; Rojas, Y.; Garcia-Ruiz, V.; Santos, A.; Martin-Puig, S.; Ruiz-Cabello, J. Metabolic Reprogramming in the Heart and Lung in a Murine Model of Pulmonary Arterial Hypertension. Front. Cardiovasc. Med 2018, 5, 110. [Google Scholar] [CrossRef]
- Piao, L.; Fang, Y.H.; Parikh, K.; Ryan, J.J.; Toth, P.T.; Archer, S.L. Cardiac glutaminolysis: A maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J. Mol. Med. 2013, 91, 1185–1197. [Google Scholar] [CrossRef]
- Egnatchik, R.A.; Brittain, E.L.; Shah, A.T.; Fares, W.H.; Ford, H.J.; Monahan, K.; Kang, C.J.; Kocurek, E.G.; Zhu, S.; Luong, T.; et al. Dysfunctional BMPR2 signaling drives an abnormal endothelial requirement for glutamine in pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 186–199. [Google Scholar] [CrossRef]
- Mey, J.T.; Hari, A.; Axelrod, C.L.; Fealy, C.E.; Erickson, M.L.; Kirwan, J.P.; Dweik, R.A.; Heresi, G.A. Lipids and ketones dominate metabolism at the expense of glucose control in pulmonary arterial hypertension: A hyperglycaemic clamp and metabolomics study. Eur. Respir. J. 2020, 55, 1901700. [Google Scholar] [CrossRef]
- Belly, M.J.; Tiede, H.; Morty, R.E.; Schulz, R.; Voswinckel, R.; Tanislav, C.; Olschewski, H.; Ghofrani, H.A.; Seeger, W.; Reichenberger, F. HbA1c in pulmonary arterial hypertension: A marker of prognostic relevance? J. Heart Lung Transpl. 2012, 31, 1109–1114. [Google Scholar] [CrossRef]
- Paulin, R.; Michelakis, E.D. The metabolic theory of pulmonary arterial hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef]
- Rai, P.R.; Cool, C.D.; King, J.A.; Stevens, T.; Burns, N.; Winn, R.A.; Kasper, M.; Voelkel, N.F. The cancer paradigm of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 558–564. [Google Scholar] [CrossRef]
- Guignabert, C.; Tu, L.; Le Hiress, M.; Ricard, N.; Sattler, C.; Seferian, A.; Huertas, A.; Humbert, M.; Montani, D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2013, 22, 543–551. [Google Scholar] [CrossRef]
- Cool, C.D.; Kuebler, W.M.; Bogaard, H.J.; Spiekerkoetter, E.; Nicolls, M.R.; Voelkel, N.F. The hallmarks of severe pulmonary arterial hypertension: The cancer hypothesis—ten years later. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1115–L1130. [Google Scholar] [CrossRef]
- Baggetto, L.G. Deviant energetic metabolism of glycolytic cancer cells. Biochimie 1992, 74, 959–974. [Google Scholar] [CrossRef]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef]
- Bos, R.; van der Hoeven, J.J.M.; van der Wall, E.; van der Groep, P.; van Diest, P.J.; ComansUrvi Joshi, E.F.I.; Semenza, G.L.; Hoekstra, O.S.; Lammertsma, A.A.; Molthoff, C.F.M. Biologic Correlates of 18Fluorodeoxyglucose Uptake in Human Breast Cancer Measured by Positron Emission Tomography. J. Clin. Oncol. 2002, 20, 379–387. [Google Scholar] [CrossRef]
- Pedersen, P.L. Warburg, me and Hexokinase 2, Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J. Bioenerg. Biomembr. 2007, 39, 211. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Diaz-Ruiz, R.; Rigoulet, M.; Devin, A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim. Biophys. Acta 2011, 1807, 568–576. [Google Scholar] [CrossRef]
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; DiFilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C.; et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347. [Google Scholar] [CrossRef]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.-I.; et al. BMPR2 Preserves Mitochondrial Function and DNA during Reoxygenation to Promote Endothelial Cell Survival and Reverse Pulmonary Hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef]
- Rehman, J.; Archer, S.L. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: The Warburg model of pulmonary arterial hypertension. Adv. Exp. Med. Biol. 2010, 661, 171–185. [Google Scholar]
- Rabinovitch, M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Investig. 2012, 122, 4306–4313. [Google Scholar] [CrossRef]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thébaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef]
- Gomez-Arroyo, J.; Mizuno, S.; Szczepanek, K.; Tassell, B.V.; Natarajan, R.; dos Remedios, C.G.; Drake, J.I.; Farkas, L.; Kraskauskas, D.; Wijesinghe, D.S.; et al. Metabolic Gene Remodeling and Mitochondrial Dysfunction in Failing Right Ventricular Hypertrophy Secondary to Pulmonary Arterial Hypertension. Circ. Heart Fail. 2013, 6, 136–144. [Google Scholar] [CrossRef]
- Nagendran, J.; Gurtu, V.; Fu, D.Z.; Dyck, J.R.; Haromy, A.; Ross, D.B.; Rebeyka, I.M.; Michelakis, E.D. A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J. Thorac. Cardiovasc. Surg. 2008, 136, 168–178.e163. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Gurtu, V.; Webster, L.; Barnes, G.; Watson, G.; Howard, L.; Cupitt, J.; Paterson, I.; Thompson, R.B.; Chow, K.; et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 2017, 9, eaao4583. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef]
- Chu, Q.S.; Sangha, R.; Spratlin, J.; Vos, L.J.; Mackey, J.R.; McEwan, A.J.; Venner, P.; Michelakis, E.D. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 603–610. [Google Scholar] [CrossRef]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef]
- Dyck, J.R.; Hopkins, T.A.; Bonnet, S.; Michelakis, E.D.; Young, M.E.; Watanabe, M.; Kawase, Y.; Jishage, K.; Lopaschuk, G.D. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 2006, 114, 1721–1728. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef]
- Häuselmann, I.; Borsig, L. Altered Tumor-Cell Glycosylation Promotes Metastasis. Front. Oncol. 2014, 4, 28. [Google Scholar] [CrossRef]
- Kamigaito, T.; Okaneya, T.; Kawakubo, M.; Shimojo, H.; Nishizawa, O.; Nakayama, J. Overexpression of O-GlcNAc by prostate cancer cells is significantly associated with poor prognosis of patients. Prostate Cancer Prostatic Dis. 2014, 17, 18–22. [Google Scholar] [CrossRef]
- Kizuka, Y.; Taniguchi, N. Enzymes for N-Glycan Branching and Their Genetic and Nongenetic Regulation in Cancer. Biomolecules 2016, 6, 25. [Google Scholar] [CrossRef]
- Mathews, M.B.; Glagov, S. Acid mucopolysaccharide patterns in aging human cartilage. J. Clin. Investig. 1966, 45, 1103–1111. [Google Scholar] [CrossRef]
- Pal-Ghosh, S.; Tadvalkar, G.; Stepp, M.A. Alterations in Corneal Sensory Nerves During Homeostasis, Aging, and after Injury in Mice Lacking the Heparan Sulfate Proteoglycan Syndecan-1. Investig. Opthalmol. Vis. Sci. 2017, 58, 4959–4975. [Google Scholar] [CrossRef]
- Kobata, A. Glycobiology in the field of aging research—Introduction to glycogerontology. Biochimie 2003, 85, 13–24. [Google Scholar] [CrossRef]
- Beeley, J.G.; Blackie, R.; Baxter, A. Glycoprotein and glycolipid changes in aged erythrocytes [proceedings]. Biochem. Soc. Trans. 1977, 5, 1725–1726. [Google Scholar] [CrossRef]
- Ovsepian, L.M.; Kazarian, G.S.; Akopdzhanian, A.A.; L’Vov, M.V. Age-dependent changes in phospholipid content and neutral lipid contents in aging. Adv. Gerontol. 2012, 25, 250–254. [Google Scholar] [CrossRef]
- Wang, W.; Gopal, S.; Pocock, R.; Xiao, Z. Glycan Mimetics from Natural Products: New Therapeutic Opportunities for Neurodegenerative Disease. Molecules 2019, 24, 4604. [Google Scholar] [CrossRef]
- Kizuka, Y.; Kitazume, S.; Fujinawa, R.; Saito, T.; Iwata, N.; Saido, T.C.; Nakano, M.; Yamaguchi, Y.; Hashimoto, Y.; Staufenbiel, M.; et al. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease. EMBO Mol. Med. 2015, 7, 175–189. [Google Scholar] [CrossRef]
- Maguire, T.M.; Gillian, A.M.; O’Mahony, D.; Coughlan, C.M.; Breen, K.C. A decrease in serum sialyltransferase levels in Alzheimer’s disease. Neurobiol. Aging 1994, 15, 99–102. [Google Scholar] [CrossRef]
- Fodero, L.R.; Sáez-Valero, J.; Barquero, M.S.; Marcos, A.; McLean, C.A.; Small, D.H. Wheat germ agglutinin-binding glycoproteins are decreased in Alzheimer’s disease cerebrospinal fluid. J. Neurochem. 2001, 79, 1022–1026. [Google Scholar] [CrossRef]
- Saito, F.; Yanagisawa, K.; Miyatake, T. Soluble derivatives of β/A4 amyloid protein precursor in human cerebrospinal fluid are both N- and O-glycosylated. Mol. Brain Res. 1993, 19, 171–174. [Google Scholar] [CrossRef]
- Griffith, L.S.; Mathes, M.; Schmitz, B. Beta-amyloid precursor protein is modified with O-linked N-acetylglucosamine. J. Neurosci. Res. 1995, 41, 270–278. [Google Scholar] [CrossRef]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef]
- Dashti, H.; Pabon Porras, M.A.; Mora, S. Glycosylation and Cardiovascular Diseases. In The Role of Glycosylation in Health and Disease; Lauc, G., Trbojević-Akmačić, I., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 307–319. [Google Scholar]
- 99Gudelj, I.; Lauc, G. Protein N-Glycosylation in Cardiovascular Diseases and Related Risk Factors. Curr. Cardiovasc. Risk Rep. 2018, 12, 16. [Google Scholar] [CrossRef]
- Wright, J.N.; Collins, H.E.; Wende, A.R.; Chatham, J.C. O-GlcNAcylation and cardiovascular disease. Biochem. Soc. Trans. 2017, 45, 545–553. [Google Scholar] [CrossRef]
- Komaromy, A.; Reider, B.; Jarvas, G.; Guttman, A. Glycoprotein biomarkers and analysis in chronic obstructive pulmonary disease and lung cancer with special focus on serum immunoglobulin G. Clin. Chim. Acta 2020, 506, 204–213. [Google Scholar] [CrossRef]
- Pavić, T.; Dilber, D.; Kifer, D.; Selak, N.; Keser, T.; Ljubičić, Đ.; Vukić Dugac, A.; Lauc, G.; Rumora, L.; Gornik, O. N-glycosylation patterns of plasma proteins and immunoglobulin G in chronic obstructive pulmonary disease. J. Transl. Med. 2018, 16, 323. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Inoue, S.; Gu, J.; Miyoshi, E.; Noda, K.; Li, W.; Mizuno-Horikawa, Y.; Nakano, M.; Asahi, M.; Takahashi, M.; et al. Dysregulation of TGF-β1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc. Natl. Acad. Sci. USA 2005, 102, 15791–15796. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Ishii, T.; Ikeda, S.; Naka-Mieno, M.; Tanaka, N.; Arai, T.; Kumasaka, T.; Gemma, A.; Kida, K.; Muramatsu, M.; et al. Association of fucosyltransferase 8 (FUT8) polymorphism Thr267Lys with pulmonary emphysema. J. Hum. Genet. 2011, 56, 857–860. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mészáros, B.; Járvás, G.; Farkas, A.; Szigeti, M.; Kovács, Z.; Kun, R.; Szabó, M.; Csánky, E.; Guttman, A. Comparative analysis of the human serum N-glycome in lung cancer, COPD and their comorbidity using capillary electrophoresis. J. Chromatogr. B 2019, 1137, 121913. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Helton, E.S.; Easter, M.; Bollenbecker, S.; Denson, R.; Zaharias, R.; Cochran, P.; Vang, S.; Harris, E.; Wells, J.M.; et al. ST6GAL1 and α2-6 Sialylation Regulates IL-6 Expression and Secretion in Chronic Obstructive Pulmonary Disease. Front. Immunol. 2021, 12, 693149. [Google Scholar] [CrossRef]
- Schulz, B.; Sloane, A.J.; Robinson, L.J.; Prasad, S.S.; Lindner, R.A.; Robinson, M.; Bye, P.T.; Nielson, D.W.; Harry, J.L.; Packer, N.H.; et al. Glycosylation of sputum mucins is altered in cystic fibrosis patients. Glycobiology 2007, 17, 698–712. [Google Scholar] [CrossRef]
- Arnold, J.N.; Saldova, R.; Galligan, M.C.; Murphy, T.B.; Mimura-Kimura, Y.; Telford, J.E.; Godwin, A.K.; Rudd, P.M. Novel Glycan Biomarkers for the Detection of Lung Cancer. J. Proteome Res. 2011, 10, 1755–1764. [Google Scholar] [CrossRef]
- Xia, B.; Royall, J.A.; Damera, G.; Sachdev, G.P.; Cummings, R.D. Altered O-glycosylation and sulfation of airway mucins associated with cystic fibrosis. Glycobiology 2005, 15, 747–775. [Google Scholar] [CrossRef]
- Davril, M.; Groux-Degroote, S.; Humbert, P.; Galabert, C.; Dumur, V.; Lafitte, J.-J.; Lamblin, G.; Roussel, P. The sialylation of bronchial mucins secreted by patients suffering from cystic fibrosis or from chronic bronchitis is related to the severity of airway infection. Glycobiology 1999, 9, 311–321. [Google Scholar] [CrossRef]
- Collum, S.D.; Chen, N.; Hernandez, A.M.; Hanmandlu, A.; Sweeney, H.; Mertens, T.C.J.; Weng, T.; Luo, F.; Molina, J.G.; Davies, J.; et al. Inhibition of hyaluronan synthesis attenuates pulmonary hypertension associated with lung fibrosis. J. Cereb. Blood Flow Metab. 2017, 174, 3284–3301. [Google Scholar] [CrossRef]
- Bjermer, L.; Lundgren, R.; Hallgren, R. Hyaluronan and type III procollagen peptide concentrations in bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. Thorax 1989, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Dittner-Moormann, S.; Lourenco, C.M.; Reunert, J.; Nishinakamura, R.; Tanaka, S.S.; Werner, C.; Debus, V.; Zimmer, K.-P.; Wetzel, G.; Naim, H.Y.; et al. TRAPγ-CDG shows asymmetric glycosylation and an effect on processing of proteins required in higher organisms. J. Med. Genet. 2021, 58, 213–216. [Google Scholar] [CrossRef]
- Toppila, S.; Paavonen, T.; Laitinen, A.; Laitinen, L.A.; Renkonen, R. Endothelial Sulfated Sialyl Lewis × Glycans, Putative L-Selectin Ligands, Are Preferentially Expressed in Bronchial Asthma but Not in Other Chronic Inflammatory Lung Diseases. Am. J. Respir. Cell Mol. Biol. 2000, 23, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Darley-Usmar, V.M.; Ball, L.E.; Chatham, J.C. Protein O-linked beta-N-acetylglucosamine: A novel effector of cardiomyocyte metabolism and function. J. Mol. Cell. Cardiol. 2012, 52, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- McClain, D.A. Hexosamines as mediators of nutrient sensing and regulation in diabetes. J. Diabetes Complicat. 2002, 16, 72–80. [Google Scholar] [CrossRef]
- Rossetti, L. Perspective: Hexosamines and nutrient sensing. Endocrinology 2000, 141, 1922–1925. [Google Scholar] [CrossRef]
- Kreppel, L.K.; Blomberg, M.A.; Hart, G.W. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J. Biol. Chem. 1997, 272, 9308–9315. [Google Scholar] [CrossRef]
- Lubas, W.A.; Frank, D.W.; Krause, M.; Hanover, J.A. O-Linked GlcNAc transferase is a conserved nucleocytoplasmic protein containing tetratricopeptide repeats. J. Biol. Chem. 1997, 272, 9316–9324. [Google Scholar] [CrossRef]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Gao, Y.; Mahoney, J.A.; Vosseller, K.; Chen, C.; Rosen, A.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Further characterization of the nucleocytoplasmic beta-N-acetylglucosaminidase, O-GlcNAcase. J. Biol. Chem. 2002, 277, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Nagel, A.K.; Ball, L.E. O-GlcNAc transferase and O-GlcNAcase: Achieving target substrate specificity. Amino Acids 2014, 46, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W. Three Decades of Research on O-GlcNAcylation—A Major Nutrient Sensor That Regulates Signaling, Transcription and Cellular Metabolism. Front. Endocrinol. 2014, 5, 183. [Google Scholar] [CrossRef]
- Ma, J.; Liu, T.; Wei, A.C.; Banerjee, P.; O’Rourke, B.; Hart, G.W. O-GlcNAcomic Profiling Identifies Widespread O-Linked beta-N-Acetylglucosamine Modification (O-GlcNAcylation) in Oxidative Phosphorylation System Regulating Cardiac Mitochondrial Function. J. Biol. Chem. 2015, 290, 29141–29153. [Google Scholar] [CrossRef]
- Zachara, N.E.; O’Donnell, N.; Cheung, W.D.; Mercer, J.J.; Marth, J.D.; Hart, G.W. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J. Biol. Chem. 2004, 279, 30133–30142. [Google Scholar] [CrossRef]
- Kamemura, K.; Hayes, B.K.; Comer, F.I.; Hart, G.W. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: Alternative glycosylation/phosphorylation of THR-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens. J. Biol. Chem. 2002, 277, 19229–19235. [Google Scholar] [CrossRef]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef]
- Lefebvre, T.; Ferreira, S.; Dupont-Wallois, L.; Bussiere, T.; Dupire, M.J.; Delacourte, A.; Michalski, J.C.; Caillet-Boudin, M.L. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins--a role in nuclear localization. Biochim. Biophys. Acta 2003, 1619, 167–176. [Google Scholar] [CrossRef]
- Wulff-Fuentes, E.; Berendt, R.R.; Massman, L.; Danner, L.; Malard, F.; Vora, J.; Kahsay, R.; Olivier-Van Stichelen, S. The human O-GlcNAcome database and meta-analysis. Sci. Data 2021, 8, 25. [Google Scholar] [CrossRef]
- Caldwell, S.A.; Jackson, S.R.; Shahriari, K.S.; Lynch, T.P.; Sethi, G.; Walker, S.; Vosseller, K.; Reginato, M.J. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM. Oncogene 2010, 29, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, C.M.; Sodi, V.L.; Reginato, M.J. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J. Mol. Biol. 2016, 428, 3282–3294. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Krause, M.W.; Love, D.C. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim. Biophys. Acta 2010, 1800, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, T.; Dehennaut, V.; Guinez, C.; Olivier, S.; Drougat, L.; Mir, A.M.; Mortuaire, M.; Vercoutter-Edouart, A.S.; Michalski, J.C. Dysregulation of the nutrient/stress sensor O-GlcNAcylation is involved in the etiology of cardiovascular disorders, type-2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta 2010, 1800, 67–79. [Google Scholar] [CrossRef]
- Slawson, C.; Housley, M.P.; Hart, G.W. O-GlcNAc cycling: How a single sugar post-translational modification is changing the way we think about signaling networks. J. Cell. Biochem. 2006, 97, 71–83. [Google Scholar] [CrossRef]
- Vaidyanathan, K.; Wells, L. Multiple tissue-specific roles for the O-GlcNAc post-translational modification in the induction of and complications arising from type II diabetes. J. Biol. Chem. 2014, 289, 34466–34471. [Google Scholar] [CrossRef]
- Wells, L.; Vosseller, K.; Hart, G.W. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell. Mol. Life Sci. 2003, 60, 222–228. [Google Scholar] [CrossRef]
- Barnes, J.W.; Tian, L.; Heresi, G.A.; Farver, C.F.; Asosingh, K.; Comhair, S.A.; Aulak, K.S.; Dweik, R.A. O-linked beta-N-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation 2015, 131, 1260–1268. [Google Scholar] [CrossRef]
- Banerjee, P.S.; Lagerlof, O.; Hart, G.W. Roles of O-GlcNAc in chronic diseases of aging. Mol. Asp. Med. 2016, 51, 1–15. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Vocadlo, D.J. O-GlcNAc and neurodegeneration: Biochemical mechanisms and potential roles in Alzheimer’s disease and beyond. Chem. Soc. Rev. 2014, 43, 6839–6858. [Google Scholar] [CrossRef]
- Ferrer, C.M.; Lynch, T.P.; Sodi, V.L.; Falcone, J.N.; Schwab, L.P.; Peacock, D.L.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol. Cell. 2014, 54, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A., 3rd; Peters, E.C.; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Vosseller, K. O-GlcNAc in cancer biology. Amino Acids 2013, 45, 719–733. [Google Scholar] [CrossRef]
- Slawson, C.; Copeland, R.J.; Hart, G.W. O-GlcNAc signaling: A metabolic link between diabetes and cancer? Trends Biochem. Sci. 2010, 35, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Dassanayaka, S.; Jones, S.P. O-GlcNAc and the cardiovascular system. Pharmacol. Ther. 2014, 142, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Zachara, N.E. The roles of O-linked beta-N-acetylglucosamine in cardiovascular physiology and disease. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1905–H1918. [Google Scholar] [CrossRef]
- Gelinas, R.; Mailleux, F.; Dontaine, J.; Bultot, L.; Demeulder, B.; Ginion, A.; Daskalopoulos, E.P.; Esfahani, H.; Dubois-Deruy, E.; Lauzier, B.; et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat. Commun. 2018, 9, 374. [Google Scholar] [CrossRef]
- Hilgers, R.H.P.; Xing, D.; Gong, K.; Chen, Y.-F.; Chatham, J.C.; Oparil, S. Acute O-GlcNAcylation prevents inflammation-induced vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H513–H522. [Google Scholar] [CrossRef]
- Barnes, J.W.; Tian, L.; Krick, S.; Helton, E.S.; Denson, R.S.; Comhair, S.A.A.; Dweik, R.A. O-GlcNAc Transferase Regulates Angiogenesis in Idiopathic Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2019, 20, 6299. [Google Scholar] [CrossRef]
- Aulak, K.S.; Barnes, J.W.; Tian, L.; Mellor, N.E.; Haque, M.M.; Willard, B.; Li, L.; Comhair, S.C.; Stuehr, D.J.; Dweik, R.A. Specific O-GlcNAc modification at Ser-615 modulates eNOS function. Redox Biol. 2020, 36, 101625. [Google Scholar] [CrossRef]
- Prisco, S.Z.; Rose, L.; Potus, F.; Tian, L.; Wu, D.; Hartweck, L.; Al-Qazazi, R.; Neuber-Hess, M.; Eklund, M.; Hsu, S.; et al. Excess Protein O-GlcNAcylation Links Metabolic Derangements to Right Ventricular Dysfunction in Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2020, 21, 7278. [Google Scholar] [CrossRef] [PubMed]
- Prisco, S.Z.; Eklund, M.; Raveendran, R.; Thenappan, T.; Prins, K.W. With No Lysine Kinase 1 Promotes Metabolic Derangements and RV Dysfunction in Pulmonary Arterial Hypertension. JACC Basic Transl. Sci. 2021, 6, 834–850. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.H.; Stanley, P.; Bertozzi, C.R.; Hart, G.W.; Etzler, M.E. Essentials in Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Lin, X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development 2004, 131, 6009–6021. [Google Scholar] [CrossRef]
- Chen, Q.; Sivakumar, P.; Barley, C.; Peters, D.M.; Gomes, R.R.; Farach-Carson, M.C.; Dallas, S.L. Potential role for heparan sulfate proteoglycans in regulation of transforming growth factor-beta (TGF-beta) by modulating assembly of latent TGF-beta-binding protein-1. J. Biol. Chem. 2007, 282, 26418–26430. [Google Scholar] [CrossRef] [PubMed]
- Kraushaar, D.C.; Yamaguchi, Y.; Wang, L. Heparan sulfate is required for embryonic stem cells to exit from self-renewal. J. Biol. Chem. 2010, 285, 5907–5916. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, A.; Germack, R.; Lortat-Jacob, H.; Muramatsu, T.; Billett, E.; Johnson, T.; Verderio, E.A. Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of transglutaminase-2. J. Biol. Chem. 2009, 284, 18411–18423. [Google Scholar] [CrossRef]
- Li, Z.; Yasuda, Y.; Li, W.; Bogyo, M.; Katz, N.; Gordon, R.E.; Fields, G.B.; Bromme, D. Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J. Biol. Chem. 2004, 279, 5470–5479. [Google Scholar] [CrossRef]
- Ghabrial, A.S. A sweet spot in the FGFR signal transduction pathway. Sci. Signal 2012, 5, pe1. [Google Scholar] [CrossRef][Green Version]
- Christensen, G.; Herum, K.M.; Lunde, I.G. Sweet, yet underappreciated: Proteoglycans and extracellular matrix remodeling in heart disease. Matrix Biol. 2019, 75–76, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Papakonstantinou, E.; Karakiulakis, G.; Tamm, M.; Perruchoud, A.P.; Roth, M. Hypoxia modifies the effect of PDGF on glycosaminoglycan synthesis by primary human lung cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L825–L834. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantinou, E.; Roth, M.; Tamm, M.; Eickelberg, O.; Perruchoud, A.P.; Karakiulakis, G. Hypoxia differentially enhances the effects of transforming growth factor-beta isoforms on the synthesis and secretion of glycosaminoglycans by human lung fibroblasts. J. Pharmacol. Exp. Ther. 2002, 301, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Ruppert, C.L.; Tozzi, C.A.; Neubauer, J.A.; Frankel, H.M.; Yu, S.Y.; Riley, D.J. Reduction of chronic hypoxic pulmonary hypertension in the rat by an inhibitor of collagen production. Am. Rev. Respir. Dis. 1987, 135, 300–306. [Google Scholar] [PubMed]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Humphries, D.E.; Lee, S.-L.; Fanburg, B.L.; Silbert, J.E. Effects of hypoxia and hyperoxia on proteoglycan production by bovine pulmonary artery endothelial cells. J. Cell. Physiol. 1986, 126, 249–253. [Google Scholar] [CrossRef]
- Figueroa, J.E.; Tao, Z.; Sarphie, T.G.; Smart, F.W.; Glancy, D.L.; Vijayagopal, P. Effect of hypoxia and hypoxia/reoxygenation on proteoglycan metabolism by vascular smooth muscle cells. Atherosclerosis 1999, 143, 135–144. [Google Scholar] [CrossRef]
- Little, P.J.; Burch, M.L.; Getachew, R.; Al-Aryahi, S.; Osman, N. Endothelin-1 stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by endothelin receptor transactivation of the transforming growth factor-β type I receptor. J. Cardiovasc. Pharmacol. 2010, 56, 360–368. [Google Scholar] [CrossRef]
- Lee, R.T.; Yamamoto, C.; Feng, Y.; Potter-Perigo, S.; Briggs, W.H.; Landschulz, K.T.; Turi, T.G.; Thompson, J.F.; Libby, P.; Wight, T.N. Mechanical strain induces specific changes in the synthesis and organization of proteoglycans by vascular smooth muscle cells. J. Biol. Chem. 2001, 276, 13847–13851. [Google Scholar] [CrossRef]
- Shimizu-Hirota, R.; Sasamura, H.; Mifune, M.; Nakaya, H.; Kuroda, M.; Hayashi, M.; Saruta, T. Regulation of Vascular Proteoglycan Synthesis by Angiotensin II Type 1 and Type 2 Receptors. J. Am. Soc. Nephrol. 2001, 12, 2609–2615. [Google Scholar] [CrossRef]
- Zimmer, B.M.; Barycki, J.J.; Simpson, M.A. Integration of Sugar Metabolism and Proteoglycan Synthesis by UDP-glucose Dehydrogenase. J. Histochem. Cytochem. 2021, 69, 13–23. [Google Scholar] [CrossRef]
- van Det, N.F.; van den Born, J.; Tamsma, J.T.; Verhagen, N.A.M.; Berden, J.H.M.; Bruijn, J.A.; Daha, M.R.; van der Woude, F.J. Effects of high glucose on the production of heparan sulfate proteoglycan by mesangial and epithelial cells. Kidney Int. 1996, 49, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Klein, D.J.; Cohen, R.M.; Rymaszewski, Z. Proteoglycan synthesis by bovine myocardial endothelial cells is increased by long-term exposure to high concentrations of glucose. J. Cell. Physiol. 1995, 165, 493–502. [Google Scholar] [CrossRef]
- Simoni, R.D.; Hill, R.L.; Vaughan, M.; Hascall, V. The discovery of hyaluronan by Karl Meyer. J. Biol. Chem. 2002, 277, e1–e2. [Google Scholar] [CrossRef]
- Armstrong, S.E.; Bell, D.R. Measurement of high-molecular-weight hyaluronan in solid tissue using agarose gel electrophoresis. Anal. Biochem. 2002, 308, 255–264. [Google Scholar] [CrossRef]
- Itano, N.; Sawai, T.; Yoshida, M.; Lenas, P.; Yamada, Y.; Imagawa, M.; Shinomura, T.; Hamaguchi, M.; Yoshida, Y.; Ohnuki, Y.; et al. Three Isoforms of Mammalian Hyaluronan Synthases Have Distinct Enzymatic Properties. J. Biol. Chem. 1999, 274, 25085–25092. [Google Scholar] [CrossRef]
- Philipson, L.H.; Schwartz, N.B. Subcellular localization of hyaluronate synthetase in oligodendroglioma cells. J. Biol. Chem. 1984, 259, 5017–5023. [Google Scholar] [CrossRef]
- Vigetti, D.; Deleonibus, S.; Moretto, P.; Karousou, E.; Viola, M.; Bartolini, B.; Hascall, V.C.; Tammi, M.; De Luca, G.; Passi, A. Role of UDP-N-Acetylglucosamine (GlcNAc) and O-GlcNAcylation of Hyaluronan Synthase 2 in the Control of Chondroitin Sulfate and Hyaluronan Synthesis. J. Biol. Chem. 2012, 287, 35544–35555. [Google Scholar] [CrossRef] [PubMed]
- Jokela, T.A.; Makkonen, K.M.; Oikari, S.; Karna, R.; Koli, E.; Hart, G.W.; Tammi, R.H.; Carlberg, C.; Tammi, M.I. Cellular content of UDP-N-acetylhexosamines controls hyaluronan synthase 2 expression and correlates with O-linked N-acetylglucosamine modification of transcription factors YY1 and SP1. J. Biol. Chem. 2011, 286, 33632–33640. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; De La Motte, C.; Lauer, M.; Hascall, V. Hyaluronan matrices in pathobiological processes. FEBS J. 2011, 278, 1412–1418. [Google Scholar] [CrossRef]
- Dentener, M.A.; Vernooy, J.H.; Hendriks, S.; Wouters, E.F. Enhanced levels of hyaluronan in lungs of patients with COPD: Relationship with lung function and local inflammation. Thorax 2005, 60, 114–119. [Google Scholar] [CrossRef]
- Galdi, F.; Pedone, C.; McGee, C.A.; George, M.; Rice, A.B.; Hussain, S.S.; Vijaykumar, K.; Boitet, E.R.; Tearney, G.J.; McGrath, J.A.; et al. Inhaled high molecular weight hyaluronan ameliorates respiratory failure in acute COPD exacerbation: A pilot study. Respir. Res. 2021, 22, 30. [Google Scholar] [CrossRef] [PubMed]
- Lauer, M.E.; Dweik, R.A.; Garantziotis, S.; Aronica, M.A. The Rise and Fall of Hyaluronan in Respiratory Diseases. International. J. Cell Biol. 2015, 2015, 712507. [Google Scholar]
- Zhu, L.; Zhuo, L.; Kimata, K.; Yamaguchi, E.; Watanabe, H.; Aronica, M.A.; Hascall, V.C.; Baba, K. Deficiency in the Serum-Derived Hyaluronan-Associated Protein-Hyaluronan Complex Enhances Airway Hyperresponsiveness in a Murine Model of Asthma. Int. Arch. Allergy Immunol. 2010, 153, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Lazrak, A.; Creighton, J.; Yu, Z.; Komarova, S.; Doran, S.F.; Aggarwal, S.; Emala, C.W.; Stober, V.P.; Trempus, C.S.; Garantziotis, S.; et al. Hyaluronan mediates airway hyperresponsiveness in oxidative lung injury. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L891–L903. [Google Scholar] [CrossRef] [PubMed]
- Lamas, A.; Marshburn, J.; Stober, V.P.; Donaldson, S.H.; Garantziotis, S. Effects of inhaled high-molecular weight hyaluronan in inflammatory airway disease. Respir. Res. 2016, 17, 123. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Noble, P.W. Regulation of Non-Infectious Lung Injury, Inflammation, and Repair by the Extracellular Matrix Glycosaminoglycan Hyaluronan. Anat. Rec. 2010, 293, 982–985. [Google Scholar] [CrossRef]
- Singleton, P.A.; Lennon, F.E. Acute Lung Injury Regulation by Hyaluronan. J. Allergy Ther. 2011, 15 (Suppl. S4), 1–9. [Google Scholar] [CrossRef]
- Cui, Z.; Liao, J.; Cheong, N.; Longoria, C.; Cao, G.; DeLisser, H.M.; Savani, R.C. The Receptor for Hyaluronan-Mediated Motility (CD168) promotes inflammation and fibrosis after acute lung injury. Matrix Biol. 2018, 78–79, 255–271. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, Y.; Xie, T.; Liu, N.; Chen, H.; Geng, Y.; Kurkciyan, A.; Mena, J.M.; Stripp, B.R.; Jiang, D.; et al. Hyaluronan and TLR4 promote surfactant-protein-C-positive alveolar progenitor cell renewal and prevent severe pulmonary fibrosis in mice. Nat. Med. 2016, 22, 1285–1293. [Google Scholar] [CrossRef]
- Aytekin, M.; Comhair, S.A.A.; de la Motte, C.; Bandyopadhyay, S.K.; Farver, C.F.; Hascall, V.C.; Erzurum, S.C.; Dweik, R.A. High levels of hyaluronan in idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L789–L799. [Google Scholar] [CrossRef]
- Lauer, M.E.; Aytekin, M.; Comhair, S.A.; Loftis, J.; Tian, L.; Farver, C.F.; Hascall, V.C.; Dweik, R.A. Modification of Hyaluronan by Heavy Chains of Inter-α-Inhibitor in Idiopathic Pulmonary Arterial Hypertension. J. Biol. Chem. 2014, 289, 6791–6798. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantinou, E.; Kouri, F.M.; Karakiulakis, G.; Klagas, I.; Eickelberg, O. Increased hyaluronic acid content in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2008, 32, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.E.; Belchenko, D.D.; Nguyen, C.M.; Colvin, K.L.; Ivy, D.D.; Stenmark, K.R. Endothelin-1, the unfolded protein response, and persistent inflammation: Role of pulmonary artery smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Hascall, V.C.; Majors, A.K.; De La Motte, C.A.; Evanko, S.P.; Wang, A.; Drazba, J.A.; Strong, S.A.; Wight, T.N. Intracellular hyaluronan: A new frontier for inflammation? Biochim. Biophys. Acta 2004, 1673, 3–12. [Google Scholar] [CrossRef]
- Ren, J.; Hascall, V.C.; Wang, A. Cyclin D3 mediates synthesis of a hyaluronan matrix that is adhesive for monocytes in mesangial cells stimulated to divide in hyperglycemic medium. J. Biol. Chem. 2009, 284, 16621–16632. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef]
- Mii, Y.; Takada, S. Heparan Sulfate Proteoglycan Clustering in Wnt Signaling and Dispersal. Front. Cell Dev. Biol. 2020, 8, 631. [Google Scholar] [CrossRef]
- Yan, D.; Lin, X. Shaping morphogen gradients by proteoglycans. Cold Spring Harb. Perspect. Biol. 2009, 1, a002493. [Google Scholar] [CrossRef]
- Gustafsson, E.; Almonte-Becerril, M.; Bloch, W.; Costell, M. Perlecan Maintains Microvessel Integrity In Vivo and Modulates Their Formation In Vitro. PLoS ONE 2013, 8, e53715. [Google Scholar] [CrossRef]
- Segev, A.; Nili, N.; Strauss, B.H. The role of perlecan in arterial injury and angiogenesis. Cardiovasc. Res. 2004, 63, 603–610. [Google Scholar] [CrossRef]
- Abdul-Salam, V.B.; Wharton, J.; Cupitt, J.; Berryman, M.; Edwards, R.J.; Wilkins, M.R. Proteomic Analysis of Lung Tissues from Patients with Pulmonary Arterial Hypertension. Circulation 2010, 122, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Tseng, C.-N.; Tannenberg, P.; Eriksson, L.; Yuan, K.; Perez, V.; Lundberg, J.; Lengquist, M.; Botusan, I.; Catrina, S.-B.; et al. Perlecan Heparan Sulfate Deficiency Impairs Pulmonary Vascular Development and Attenuates Hypoxic Pulmonary Hypertension. Cardiovasc. Res. 2015, 107, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Lipke, D.W.; Arcot, S.S.; Gillespie, M.N.; Olson, J.W. Temporal alterations in specific basement membrane components in lungs from monocrotaline-treated rats. Am. J. Respir. Cell Mol. Biol. 1993, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Vyas-Somani, A.C.; Aziz, S.M.; Arcot, S.A.; Gillespie, M.N.; Olson, J.W.; Lipke, D.W. Temporal alterations in basement membrane components in the pulmonary vasculature of the chronically hypoxic rat: Impact of hypoxia and recovery. Am. J. Med. Sci. 1996, 312, 54–67. [Google Scholar] [CrossRef]
- Lemire, J.M.; Merrilees, M.J.; Braun, K.R.; Wight, T.N. Overexpression of the V3 variant of versican alters arterial smooth muscle cell adhesion, migration, and proliferation in vitro. J. Cell. Physiol. 2002, 190, 38–45. [Google Scholar] [CrossRef]
- Sheng, W.; Wang, G.; Wang, Y.; Liang, J.; Wen, J.; Zheng, P.-S.; Wu, Y.; Lee, V.; Slingerland, J.; Dumont, D.; et al. The Roles of Versican V1 and V2 Isoforms in Cell Proliferation and Apoptosis. Mol. Biol. Cell 2005, 16, 1330–1340. [Google Scholar] [CrossRef]
- Chang, Y.T.; Chan, C.K.; Eriksson, I.; Johnson, P.Y.; Cao, X.; Westoo, C.; Norvik, C.; Andersson-Sjoland, A.; Westergren-Thorsson, G.; Johansson, S.; et al. Versican accumulates in vascular lesions in pulmonary arterial hypertension. Pulm. Circ. 2016, 6, 347–359. [Google Scholar] [CrossRef]
- Jandl, K.; Marsh, L.M.; Hoffmann, J.; Mutgan, A.C.; Baum, O.; Bloch, W.; Thekkekara-Puthenparampil, H.; Kolb, D.; Sinn, K.; Klepetko, W.; et al. Basement Membrane Remodeling Controls Endothelial Function in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2020, 63, 104–117. [Google Scholar] [CrossRef]
- Hassan, H.; Greve, B.; Pavao, M.S.; Kiesel, L.; Ibrahim, S.A.; Götte, M. Syndecan-1 modulates β-integrin-dependent and interleukin-6-dependent functions in breast cancer cell adhesion, migration, and resistance to irradiation. FEBS J. 2013, 280, 2216–2227. [Google Scholar] [CrossRef]
- Li, W.; Wang, W. Membrane tension regulates syndecan-1 expression through actin remodelling. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129413. [Google Scholar] [CrossRef]
- Mochizuki, M.; Güç, E.; Park, A.J.; Julier, Z.; Briquez, P.S.; Kuhn, G.A.; Müller, R.; Swartz, M.A.; Hubbell, J.A.; Martino, M.M. Growth factors with enhanced syndecan binding generate tonic signalling and promote tissue healing. Nat. Biomed. Eng. 2020, 4, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zuo, D.; Chen, Y.; Li, W.; Liu, R.; He, Y.; Ren, L.; Zhou, L.; Deng, T.; Ying, G.; et al. Shed Syndecan-1 is involved in chemotherapy resistance via the EGFR pathway in colorectal cancer. Br. J. Cancer 2014, 111, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Blaine, S.A.; Qiao, D.; Friedl, A. Membrane type 1 matrix metalloproteinase-mediated stromal syndecan-1 shedding stimulates breast carcinoma cell proliferation. Cancer Res. 2008, 68, 9558–9565. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Bai, Y.; Kitagawa, H. Cleavage of Syndecan-1 Promotes the Proliferation of the Basal-Like Breast Cancer Cell Line BT-549 Via Akt SUMOylation. Front. Cell Dev. Biol. 2021, 9, 659428. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Z.C.; Liu, Y. Attenuating Pulmonary Hypertension by Protecting the Integrity of Glycocalyx in Rats Model of Pulmonary Artery Hypertension. Inflammation 2019, 42, 1951–1956. [Google Scholar] [CrossRef]
- Arvidsson, M.; Ahmed, A.; Bouzina, H.; Radegran, G. Plasma proteoglycan prolargin in diagnosis and differentiation of pulmonary arterial hypertension. ESC Heart Fail. 2021, 8, 1230–1243. [Google Scholar] [CrossRef]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131, jcs208884. [Google Scholar] [CrossRef]
- Hughes, R.C. Galectins. In Encyclopedia of Biological Chemistry; Lennarz, W.J., Lane, M.D., Eds.; Elsevier: New York, NY, USA, 2004; pp. 171–174. [Google Scholar]
- Liu, F.T.; Hsu, D.K. The role of galectin-3 in promotion of the inflammatory response. Drug News Perspect. 2007, 20, 455–460. [Google Scholar] [CrossRef]
- Pang, J.; Rhodes, D.H.; Pini, M.; Akasheh, R.T.; Castellanos, K.J.; Cabay, R.J.; Cooper, D.; Perretti, M.; Fantuzzi, G. Increased Adiposity, Dysregulated Glucose Metabolism and Systemic Inflammation in Galectin-3 KO Mice. PLoS ONE 2013, 8, e57915. [Google Scholar] [CrossRef]
- Thurston, T.L.M.; Wandel, M.P.; von Muhlinen, N.; Foeglein, Á.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef]
- Thijssen, V.L.J.L.; Postel, R.; Brandwijk, R.J.M.G.E.; Dings, R.P.M.; Nesmelova, I.; Satijn, S.; Verhofstad, N.; Nakabeppu, Y.; Baum, L.G.; Bakkers, J.; et al. Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 15975–15980. [Google Scholar] [CrossRef] [PubMed]
- Vasta, G.R. Chapter 8—Lectins as Innate Immune Recognition Factors: Structural, Functional, and Evolutionary Aspects. In The Evolution of the Immune System; Malagoli, D., Ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 205–224. [Google Scholar]
- Chou, F.-C.; Chen, H.-Y.; Kuo, C.-C.; Sytwu, H.-K. Role of Galectins in Tumors and in Clinical Immunotherapy. Int. J. Mol. Sci. 2018, 19, 430. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-c.; Li, J.; Gao, J. Functions of Galectin-3 and Its Role in Fibrotic Diseases. J. Pharmacol. Exp. Ther. 2014, 351, 336–343. [Google Scholar] [CrossRef]
- Markowska, A.I.; Liu, F.T.; Panjwani, N. Galectin-3 is an important mediator of VEGF- and bFGF-mediated angiogenic response. J. Exp. Med. 2010, 207, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Croci Diego, O.; Cerliani Juan, P.; Dalotto-Moreno, T.; Méndez-Huergo Santiago, P.; Mascanfroni Ivan, D.; Dergan-Dylon, S.; Toscano Marta, A.; Caramelo Julio, J.; García-Vallejo Juan, J.; Ouyang, J.; et al. Glycosylation-Dependent Lectin-Receptor Interactions Preserve Angiogenesis in Anti-VEGF Refractory Tumors. Cell 2014, 156, 744–758. [Google Scholar] [CrossRef]
- Zhong, X.; Qian, X.; Chen, G.; Song, X. The role of galectin-3 in heart failure and cardiovascular disease. Clin. Exp. Pharmacol. Physiol. 2019, 46, 197–203. [Google Scholar] [CrossRef]
- Chou, R.-H.; Huang, S.-S.; Kuo, C.-S.; Wang, S.-C.; Tsai, Y.-L.; Lu, Y.-W.; Chang, C.-C.; Huang, P.-H.; Lin, S.-J. Galectin-1 is associated with the severity of coronary artery disease and adverse cardiovascular events in patients undergoing coronary angiography. Sci. Rep. 2020, 10, 20683. [Google Scholar] [CrossRef]
- He, J.; Li, X.; Luo, H.; Li, T.; Zhao, L.; Qi, Q.; Liu, Y.; Yu, Z. Galectin-3 mediates the pulmonary arterial hypertension–induced right ventricular remodeling through interacting with NADPH oxidase. J. Am. Soc. Hypertens. 2017, 11, 275–289.e272. [Google Scholar] [CrossRef]
- Shen, Q.; Chen, W.; Liu, J.; Liang, Q. Galectin-3 aggravates pulmonary arterial hypertension via immunomodulation in congenital heart disease. Life Sci. 2019, 232, 116546. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.M.; Zeng, X.X.; Wang, X.Y.; Chen, S.K.; Gui, L.X.; Lin, M.J. Galectin-3- Mediated Transdifferentiation of Pulmonary Artery Endothelial Cells Contributes to Hypoxic Pulmonary Vascular Remodeling. Cell. Physiol. Biochem. 2018, 51, 763–777. [Google Scholar] [CrossRef]
- Fulton, D.J.R.; Li, X.; Bordan, Z.; Wang, Y.; Mahboubi, K.; Rudic, R.D.; Haigh, S.; Chen, F.; Barman, S.A. Galectin-3, A Harbinger of Reactive Oxygen Species, Fibrosis, and Inflammation in Pulmonary Arterial Hypertension. Antioxid. Redox Signal 2019, 31, 1053–1069. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Irwin, D.; Ivester, C.; Harral, J.; Morris, K.; Imamura, M.; Roedersheimer, M.; Patterson, A.; Carr, M.; Hagen, M.; et al. Mice deficient in galectin-1 exhibit attenuated physiological responses to chronic hypoxia-induced pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2007, 292, L154–L164. [Google Scholar] [CrossRef] [PubMed]
- Morrow, R.; Cioffi, E.; Murphy, F.; Cioffi, D. Changes in IgG sialylation and glycosylation in pulmonary arterial hypertension (1089.3). FASEB J. 2014, 28, 1089.3. [Google Scholar] [CrossRef]
- Morrow, R.; Cioffi, E.; Cioffi, D. Change in Sialic Acid in Pulmonary Arterial Hypertension. FASEB J. 2013, 27, lb215. [Google Scholar] [CrossRef]
- Kawut, S.M.; Horn, E.M.; Berekashvili, K.K.; Widlitz, A.C.; Rosenzweig, E.B.; Barst, R.J. von Willebrand Factor Independently Predicts Long-term Survival in Patients With Pulmonary Arterial Hypertension. Chest 2005, 128, 2355–2362. [Google Scholar] [CrossRef]
- Lopes, A.A.; Ferraz de Souza, B.; Maeda, N.Y. Decreased sialic acid content of plasma von Willebrand factor in precapillary pulmonary hypertension. Thromb. Haemost. 2000, 83, 683–687. [Google Scholar] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vang, S.; Cochran, P.; Sebastian Domingo, J.; Krick, S.; Barnes, J.W. The Glycobiology of Pulmonary Arterial Hypertension. Metabolites 2022, 12, 316. https://doi.org/10.3390/metabo12040316
Vang S, Cochran P, Sebastian Domingo J, Krick S, Barnes JW. The Glycobiology of Pulmonary Arterial Hypertension. Metabolites. 2022; 12(4):316. https://doi.org/10.3390/metabo12040316
Chicago/Turabian StyleVang, Shia, Phillip Cochran, Julio Sebastian Domingo, Stefanie Krick, and Jarrod Wesley Barnes. 2022. "The Glycobiology of Pulmonary Arterial Hypertension" Metabolites 12, no. 4: 316. https://doi.org/10.3390/metabo12040316
APA StyleVang, S., Cochran, P., Sebastian Domingo, J., Krick, S., & Barnes, J. W. (2022). The Glycobiology of Pulmonary Arterial Hypertension. Metabolites, 12(4), 316. https://doi.org/10.3390/metabo12040316